

Background: Huntington disease is characterized by the generation of mutant huntingtin fragments, which correlate with disease progression.
Results: The transgenic mice N171-Q148 and N552-Q148 display a significantly accelerated phenotype and shortened life span when compared with N463-Q148, N536-Q148, and N586-Q148 transgenic mice.
Conclusion: Some HTT proteolysis fragments have distinct neurotoxicity.
Significance: Reducing proteolysis of huntingtin is a viable therapeutic treatment for Huntington disease.
Keywords: caspase, neurodegenerative disease, polyglutamine disease, proteolysis, transgenic mice
Abstract
The cascade of events that lead to cognitive decline, motor deficits, and psychiatric symptoms in patients with Huntington disease (HD) is triggered by a polyglutamine expansion in the N-terminal region of the huntingtin (HTT) protein. A significant mechanism in HD is the generation of mutant HTT fragments, which are generally more toxic than the full-length HTT. The protein fragments observed in human HD tissue and mouse models of HD are formed by proteolysis or aberrant splicing of HTT. To systematically investigate the relative contribution of the various HTT protein proteolysis events observed in vivo, we generated transgenic mouse models of HD representing five distinct proteolysis fragments ending at amino acids 171, 463, 536, 552, and 586 with a polyglutamine length of 148. All lines contain a single integration at the ROSA26 locus, with expression of the fragments driven by the chicken β-actin promoter at nearly identical levels. The transgenic mice N171-Q148 and N552-Q148 display significantly accelerated phenotypes and a shortened life span when compared with N463-Q148, N536-Q148, and N586-Q148 transgenic mice. We hypothesized that the accelerated phenotype was due to altered HTT protein interactions/complexes that accumulate with age. We found evidence for altered HTT complexes in caspase-2 fragment transgenic mice (N552-Q148) and a stronger interaction with the endogenous HTT protein. These findings correlate with an altered HTT molecular complex and distinct proteins in the HTT interactome set identified by mass spectrometry. In particular, we identified HSP90AA1 (HSP86) as a potential modulator of the distinct neurotoxicity of the caspase-2 fragment mice (N552-Q148) when compared with the caspase-6 transgenic mice (N586-Q148).
Introduction
Huntington disease (HD)2 is a dominant autosomal genetic disorder affecting approximately 30,000 people in the United States. HD is caused by expansion of a CAG trinucleotide repeat in the first exon of the huntingtin (HTT) gene and the translated protein has an enlarged CAG-encoded polyglutamine (polyQ) tract at the N-terminal region of the protein (1). HD is characterized by progressive loss of medium spiny neurons and cortical neurons during aging (2, 3). Neurodegeneration in HD results in neurocognitive decline, personality changes, chorea, dementia, and death (4). Contributing to the toxicity of mutant HTT protein is the production of protein fragments (5, 6). These fragments begin to accumulate and aggregate in neurons during aging and may be necessary to lead to the neurodegeneration that causes HD (5, 7–21).
The HTT locus on chromosome 4 at 4p16.3 encodes a 13.4-kb mRNA transcribed from a genomic region of 180 kb and containing 67 exons. The protein contains several known domains, including the polyQ domain, nuclear export signals (22–24), and proteolysis-susceptibility domains (Fig. 1A). HTT protein is extensively modified after translation, and many of these modifications have been recognized as important for progression of the disease (25–33). In addition to phosphorylation and acetylation (30, 34, 35), which have been shown to modulate mutant HTT toxicity, proteolysis plays a role in HD neurodegeneration. HTT is a target of several protease families, including caspases, calpains, and matrix metalloproteases (Fig. 1A) (8). Calpain and caspase cleavage products have been shown to be present in HD post-mortem tissue (13, 20, 36). Careful analysis of HD post-mortem tissue shows extensive proteolysis (37). The resulting N-terminal fragments of HTT have been shown to be toxic to neurons in mice and flies. Inhibition of protease cleavage by genetic means at specific sites in mouse models of the disease has been demonstrated to reduce progression of the disease; thus, the N-terminal HTT fragments are sufficient to cause disease progression in mice. These observations form the basis of the “toxic fragment” hypothesis for HD neurodegeneration.
FIGURE 1.
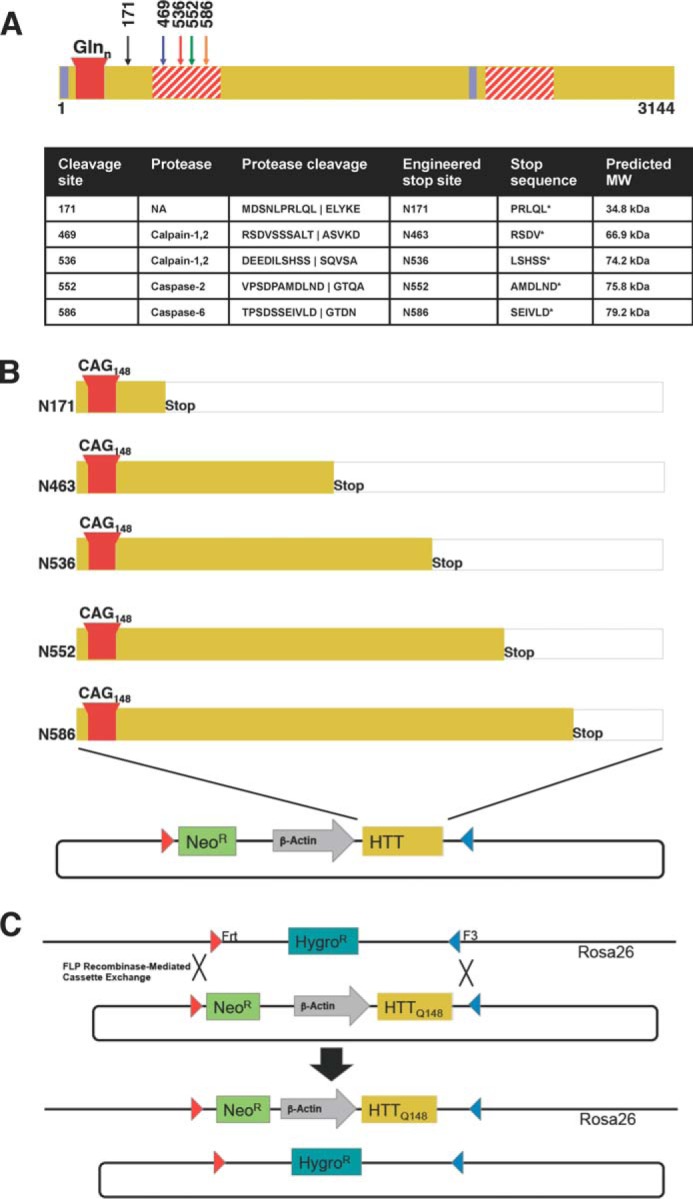
Schematic of proteolysis of HTT and the generation of Rosa-stop HD transgenic mice. A, schematic and table summary of the proteolysis events occurring in the HTT protein. Only the cleavage sites for fragments investigated in this paper are shown. The proteolytic susceptibility domains (red striped), nuclear export signals (blue), and polyQ stretch (red) are noted. HTT is cleaved by calpains-1 and -2 at amino acids 469 and 536, by caspase-2 at amino acid 552, and by caspase-6 at amino acid 586. B, design of HTT N-terminal fragment transgenic HD mouse models. mHTT N-terminal cleavage sites were modeled by engineering of a stop codon after the sites indicated, and these sites result in truncated protein fragments that are identical to caspase or calpain cleavage products with the exception of N463, which mimics cleavage site at an amino acid just before amino acid 469. Individual N-terminal fragment constructs in a donor vector flanked by Frt and F3 sites and engineered for recombinase-mediated cassette exchange in C57BL/6-Gt(ROSA)26Sortm596Arte mESCs are shown. C, schematic of the Flp recombinase-mediated cassette exchange in mESCs. Hygromycin resistant mESCs were transfected with recombinase and donor and selected for resistance to neomycin. Resulting clones were verified and used to generate chimeric mice with BALB/c embryos. Chimerism was assessed by coat color, and highly chimeric males were bred to C57BL/6 female mice to generate founders. Predicted average molecular masses of the fragments, calculated in the ExPASy Compute pI/Mw tool (81) are as follows: N171 Q145, 34.8 kDa; N463 Q145, 66.9 kDa; N536 Q145, 74.2 kDa; N552 Q145, 75.8 kDa; N586 Q145, 79.2 kDa. The fragments run higher than their calculated molecular masses as the expanded polyQ region reduces mobility in SDS-PAGE (82).
A lingering question associated with the toxic fragment hypothesis is how the various fragments of HTT differ in toxicity. Factors such as subcellular localization, protein interactions, aggregation propensity, and size of the fragment are thought to determine disease progression and phenotype. The analysis of this hypothesis has been addressed in mouse models of HD with a number of different approaches, including the generation of noncleavable full-length HD models and fragment models (10, 12, 38–43). Mutation of cleavage sites does not directly compare the effect of fragments expressed at equal levels of expression on neuropathology, behavior, biochemistry, and life span. One approach to understand the relative significance of these fragments, particularly in proteolysis events, is to compare mouse models of HD. However, in most cases comparisons cannot be made between transgenic HD mouse models because copy number, transgene structure upon integration, and integration site, all impact, among other things, the level of expression of HTT. The goal of our study was to utilize a systematic approach to understand the individual contribution of five of the major HTT proteolysis products to neurodegeneration and to identify the mechanisms that lead to their toxicity in vivo.
Experimental Procedures
Generation of Transgenic HTT Fragment Mice at the ROSA26 Locus
We generated the mouse transgenic lines described in this work with Taconic/Artemis (Cologne, Germany). The recombination-mediated cassette exchange ES cell line (derived from mouse strain C57BL/6-Gt(ROSA)26S or tm596Arte) is grown on a mitotically inactivated feeder layer composed of mouse embryonic fibroblasts in DMEM high glucose medium containing 20% FBS and 1200 μg/ml leukemia inhibitory factor (Millipore). For manipulation, 2 × 105 ES cells were plated on 3.5-cm dishes in 2 ml of medium. For the HTT Q148 cassette exchange, FuGENE 6 reagent (3 μl, Roche Applied Science) was mixed with serum-free medium (100 μl of OptiMEM I with GlutaMAX I; Invitrogen) and incubated for 5 min at room temperature. FuGENE/OptiMEM solution (100 μl) was added to the DNA mixture containing circular cassette vector (2 μg) and CAGGS-Flp plasmid to drive the targeted recombination (2 μg). This transfection complex was incubated for 20 min at room temperature and then added dropwise to the cells. From day 2 onward, the medium was replaced daily with medium containing 250 μg/ml G418 (Geneticin, Invitrogen). Seven days later, single clones were isolated, expanded, and analyzed by Southern blotting according to standard procedures. After administration of hormones, superovulated BALB/c female mice were mated with BALB/c males. Blastocysts were isolated from the uterus at 3.5 d post-coitum. For microinjection, blastocysts were placed in a drop of DMEM with 15% FCS under mineral oil. A flat tip, piezo-actuated microinjection pipette with an internal diameter of 12–15 μm was used to inject 10–15 targeted C57BL/6 N.tac ES cells into each blastocyst. After recovery, eight injected blastocysts were transferred to each uterine horn at 2.5 days post-coitum in pseudopregnant NMRI females. Chimerism was measured in chimeras (G0) by coat color contribution of ES cells to the BALB/c host (black/white). Highly chimeric mice were bred to strain C57BL/6 females. The C57BL/6 mating partners are nonmutant or mutant for the presence of a recombinase gene. Germ line transmission was identified by the presence of black, strain C57BL/6, offspring (G1).
PCR Genotyping for Transgenic Mice
The Buck Institute for Research on Aging animal facility is an AAALAC international accredited institution (Unit Number 001070). All procedures were approved by the Institutional Animal and Use Committee (A4213-01). Mice were genotyped by PCR analysis with GoTaq Green (Promega). DNA extraction from isolated tail snips of 3-week-old mice was performed following the manufacturer's protocol (Qiagen, DNeasy Blood and Tissue kit). 10 pmol of PCR control (F, 5′-GAGACTCTGGCTACTCATCC-3′, and R, 5′-CCTTCAGCAAGAGCTGGGGA-3′) and sequence-specific primers (IDT) N171 (F, 5′-ACGCAGAGTCAGATGTCAGG-3′, and R, 5′-CTGGATCTCGAGGCTG-ACG-3′), N536 (F, 5′-AGCAGCTCTGCCTTAACAGC-3′, and R, 5′-CTGGATCTCGAGGTCGACG-3′), N552 (F, 5′-GGATGAGATCAGTGGAGAGC-3′, and R, 5′-CTGGATCTCGAGGTCGACG-3′), and N586 (F, 5′-GCAGCTCTGCCTTAACAGC-3′, and R, 5′-CTGGA-TCTCGAGGTCGACG-3′) were used to amplify genomic DNA (1 μg). Cycle conditions were as follows: 95 °C for 5 min, 35 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min followed by a 10-min incubation at 72 °C. N463-Q148 mice were analyzed with 5 pmol of PCR control primer (F, 5′-GTGGCACGGAACTTCTAGTC-3′, and R, 5′-CTTGTCAAGTAGCAGGAAGA-3′) and 200 pmol of sequence-specific primer (F, 5′-AGCCTGAAAGGCAGCTTCG-3′, and R, 5′-CTGGATCTCGAGGTCGACG-3′). Cycle conditions were as follows: 95 °C for 5 min, 35 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min followed by a 10-min incubation at 72 °C. Positive mice yielded reaction products of the following lengths: N463-Q148, control = 357 and positive = 428; N536-Q148, control = 585 and positive = 248; N552-Q148, control = 585 and positive = 267; and N586-Q148, control = 585 and positive = 397.
Rotarod
A rotarod device was used to evaluate motor coordination (Mouse Rota-Rod Ugo Basile) during the light cycle. Each group consisted of at least n = 5–16 animals (smaller n at 6 months of age) and were tested for rotarod performance in a longitudinal fashion every 28 days for 3 days in a row starting at 4 months of age. Each measurement consisted of training and trial portions (three trials per day at 6 min). The training and each trial session were separated by at least a 30-min resting time interval. During the training session, mice were accustomed to the device for 5 min at a constant speed (4 rpm). For each trial session, the rotarod device was set to increase from 4 to 40 rpm during a 6-min period. The latency to fall from the apparatus was recorded, and the average from all three trials was used to evaluate motor coordination.
Open Field Measurements
Recording of 22 parameters during the open-field test in the floor and vertical plane was monitored to evaluate spontaneous locomotor activity. Animals, n = 5–16, were tested at 14 weeks for 30 min during the dark cycle. Each mouse was individually placed in the center of a square chamber (dimensions: 26 inches deep × 25 inches high × 26 inches wide) equipped with infrared photobeam sensors (E-63–12, Coulbourn Instruments, Whitehall, PA) and a camera coupled to TruScan99 software (Version 2.02, Coulbourn Instruments).
Survival Curve
Survival was monitored daily for each transgenic mouse in this study. Survival data were analyzed using Kaplan-Meier survival curves (N171-Q148, n = 158; N463-Q148, n = 104; N536-Q148, n = 72; N552-Q148, n = 96; and N586-Q148, n = 93 per group). The mean survival time was tested for significance by two-way ANOVA.
Statistical Comparisons
Statistical significances for rotarod, open field, and weight between the different groups were performed by two-way ANOVA Graphpad Prism software (La Jolla, CA), and comparisons were significant as follows: *, p < 0.05; **, p < 0.005; ***, p < 0.001, and ****, p < 0.0005.
Tissue Isolation and Homogenization
Mice were anesthetized with isoflurane (Butler Schein) prior to cervical dislocation. Whole brain was dissected in an adult brain matrix with 1-mm coronal section slice intervals at 4 °C. Slices were used to microdissect cortical and striatal regions spanning the rostral to the caudal portions of the brain. Samples were stored immediately at −80 °C until tissue homogenization. TPER (10 ml, Thermo Scientific) supplemented with protease inhibitors (Roche Applied Science, 1 tablet/10 ml, Complete Mini, EDTA-free), DNase (1 μl, Invitrogen), MgCl2 (1.2 mm, Fluka), epoxomycin (1 μm, Sigma), phosphatase inhibitor mixture II (100 μl, Calbiochem PPI II), trichostatin A (50 μm Sigma), nicotinamide (30 mm, Sigma), and sodium butyrate (30 μm, Sigma) were used for lysis. Cortical samples were resuspended at a ratio of 5 ml/g (volume of tissue protein extraction reagent (TPER, Thermo Scientific, catalog no. 78510) buffer, ml/weight of tissue/g) and striatal samples at 10 ml/g. A 2-ml Dounce homogenizer was used for tissue lysis on ice (2 × 60 pumps with a 30-s interval). Samples were stored at −80 °C prior to sonication. Tissue lysates were sonicated with continuous 40 mA pulses for 5 s five times on ice. Sonicated samples were centrifuged at 12,000 × g at 4 °C for 20 min. Supernatant was collected for Western blot analysis.
Western Blotting for HD Transgenic Mice
Cortical and striatal lysate protein concentrations were determined by BCA protein assay (Thermo Scientific). Lysates (40 μg) were prepared in 1× LDS buffer (Invitrogen) and 0.05 m DTT. Samples were boiled for 10 min at 100 °C and separated by one-dimensional SDS-PAGE on a 4–12% BisTris gel (Invitrogen) utilizing the Invitrogen Novex gel running apparatus at 200 V for 1 h in MES running buffer (Invitrogen) on ice. Overnight transfer was performed at 20 V constant for 14 h onto a 0.45-μm nitrocellulose membrane (Whatman) in NuPAGE transfer buffer (Invitrogen) at 4 °C. For smaller molecular weight fragments of HTT, a 12% BisTris gel was used at 200 V for 50 min in MOPS running buffer (Invitrogen). Transfer was performed at 350 mA constant for 1 h in transfer buffer (Invitrogen) with 10% methanol onto a 0.2-μm PVDF membrane (Millipore). After blocking in 5% milk in TBST, primary antibody (Millipore, MAB2166, 1:1000; MAB5374, EM48, 1:500; MAB5492(1–82), 1:100; MAB5490(115–129), 1:500; Sigma HTT N terminus(3–16), H7540, 1:200; polyQ 1C2, MAB1574, 1:1000; or Fitzgerald GAPDH 10R-G109A, 1:5000) was incubated overnight at 4 °C. Membranes were incubated with secondary HRP-coupled antibodies (1:3000, GE Healthcare) at room temperature for 1 h in blocking solution. Protein bands were detected by chemiluminescence (Pierce ECL Thermo Scientific, catalog no. 32106). ImageQuant TL (version 2005, Amersham Biosciences) was used for densitometry analysis.
IHC Staining of HD Mouse Brains
Paraffin-embedded coronal mouse brain sections from HD and wild-type mice were stained using the following immunohistochemistry methods. Sections were deparaffinized by soaking in xylene (2 times for 7 min each), rehydrated in ethanol (4 min with 100% EtOH, 4 min with 95% EtOH, 4 min with 80% EtOH, and 4 min with 70% EtOH), and rinsed in H2O. Sections were washed in TBS for 10 min. Antigen retrieval was performed by microwaving sections in 10 mm citrate buffer for 5 min at 40% power in an 1100-watt microwave oven. Sections were allowed to cool for 20 min and then washed once in TBS for 10 min. Sections were blocked for 1 h at room temperature in 10% normal goat serum diluted in TBS in a humidified chamber. Chicken anti-mouse IgG was added at 1:500 dilutions to the blocking buffer to allow for future probing with a primary mouse antibody on mouse tissue. After blocking, primary antibody was diluted in 1% BSA in TBS, and sections were incubated overnight at 4 °C in a humidified chamber. The following day, sections were washed in TBS three times for 10 min. Secondary antibodies were diluted in 1% BSA in TBS, and sections were incubated in secondary for 1 h at room temperature in the dark. Sections were then washed three times for 10 min in TBS and then allowed to air-dry for 10 min in the dark. Sections were mounted using ProlongGold and allowed to set overnight in the dark. For IHC staining of HD and wild-type mouse brains, the following primary antibodies were used: mouse α-EM48 (Millipore, MAB5374, 1:50) and rabbit α-DARPP-32 (Abcam, ab40801, 1:50). Alexa-fluor goat α-rabbit 555 and goat α-mouse 488 secondary antibodies (1:1000) were used.
FRET Analysis of HD Transgenic Mice
For TR-FRET analysis, tissue was lysed in PBS (Cellgro) supplemented with protease inhibitors (Roche Applied Science, 1 tablet/10 ml, Complete Mini, EDTA-free), phosSTOP (Roche Applied Science, 1 tablet/10 ml, phosphatase inhibitor mixture), and 1% Triton. Samples were lysed in a ratio of 10 ml/g. A 2-ml Dounce homogenizer was used for tissue lysis on ice (2 × 60 pumps with a 30-s interval). Samples were stored at −80 °C prior to sonication. Tissue lysates were sonicated with continuous 40 mA pulses for 5 s five times on ice and stored at −80 °C. Measurements were made as described previously (20, 44–46).
Coimmunoprecipitation with C-terminal HTT Antibody
C-terminal antibody was produced at Open Biosystems (Thermo Fisher) using the following HTT C-terminal peptide sequence: NH2-SPYHRLLTCLRNVHKVTTCKHL to immunize rabbits. The peptide sequence is against human HTT: NH2-SPYHRLLTCLRNVHKVTTCKHL. The mouse sequence is highly similar, containing two distinct amino acids (Thr to Ala and Arg to Gln) in the middle of the peptide. SPYHRLLACLQNVHKVTTC. Two rabbit polyclonal C-terminal antibodies generated from this peptide cross-react with mouse HTT by Western blot and pull down the full-length protein. The resulting polyclonal antibody was purified using a peptide column. Coimmunoprecipitation (coIP) of the FL-HTT was carried out with cortical lysates (500 μg, see above for lysis conditions) with purified antibody (4 μg). Samples were incubated with antibody at 4 °C overnight followed by 120 min binding with protein G-Sepharose (30 μl, GE Healthcare). Samples were eluted by boiling in 2× LDS loading buffer (30 μl, Life Technologies, Inc.).
Preparation of Protein Complexes for Mass Spectrometry
HTT MAB2166 (25 μl) or control preimmune mouse IgG (25 μg, Santa Cruz Biotechnology) was cross-linked to the fast flow protein G-Sepharose beads (50 μl, GE Healthcare). Beads were equilibrated in PBS (Corning) and incubated with the appropriate antibody overnight at 4 °C. Beads were washed twice with PBS, 550 mm NaCl to remove nonspecific proteins and washed with PBS to remove excess NaCl. Beads were then cross-linked in a 1:1 mixture of 0.1 m triethylamine, pH 9, in PBS, fresh dimethyl pimelimidate (13 mg/ml, Pierce) dissolved in anhydrous DMSO (Sigma) (pH of final mixture is ∼8.5) for 30 min at room temperature. This procedure was repeated for a total of three 30-min cross-linking reactions. The final cross-linking reaction was quenched with 50 mm ethanolamine, and the beads were washed three times in PBS. For coIP experiments, cortical lysates (1 mg) from three 4.5-month-old N552-Q148 and N586–148Q mice were processed in TPER (Pierce) with inhibitors (as in Western protocol) at a final concentration of 2.5 μg/μl (400 μl total volume). Samples were spun at 16,000 × g for 10 min (4 °C), precleared with purified mouse preimmune IgG (25 μg, Santa Cruz Biotechnology), and rotated 1 h (4 °C) followed by 3 h of incubation with protein G (50 μl, GE Healthcare) to remove IgG. Precleared samples were then incubated with cross-linked antibody/protein G beads overnight (4 °C). Samples were washed three times for 15 min in TPER and inhibitors and then twice at 15 min in PBS (4 °C). Samples were eluted in 1× SDS LB (Life Technologies, Inc.) or Tris, 0.5% SDS and subjected to either in-gel or in-solution digestion prior to mass spectrometry.
In-gel digestion was performed. Immunoprecipitated samples from cortex were boiled in LDS loading buffer with 0.05 m DTT and separated by SDS-PAGE on 4–12% NuPAGE gels (Life Technologies) in 1× MES for 10 min. Samples were stained with Sypro Ruby (Life Technologies, Inc.), and two slices covering all proteins were excised and digested by in-gel digestion (34). Briefly, gel slices were heated in 50 mm DTT at 55 °C for 30 min, followed by alkylation with iodoacetamide (50 mm) at room temperature in the dark for 45 min. Samples were then digested overnight with 200 ng of trypsin (Promega). In parallel, in-solution digestion was performed as follows. Samples were boiled in 10 mm DTT at 55 °C for 30 min followed by alkylation with 50 mm iodoacetamide in the dark for 45 min. Detergent was then removed from samples using the Filter-aided Sample Prep protocol (47). Samples were placed in a 30-kDa cutoff spin filters (Vivicon) and diluted with freshly prepared 8 m urea, 100 mm Tris, pH 7.5. Samples were spun at 18 °C for 30 min. Samples were then washed three times in 8 m urea, 100 mm Tris, pH 7.5 (400 μl), to ensure complete removal of SDS from the sample. Finally, samples were washed twice in 25 mm ammonium bicarbonate and digested with 200 ng of trypsin in 25 mm ammonium bicarbonate, 10% acetonitrile for 18 h at 37 °C. Digested peptides were eluted in 10% acetonitrile/ammonium bicarbonate. Samples were desalted with a C18 Zip Tip (Millipore).
Mass Spectrometry, Acquisition, and Database Searches
Samples were analyzed by reverse-phase HPLC-ESI-MS/MS using an Eksigent Ultra Plus nano-LC two-dimensional HPLC system (Dublin, CA), which was directly connected to a new generation quadrupole time-of-flight TripleTOF 5600 mass spectrometer (SCIEX, Concord, Canada) as described previously (48). Briefly, after injection, peptide mixtures were transferred onto the analytical C18-nanocapillary HPLC column (C18 Acclaim PepMap100, 75-μm inner diameter × 15 cm, 3-μm particle size, 100 Å pore size, Dionex, Sunnyvale, CA) and eluted at a flow rate of 300 nl/min using the following gradient: 3% solvent B in A (from 0 to 13 min); 3–7% solvent B in A (from 13 to 16 min); 7–25% solvent B in A (from 16 to 48 min); 25–40% solvent B in A (from 48 to 65 min); 40–90% solvent B in A (from 65 to 75 min); and 90% solvent B in A (from 75 to 85 min), with a total run time of 120 min, including mobile phase equilibration. Solvents were prepared as follows: mobile phase A: 2% acetonitrile, 98% of 0.1% formic acid (v/v) in water; mobile phase B: 98% acetonitrile, 2% of 0.1% formic acid (v/v) in water. Mass spectra and tandem mass spectra were recorded in positive ion and “high sensitivity” mode with a resolution of ∼35,000 full-width half-maximum. In data-dependent acquisition mode, 1 MS1 survey scan (m/z 400–1500) was followed by 30 MS/MS scans per 1.8-s acquisition cycle. Additional data sets were recorded in data-independent mode using SWATH-MS2 acquisitions. In the SWATH-MS2 acquisition, a wider window of 25 m/z was passed in incremental steps over the mass range from m/z 400 to 1000. Mass spectrometric data were searched using the database search engine ProteinPilot (49) (SCIEX Beta 4.0) with the Paragon algorithm (4.0.0.0,148083). The search parameters were set as follows: trypsin digestion, cysteine alkylation set to iodoacetamide, trypsin specificity, and species Mus musculus. All data files were searched using the SwissProt 2011_08 database) with 16,671 M. musculus protein sequences. For database searches, a cutoff peptide confidence value of 95 was chosen. For database searches, the Protein Pilot false discovery rate analysis tool (PSPEP) algorithm (49) provided a global false discovery rate of ≤1% in all cases. A peptide confidence value of 95 was chosen as a cutoff.
Quantification by Mass Spectrometry
Protein abundances of interacting proteins from HTT coIP experiments (in-solution digestion) were quantitatively compared specifically between the N552-Q148 and N586-Q148 transgenic mice and the wild-type littermate mouse using a Skyline MS1 Filtering approach as reported recently (48). Quantitative data for each of the mice was obtained using three technical (MS acquisition) replicates. Briefly, the search results from data-dependent searches were used to build peptide libraries in Skyline 2.5 (48). Peptide peaks were identified and confirmed by cross-referencing retention time, mass error, and comparison of MS2 scan profile. Quantitative analysis was based on extracted ion chromatograms and the resulting precursor ion peak areas for M, M + 1, and M + 2 for all peptides; the final quantitative measurements were typically based on the highest ranked (naturally most abundant) precursor ion. Peptide peak areas were integrated in Skyline with results exported to Excel. For proteins with multiple peptides, peak area measurements of separate peptides were summed. Not all peptides linked to a protein were chosen for quantitation; some peptides were excluded due to ragged ends or artificial modifications likely introduced during sample workup, such as methionine oxidation. In general, the proteins were quantified using at least three nonredundant peptides if available. HTT was quantified using 13 distinct tryptic peptides, and HSP86 was quantified using measurements from the three peptides also not present in HSP84. Statistical significance was assessed using Student's t test to test for significant differences between N552-Q148 IP samples and N586-Q148 IP samples. For the purpose of graphing, peak area data were normalized to the N552 IP sample for a consistent scale of the y axis.
Two-dimensional Blue Native/SDS-PAGE
Dissected tissues from transgenic mice were processed in TPER with inhibitors for use in BN-PAGE analysis as described above. Lysates (50 μg) were diluted to a final volume of 10 μl in H2O, 5× loading buffer (2 μl, 50% glycerol in TPER, 0.5% Coomassie Blue G-250), 5% digitonin (4 μl, Life Technologies, Inc.). Samples were mixed and spun at 4 °C and incubated on ice for 1–2 h. Samples were spun at 16,000 × g for 1 h prior to loading of the supernatant on BN-PAGE gels. Native-PAGE (Invitrogen) 3–12% gels (1.0 mm) were pre-run for 5 min with Anode buffer (400 ml, 1.1× Native-PAGE (Invitrogen), 0.1% Triton X-100) in the outer chamber and Cathode buffer (200 ml, 1.1 Native-PAGE (Invitrogen) buffer, 0.1% Triton X-100, 1.1× Cathode Additive (Invitrogen)) in the inner chamber. Samples were run at 150 V on ice for 45 min. Cathode buffer was then removed and replaced with Anode buffer. Samples were run at 250 V for 45 min or until low current <3 mA caused the power supply to shut down. After BN-PAGE was complete, the ladder was fixed in 50% methanol and 7% acetic acid and stained in Simply Safe stain (Invitrogen). Whole BN-PAGE lanes were excised, placed on thin clear plastic supports, and heated in 1% SDS, 50 mm Tris, pH 7.5, and 10 mm DTT at 65 °C for 15 min. After boiling, gel slices were placed into 1.5 mm NuPAGE 4–12% two-dimensional gels, and NuPAGE ladder was placed in the ladder lane. Samples were separated at 200 V on ice in MES buffer for 1 h. Gels were transferred overnight in NuPAGE transfer buffer at 20 V for 14 h, blocked in milk (5%, TBST), and analyzed by Western blotting with MAB2166 (Chemicon, 1:1000) as the primary antibody, followed by anti-mouse HRP secondary (GE Healthcare, 1:3000). Pierce Femto ECL was used for detection of HTT protein.
coIP/Western Blot for HSP86
Mouse cortical lysates were generated from 4.5- to 5- month-old mice (n = 3) as described above. Fresh aliquots were used to prepare 500 μg of lysate in 250 μl of MPER plus inhibitors for pulldown. Lysates were precleared with Sepharose beads for 1 h. Sepharose beads (25 μl, GE Healthcare) for IP were blocked with 3% BSA in PBS for 1 h followed by washing and incubation with either MAB2166 (5 μl) or preimmune mouse IgG (5 μl). The resulting beads were then added to 500 μg of precleared lysate and incubated on a rotator at 4 °C overnight. Samples were then washed twice for 1 h with MPER buffer (500 μl), 3% BSA for 1 h with PBS/Triton X-100 and 3% BSA and once for 15 min with MPER. Beads were boiled for 15 min in 2× LDS sample buffer, 50 mm DTT and stored at −80 °C prior to Western blot analysis.
Control samples were loaded such that an equal percentage (30%) of IP was present in the gel. The coIP samples were adjusted to load equal amounts of FL-HTT per well. Samples were run for 1 h at 200 V on a 10-well 4–12% NuPAGE BisTris gel with MES buffer followed by a 14-h transfer at 20 V at 4 °C. Samples were probed overnight for HSP86 (Affinity BioReagents, PA3-013, 1:1000), anti-rabbit secondary (Amersham Biosciences, 1:3000), and detected with femto-ECL (Pierce). Blots were then stripped and reprobed for HTT using MAB2166 (1:1000).
Human brain (striatal, HD grade 3 and control) lysates were processed in TPER plus inhibitors as above. Aliquots were used to prepare 500 μg of lysate in 1000 μl of TPER plus inhibitors for pulldown. Lysates were precleared with protein G-Sepharose beads for 1 h. Sepharose beads (25 μl GE; Healthcare) for IP were blocked with 3% BSA in TPER for 1 h followed by washing and incubation with MAB2166 (5 μl) followed by two washes in PBS, 350 mm NaCl and one wash TPER with inhibitors. The resulting beads were then added to 500 μg of precleared lysate and incubated on a rotator at 4 °C overnight. Samples were then washed three times for 1 h with TPER buffer (500 μl) and on time for 15 min with TPER. Beads were boiled for 15 min in 2× LDS sample buffer, 50 mm DTT and stored at −80 °C prior to Western blot analysis. Western blot was performed as described above.
Caspase Activity Assay
Mouse HdhQ7/Q7 striatal cells (2 × 106) were electroporated with a total of 6.5 μg of DNA (0.5 μg of GFP, 2 μg of pTet-tetracycline transactivator, 2 μg of HTT fragment or pCDNA3.1, and 2 μg of pCDNA3.1 or HSP86 cDNA) in an Amaxa nucleofector using program T-030. Cells were harvested with trypsin for 5 min, pelleted for 5 min at 1000 × g, and resuspended at 2 × 107 cells/ml in 5 mm KCl, 15 mm MgCl2, 15 mm HEPES, 150 mm Na2HPO4·NaH2PO4, 50 mm mannitol, pH 7.2. 100 μl of cell/DNA suspension was electroporated followed by recovery in RPMI 1640 medium, 10% FBS for 30 min. Recovered cells were plated on 6- and 12-well plates (400,000/well and 200,000/well, respectively). 6-well plates were used for Western analysis and were harvested at 72 h post-electroporation. At 24 h, 12-well plates were washed with PBS and treated with serum-free DMEM for 48 h. Cells were harvested by adding 1 volume of APO3/7 lysis buffer to the wells followed by scraping. Caspase-3/7 activity was assessed with using the caspase-3/7 HTS Z-DEVD kit (Cell Technology, Inc.). Briefly, three technical replicates of each biological replicate were assessed by loading 100 μl of lysate per well in a black-walled 96-well plate. Two 20-μl aliquots were removed for protein quantitation by BCA assay. 37.5 mm DTT (40 μl) and 2.5× Z-DEVD substrate were added for a final concentration of 1× Z-DEVD and 15 mm DTT. Plates were assayed on a fluorescent plate reader at 37 °C. Caspase-3/7 activity was assessed by subtracting the final signal from the initial signal, normalizing to protein content and divided by the elapsed assay time. This yielded the caspase-3/7 activity in relative fluorescence units/μg/min. Technical replicates of each biological replicate were averaged to obtain a mean activity for each biological replicate. Significance was assessed by ANOVA using Bonferroni's post test of indicated pairs in Prism. Samples from 6-well plates were assessed for electroporation efficiency by Western blot analysis. Protein (25 μg) was run on a 4–12% BisTris gel in MES buffer followed by a 14-h transfer at 20 V at 4 °C. Samples were probed for HTT (MAB2166, 1:1000) and rabbit β-actin (Cell Signaling, catalog no. 4968S, 1:500).
TUNEL Assay
Mouse HdhQ7/Q7 and HdhQ111/Q111 striatal cells were transfected with Amaxa Cell Line Nucleofection Kit L (Lonza) with siRNA against HSP86 (Dharmacon, GE Healthcare, M-055680-01-0005) or nontargeting siRNA. 4 μg of siRNA along with 0.5 μg of pEGFP-N1 was transfected in 2.5 × 106 cells with program T-030. Cells were plated on 24-well plates containing glass coverslips coated in collagen or 6-well plates for Western blot. Cells were cultured in serum for 72 h before fixation in 4% paraformaldehyde for 20 min at room temperature. TUNEL assay was done with CLICK-iT TUNEL Alexa Kit (Life Technologies, Inc.) following the manufacturer's instructions. Briefly, fixed cells were permeabilized with 0.25% Triton X-100 for 20 min at room temperature and equilibrated with terminal deoxynucleotidyltransferase reaction buffer for 10 min at room temperature. Terminal deoxynucleotidyltransferase reaction mixture consisting of buffer, EdUTP, and terminal deoxynucleotidyltransferase was applied to cells followed by 1 h of incubation at 37 °C. The CLICK-iT reaction was then performed by adding CLICK-iT reaction mixtures (buffer and buffer additive) to cells and incubating at room temperature for 30 min. After washing, coverslips were mounted on slides with ProLong Gold with DAPI (Life Technologies, Inc.). Striatal cells were harvested in MPER protein extraction buffer (Thermo) containing inhibitors 72 h post-electroporation and analyzed by Western blot. 25 μg of protein was loaded on NuPAGE 4–12% gels (Life Technologies, Inc.) and separated in MES (Life Technologies, Inc.) running buffer at 200 V for 1 h. Protein was transferred to nitrocellulose membranes (GE Healthcare) via overnight wet transfer (20 V, 14 h). Membranes were then blocked in 5% milk in TBST and probed with anti-HSP86, rabbit β-actin (Cell Signaling, catalog no. 4968S, 1:500) and anti-HTT MAB2166 (1:1000.) All primary antibodies were detected with 1:5000 dilutions of anti-rabbit or anti-mouse HRP conjugated secondary (GE Healthcare).
293F Cell Culture and Lipofection
293F cells were cultured in DMEM, 10% FBS, penicillin-streptomycin (10 IU, 10 μg/ml) and transfected with LF2000 (3 μl, Life Technologies, Inc., 1.5 μg of DNA). For HSP90 inhibition, cells received 1.5 μg of HTT cDNA, 0.1 μg of GFP for HSP86 overexpression, and cells received 0.5 μg of HTT cDNA, 1 μg of HSP86 or empty pcDNA3.1 vector, and 0.1 μg of GFP. Cells were cultured for 72 h prior to harvest with MPER with protease inhibitor (Roche Applied Science). HSP90 inhibition was performed at 48 h post-transfection, when cells were treated for 24 h with 500 nm 17-DMAG (Invitrogen) or DMSO vehicle alone. Samples were assessed by Western blot analysis. All experiments were performed in triplicate. Protein samples (25 μg) were run on a 4–12% BisTris gel in MES buffer followed by 14-h wet transfer at 20 V at 4 °C. Samples were probed with antibody top HTT (MAB2166), HSP86, and rabbit β-actin (Cell Signaling, catalog no. 4968S, 1:500). Protein levels were measured by densitometry using ImageQuant TL software (GE Healthcare).
Results
Generation of ROSA26 HTTQ148 Fragment Mice
To characterize the contribution of individual N-terminal HTT fragments to neurodegeneration in HD, we generated transgenic mice that express defined human HTT fragments in a C57B6 background. We designed HTT truncation constructs representing proteolysis cleavage products of HTT (Fig. 1A). HTT is cleaved by calpains-1 and -2 at amino acids 469 and 536, by caspase-2 at amino acid 552, and by caspase-6 at amino acid 586 (13, 18, 36). A protein product at N586 exists in mouse and human post-mortem tissue; therefore, we include this fragment. In addition, there is evidence for proteolysis or aberrant splicing occurring for protein products ending at amino acid 171 or smaller (50–52). We attempted to generate N92-Q148 (exon 1), but the founder animals failed to survive until weaning. As shown in Fig. 1, B and C, all five mouse models contain a single integration at the ROSA26 locus, with the expression of the HTT fragments driven by the chicken β-actin promoter. We generated transgenic mouse models by knock-in of a single copy of the polyQ-expanded HTT fragments with 148 polyQ repeats at the ROSA26 locus using recombinase-mediated cassette exchange ensuring that positional effects and copy number do not affect expression (Fig. 1C). The polyQ repeats were encoded by repeating mix of CAG/CAA triplets to stabilize the intergenerational and somatic polyQ size. Transgenic mice were produced from mESCs and characterized for HTT protein levels in regions of the brain. Modeling HD in mouse models requires a longer CAG repeat size than that found in humans (typically 38–80). Therefore, we used a polyQ of 148 repeats.
Expression Levels of the ROSA26 HTT148Q Fragment Mice
The expression of the individual HTT fragments of the ROSA26 HTTQ148 fragment mice was measured by Western blot analysis in the cortex and striatum at 4.5 months (Fig. 2A). HD transgenic mice expressing N-terminal HTT fragments ending at amino acids Ala-463, Ser-536, Asp-552, and Asp-586 are present at roughly proportional levels in cortex and striatum (Fig. 2A). Similar results were found in other regions of the brain. Expression of the fragment ending at Glu-171 was not detected, as it was not immunoreactive to the HTT antibody MAB2166. The migration of the fragment proteins are as predicted accounting for the slower migration of the HTT with a length of 148 polyQ. For quantitative analysis of soluble HTT fragment protein levels in the cortex and midbrain, we used a TR-FRET-based assay (20, 44–46). TR-FRET-based assays represent sensitive immunoassays to quantify proteins of interest and in our case use antibodies specific for different N-terminal HTT epitopes. Analysis of soluble HTT revealed similar levels of soluble HTT fragments in the cortex and midbrain (Fig. 2, B and C). We also measured insoluble HTT with TR-FRET at this same time point and found similar levels (data not shown). Littermate control lysates served as a negative control as the antibody MW1 recognizes only the transgene. Our results indicate that with the exception of N171 HQ0021 (HD_N171 (TT), N463 HQ0020 (HD_N463(TT)), N536 HQ0019 (HD_N536(TT)), N552 HQ0017 (HD_N552 (TT)), and N586 HQ0016 (HD_N586(TT)) expressed similar levels of protein. Thus, any differences in motor and behavioral deficits can be directly attributed to specific properties of the specific HTT fragment.
FIGURE 2.

Measurement of protein levels in the ROSA stop transgenic mice. A, Western blot analysis of cortical and striatal lysates of the N463-Q148, N536-Q148, N552-Q148, and N586-Q148 transgenic ROSA HD transgenic mouse models. * kDa is estimated on striatal Western blot from the same samples run on a 10-well gel. B, TR-FRET analysis of transgenic HTT protein levels compared with littermate controls in cortex. C, TR-FRET analysis of transgenic HTT protein levels compared with littermate controls in midbrain.
Life Span of the ROSA26 HTTQ148 Fragment Transgenic HD Mice
Transgenic HD mice expressing each of the five HTT N-terminal fragments die sooner than wild-type C57BL/6 littermate control mice (Fig. 3A). The Jackson Laboratory reports median life span of the C57BL/6 mice as 767 days (53). Mice expressing the smallest fragment, N171-Q148, live for a median period of 96 days and die significantly sooner than other fragment mice (****, p < 0.0001 in all pairwise comparisons). The N552-Q148 mice live for a median period of 204 days and die significantly sooner (p < 0.0001 in all pairwise comparisons) than the three other HD transgenics, N463-Q148 (median survival = 255 days), N536-Q148 (median survival = 289 days), and N586-Q148 (median survival = 277 days), which have roughly comparable life spans to each other (Fig. 3A). All five transgenic mice have statistically distinct life spans (****, p < 0.0001). Therefore, the shortest lived N-terminal fragment HD mouse model is N171-Q148. Comparison of N463, N536, N552, and N586 indicates that the median life span of the caspase-2 transgenic mouse model, N552-Q148, is 10 weeks (26%) shorter than the caspase-6 transgenic mouse model N586-Q148, despite similar levels of HTT fragment protein present in the cortex, striatum, and midbrain of the two models.
FIGURE 3.
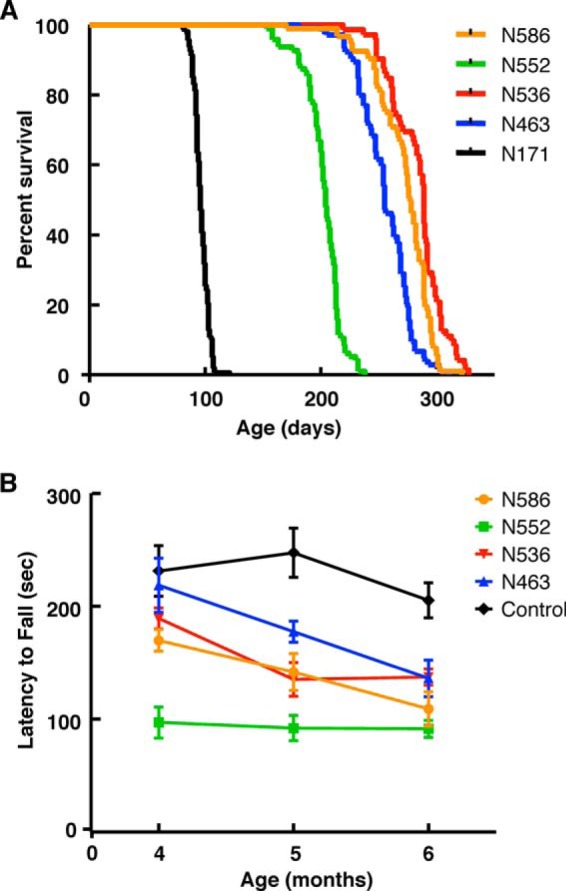
Life span and rotarod measurements in the ROSA-stop transgenic mice. A, life span of the N171-Q148, N463-Q148, N536-Q148, N552-Q148, and N586-Q148 transgenic ROSA HD transgenic mouse models was measured. B, Rotarod measurements of N463-Q148, N536-Q148, N552-Q148, and N586-Q148 transgenic ROSA HD transgenic mouse models demonstrated that the N552-Q148 had statistically significant impaired rotarod performance when compared with the littermate controls, N536-Q148, N552-Q148, and N586-Q148.
Rotarod Evaluation of the ROSA26 HTTQ148 Fragment Mice
The CAG expansion in HTT leads to the primary deficits of motor dysfunction, combined with behavioral deficits in cognition and psychiatric conditions. Some aspects of this can be modeled in HD mouse models. To further characterize the N-terminal fragment mice, we measured rotarod performance of the N463-Q148, N536-Q148, N552-Q148, and N586-Q148 mice at 4–6 months of age. As shown in Fig. 3B, the N552-Q148 HD transgenic mice have impaired rotarod performance compared with the other HD transgenic mice analyzed at 4 months of age (***, p < 0.001). At 6 months of age, the N552-Q148 performed poorly compared with N463-Q148, N536-Q148, and N586-Q148. However, the number of N552-Q148 mice surviving at 6 months of age limited the statistical significance at this time point. N171-Q148 mice were not included in these time points as they were not alive.
Open Field Activity of the ROSA26 HTT148Q Fragment Mice
Next, we assessed open field activity at 4 and 5 months of age measuring 22 variables in these assessments. Open field measures general activity, exploration, and anxiety (54). The N-terminal HD transgenic mouse models displayed significantly reduced rearing at 4 and 5 months relative to C57BL/6 control mice as measured by the number of entries into the vertical plane, time spent in the vertical plane, and distance traveled in the vertical plane (not significant in N463-Q148, N536-Q148, and N586-Q148 mice at 4 months) lower relative to C57BL/6 mice in the open field at 4 months of age (Fig. 4A). N552-Q148 mice demonstrate both the greatest numerical and the most statistically significant reduction in rearing relative to C57BL/6 control mice at both 4 and 5 months; however, the differences relative to other fragment models do not reach statistical significance (Fig. 4A). At 5 months, all four transgenic mouse models showed low levels of rearing relative to control mice (Fig. 4B). We also assessed floor plane move time at 4 and 5 months of age. Although there was a slight trend toward reduced movement events, distance traveled, and time spent moving at 5 months, the only parameter that displayed a reduction relative to wild-type animals that reached statistical significance was the number of floor plan moves (N552-Q148, N586-Q148, and N536-Q148) at 5 months of age. As with other parameters, N552-Q148 mice displayed both the lowest number of floor plane moves and the highest statistical confidence relative to control animals at 5 months.
FIGURE 4.

Open field testing in the ROSA-stop transgenic mice. The open field parameters for the N463-Q148, N536-Q148, N552-Q148, and N586-Q148 transgenic ROSA HD transgenic mouse models were measured at 4 months of age (A) and 5 months of age (B). Recordings of 22 parameters during the open-field test in the floor and vertical plane were monitored to evaluate spontaneous locomotor activity. The measurements shown are noted in the panels. An n = 5–16 animals were tested at 14 weeks for 30 min during the dark cycle. Statistical significance is noted in the graphs.
Pathological Analysis of ROSA26 HTT148Q Fragment Mice
Given the observed life span and motor phenotypes in the fragment mice, we set out to determine whether there is evidence of neuronal pathology in the mice (Fig. 5). Striatal lysates from B6, N463-Q148, N536-Q148, and N586-Q148 mice at 4 months were analyzed by Western blot analysis for loss of DARPP-32, a key marker of medium spiny neurons. In models of HD, the change in signaling for DARPP-32 occurs well before loss of neurons and therefore represents early events in neuronal dysregulation and dysfunction (55–62). DARPP-32 expression in the striatum of N552-Q148 mice was significantly reduced (p < 0.01) when compared with B6, N463-Q148, N536-Q148, and N586-Q148 mice (Fig. 5A). IHC supports the Western-based quantitation showing reduced DARPP-32 levels in N552-Q148 mice when compared with N586-Q148 mice (Fig. 5B). Another hallmark of neuronal pathology in HD is aggregation of mHTT protein and fragments. IHC analysis of N552-Q148 and N586-Q148 mice revealed aggregation of HTT in the striatum and cortex (Fig. 5C). Aggregation was sparse in these fragment mice when compared with the exon 1 transgenic model R6/2 mice at 3 months of age.
FIGURE 5.

Neural pathology is present in brains of N552 mice. A, Western analysis of DARPP-32 levels in 4.5-month-old mouse striatal lysates identifies a significant reduction (p > 0.01) in DARPP-32 protein of N552-Q148 mice relative to wild-type brains. B, IHC analysis of from 6-month-old mice reveals altered localization of DARPP-32 in N552 mice relative to wild-type mice. The settings for exposure time were the same for all panels. C, although HTT aggregates, identified by EM48 staining, are present in the brains of N552 and N586 mice (4.5 months), they are less readily apparent than in R6/2 (positive control) mice (3 months).
Proteolysis of the Transgenic Rosa HTTQ148 Fragment Mice
There is growing evidence that protein products are generated from the HTT gene either by aberrant splicing or proteolysis after translation with protein products ending between amino acids 90 and 171 (19, 20, 51, 52, 63). Taking advantage of the fact that the HTT cDNA generated to make these new models is not likely to be aberrantly spliced, we analyzed the role of proteolytic processing in these new transgenic mouse models of HD. Cortical lysates were analyzed by Western blot analysis to assess the presence of HTT fragments. As shown in Fig. 6A (2nd panel), all five transgenic mice generate a protein product at 52 kDa detected by antibodies directed at the polyQ and polyproline stretch, respectively (1C2, EM48), when compared with the B6 controls (Fig. 6, A and B). This cleavage product is also detected by MAB5492 (HTT(1–82)) and MAB5490 (HTT(115–129)) (Fig. 6, A and B). Therefore, an N-terminal fragment is produced by cleavage at an amino acid above the epitope ending at amino acid 129. The protein produced at 52 kDa does not appear to contain the N terminus as the protein is not detected by the HTT N-terminal antibody recognizing amino acids 3–17 of HTT (Fig. 6A, MAB5492) despite detection of the N terminus in the intact transgenic proteins. If the transgenic HD mouse models are subject to cleavage in a region of ∼120 amino acids, then the corresponding protein products on the C-terminal end should be present (Fig. 6B, see fragments generated). These fragments would have molecular weights corresponding to amino acids ∼120–463, ∼120–536, ∼120–552, and ∼120–586 (Fig. 6B, denoted with a red *). These products are indeed detected with the HTT MAB 2166 antibody as shown in Fig. 6B. The exact mechanism behind the loss of the N terminus is unknown but could be the result of disease-associated proteolysis or other mechanisms (64, 65). Fig. 6B schematic summarizes the protein products generated from the transgenic ROSA26 HTTQ148 fragment mice, what region the antibodies detect, and a putative map of the protein products observed. The amount of the 52-kDa product was similar in the different HD fragment mouse models and therefore is not likely to explain the differential phenotypes observed in these models. The 52-kDa product does accumulate with age, which is consistent with other studies (16).
FIGURE 6.

Western blot analysis of ROSA stop transgenic mice. A, Western blot analysis of 4.5-month-old lysates from N171-Q148, N463-Q148, N536-Q148, N552-Q148, and N586-Q148 transgenic ROSA HD transgenic mouse models using distinct HTT antibodies to distinct regions of the protein. The open arrowhead indicates where the cleavage product should be detected. The product is absent. B, schematic of the proteolysis or fragments of HTT present in cortical lysates of the transgenic mouse models. The lower Western blot demonstrates the appearance of C-terminal cleavage fragments detected with HTT MAB2166.
Molecular Mechanism for Accelerated Phenotype in N552-Q148 Transgenic Mice
We hypothesized that changes in protein interactions are responsible for differences in HTT fragment and behavioral phenotypes of the caspase-2 N552-Q148 mice. We focused on comparison of the N552-Q148 and N586-Q148 transgenic mice because although the observed differences in motor, life span, and behavioral phenotypes are distinct, they differ in length by only 34 amino acids and because they represent caspase cleavage products that have been implicated as highly significant in HD disease progression and pathology (5, 7, 9, 12). To explore this hypothesis, interaction studies used two approaches, coIP of full-length HTT (FL-HTT) and two-dimensional BN-PAGE/SDS-PAGE. We reasoned that the differences observed in neurodegenerative phenotypes caused by different HTT fragments are due to differences in biochemical properties of these fragments. Specifically, we suspected that fragments displayed differential interaction or conformation with protein complexes containing endogenous FL-HTT. To characterize the properties of these fragments, we used a custom antibody against the C terminus of FL-HTT, an epitope that is absent in the fragments to coIP FL-HTT containing complexes from cortical lysates of 4.5-month-old mice. coIP demonstrated that FL-HTT interacts with both HTT fragments (Fig. 7A). However, despite similar levels of the fragments in lysates, HTT N552 interacts with FL-HTT complexes more robustly than HTT N586 at 4.5 months (Fig. 7A). Control beads did not immunoprecipitate HTT.
FIGURE 7.

Separation of supramolecular HTT complexes by two-dimensional Blue Native/SDS-PAGE in mouse models of HD. A, analysis of HTT fragment interaction with endogenous HTT. The coIP with a C-terminal antibody to full-length endogenous HTT demonstrates a stronger interaction with the N552 fragment when compared with N586 by coIP. HTT fragments positions on the gel are indicated with an arrow. The Western blot was probed with HTT antibody 2166. B, cortical lysates (500 μg) of the indicated mouse strain were analyzed by Blue Native PAGE (BN PAGE), followed by SDS-PAGE. The HTT complexes were visualized by Western blotting the transferred proteins with HTT antibody MAB2166. The horizontal axis shows the separation by molecular mass of intact complexes (>1.2 megadaltons, 140 kDa via BN-PAGE). The vertical axis shows the separation of BN-PAGE separated lanes by SDS-PAGE. In all cases endogenous mouse HTT is present in a complex of >750 kDa (horizontal arrows). All lysates, including wild-type, also contained small (∼100 kDa) fragments present in complexes smaller than 750 kDa (vertical arrows, white stars). In contrast with D586 and S536 mice, D552 mice display a series of ∼100-kDa fragments present in complexes that are the same molecular mass as endogenous HTT and diminished native complex. Black * denotes a nonspecific protein.
A second method to measure incorporation of fragments into FL-HTT complexes, two-dimensional PAGE, was utilized to confirm distinct incorporation of fragments into FL-HTT complexes. In the first dimension, BN-PAGE was used to separate intact protein complexes by molecular weight (Fig. 7B). For the second dimension, components of the complexes were separated by size using SDS-PAGE. This technique allows the measurement of fragment incorporation into protein complexes by Western blot analysis of the two-dimensional PAGE separated proteins. As was seen in the coIP with the C-terminal anti-HTT antibody, more HTT fragments are present in the large complex that contains FL-HTT in N552-Q148 mice as compared with N586-Q148 or C57BL/6 mice at 4.5 months (compare the amount of the 100-kDa fragment protein directly below the FL-HTT signal versus amount of 100-kDa protein to the right of the FL-HTT signal Fig. 7B).
Mass Spectrometry Identifies HTT-interacting Proteins
Given the different interactions of the N552 and N586 with endogenous HTT, we set out to generate an interactome data set from the cerebral cortex of the N552-Q148 and N586-Q148 mice at 4.5 months, a time point when there is a substantial difference in physical interaction with endogenous HTT and a significant difference in symptoms of motor dysfunction between N552 and N586 mice. Three biological replicates of coIP and discovery mass spectrometry on cortical lysates from male mice were analyzed. coIP was performed with the HTT antibody MAB2166, which recognizes full-length and the N552 and N586 fragments of HTT. A mouse preimmune IgG was used as control for the coIP. coIP of HTT was confirmed by Western blotting in all three biological replicates prior to analysis on a SCIEX TripleTOF 5600 mass spectrometer after tryptic digestion. In both Western blot and mass spectrometry runs, HTT was only identified in the HTT MAB2166 antibody pulldowns and not IgG controls (Table 1). Proteins with >2 peptides identified at 95% confidence in coIPs, with none identified in control IPs, were considered to be putative HTT-interacting proteins when identified in more than two samples. Using these criteria, 106 putative HTT-interacting proteins were identified. Proteins identified in four or more of the 12 mice analyzed were considered as strong HTT interaction candidates and are presented in Table 1.
TABLE 1.
HTT interacting proteins identified by mass spectrometry
The coIP with MAB2166 and mIgG followed by mass spectrometry was performed using two methods (in-gel and in-solution) on a total of 12 mice. The coIP and control samples from N552 (n = 3) and N586 (n = 3) mice were analyzed after in-gel digestion. An additional N552 (n = 2), N586 (n = 2) and WT (n = 2) mice were analyzed by in-solution digestion. Proteins were considered to be identified in samples if 2 or more peptides were identified at >95% confidence. Proteins identified only in IP samples are reported in this table. HTT protein was identified 6 of 6 CoIPs in both in-gel and in-solution samples and 0 of 6 control samples in both in-gel and in-solution digestion. For in-gel digestion a total of 68 proteins were identified exclusively in at least one CoIP sample, here we present four proteins that were identified in at least four CoIP samples with none identified in control samples. For in-solution digestion a total of 232 proteins were identified exclusively in at least one CoIP sample, here we present 30 proteins that were identified in at least four CoIP samples with none identified in control samples. These proteins were cross-referenced to published data sets from three other groups as follows: Shirasaki et al. (68); Culver et al. (69); and Kaltenbach et al. (66). All but one have previously been reported as primary or secondary HTT interacting proteins. Proteins identified as secondary interactors are indicated as 2°. Finally, the proteins from one biological replicate (three injections) of the in-solution digestion were quantified by mass spectrometry in N552, N586 and a WT mouse using MS1 filtering in Skyline (48) to screen for proteins whose levels putatively differ between N552 and N586 mice. These levels were quantified by integration of the peak area of the identified peptides (number of peptides used for quantification is indicated). Peak areas were then summed and N552/N586 ratio, standard deviation, and p values were calculated.
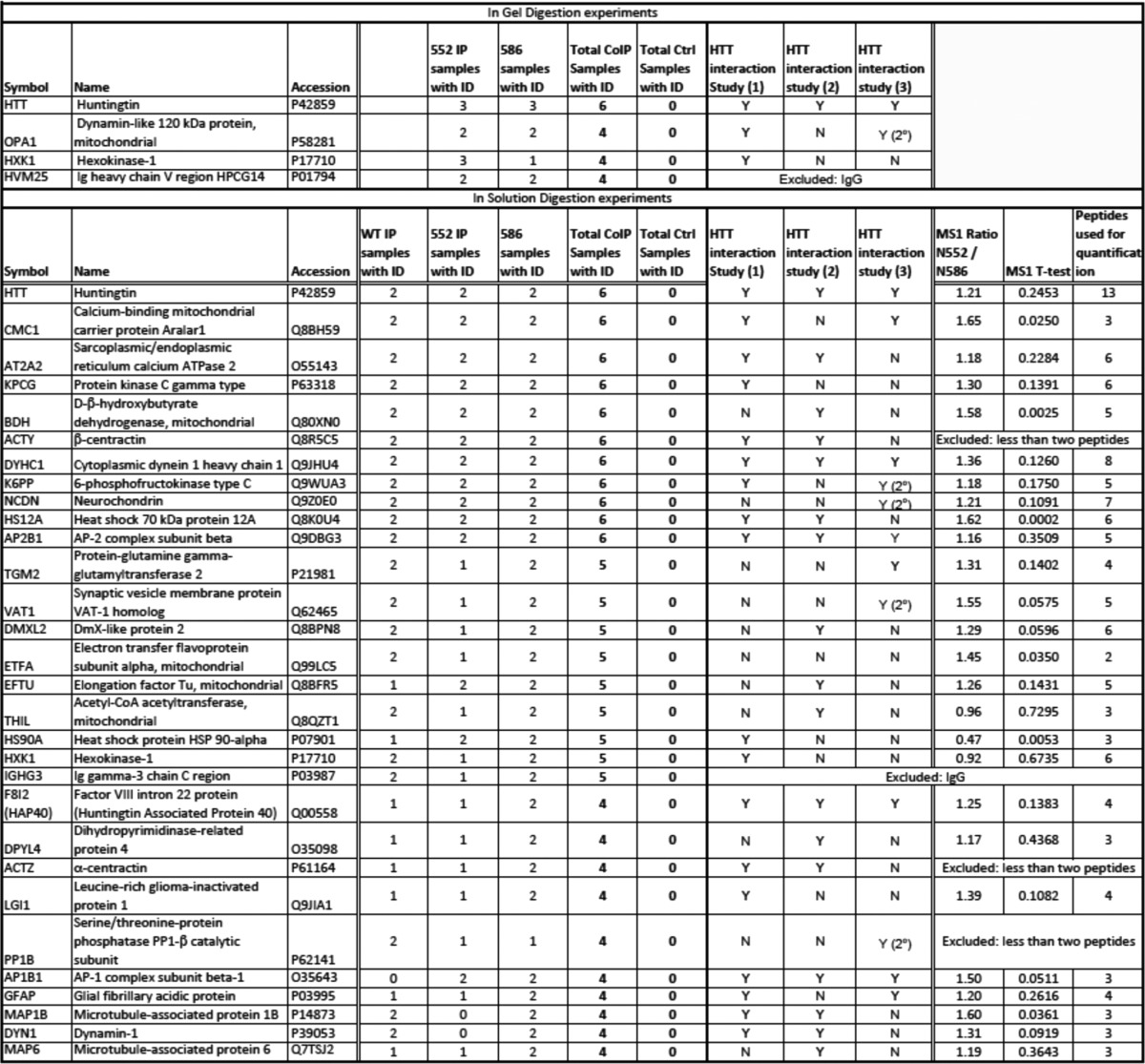
The majority of the proteins identified in our study have been published in previous HTT interaction data sets (22 of the 27, see Table 1). For example, a well characterized HTT interaction protein is F8l2, better known as HAP40 (Table 1) (66–69). To determine whether there was a difference in interaction with the N552 and N586 fragments with the top 27 interacting proteins mass spectrometric, the MS1 chromatogram-based quantification of one biological replicate processed by in-solution digestion was carried out using MS1 filtering in Skyline 2.5 (Fig. 8 and Table 1) (48). Briefly, for the three injection replicates of each IP or control sample, extracted ion chromatograms from MS1 scan data of three isotopic (M, M + 1, and M + 2) forms of peptides identified in discovery experiments were imported into Skyline 2.5 and used for quantitation of peak area as a measure of peptide abundance. Peak areas were exported to Excel, and peak areas of multiple peptides constituting a protein were summed to give a measure of protein abundance.
FIGURE 8.
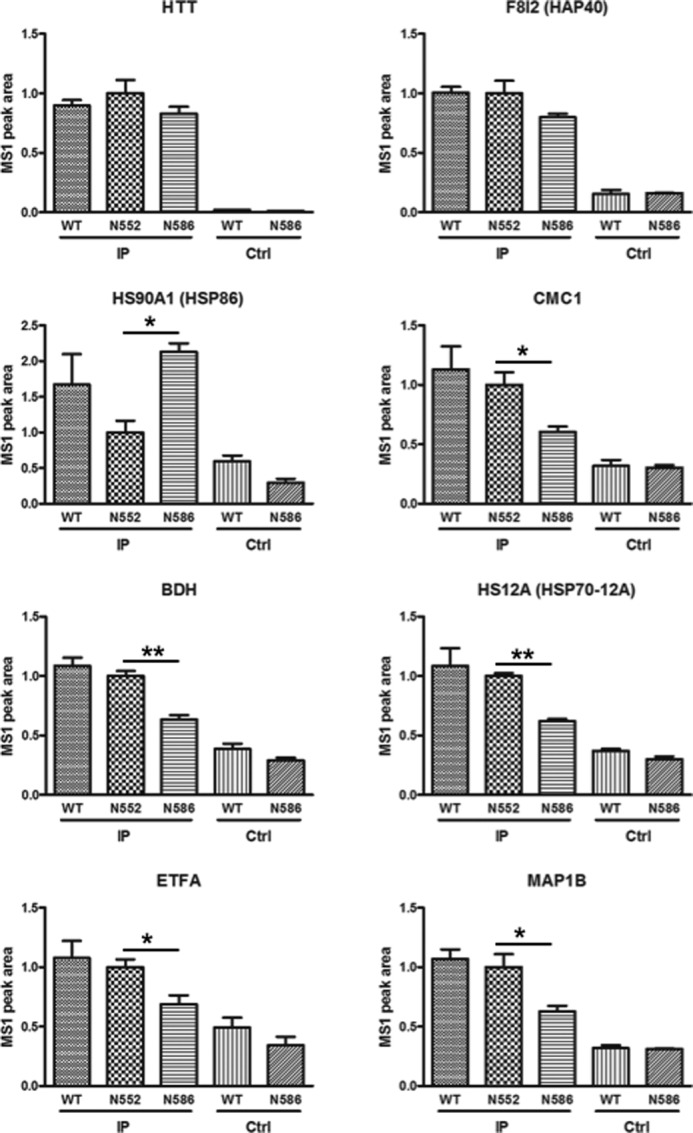
MS1 filtering identifies candidate HTT interacting proteins with differential interaction between N552 and N586 mice. The coIP protein from mouse cortex was processed by in-solution digestion and analyzed by mass spectrometry using three injection replicates. HTT-interacting proteins identified in Table 1 were quantified using the MS1 filtering function in Skyline 2.5. Briefly, MS1 peaks for peptides of these proteins were identified and quantified. The resulting peptide measurements were summed. The six proteins whose levels significantly differed between N552 and N586 mice are presented here. HTT and HSP40 are presented as positive controls (Ctrl). The control is a sample under the IP conditions without primary antibody. Statistical significance is noted.
The majority of the identified HTT-interacting proteins showed lower abundance in the N586 protein lysates when compared with the N552 protein lysates consistent with the N552 protein integration into the native HTT complex (Fig. 7B). Of the 27 interacting proteins, six showed a significant difference in levels between N552-Q148 and N586-Q148 IPs by t test, but only one showed a 2-fold difference in protein abundance between N552-Q148 and N586-Q148 mice (Fig. 8 and Table 1). HSP86, which met both criteria, gave a 2.1-fold increase in N586 mice relative to N552-Q148 mice (Fig. 8, 2nd panel, left side).
HSP86 Interaction Is Altered in the N586 Mice
HSP86 (HSP90AA1) is an HSP90 family member that has been previously shown to be an HTT-interacting protein (66–69). Further investigation using coIP followed by Western blot on separate mice (n = 3 per condition) confirmed the interaction and identified an increase in the HSP86 interaction with HTT in the N586-Q148 mice relative to N552-Q148 (1.6-fold increase) and WT mice (littermate controls) (Fig. 9, A and B) consistent with the mass spectrometry quantification (Fig. 8). These differences did not reach statistical significance by ANOVA for the Western blot. coIP of HSP86 upon pulldown of HTT was confirmed in human post-mortem brain tissue by IP with MAB2166 followed by Western blot. IP from tissue from an HD patient and an age-matched wild-type control brain confirms the protein interaction (Fig. 8C).
FIGURE 9.
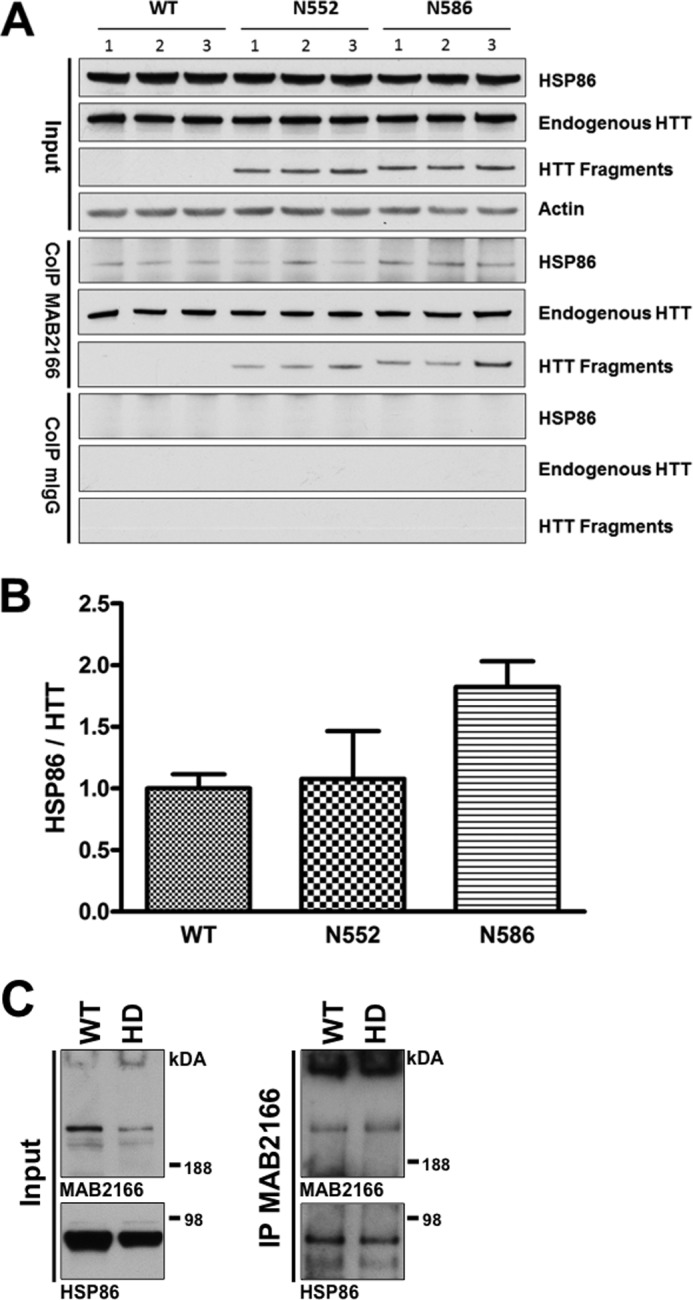
HSP86 interacts with HTT. A, to further investigate the interaction between HSP86 and HTT, we performed coIP and Western blot on 4.5-month-old mice from WT, N552-Q148, and N586-Q148 backgrounds. The coIP with MAB2166 confirmed HTT and HSP86 specifically interact. B, analysis of the resulting HSP86 by densitometry showed an increase in HSP86 pulled down in N586-Q148 mice relative to wild-type mice when normalized to full-length HTT. p < 0.05 Student's t test but is not significant by ANOVA. The only other bands present in the gel image were due to IgG immunoreactivity. C, mouse results were confirmed by coIP of HTT from post-mortem human striatal tissue (HD grade 3 and nondisease control) using MAB2166 followed by Western blot of HSP86.
HSP86 Modifies HTT Fragment Levels in Cell Culture
Overexpression of HSP86 led to a larger-fold increase in N586 (2.6×) relative to N552 (1.4×) levels (Fig. 10A). The levels of endogenous HTT were unchanged in this same experiment (Fig. 10B). This same trend in levels of HTT fragments was found in the HdhQ7/Q7 mouse striatal cell line, although it was nonsignificant in those cells (see below and Fig. 11B) perhaps due to the lower transfection efficiency of the striatal cells. To understand how inhibition of HSP86 and its family members affects stability of HTT fragments, the pan-HSP90 inhibitor 17-DMAG was used. Inhibition of HSP90 family members, including HSP86, resulted in a significant reduction in the N552 and N586 protein levels as well as endogenous HTT (Fig. 10, C and D). Inhibition of HSP90 with 17-DMAG led to a similar overall reduction in both endogenous and HTT fragment levels (2.0-fold reduction in N552 and 2.4-fold reduction in N586; 1.9- and 1.9-fold reduction of endogenous HTT in N552- and N586-transfected cells, respectively). We found that 17-DMAG treatment of HdhQ7/Q7 mouse striatal cells caused differentiation and therefore did not evaluate cytotoxicity under these conditions (data not shown).
FIGURE 10.

HSP86 stabilizes HTT. A, 293T cells were cotransfected with HTT fragment cDNA and either empty vector or HSP86 cDNA. After 72 h, cells cotransfected with HSP86 expressed higher levels of the fragments as assessed by Western blot. Intriguingly, N586-transfected cells displayed a 2.6× increase in fragment levels, and N552 transfected cells displayed a 1.4-fold increase. B, this increase in fragment levels did not occur with endogenous HTT in the same cells. C, inhibition of HSP90 by 17-DMAG leads to a significant decrease in HTT fragment levels. Cells were transfected and cultured for 48 h and then treated with pan-HSP90 inhibitor 17-DMAG (500 nm) or DMSO vehicle control for 24 h. The N552-transfected cells displayed a 2× reduction in fragment levels upon HSP90 inhibition, and the N586 cells displayed a 2.4× reduction upon HSP90 inhibition. D, inhibition of HSP90 by 17-DMAG leads to significant reduction of endogenous HTT in transfected 293T cells. Quantitation of endogenous HTT levels from cells treated in C is shown. In contrast with overexpression, transient inhibition of HSP90 is sufficient to reduce endogenous HTT by 2-fold.
FIGURE 11.

HSP86 modifies the toxicity of HTT fragments. A, HSP86 overexpression protects against toxicity of HTT fragments in mouse HdhQ7/Q7 striatal cells. To test the effect of altered HSP86-HTT interaction, we tested the ability of HSP86 to alter caspase-3/7 activity in cells overexpressing either N552 or N586 fragments of mHTT. Overexpression of HSP86 was generally protective and led to a significant reduction of caspase-3/7 activity in N552- and N586-expressing cells after 48 h of serum starvation and 72 h post-electroporation. B, Western analysis of cells used for caspase activity assays confirms HSP86 does not reduce toxicity by reducing fragment levels. C, mouse striatal HdhQ7/Q7 and HdhQ111/Q111 cells were transfected with siRNA to HSP86, and apoptosis was measured using TUNEL. Statistical significance was determined with ANOVA. D, Western blot analysis of mouse striatal cells HdhQ7/Q7 and HdhQ111/Q111 lysates confirm knockdown of HSP86.
HSP86 Modifies HTT Cytotoxicity in HD Mouse Striatal Cells
To determine whether the HTT-HSP86 interaction may contribute to the observed acceleration of the N552 relative to the N586 motor, life span, and behavioral phenotypes, a cell-based toxicity assay was used to assess caspase-3/7 activity in HdhQ7/Q7 mouse striatal cells (Fig. 11A). We have previously published that the caspase-3/7 activity in HdhQ111/Q111 cells occurs after 24 h of serum withdrawal, whereas the cell death measured by Hoechst or other nuclear dyes is detectable after 48 h (8). Therefore, the caspase activity precedes cell death but correlates with the induction. HdhQ7/Q7 cells were electroporated with Htt143Q(1–552) or Htt144Q(1–586) constructs with either vector control or HSP86 cDNA. After culturing for 24 h, serum was withdrawn for 48 h. In agreement with our in vivo work, expression of the Htt143Q(1–552) protein displayed greater toxicity than the Htt144Q(1–586) protein (1.4-fold, Fig. 11A). Transfection with HSP86 cDNA led a reduction of caspase-3/7 activity in both the Htt143Q(1–552)- and the Htt144Q(1–586)-transfected cells. Western blot demonstrated that the protein expression was equivalent for the fragments without HSP86 (Fig. 11B). Rather than lowering HTT levels, cells transfected with HSP86 showed a trend toward increasing levels of HTT fragments (Fig. 11B). This same effect was found in 293T cells, which were cotransfected with HSP86 and fragments. We also evaluated the effected of HSP86 siRNA knockdown (pool) on cellular toxicity of striatal HdhQ7/Q7 and HdhQ111/Q111 cells using a TUNEL assay in cultured cells under normal growth conditions (Fig. 11, C and D). The knockdown of HSP86 increased TUNEL staining suggesting increased cytotoxicity. Knockdown of HSP86 was confirmed by Western blot analysis (Fig. 11D).
Discussion
We set out to directly compare life span, rate of neurodegeneration, and behavioral phenotypes for a number of well characterized HTT proteolysis fragments identified in mouse models and HD post-mortem tissue (5–14, 16–18, 20, 21, 36). We used a novel approach in which we generated each mouse model with a single integration at the ROSA26 locus, with expression of the fragments driven by the chicken β-actin promoter at nearly identical levels and in identical genetic background B6. Although several proteases have been implicated in the formation of N-terminal fragments during pathogenesis and neurodegeneration in human HD patients and mouse models of HD, there is still controversy regarding the relative contributions of the proteases and their cleavage products to disease. Caspase-2 and -6 and calpains-1 and -2 have been shown to be activated in HD patients and to produce distinct N-terminal HTT cleavage products (5, 7, 13, 14, 18, 20, 25, 36). Caspase-6, and by implication its product N586, was initially reported to be activated in mouse models and post-mortem tissue and to cause HD-like pathology in YAC128 mice (5, 12). Genetic approaches making mouse models resistant to cleavage at caspase-3 and -2 suggested these sites were less significant to HD pathology and progression than the mouse model resistant to caspase-6 cleavage (12). However, subsequent work in BAC Huntington disease mice suggested that caspase-6 knock out modified disease progression through alteration in HTT clearance pathways rather than blocking proteolysis of mutant HTT. These results may reflect compensatory pathways evoked by caspase-6 knock out during development (7). By contrast knock of caspase-2 was shown to reduce behavioral deficits such as performance on the rotarod and forced swim test in YAC128 mice but failed to prevent the reductions in brain weight and changes in volume of subregions of the brain associated with HTT pathology in YAC128 mice (9). The direct impact on HTT proteolysis at the caspase-2 site was not fully elucidated.
There do exist mouse models that express N93, N171, and N586 fragments in mice, all of which show deficits in survival and motor function (39, 42, 70). However, differences in expression levels and site of integration and promoters make a direct comparison of the fragment models problematic. Because proteolysis can occur in a cascade, the generation of a particular fragment that is more toxic and generated after a particular initial cleavage event may not be generated at the same levels as other fragments. Therefore, abolition of cleavage sites does not directly compare the effect of fragments expressed at equal levels of expression on neuropathology, behavior, biochemistry, and life span. To enable comparison of relative toxicity of fragments in vivo, we produced a series of mouse models harboring a single integration of various N-terminal fragments at the ROSA26 locus that express similar levels of these fragments. As a result, direct comparison of the relative toxicity of these fragments in vivo is possible. The most toxic of the fragments tested is N171, which reduces life span of the mice to 100 days. This life span phenotype is similar to that of R6/2 mice, which express exon 1 of HTT (40), and it is similar to the phenotype of line 77 of the N171–82Q mice, which were initially reported to live for ∼2.5 months (42). By contrast, other FL-HTT mouse models live for nearly normal life spans and measurable behavioral deficits occur after 2–4 months of age (71).
The four fragments that are produced by known proteases, N463 (calpains-1,2), N536 (calpains-1,2), N552 (caspase-2), and N586 (capase-6), all resulted in life spans that are shorter than is typical for C57BL/6 mice (JAX data), as well as deficits in motor performance measured by rotarod and exploratory rearing behavior in the open field test. Therefore, it is clear that all fragments tested are sufficient to cause neurodegeneration and disease progression in vivo. However, there are significant differences in life span and motor phenotypes between these mice. Mice expressing the caspase-2 fragment, N552-Q148, displayed a significantly reduced life span relative to the N463-Q148, N536-Q148, and N586-Q148 mice, as well as accelerated deficits in the motor behaviors mentioned above. Therefore, we conclude that the N552-Q148 fragment is inherently more toxic than the other three fragments studied, although less toxic than the N171-Q148 fragment.
We also evaluated the proteolysis of the N463-Q148, N536-Q148, N552-Q148, and N586-Q148 mice. We found they all generated similarly sized fragments with the prominent proteolysis product of 52-kDa. Notably, this fragment did not contain the N terminus perhaps suggesting further processing of the protein at both the C and N terminus of the protein. The putative protease of this fragment remains unknown; but recent work has identified the cause of the exon 1 fragment as an aberrant splicing event (19, 52). However, it should be noted that our transgenic mouse models produce a product containing less than 120 amino acids; but as they are derived from cDNA, they do not contain the introns that could cause an aberrant splicing event. This may suggest that both proteolysis and aberrant splicing can produce smaller fragments (less than 120 amino acids). We did not detect differences in the levels of the 52-kDa fragment in the different calpain/caspase fragment models, and therefore, it is not likely that the accelerated phenotype of the N552-Q148 mice is a result of this smaller fragment. However, it is likely to contribute to disease progression of all the mice and increases in levels with age. Our studies do not address the cascade of proteolysis events that result in the formation of the fragments from full-length HTT protein. This could occur by cascade of sequential proteolytic events or simultaneously. It is noteworthy that each of caspase/calpain fragment models appeared to generate N-terminal fragments with the same pattern suggesting the same processing occurs across the models.
To understand the potential mechanisms for the accelerated phenotype of the N552-Q148 mice, we hypothesized that the protein may have a distinct HTT interaction network. Consistent with this, the N552-Q148 fragment physically interacts more readily with the FL-HTT than the N586-Q148 fragment mice at 4.5 months of age. Whether this interaction of fragments with the full-length protein is polyQ-dependent or -independent remains an open question. This interaction also correlated with a disruption of the native wild-type HTT complex as detected by two-dimensional BN-PAGE analysis. Additionally, IP of HTT in N586-Q148 mice shows a stronger physical interaction with the chaperone HSP86 relative to the wild-type or N552-Q148 mice. We have presented evidence that HSP86 overexpression can protect striatal cells overexpressing either N552 or N586 fragments; therefore, this increased interaction with the chaperone may be one mechanism by which cells protect against the detrimental effects of HTT N-terminal fragments and a mechanism that seems to be less effective in the N552-Q148 mice. Interestingly, the expression of the HSP86 affects the levels of overexpressed fragments and not the FL-HTT protein, suggesting selectivity in the HSP86 mechanism.
Identification of HSP86 as a protective factor in the N586 fragment-expressing mice points to a novel target in HD. Previous studies have tested inhibitors of HSP90 in R6/2 mice, based on the ability of these inhibitors to reduce mutant HTT protein. However, these studies failed to identify a long term benefit of HSP90 inhibition (72). We propose that although HSP86 is capable of stabilizing soluble HTT, it is capable of maintaining it in a form that is less detrimental than the form that the protein takes in its absence. This is likely due to a unique conformation of the N552-Q148 protein. In a related neurodegenerative disease, Parkinson disease, HSP90 has been shown to prevent the aggregation of the pathological α-synuclein A53T, and this is accomplished by forming a strong complex with the transiently populated oligomeric α-synuclein A53T species (73). HSP90 family members have a limited number of client proteins, which they are capable of stabilizing. Inhibition of HSP90 leads to loss of this chaperone function, turnover of clients, and up-regulation of other heat shock proteins such as HSP70 via HSF1 activation (74). Both wild-type and mutant HTT are clients of HSP86, and it has been proposed as a target to reduce mutant HTT levels in vivo. Our results may aid in explaining these observations. Although inhibition of HSP90 leads to rapid and significant reduction of overall HTT levels, HSP86 seems to be sufficient to significantly reduce the toxicity of mutant HTT fragments even as it increases levels assessed by Western blot. This may indicate the HTT fragments are misfolded, but not the FL-HTT, because the overexpression of HSP86 appears to effect the levels of fragments and not the FL-HTT. Given the paradoxical lack of increase in FL-HTT levels after transient HSP86 overexpression and strong reduction in FL-HTT upon 24 h inhibition with 17-DMAG, we propose that HSP90 proteins are actively interacting with HTT and is necessary to maintain its stability, such that loss of this chaperone activity is sufficient to reduce protein levels by 2-fold in 24 h. Consistent with our studies, inhibition of HSP90 reduces full-length mutant HTT levels in HdhQ111/Q111 striatal cells by a similar amount (75).
In conclusion, we found the transgenic mice N171-Q148 and N552-Q148 display a significantly accelerated phenotype and shortened life span when compared with N463-Q148, N536-Q148, and N586-Q148 transgenic mice. We found that the accelerated phenotype was due to an altered HTT protein interactions/complexes that accumulate with age. We found evidence for altered HTT complexes in caspase-2 fragment transgenic mice (N552-Q148) and a stronger interaction with the endogenous HTT protein. All fragment mice had accelerated onset of motor and behavioral phenotypes relative to WT mice as well as a reduced life span. Finally, we identified HSP90AA1 (HSP86) as a protein interactor that may modulate the distinct neurotoxicity of the caspase-2 fragment (N552-Q148) when compared with the caspase-6 fragment (N586-Q148). It is known that overexpression of the wild-type protein has some protective properties in vivo, and several groups have reported in culture models that the fragments of HTT expressed with normal polyQ length are neuroprotective or have no effect (5, 18, 21, 76–80). Our studies do address the impact of expressing normal HTT fragments that may affect the mouse phenotypes in vivo or how the polyQ-expanded HTT fragments affect normal HTT function. Future work will evaluate the localization of the various fragments as their localization pattern and different interaction domains may reflect on the mechanisms of neuropathogenesis.
Author Contributions
R. O., F. D., J. H., A. B., K. L. R., N. Z., K. Z., A. W., B. S., S. C., L. M. E. carried out experiments. A. W., R. O., N. Z., B. L., B. W. G., and L. M. E. designed experiments. R. O., F. D., J. H., A. B., K. L. R., N. Z., K. Z., A. W., B. S., S. C., and L. M. E. performed analysis of the data sets. R. O., S. K., and L. M. E. wrote the paper. All authors edited and contributed to the manuscript preparation. S. K. and L. M. E. conceived the study.
This work was supported, in whole or in part, by National Institutes of Health Grant T32 AG000266 (to L. M. E., R. O., and K. L. R.), Training Grant F32 NS080551 (to R. O.), and NCRR Shared Instrumentation Program Grant 1S10 OD016281 for TripleTOF 5600 (to B. W. G.). This work was also supported by CHDI (to L. M. E.). The authors declare that they have no conflicts of interest with the contents of this article.
- HD
- Huntington disease
- HTT
- huntingtin
- ANOVA
- analysis of variance
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- BN-PAGE
- blue native PAGE
- F
- forward
- R
- reverse
- coIP
- coimmunoprecipitation
- IP
- immunoprecipitation
- Z
- benzyloxycarbonyl
- polyQ
- polyglutamine
- FL-HTT
- full-length HTT
- TR-FRET
- time-resolved Forster energy transfer
- TPER
- tissue protein extraction reagent
- IHC
- immunohistochemistry
- 17-DMAG
- 17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride
- mESC
- mouse embryonic stem cell.
References
- 1. The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Kiyama H., Seto-Ohshima A., Emson P. C. (1990) Calbindin D28K as a marker for the degeneration of the striatonigral pathway in Huntington's disease. Brain Res. 525, 209–214 [DOI] [PubMed] [Google Scholar]
- 3. Hedreen J. C., Peyser C. E., Folstein S. E., Ross C. A. (1991) Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci. Lett. 133, 257–261 [DOI] [PubMed] [Google Scholar]
- 4. Cardoso F. (2014) Differential diagnosis of Huntington's disease: what the clinician should know. Neurodegener. Dis. Manag. 4, 67–72 [DOI] [PubMed] [Google Scholar]
- 5. Hermel E., Gafni J., Propp S. S., Leavitt B. R., Wellington C. L., Young J. E., Hackam A. S., Logvinova A. V., Peel A. L., Chen S. F., Hook V., Singaraja R., Krajewski S., Goldsmith P. C., Ellerby H. M., et al. (2004) Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ. 11, 424–438 [DOI] [PubMed] [Google Scholar]
- 6. Mende-Mueller L. M., Toneff T., Hwang S. R., Chesselet M. F., Hook V. Y. (2001) Tissue-specific proteolysis of Huntingtin (htt) in human brain: evidence of enhanced levels of N- and C-terminal htt fragments in Huntington's disease striatum. J. Neurosci. 21, 1830–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gafni J., Papanikolaou T., Degiacomo F., Holcomb J., Chen S., Menalled L., Kudwa A., Fitzpatrick J., Miller S., Ramboz S., Tuunanen P. I., Lehtimäki K. K., Yang X. W., Park L., Kwak S., et al. (2012) Caspase-6 activity in a BACHD mouse modulates steady-state levels of mutant huntingtin protein but is not necessary for production of a 586-amino acid proteolytic fragment. J. Neurosci. 32, 7454–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller J. P., Holcomb J., Al-Ramahi I., de Haro M., Gafni J., Zhang N., Kim E., Sanhueza M., Torcassi C., Kwak S., Botas J., Hughes R. E., Ellerby L. M. (2010) Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington's disease. Neuron 67, 199–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carroll J. B., Southwell A. L., Graham R. K., Lerch J. P., Ehrnhoefer D. E., Cao L. P., Zhang W. N., Deng Y., Bissada N., Henkelman R. M., Hayden M. R. (2011) Mice lacking caspase-2 are protected from behavioral changes, but not pathology, in the YAC128 model of Huntington disease. Mol. Neurodegener. 6, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Graham R. K., Deng Y., Carroll J., Vaid K., Cowan C., Pouladi M. A., Metzler M., Bissada N., Wang L., Faull R. L., Gray M., Yang X. W., Raymond L. A., Hayden M. R. (2010) Cleavage at the 586 amino acid caspase-6 site in mutant huntingtin influences caspase-6 activation in vivo. J. Neurosci. 30, 15019–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Warby S. C., Doty C. N., Graham R. K., Carroll J. B., Yang Y. Z., Singaraja R. R., Overall C. M., Hayden M. R. (2008) Activated caspase-6 and caspase-6-cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum. Mol. Genet. 17, 2390–2404 [DOI] [PubMed] [Google Scholar]
- 12. Graham R. K., Deng Y., Slow E. J., Haigh B., Bissada N., Lu G., Pearson J., Shehadeh J., Bertram L., Murphy Z., Warby S. C., Doty C. N., Roy S., Wellington C. L., Leavitt B. R., et al. (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125, 1179–1191 [DOI] [PubMed] [Google Scholar]
- 13. Wellington C. L., Ellerby L. M., Gutekunst C. A., Rogers D., Warby S., Graham R. K., Loubser O., van Raamsdonk J., Singaraja R., Yang Y. Z., Gafni J., Bredesen D., Hersch S. M., Leavitt B. R., Roy S., et al. (2002) Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J. Neurosci. 22, 7862–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wellington C. L., Ellerby L. M., Hackam A. S., Margolis R. L., Trifiro M. A., Singaraja R., McCutcheon K., Salvesen G. S., Propp S. S., Bromm M., Rowland K. J., Zhang T., Rasper D., Roy S., Thornberry N., et al. (1998) Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem. 273, 9158–9167 [DOI] [PubMed] [Google Scholar]
- 15. Bhat K. P., Yan S., Wang C. E., Li S., Li X. J. (2014) Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc. Natl. Acad. Sci. U.S.A. 111, 5706–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang C. E., Tydlacka S., Orr A. L., Yang S. H., Graham R. K., Hayden M. R., Li S., Chan A. W., Li X. J. (2008) Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum. Mol. Genet. 17, 2738–2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 [DOI] [PubMed] [Google Scholar]
- 18. Gafni J., Hermel E., Young J. E., Wellington C. L., Hayden M. R., Ellerby L. M. (2004) Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 279, 20211–20220 [DOI] [PubMed] [Google Scholar]
- 19. Gipson T. A., Neueder A., Wexler N. S., Bates G. P., Housman D. (2013) Aberrantly spliced HTT, a new player in Huntington's disease pathogenesis. RNA Biol. 10, 1647–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Landles C., Sathasivam K., Weiss A., Woodman B., Moffitt H., Finkbeiner S., Sun B., Gafni J., Ellerby L. M., Trottier Y., Richards W. G., Osmand A., Paganetti P., Bates G. P. (2010) Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 285, 8808–8823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wellington C. L., Singaraja R., Ellerby L., Savill J., Roy S., Leavitt B., Cattaneo E., Hackam A., Sharp A., Thornberry N., Nicholson D. W., Bredesen D. E., Hayden M. R. (2000) Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J. Biol. Chem. 275, 19831–19838 [DOI] [PubMed] [Google Scholar]
- 22. Xia J., Lee D. H., Taylor J., Vandelft M., Truant R. (2003) Huntingtin contains a highly conserved nuclear export signal. Hum. Mol. Genet. 12, 1393–1403 [DOI] [PubMed] [Google Scholar]
- 23. Zheng Z., Li A., Holmes B. B., Marasa J. C., Diamond M. I. (2013) An N-terminal nuclear export signal regulates trafficking and aggregation of Huntingtin (Htt) protein exon 1. J. Biol. Chem. 288, 6063–6071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maiuri T., Woloshansky T., Xia J., Truant R. (2013) The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal. Hum. Mol. Genet. 22, 1383–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schilling B., Gafni J., Torcassi C., Cong X., Row R. H., LaFevre-Bernt M. A., Cusack M. P., Ratovitski T., Hirschhorn R., Ross C. A., Gibson B. W., Ellerby L. M. (2006) Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J. Biol. Chem. 281, 23686–23697 [DOI] [PubMed] [Google Scholar]
- 26. Humbert S., Bryson E. A., Cordelières F. P., Connors N. C., Datta S. R., Finkbeiner S., Greenberg M. E., Saudou F. (2002) The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2, 831–837 [DOI] [PubMed] [Google Scholar]
- 27. Anne S. L., Saudou F., Humbert S. (2007) Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 27, 7318–7328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luo S., Vacher C., Davies J. E., Rubinsztein D. C. (2005) Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J. Cell Biol. 169, 647–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Havel L. S., Wang C. E., Wade B., Huang B., Li S., Li X. J. (2011) Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum. Mol. Genet. 20, 1424–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson L. M., Aiken C. T., Kaltenbach L. S., Agrawal N., Illes K., Khoshnan A., Martinez-Vincente M., Arrasate M., O'Rourke J. G., Khashwji H., Lukacsovich T., Zhu Y. Z., Lau A. L., Massey A., Hayden M. R., et al. (2009) IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187, 1083–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Pardo A., Maglione V., Alpaugh M., Horkey M., Atwal R. S., Sassone J., Ciammola A., Steffan J. S., Fouad K., Truant R., Sipione S. (2012) Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci. U.S.A. 109, 3528–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Atwal R. S., Desmond C. R., Caron N., Maiuri T., Xia J., Sipione S., Truant R. (2011) Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 7, 453–460 [DOI] [PubMed] [Google Scholar]
- 33. Gu X., Greiner E. R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J. S., Thompson L. M., Wetzel R., Yang X. W. (2009) Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cong X., Held J. M., DeGiacomo F., Bonner A., Chen J. M., Schilling B., Czerwieniec G. A., Gibson B. W., Ellerby L. M. (2011) Mass spectrometric identification of novel lysine acetylation sites in huntingtin. Mol. Cell. Proteomics 10, M111.009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jeong H., Then F., Melia T. J., Jr., Mazzulli J. R., Cui L., Savas J. N., Voisine C., Paganetti P., Tanese N., Hart A. C., Yamamoto A., Krainc D. (2009) Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137, 60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gafni J., Ellerby L. M. (2002) Calpain activation in Huntington's disease. J. Neurosci. 22, 4842–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Toneff T., Mende-Mueller L., Wu Y., Hwang S. R., Bundey R., Thompson L. M., Chesselet M. F., Hook V. (2002) Comparison of huntingtin proteolytic fragments in human lymphoblast cell lines and human brain. J. Neurochem. 82, 84–92 [DOI] [PubMed] [Google Scholar]
- 38. Tebbenkamp A. T., Green C., Xu G., Denovan-Wright E. M., Rising A. C., Fromholt S. E., Brown H. H., Swing D., Mandel R. J., Tessarollo L., Borchelt D. R. (2011) Transgenic mice expressing caspase-6-derived N-terminal fragments of mutant huntingtin develop neurologic abnormalities with predominant cytoplasmic inclusion pathology composed largely of a smaller proteolytic derivative. Hum. Mol. Genet. 20, 2770–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Waldron-Roby E., Ratovitski T., Wang X., Jiang M., Watkin E., Arbez N., Graham R. K., Hayden M. R., Hou Z., Mori S., Swing D., Pletnikov M., Duan W., Tessarollo L., Ross C. A. (2012) Transgenic mouse model expressing the caspase 6 fragment of mutant huntingtin. J. Neurosci. 32, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bates G. P., Mangiarini L., Mahal A., Davies S. W. (1997) Transgenic models of Huntington's disease. Hum. Mol. Genet. 6, 1633–1637 [DOI] [PubMed] [Google Scholar]
- 41. Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S. W., Bates G. P. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 [DOI] [PubMed] [Google Scholar]
- 42. Schilling G., Becher M. W., Sharp A. H., Jinnah H. A., Duan K., Kotzuk J. A., Slunt H. H., Ratovitski T., Cooper J. K., Jenkins N. A., Copeland N. G., Price D. L., Ross C. A., Borchelt D. R. (1999) Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 8, 397–407 [DOI] [PubMed] [Google Scholar]
- 43. Wheeler V. C., Auerbach W., White J. K., Srinidhi J., Auerbach A., Ryan A., Duyao M. P., Vrbanac V., Weaver M., Gusella J. F., Joyner A. L., MacDonald M. E. (1999) Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum. Mol. Genet. 8, 115–122 [DOI] [PubMed] [Google Scholar]
- 44. Marcellin D., Abramowski D., Young D., Richter J., Weiss A., Marcel A., Maassen J., Kauffmann M., Bibel M., Shimshek D. R., Faull R. L., Bates G. P., Kuhn R. R., Van der Putten P. H., Schmid P., Lotz G. P. (2012) Fragments of HdhQ150 mutant huntingtin form a soluble oligomer pool that declines with aggregate deposition upon aging. PloS One 7, e44457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mielcarek M., Landles C., Weiss A., Bradaia A., Seredenina T., Inuabasi L., Osborne G. F., Wadel K., Touller C., Butler R., Robertson J., Franklin S. A., Smith D. L., Park L., Marks P. A., et al. (2013) HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 11, e1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weiss A., Abramowski D., Bibel M., Bodner R., Chopra V., DiFiglia M., Fox J., Kegel K., Klein C., Grueninger S., Hersch S., Housman D., Régulier E., Rosas H. D., Stefani M., et al. (2009) Single-step detection of mutant huntingtin in animal and human tissues: a bioassay for Huntington's disease. Anal. Biochem. 395, 8–15 [DOI] [PubMed] [Google Scholar]
- 47. Wiśniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 48. Schilling B., Rardin M. J., MacLean B. X., Zawadzka A. M., Frewen B. E., Cusack M. P., Sorensen D. J., Bereman M. S., Jing E., Wu C. C., Verdin E., Kahn C. R., Maccoss M. J., Gibson B. W. (2012) Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in Skyline: application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007) The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 50. Ratovitski T., Gucek M., Jiang H., Chighladze E., Waldron E., D'Ambola J., Hou Z., Liang Y., Poirier M. A., Hirschhorn R. R., Graham R., Hayden M. R., Cole R. N., Ross C. A. (2009) Mutant huntingtin N-terminal fragments of specific size mediate aggregation and toxicity in neuronal cells. J. Biol. Chem. 284, 10855–10867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lunkes A., Lindenberg K. S., Ben-Haïem L., Weber C., Devys D., Landwehrmeyer G. B., Mandel J. L., Trottier Y. (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 10, 259–269 [DOI] [PubMed] [Google Scholar]
- 52. Sathasivam K., Neueder A., Gipson T. A., Landles C., Benjamin A. C., Bondulich M. K., Smith D. L., Faull R. L., Roos R. A., Howland D., Detloff P. J., Housman D. E., Bates G. P. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 110, 2366–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Harrison D. E. (2008) Baseline life span data: twelve strains of commonly used laboratory mice (Study 2) [Google Scholar]
- 54. Prut L., Belzung C. (2003) The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur. J. Pharmacol. 463, 3–33 [DOI] [PubMed] [Google Scholar]
- 55. Young D., Mayer F., Vidotto N., Schweizer T., Berth R., Abramowski D., Shimshek D. R., van der Putten P. H., Schmid P. (2013) Mutant huntingtin gene-dose impacts on aggregate deposition, DARPP32 expression and neuroinflammation in HdhQ150 mice. PLoS One 8, e75108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heikkinen T., Lehtimäki K., Vartiainen N., Puoliväli J., Hendricks S. J., Glaser J. R., Bradaia A., Wadel K., Touller C., Kontkanen O., Yrjänheikki J. M., Buisson B., Howland D., Beaumont V., Munoz-Sanjuan I., Park L. C. (2012) Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington's disease. PLoS One 7, e50717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Menalled L. B., Kudwa A. E., Miller S., Fitzpatrick J., Watson-Johnson J., Keating N., Ruiz M., Mushlin R., Alosio W., McConnell K., Connor D., Murphy C., Oakeshott S., Kwan M., Beltran J., et al. (2012) Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS One 7, e49838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stanek L. M., Yang W., Angus S., Sardi P. S., Hayden M. R., Hung G. H., Bennett C. F., Cheng S. H., Shihabuddin L. S. (2013) Antisense oligonucleotide-mediated correction of transcriptional dysregulation is correlated with behavioral benefits in the YAC128 mouse model of Huntington's disease. J. Huntington's Dis. 2, 217–228 [DOI] [PubMed] [Google Scholar]
- 59. Jia H., Pallos J., Jacques V., Lau A., Tang B., Cooper A., Syed A., Purcell J., Chen Y., Sharma S., Sangrey G. R., Darnell S. B., Plasterer H., Sadri-Vakili G., Gottesfeld J. M., et al. (2012) Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington's disease. Neurobiol. Dis. 46, 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hickey M. A., Zhu C., Medvedeva V., Lerner R. P., Patassini S., Franich N. R., Maiti P., Frautschy S. A., Zeitlin S., Levine M. S., Chesselet M. F. (2012) Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington's disease. Mol. Neurodegener. 7, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pouladi M. A., Stanek L. M., Xie Y., Franciosi S., Southwell A. L., Deng Y., Butland S., Zhang W., Cheng S. H., Shihabuddin L. S., Hayden M. R. (2012) Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum. Mol. Genet. 21, 2219–2232 [DOI] [PubMed] [Google Scholar]
- 62. Brown T. B., Bogush A. I., Ehrlich M. E. (2008) Neocortical expression of mutant huntingtin is not required for alterations in striatal gene expression or motor dysfunction in a transgenic mouse. Hum. Mol. Genet. 17, 3095–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ratovitski T., Chighladze E., Waldron E., Hirschhorn R. R., Ross C. A. (2011) Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant huntingtin. J. Biol. Chem. 286, 12578–12589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cleary J. D., Ranum L. P. (2013) Repeat-associated non-ATG (RAN) translation in neurological disease. Hum. Mol. Genet. 22, R45–R51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Reddy K., Pearson C. E. (2013) RAN translation: fragile X in the running. Neuron 78, 405–408 [DOI] [PubMed] [Google Scholar]
- 66. Kaltenbach L. S., Romero E., Becklin R. R., Chettier R., Bell R., Phansalkar A., Strand A., Torcassi C., Savage J., Hurlburt A., Cha G. H., Ukani L., Chepanoske C. L., Zhen Y., Sahasrabudhe S., et al. (2007) Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 3, e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tourette C., Li B., Bell R., O'Hare S., Kaltenbach L. S., Mooney S. D., Hughes R. E. (2014) A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J. Biol. Chem. 289, 6709–6726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shirasaki D. I., Greiner E. R., Al-Ramahi I., Gray M., Boontheung P., Geschwind D. H., Botas J., Coppola G., Horvath S., Loo J. A., Yang X. W. (2012) Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 75, 41–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Culver B. P., Savas J. N., Park S. K., Choi J. H., Zheng S., Zeitlin S. O., Yates J. R., 3rd, Tanese N. (2012) Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J. Biol. Chem. 287, 21599–21614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hockly E., Woodman B., Mahal A., Lewis C. M., Bates G. (2003) Standardization and statistical approaches to therapeutic trials in the R6/2 mouse. Brain Res. Bull. 61, 469–479 [DOI] [PubMed] [Google Scholar]
- 71. William Yang X., Gray M. (2011) in Neurobiology of Huntington's Disease: Applications to Drug Discovery (Lo D. C., Hughes R. E., eds) pp. 1–33, CRC Press, Inc., Boca Raton, FL: [PubMed] [Google Scholar]
- 72. Labbadia J., Cunliffe H., Weiss A., Katsyuba E., Sathasivam K., Seredenina T., Woodman B., Moussaoui S., Frentzel S., Luthi-Carter R., Paganetti P., Bates G. P. (2011) Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Invest. 121, 3306–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Daturpalli S., Waudby C. A., Meehan S., Jackson S. E. (2013) Hsp90 inhibits alpha-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 425, 4614–4628 [DOI] [PubMed] [Google Scholar]
- 74. Pratt W. B., Morishima Y., Gestwicki J. E., Lieberman A. P., Osawa Y. (2014) A model in which heat shock protein 90 targets protein-folding clefts: Rationale for a new approach to neuroprotective treatment of protein folding diseases. Exp. Biol. Med. 239, 1405–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Baldo B., Weiss A., Parker C. N., Bibel M., Paganetti P., Kaupmann K. (2012) A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. J. Biol. Chem. 287, 1406–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pouladi M. A., Xie Y., Skotte N. H., Ehrnhoefer D. E., Graham R. K., Kim J. E., Bissada N., Yang X. W., Paganetti P., Friedlander R. M., Leavitt B. R., Hayden M. R. (2010) Full-length huntingtin levels modulate body weight by influencing insulin-like growth factor 1 expression. Hum. Mol. Genet. 19, 1528–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang Y., Leavitt B. R., van Raamsdonk J. M., Dragatsis I., Goldowitz D., MacDonald M. E., Hayden M. R., Friedlander R. M. (2006) Huntingtin inhibits caspase-3 activation. EMBO J. 25, 5896–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang Y., Li M., Drozda M., Chen M., Ren S., Mejia Sanchez R. O., Leavitt B. R., Cattaneo E., Ferrante R. J., Hayden M. R., Friedlander R. M. (2003) Depletion of wild-type huntingtin in mouse models of neurologic diseases. J. Neurochem. 87, 101–106 [DOI] [PubMed] [Google Scholar]
- 79. Rigamonti D., Sipione S., Goffredo D., Zuccato C., Fossale E., Cattaneo E. (2001) Huntingtin's neuroprotective activity occurs via inhibition of procaspase-9 processing. J. Biol. Chem. 276, 14545–14548 [DOI] [PubMed] [Google Scholar]
- 80. Rigamonti D., Bauer J. H., De-Fraja C., Conti L., Sipione S., Sciorati C., Clementi E., Hackam A., Hayden M. R., Li Y., Cooper J. K., Ross C. A., Govoni S., Vincenz C., Cattaneo E. (2000) Wild-type huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 20, 3705–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 82. Iuchi S., Hoffner G., Verbeke P., Djian P., Green H. (2003) Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc. Natl. Acad. Sci. U.S.A. 100, 2409–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]