

Background: Interleukin-35 is a novel inhibitory cytokine.
Results: Interleukin-35 inhibits vascular endothelial cell activation by suppressing MAPK-AP1-mediated VACM-1 expression in LPS-induced acute inflammation.
Conclusion: Interleukin-35 suppresses acute vascular endothelium response.
Significance: Interleukin-35 may be an attractive reagent for anti-inflammatory therapy.
Keywords: cytokine, endothelial cell, inflammation, lipopolysaccharide (LPS), sepsis, endothelial cell activation, interleukin-35, vascular cell adhesion molecule-1 (VCAM-1), vascular inflammation
Abstract
Vascular response is an essential pathological mechanism underlying various inflammatory diseases. This study determines whether IL-35, a novel responsive anti-inflammatory cytokine, inhibits vascular response in acute inflammation. Using a mouse model of LPS-induced acute inflammation and plasma samples from sepsis patients, we found that IL-35 was induced in the plasma of mice after LPS injection as well as in the plasma of sepsis patients. In addition, IL-35 decreased LPS-induced proinflammatory cytokines and chemokines in the plasma of mice. Furthermore, IL-35 inhibited leukocyte adhesion to the endothelium in the vessels of lung and cremaster muscle and decreased the numbers of inflammatory cells in bronchoalveolar lavage fluid. Mechanistically, IL-35 inhibited the LPS-induced up-regulation of endothelial cell (EC) adhesion molecule VCAM-1 through IL-35 receptors gp130 and IL-12Rβ2 via inhibition of the MAPK-activator protein-1 (AP-1) signaling pathway. We also found that IL-27, which shares the EBI3 subunit with IL-35, promoted LPS-induced VCAM-1 in human aortic ECs and that EBI3-deficient mice had similar vascular response to LPS when compared with that of WT mice. These results demonstrated for the first time that inflammation-induced IL-35 inhibits LPS-induced EC activation by suppressing MAPK-AP1-mediated VCAM-1 expression and attenuates LPS-induced secretion of proinflammatory cytokines/chemokines. Our results provide insight into the control of vascular inflammation by IL-35 and suggest that IL-35 is an attractive novel therapeutic reagent for sepsis and cardiovascular diseases.
Introduction
Atherosclerosis is a chronic inflammatory disorder of the arteries that leads to cardiovascular diseases and cerebrovascular diseases, which are the leading causes of mortality and morbidity in the world. In addition, sepsis caused by bacterial infection, in which vascular pathological response also plays a critical role, strikes about 700,000 people annually (1, 2). Aside from eliciting acute infection, endotoxin at levels as low as 50 pg/ml constitutes a strong risk factor for the development of atherosclerosis (3), suggesting that LPS-induced vascular response is pathologically related to cardiovascular diseases. In the hope of identifying novel immunosuppressive mechanisms of vascular inflammation, our laboratory and others have reported that diminished CD4+CD25+ regulatory T cell (Treg)2 population via apoptosis promotes vascular inflammation (4–6). However, the underlying mechanisms of how Tregs inhibit vascular response during inflammatory response are still not fully understood.
After exposure to foreign pathogens and metabolite-related endogenous danger signals in the bloodstream (7), endothelial cells (ECs) lining the innermost layer of blood vessel are activated and initiate vascular inflammatory response, contributing to atherosclerosis development (8). EC activation is characterized by up-regulated expression of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), which mediate leukocyte adhesion to endothelial cells. Although Tregs can inhibit EC activation via cell-cell interaction (9), an important question remains concerning whether Tregs can suppress EC activation by secretion of soluble factors.
IL-35 is a newly identified anti-inflammatory cytokine of IL-12 heterodimeric cytokine family, which includes four other members, IL-12, IL-23, IL-27, and IL-27p28/IL-12p40 (10). IL-35 is formed by pairing IL-12α (also known as p35) and Epstein-Barr virus-induced gene 3 (EBI3). It shares the IL-12α subunit with IL-12, and shares the EBI3 subunit with IL-27. IL-35 is secreted by Tregs, regulatory B cells (Bregs) (11), dendritic cells (12), and to a lesser extent, by endothelial cells, smooth muscle cells, and monocytes (13). It can suppress several types of chronic inflammatory diseases such as inflammatory bowel disease (14–16). Mechanistically, IL-35 signals in the target cells by binding to heterodimeric (gp130-IL-12Rβ2) or homodimeric (gp130-gp130 or IL-12Rβ2-IL-12Rβ2) receptors to inhibit immunological responses (17–19). We have shown previously that in contrast to another immunosuppressive cytokine, TGF-β, IL-35 is not a constitutively expressed cytokine but rather a responsive cytokine induced by inflammatory stimuli (17). In addition, the genes encoding IL-35 subunits could also be induced in endothelial cells, smooth muscle cells, and monocytes after activation of proinflammatory stimuli (17). However, the effects of IL-35 in non-lymphocytes such as vascular endothelial cells are not known.
In this study, we hypothesized that IL-35 could inhibit acute vascular inflammation by suppressing EC activation. To examine this hypothesis, we studied the role of IL-35 in a commonly used, pathologically relevant murine sepsis model and in vitro LPS-treated human primary ECs. We found that IL-35 was increased in the plasma of WT mice after LPS challenge and that IL-35 was also elevated in the plasma samples from sepsis patients when compared with healthy controls. In addition, we found that IL-35 significantly reduced leukocyte adhesion to the endothelium in both lung and cremaster muscle vessels, and decreased leukocyte exodus into bronchoalveolar lavage fluid (BALF) and connective tissue. We further demonstrated that IL-35 suppressed LPS-induced leukocyte adhesion by inhibiting MAPK-AP-1-mediated up-regulation of EC adhesion molecule VCAM-1 in human aortic ECs (HAECs) and mouse aortic endothelium. Furthermore, we found that IL-35 suppressed LPS-induced secretion of pro-inflammatory cytokines and chemokines in the plasma of mice. Thus, our findings suggest that IL-35 inhibits acute inflammatory response via the suppression of vascular EC activation, which implies a therapeutic potential for IL-35 in sepsis and cardiovascular diseases.
Experimental Procedures
Mice and Human Plasma Samples
Wild type C57BL/6 mice and EBI3−/− mice (B6.129X1-Ebi3/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). All the mice were kept under pathogen-free conditions in a temperature-controlled environment. 16-week-old male mice were used for all the experiments. The protocols for all experiments were approved by the Temple University Institutional Animal Care and Use Committee (IACUC), which confirmed to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. In accordance with a protocol approved by the Institutional Review Board at Temple University, plasma samples of de-identified patients and healthy controls were obtained from BioreclamationIVT (East Meadow, NY), which confirmed the Declaration of Helsinki and preparation of informed consent forms.
LPS-induced Acute Inflammation Mouse Model and Intravital Microscopy
Mice were intraperitoneally injected with LPS (20 μg/g of body weight, Sigma, Escherichia coli, O127:B8) or PBS alone, with or without pretreatment of recombinant IL-35 (rIL-35) (0.3 μg/g of body weight or 70 pmol, Enzo Life Sciences, Farmingdale, NY) for 30 min. The dosage of IL-35 is comparable with one previous study, which investigated the effects of anti-inflammatory cytokine IL-10 in a murine sepsis model (single intraperitoneal injection of 54 pmol of IL-10) (20). Four hours after LPS injection, the mice were anesthetized with a mixture of ketamine (75 mg/kg ketamine, 0.74 mg/kg acepromazine, 1.48 mg/kg xylazine) and then subjected to leukocyte adhesion analysis using intravital microscopy. Cremaster muscles of the mice were prepared on a Plexiglas microscope stage and viewed with the Olympus BX51 microscope (Tokyo, Japan). The image was captured with a video camera (Photometrics QuantEM, Tucson AZ) and recorded for 1 min with video acquisition software (MetaMorph, Molecular Devices, Sunnyvale, CA). Post-capillary venules within the following criteria were included in the study: diameter 40–50 μm, velocity 2.0–4.9 mm/s, and shear rates 200–700.
Mouse Plasma Cytokine Array
Mouse plasma cytokine antibody array analyses were carried out according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
BALF and Cell Counting
Four hours after treatment with LPS in combination with or without rIL-35, BALF was collected by cannulating the upper part of the trachea and lavaging with 1.5 ml of PBS. The fluid recovery rate was more than 90%. Total cells were then counted with a Scepter handheld automated cell counter (EMD Millipore).
Immunohistochemistry
5-micrometer-thick fixed paraffin-embedded lung sections were prepared from WT and EBI3−/− mice at 4 h after treatment with LPS in combination with or without rIL-35. H&E staining was applied for histochemical analysis. Myeloperoxidase (MPO) staining was applied according to the manufacturer's instructions (HRP-DAB (where DAB is 3,3′-diaminobenzidine) system, R&D Systems).
Leukocyte Adhesion Assay
HAECs (Lonza, Allendale, NJ) were cultured in gelatin-coated dishes. rIL-35 (10, 20, and 40 ng/ml) was then added 18 h before treating cells with LPS (3 ng/ml) for 4 h. The HAECs were then rinsed, and 1 × 105/well (48-well dish) calcein AM (Invitrogen)-labeled human monocytic THP1 cells (ATCC, Manassas, VA) or 5 × 105/well (24-well dish) calcein AM-labeled human peripheral blood mononuclear cells (PBMCs) (Temple University Thrombosis Research Center with a protocol approved by the Institutional Review Board at Temple University) were added and incubated at 37 °C for 20 min. Finally, the unbound cells were removed, and fluorescence density was determined with a fluorometer.
Flow Cytometry Analysis
HAECs were pretreated with rIL-35 (40 ng/ml) for 18 h, and then the cells were treated with LPS (10 ng/ml) for 6 h (ICAM-1) or 16 h (VCAM-1). The expression of these surface proteins was determined by the FACSCalibur (BD Biosciences). For study of IL-27, rIL-27 (R&D Systems, 40 ng/ml) was added with LPS (10 ng/ml). FITC-mouse anti-human CD108 and APC (allophycocyanin) mouse anti-human CD54 were obtained from BD Biosciences.
Electrophoretic Mobility Shift Assay
HAECs cultured in 10-cm dishes were pretreated with rIL-35 (indicated dose) for 18 h. Next, the cells were treated with LPS (100 ng/ml) for 1.5 h. The assay was then carried out according to the manufacturer's instructions (LI-COR Odyssey infrared EMSA kit, Lincoln, NE) using isolated nuclear proteins. IRDye 700-labeled consensus-binding sequences for transcription factors AP-1 and NF-κB oligonucleotides were purchased from LI-COR Inc. The consensus-binding sequence for AP-1 is 5′-CGC TTG ATG ACT CAG CCC GAA-3′, whereas the consensus-binding sequence for NF-κB is 5′-AGT TGA GGG GAC TTT CCC AGG C-3′. The consensus competitor oligonucleotides and mutant competitor oligonucleotides were purchased from Santa Cruz Biotechnology (Dallas, TX). The mutant competitor sequence for AP-1 is 5′-CGC TTG ATG ACT TGG CCG GAA-3′and the mutant competitor sequence for NF-κB is 5′-AGT TGA GGC GAC TTT CCC AGG-3′. 5 μg of nuclear extracts were used in binding reaction. The antibodies used in the supershift assay were from Santa Cruz Biotechnology: c-Fos (sc-253X), phospho-c-Jun (sc-822X), p65 (sc-372X), and p50 (sc-7178X).
Antibody Blocking Assay
HAECs were preincubated with anti-gp130 (0.1 μg/ml), anti-IL-12Rβ2(0.005 μg/ml), mouse IgG1 (0.1 μg/ml), and goat IgG (0.005 μg/ml) for 1 h. rIL-35 was then added 18 h before treating cells with LPS (10 ng/ml) for 16 h. VCAM-1 expression was determined with fluorescence-labeled specific antibody analyzed using the FACSCalibur (BD Biosciences).
Western Blotting
Protein expression was detected by Western blotting using cell lysates (100 μg) from HAECs or fresh aortas. We used the following antibodies. Antibodies from Cell Signaling Technology (Danvers, MA) include anti-phospho-SAPK/JNK (where SAPK is stress-activated protein kinase) (Thr-183/Tyr-185), anti-SAPK/JNK, anti-phospho-p44/42MAPK (Erk1/2) (Thr-202/Tyr-204, 20G11), anti-p44/42MAPK, anti-phospho-p38MAPK (Thr-180/Tyr-182), anti-p38MAPK, phospho-IκBα (Ser-32, 14D4), anti-IκBα (L32A5), and anti-β-actin. Antibodies from Santa Cruz Biotechnology include anti-ICAM-1, anti-VCAM-1, and anti-gp130. Anti-12Rβ2 is from R&D Systems.
ELISA
Mouse IL-35 ELISA kit (USCN Life Science Inc., Atlanta, GA) was used to determine mouse plasma IL-35 levels. Mouse IL-27p28 Quantikine ELISA kit (R&D Systems) and mouse IL-12/IL-23 total p40 ELISA kit were used to determine mouse plasma p28 and p40 levels, respectively, according to the manufacturer's instructions.
Statistical Analysis
Data were analyzed using the Prism software (GraphPad, La Jolla, CA). The results were expressed as the means ± S.E. Statistical comparison of single parameters between two groups was performed by paired Student's t test. One-way analysis of variance was used to compare the means of multiple groups. Data were considered statistically significant if p was <0.05.
Results
Plasma Levels of IL-35 Are Induced in LPS-induced Murine Sepsis and Human Sepsis Patients
To determine the pathophysiological relevance of IL-35 in acute inflammatory vascular responses, we examined the plasma concentrations of IL-35 after LPS challenge in mice. We observed that intraperitoneal injection of LPS significantly increased plasma IL-35 levels from 18.7 ± 9.1 pg/ml at 0 h to 804.6 ± 103.3 pg/ml at 1.5 h, 800 pg/ml at 4 h, and 701.7 pg/ml at 24 h as determined by ELISA (Fig. 1, A and B). Because IL-35 shares its p35 subunit with IL-12 (18) and shares its EBI3 subunit with IL-27 (18), we also performed ELISAs for IL-12 (IL-12p40) and IL-27 (IL-27p28) as controls (Fig. 1, C and D) to verify that our ELISA results were specific for IL-35. The results showed that IL-35 levels at all four time points were statistically different from the levels of IL-12p40 and different from IL-27p28 levels after 4- and 24-h treatment (Fig. 1E). To further confirm the pathophysiological relevance of IL-35 in human, we also performed ELISA to detect IL-35 in the plasma of sepsis patients and healthy controls (Fig. 2). The result in Fig. 2A showed that IL-35 levels were also significantly increased to 31.7 ± 11.6 pg/ml in the plasma of patients with sepsis from 19.2 ± 12.2 pg/ml in that of healthy controls.
FIGURE 1.

Plasma levels of IL-35 are induced in mice with LPS-induced endotoxemia. A, schematic representation of LPS-induced endotoxemia model (LPS) with intraperitoneal (ip) injection (20 μg/g of body weight)). B–D, detection of IL-35, IL-12p40, and IL-27p28 by ELISAs in the plasma of wild type mice after LPS challenge after 0, 1.5, 4, and 24 h, n = 9–10 in each group. mIL, mouse IL. E, comparison of plasma levels of IL-35, IL-12p40, and IL-27p28 in each time point to further demonstrate the specificity of IL-35 detection. n = 9–10 in each group. The results are expressed as the means ± S.E. NS, not significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 2.
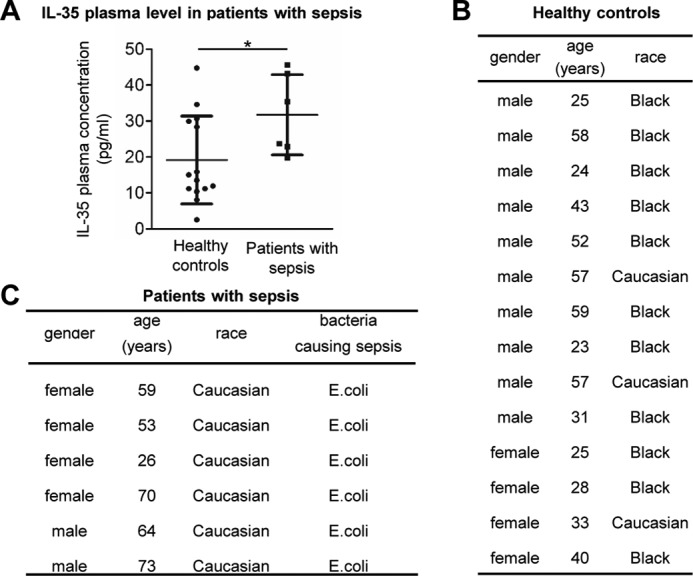
Plasma levels of IL-35 are induced in patients with sepsis. A, human IL-35 levels in the plasma samples from patients with sepsis (n = 6) and healthy controls (n = 14). C, general information about patients with sepsis. B, general information of healthy controls. The results are expressed as the means ± S.E. *, p < 0.05.
IL-35 Suppresses Pro-inflammatory Cytokine and Chemokine Secretion in the Plasma of Mice with Endotoxemia
To determine whether IL-35 could inhibit LPS-induced inflammatory responses in mice, we performed cytokine array analysis with mouse plasma samples collected from the mice challenged with LPS or the mice co-treated with LPS and rIL-35 (Fig. 3A). The result showed that LPS induced secretion of 14 out of 40 cytokines and chemokines in the plasma of mice. Upon rIL-35 co-treatment, the levels of 10 out of 14 LPS-induced proinflammatory cytokines and chemokines including IL-6, M-CSF, chemokine CCL1, CXCL-10, CXCL-1, CXCL-9, CCL-3, CCL-4, CXCL-2, and CCL5 were significantly decreased. In contrast, G-CSF, CCL-2, and tissue inhibitor of metalloproteinase-1 (TIMP-1) were not suppressed by IL-35 (Fig. 3, B and C). These results indicate that LPS increases plasma IL-35 secretion in response to acute inflammation, which correlates well with our previous study on the up-regulation of IL-35 mRNA transcripts in response to stimuli (17). We also noticed that LPS did not induce secretion of plasma IL-10 (Fig. 3A). Thus, our data further emphasize that LPS induces IL-35 instead of IL-10 and that IL-35 suppresses proinflammatory cytokines and chemokines in the plasma of mice with endotoxemia, suggesting that in LPS-induced inflammation, IL-35 plays more important roles than IL-10.
FIGURE 3.

IL-35 suppresses cytokine and chemokine expression in the plasma of wild type mice challenged with LPS. A, in the left panel, mouse cytokine/chemokine arrays were performed to detect the expression changes of 40 inflammatory cytokines/chemokines in the plasma from mice treated with LPS, LPS plus rIL-35, and WT control mice. Plasma samples in each group were pooled from three mice. In the right panel, the arrangement of the mouse cytokine/chemokine array was shown. Cytokines and chemokines that were induced by LPS are in bold. rIL-35-inhibited cytokines and chemokines are labeled with asterisks. B and C, quantification of cytokine and chemokine expressions. The variations of the manufacturer's designated positive control spots between each array were used to determine the confidence interval of nonspecific variations between samples. *, p < 0.05.
IL-35 Inhibits LPS-induced Leukocyte Adhesion to the Endothelium in the Mouse Cremaster Venules and Lung Vessels, and Reduces Trans-endothelial Exodus of Inflammatory Cells into BALF
We reasoned that because IL-35 inhibits LPS-induced secretion of proinflammatory cytokines and chemokines into plasma, IL-35 should be able to suppress leukocyte adhesion to ECs lining the vascular wall. Thus, we examined the effects of rIL-35 on LPS-induced leukocyte adhesion to the vascular wall in cremaster muscle vessels and lung vessels in mice. Using intravital microscopy, we found that LPS significantly increased leukocyte adhesion to the endothelium on the post-capillary venule wall of mouse cremaster muscle by 4.5-fold after 4 h of LPS treatment. More importantly, IL-35 significantly inhibited leukocyte adhesion to the endothelium on the post-capillary venule wall (Fig. 4, A and B, supplemental Movies 1–3). In addition, we performed histological analysis of the lungs collected from these mice and found that IL-35 significantly inhibited LPS-induced inflammatory cell adhesion to the lung microvascular ECs (Fig. 4C). Of note, the therapeutic dose of rIL-35 used in this study (0.3 μg/g) was lower than the therapeutic dose of the anti-inflammatory cytokine IL-10 (0.5–1 μg/g) used in a previous study (20). Moreover, using neutrophil-specific MPO staining, we determined whether IL-35 can suppress MPO+ neutrophil/granulocyte adhesion to the endothelium of lung vessels. The results showed that IL-35 significantly suppressed LPS-induced adherence of MPO+ inflammatory cells to the lung microvessel ECs and the alveolar wall of mice (Fig. 4D). Finally, we determined whether IL-35 co-treatment with LPS could decrease the exodus of LPS-induced inflammatory cells into BALF. The results in Fig. 4E showed that IL-35 significantly decreased LPS-induced inflammatory cell numbers in BALF, suggesting that IL-35 not only inhibits inflammatory cell adhesion to the endothelium but also suppresses trans-endothelium infiltration of inflammatory cells. Taken together, our results demonstrate that IL-35 inhibits EC activation as judged by decreasing inflammatory cell adhesion to ECs, lung inflammation scale, and inflammatory cell trans-endothelial infiltration into BALF.
FIGURE 4.

IL-35 inhibits LPS-induced leukocyte adhesion in vivo and in vitro. A, leukocyte adhesion to cremaster post-capillary venules of control, LPS-treated, and LPS plus rIL-35 co-treated mice after 4-h treatment was visualized using intravital microscopy (n = 6 in each group, scale bar = 20 μm). B, quantitative analysis was performed by enumerating rhodamine 6G-labeled leukocytes that adhered to mouse cremaster muscle post-capillary venule endothelium (n = 6 in each group). C, mouse lung histological analysis in control, LPS-treated, and LPS plus rIL-35 co-treated mice was performed after hematoxylin and eosin (HE) staining (scale bar in upper panel = 200 μm; scale bar in lower panel = 20 μm), and the inflammatory cell adhesion to endothelium was further indicated by the arrows (n = 6 in each group). D, the adhesion of MPO-positive neutrophil granulocytes and macrophages to the vascular wall was detected in the lung paraffin-embedded sections of the control mice (left panel), LPS-treated mice (middle panel), and LPS and rIL-35 co-treated mice (right panel) after 4-h treatment (n = 6 mice in each group). Scale bars: 200 μm (upper panel), 20 μm (lower panel) (n = 6 in each group). E, exodus of inflammatory cells into the mouse lungs was counted in the BALF of mice (n = 6 in each group). F, schematic representation of monocyte adhesion assay. G and H, the adhesion of non-stimulated, fluorescence-labeled human peripheral blood mononuclear cells/THP1 cells to HAECs stimulated by LPS and LPS plus rIL-35 was quantified. The result shown is representative of 3 independent experiments. untreat, untreated. The results are expressed as the means ± S.E. *, p < 0.05; ***, p < 0.001.
IL-35 Inhibits Primary Monocyte Adhesion to LPS-activated Human Aortic ECs in Vitro
To dissect the cellular mechanism underlying the suppression of inflammatory cell adhesion to the endothelium by IL-35, we hypothesized that IL-35 can inhibit LPS-induced EC activation. To test this hypothesis, we performed a monocyte adhesion assay. The results showed that after treatment of HAECs with LPS, the adhesion of non-activated human PBMCs (Fig. 4, F and G) and human monocytic THP-1 cells (Fig. 4H) to ECs was significantly increased. However, IL-35, as low as 10 ng/ml, significantly inhibited the adhesion of both non-activated human PBMCs and THP-1 monocytic cells to LPS-activated HAECs. Of note, the inhibitory effect of IL-35 was more pronounced in the binding of human PBMCs to HAECs than that of THP-1 cells. Our results demonstrate that IL-35 inhibits LPS-induced EC activation as judged by reduced monocyte adhesion to HAECs in vitro.
IL-35 Inhibits LPS-induced VCAM-1 in Human Aortic ECs through Its Receptor
Next, we hypothesized that IL-35 could inhibit LPS-induced monocyte adhesion to ECs by suppressing EC adhesion molecules including ICAM-1 and VCAM-1, which mediate the tight adhesion of neutrophils/granulocytes and monocytes to ECs during LPS-induced acute inflammatory response (21). To examine this hypothesis, we first examined the expression of VCAM-1 and ICAM-1 in the aortas and lungs collected from LPS-challenged mice using Western blot analysis. The results showed that IL-35 significantly inhibited VCAM-1 but not ICAM-1 up-regulation in the aortas and lungs from LPS-challenged mice when compared with that from control mice (Fig. 5, A and B). Similar to the results from mouse, we found that rIL-35 significantly inhibited LPS-induced up-regulation of VCAM-1 but not ICAM-1 of HAECs in vitro (Fig. 5, C and D). It has been reported that IL-35 has three receptor formats in Treg including a homodimer of IL-12 receptor β2 (IL-12Rβ-IL-12Rβ), a homodimer of gp130 (gp130-gp130), and a heterodimer of IL-12Rβ-gp130 (Fig. 5G) (19). We found that IL-35 receptor subunits IL-12Rβ and gp130 were expressed in HAEC (Fig. 5E). In addition, the results of the antibody blocking experiment showed that either the IL-12Rβ antibody or the gp130 antibody reversed the inhibitory effects of IL-35 on LPS-induced VCAM-1 up-regulation in HAECs (Fig. 5F), suggesting that the heterodimer of IL-12Rβ and gp130 serves as a functional IL-35 receptor in HAECs (Fig. 5G). Taken together, these results indicate that IL-35 specifically inhibits LPS-induced VCAM-1 up-regulation through IL-35 heterodimeric receptor in HAECs.
FIGURE 5.

IL-35 inhibits LPS-induced up-regulation of VCAM-1 in vivo and in vitro. A, upper panel, the expressions of endothelial cell adhesion molecules VCAM-1 and ICAM-1 in mouse aortas were determined by Western blot analyses after control, LPS, and LPS plus rIL-35 treatments. VCAM-1 and ICAM-1 quantifications were presented in the lower panel. (n = 4 in each group). B, upper panel, VCAM-1 and ICAM-1 expressions in mouse lungs were determined by Western blot analyses. The quantitative data of VCAM-1 and ICAM-1 were presented in the lower panel (n = 3 in each group). C and D, VCAM-1 and ICAM-1 expressions in non-treated, LPS-treated, and LPS plus IL-35 co-treated HAECs were quantified by using flow cytometry. The quantitative data of the -fold changes of VCAM-1+ cell percentage and ICAM-1+ cell percentage in each treatment group over the control group are presented to the right of representative flow cytometry data. The result shown is representative of 3 independent experiments. E, the expressions of IL-35 receptor subunits gp130 and IL-12Rβ2 in HAECs were determined by Western blot analysis. F, inhibition of LPS up-regulation of VCAM-1 by IL-35 in HAECs was reversed by anti-gp130 antibody and anti-IL-12Rβ2 antibody but not by IgG controls. The results shown are representative of 3 independent experiments. G, schematic representation of IL-35 receptor formats including two types of homodimers, IL-12Rβ2-IL-12Rβ2 and gp130-gp130, and a heterodimer of IL-12Rβ2-gp130 (upper panel). IL-35 receptor formats can be blocked by anti-IL-12Rβ2 antibody and anti-gp130 antibody (lower panel). The results are expressed as the means ± S.E. NS, not significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
IL-35 Inhibits LPS-induced VCAM-1 Up-regulation by Suppression of MAPK-AP-1
It has been recently reported that MAPK plays a critical role in mediating LPS-induced EC activation (22). We examined whether IL-35 could inhibit the activation of three pathways of MAPK family including JNK, ERK, and p38. The results showed that IL-35 inhibited the phosphorylations of JNK and ERK at 5 min after LPS stimulation as well as the phosphorylation of p38 at 30 min after LPS stimulation (Fig. 6A). AP-1 serves as the downstream proinflammatory transcription factor of MAPK because the inhibition of MAPK pathways attenuated transcription factor AP-1 binding to the specific site on the promoters of target genes (23). In addition, a putative binding site for the proinflammatory transcription factor AP-1 has been identified at position −495 bp on the promoter of human VCAM-1 (Fig. 6D) (24). Based on these analyses, we determined whether IL-35 inhibits LPS-induced VCAM-1 by suppressing AP-1 binding in the nucleus using EMSA. The results in Fig. 6B showed that LPS induced AP-1 nuclear binding in HAECs, which was significantly inhibited by IL-35. The specificity of AP-1 binding was verified using non-labeled AP-1 consensus oligonucleotide and mutant AP-1 oligonucleotide as well as supershift assay with antibodies for AP-1 subunits c-Fos and c-Jun (Fig. 6C). Moreover, because it has also been shown that LPS also up-regulates VCAM-1 through activating NF-κB signaling (25), we examined the expressions of phosphorylated IκBα, total IκBα, and β-actin using Western blots and NF-κB nucleus bindings using EMSA. The results showed that IL-35 slightly inhibited phosphorylation of IκBα at 5 and 30 min (Fig. 6E), but IL-35 did not inhibit NF-κB nuclear translocation and binding (Fig. 6F). The specificity of NF-κB binding was verified by the following methods: (a) successful competition blocking using NF-κB consensus oligonucleotide, (b) failure in blocking with mutant NF-κB oligonucleotide, and (c) successful supershift using anti-p65 and p50 subunits of NF-κB (Fig. 6G). Taken together, our results suggest that IL-35 inhibits LPS-induced VCAM-1 up-regulation by suppressing the MAPK-AP-1 pathway. Of note, in addition to VCAM-1, we also found that the expressions of all the LPS-induced 11 cytokines/chemokines, which were suppressed by IL-35 except CXCL1 and CXCL9 (Fig. 3), were also the target of AP-1 genes (Fig. 6D). These results suggest that IL-35 transcriptionally down-regulates VCAM-1 and other inflammatory cytokines/chemokines induced by LPS through suppression of the MAPK-AP1 pathway, but not the NF-κB pathway. Of note, LPS induced secretion of two additional proinflammatory cytokines including IL-6 and M-CSF in plasma (Fig. 3), which were suppressed by IL-35. These two proinflammatory cytokines may further amplify the inflammation response by their own signaling. Because IL-6 and M-CSF may promote inflammation via MAPK pathways (26), our results reported here also suggest that IL-35 may suppress LPS-induced secondary pathways, such as IL-6- and M-CSF-activated MAPK pathways.
FIGURE 6.

IL-35 inhibits LPS-induced activation of MAPK-AP1 pathway, which mediates LPS-induced VCAM-1 up-regulation in human aortic endothelial cells. A, the expressions of proteins involved in the MAPK pathway were analyzed with Western blots in HAECs stimulated by LPS and LPS plus IL-35 treatments for 5, 15, and 30 min. The relative mean pixel density ratios of phosphorylated (p) kinase over total kinase proteins were shown. B, HAECs were treated with LPS for 1.5 h with or without IL-35 pretreatment for 18 h, and EMSA was used to detect the binding of transcription factor AP-1 in the nuclear extracts. C, EMSA was used to determine the specificity of AP-1 binding to the AP-1 consensus oligonucleotides using consensus competitor oligonucleotides, mutant competitor oligonucleotides, and supershift antibodies for AP-1 subunits c-Fos and c-Jun. D, AP-1-binding sites were found in the promoters of VCAM-1 gene, proinflammatory cytokine genes, and chemokine genes that were suppressed by IL-35. N.D., not determined; N.F., not found; PMID, PubMed IDs of the studies. E, protein expressions of phosphorylated (p)-IκBα, total IκBα, and β-actin were analyzed using Western blots. The relative mean pixel density ratios of phosphorylated over total proteins were shown. F, EMSAs were used to detect the binding of transcription factor NF-κB in the nuclear extracts in HAECs. G, EMSA was used to determine the specificity of NF-κB binding to the NF-κB consensus oligonucleotide using competitor oligonucleotides, mutant oligonucleotides, and supershift antibodies for NF-κB subunits p50 and p65. The result shown is representative of 3 independent experiments.
IL-35 Inhibits LPS-induced Monocyte Adhesion to ECs in EBI3-deficient Mice
To identify the endogenous effects of IL-35/IL-27 in LPS-induced vascular inflammation, we used EBI3 knock-out mice. Deficiency of EBI3 (EBI3−/−) in mice results in the deficiency of both functional IL-27 and IL-35 (18). So far, the studies on the roles of IL-27 in vascular inflammation have been controversial. IL-27 was shown to promote EC activation and inflammation (27), but IL-27 was also demonstrated to inhibit atherosclerosis (28). We reasoned that if IL-27 and IL-35 have synergistic effects in inhibiting vascular inflammation, EBI3−/− mice would have exacerbated proinflammatory response after LPS stimulation (Fig. 7A, top panel). In contrast, if IL-27 promotes inflammation, whereas IL-35 inhibits inflammation (Fig. 7A, lower panel), EBI3−/− mice would have the antagonism of IL-27 and IL-35, resulting in neither spontaneous proinflammatory nor anti-inflammatory status after LPS stimulation. To examine this issue, we performed several experiments. First, we found that EBI3−/− mice and WT mice showed similar levels of leukocyte adhesion in the post-capillary venules of the cremaster muscle (Fig. 7B). Second, EBI3−/− mice had similar lung structure and leukocyte adhesion on the endothelium in mouse lungs with that of WT mice (Fig. 7, C and D). Third, blood leukocyte numbers in EBI3−/− mice were not different from those of WT control mice (Fig. 7E). These findings also correlated well with the results in Fig. 1, which showed undetectably low levels of IL-35 and IL-27 in the plasma of mice without LPS stimulation. Taken together, these findings suggest that EBI3, IL-27, and IL-35 do not play significant roles in regulating vascular EC activation under physiological conditions. This finding is in line with our previous study, which indicated that IL-35 and IL-27 are not housekeeping inflammation modulatory cytokines (17), unlike the housekeeping inhibitory roles of TGF-β (17). We then determined the vascular response after LPS challenge in EBI3−/− mice. The results in Fig. 7F showed that LPS induced leukocyte adhesion to the cremaster muscle post-capillary venule endothelium in EBI3−/−mice similar to that in WT mice. In addition, IL-35 was able to inhibit leukocyte adhesion in EBI3−/−mice (Fig. 7F). Moreover, we found that LPS induced inflammatory cell adhesion, lung inflammation, and neutrophil-specific MPO staining in EBI3−/−mice similarly to that in WT control mice (Fig. 7, G and H). Furthermore, IL-35 was able to inhibit leukocyte adhesion to lung microvessel endothelium, lung inflammation (Fig. 7G,) and neutrophil/granulocyte adhesion to lung vascular endothelium in EBI3−/−mice (Fig. 7H). These results suggested that EBI3 has neither proinflammatory nor anti-inflammatory roles on acute vascular response to LPS stimulation.
FIGURE 7.

IL-35 inhibits LPS-induced monocyte adhesion to endothelial cells in EBI3-deficient mice in vivo. A, schematic representation on two possibilities concerning whether EBI3−/− mice have increased LPS-induced EC activation. B, leukocyte adhesion to cremaster post-capillary venules in living WT mice and EBI3−/− mice was visualized using intravital microscopy in the upper panel (n = 3 male mice for each group, scale bar = 20 μm). Quantification was shown in the lower panel. Data are representative of 3 independent experiments. C, mouse lung histological analyses were performed by hematoxylin and eosin (HE) staining in WT control mice and EBI3−/− mice (scale bar in upper panel = 200 μm; scale bar in lower panel = 20 μm) in non-stimulated conditions. D, MPO-positive adhesion of neutrophil/granulocyte and macrophages to the vascular wall in lung was examined (scale bar in upper panel = 200 μm; scale bar in lower panel = 20 μm) in non-stimulated conditions. E, leukocyte numbers in the peripheral blood of WT mice and EBI3−/− mice were quantified (WT mice, n = 11; EBI3−/− mice, n = 12). F, leukocyte adhesion to cremaster post-capillary venules in live WT mice and EBI3−/− mice challenged with LPS was examined using intravital microscopy in the upper panel. The pictures were captured from the venules of LPS-challenged WT mice, LPS-challenged EBI3−/− mice, and LPS plus rIL-35 co-treated EBI3−/− mice. Scale bar = 20 μm. In the lower column graph, quantification was performed by enumerating rhodamine 6G-labeled leukocytes that adhered to mouse cremaster muscle post-capillary venule endothelium per minute (n = 6 in each group). G, mouse lung histological analysis was performed by H&E staining in LPS-treated WT control mice (left panel), LPS-treated EBI3−/− mice (middle panel), and LPS plus IL-35 co-treated EBI3−/− mice (right panel). Upper pictures, scale bar = 200 μm; lower pictures, scale bar = 20 μm. The adhesion of inflammatory cells to endothelium is indicated by the arrows (n = 6 in each group). H, MPO-positive neutrophil granulocyte and macrophage adhesions to the vascular wall in lung were examined. Scale bar = 200 μm (top panel), scale bar = 20 μm (bottom panel) (n = 6). I and J, VCAM-1 and ICAM-1 expressions in non-treated, LPS-treated, and LPS plus IL-27 co-treated HAECs were quantified using flow cytometry. The quantitative data of the -fold changes of VCAM-1+ cell percentage and ICAM-1+ cell percentage in each treatment group over the control group are presented to the right of representative flow cytometry data. The results shown are representative of 3 independent experiments. The results are expressed as the means ± S.E. NS, not significant. *, p < 0.05; ***, p < 0.001.
We further hypothesized that IL-35 and IL-27 may be co-up-regulated in response to LPS stimulation and counteract with each other in modulating EC activation (Fig. 7A, lower panel). To test this hypothesis, we performed ELISA to determine IL-27p28, the unique subunit of IL-27. The results showed that LPS-induced plasma levels of IL-27 were similar to the level of IL-35 at 1.5 h, but lower than the IL-35 plasma levels at 4 and 24 h (Fig. 1E). Next, we examined the effects of IL-27 in LPS-induced up-regulation of EC adhesion molecules. We found that in contrast to IL-35, IL-27 further enhanced LPS-induced VCAM-1 and ICAM-1 (Fig. 7, I and J). Taken together, our results suggest that, first, EBI3 itself is not an anti-inflammatory protein, at least in LPS-induced acute vascular inflammation, because EBI3−/− mice and WT mice showed similar levels of leukocyte adhesion to the endothelium in the presence or absence of LPS. Second, IL-27 promotes LPS-induced up-regulation of VCAM-1 and ICAM-1 in HAECs, which supports our hypothesis that EBI3−/− mice have similar inflammatory response as WT mice under LPS stimulation as a result of the antagonism between proinflammatory effects of IL-27 and anti-inflammatory effects of IL-35 (Fig. 7A, lower panel). Third, EBI3−/− mice have no deficiency in IL-35 receptor signaling because IL-35 can similarly suppress LPS-induced vascular response in EBI3−/− mice similarly as that in WT mice. Fourth, the inhibitory effects of IL-35 in EC activation are IL-27-independent.
Discussion
Sepsis has been estimated to lead to the death ∼28–50% of its victims (2). In regard to the alarming death rates, a major question is whether the host's immune system has constitutive housekeeping anti-inflammatory defense(s) to counteract inflammation onset or whether the host needs to generate and secrete anti-inflammatory cytokines during inflammation de novo. Our recent study reported that the newly identified anti-inflammatory cytokine IL-35 is a responsive cytokine up-regulated in response to inflammation, but not a constitutively expressed housekeeping cytokine (17). In this current work, we hypothesize that IL-35 inhibits LPS-induced acute vascular inflammation by suppressing EC activation. We report the following findings. 1) LPS treatment significantly increases plasma IL-35 levels. 2) IL-35 decreases LPS-induced secretion of inflammatory cytokines and chemokines in the plasma of WT mice. 3) IL-35 inhibits LPS-induced leukocyte adhesion to the endothelium in lung and cremaster muscle in vivo, and reduces inflammatory cell trans-endothelial infiltration. 4) Mechanistically, IL-35 inhibits human primary monocyte adhesion to LPS-activated HAECs in vitro. 5) IL-35 inhibits LPS-induced VCAM-1 but not ICAM-1 in HAECs via binding to its receptor. 6) IL-35 inhibits LPS-induced activation of the MAPK-AP1 pathway, which mediates LPS-induced up-regulation of VCAM-1. 7) EBI3−/− mice display vascular inflammation response similar to that in WT mice after LPS stimulation. IL-27 promotes LPS-induced EC activation in HAECs, whereas IL-35 inhibits LPS-induced monocyte adhesion to ECs in EBI3−/− mice in vivo. These results suggest that (a) IL-35 inhibition of vascular inflammation is IL-27-independent although they share EBI3 and (b) IL-35 receptor is functional in EC of EBI3-deficient mice.
The reported role of IL-27 in vascular inflammation has been controversial. IL-27 enhances LPS-induced proinflammatory cytokine expression in primary human monocytes (29), and also promotes peritonitis (27), but IL-27Rα signaling has also been shown to inhibit atherosclerosis (28). Our results showed that IL-27 enhanced LPS-induced up-regulation of VCAM-1 and ICAM-1 in HAECs, suggesting that IL-27 promotes LPS-induced EC activation and vascular inflammation. Of note, Vignali and Kuchroo (18) claimed that IL-27 is a relatively weak immunosuppressive cytokine but IL-35 is the strongest immunosuppressive cytokine in the IL-12 family. Our data showed that under physiological conditions, EBI3−/− mice, deficient in both IL-27 and IL-35, had no adverse inflammatory response in contrast to the findings in TGF-β−/− mice (17), which clearly indicates that both IL-35 and IL-27 are inflammation-responsive cytokines but not housekeeping cytokines. Our data also showed that EBI3−/− mice had a similar scale of LPS-induced vascular response to that in WT mice, whereas others showed that EBI3 deficiency exacerbated experimental abdominal aortic aneurysms.3 Taken together, these results suggest that the effects of EBI3 on inhibiting autoimmune and inflammation may be relevant, if at all, in a chronic disease setting instead.
Most anti-inflammatory mechanisms in the vascular wall, including IL-10, TGF-β, high density lipoprotein, and peroxisome proliferator-activated receptor α activators, are mediated by inhibition of the NF-κB pathway (31). IL-10 decreases leukocyte vein extravasation through inhibition of endothelial expression of P-selectin, E-selectin, and ICAM-1 (32). However, our data showed that IL-35 inhibits LPS-induced VCAM-1 by suppressing MAPK-AP1 but not the NF-κB pathway, suggesting the unique inhibitory effects of IL-35 in EC activation. The differential response of IL-35 on LPS-induced VCAM-1 and ICAM-1 is similar to what has been previously reported for peroxisome proliferator-activated receptor (PPAR) activators (33), phosphatase, and tensin homologue activator CKD712 (34). VCAM-1 is an inducible adhesion molecule expressed on ECs. It has been shown to play a key role in vascular inflammation. Endothelial VCAM-1 binds to very late antigen 4 (VLA-4) on leukocytes, and thus mediates leukocyte trans-endothelial migration into the sub-endothelial compartment of the vessel. In atherosclerosis, a chronic inflammatory vascular disease, truncated non-functional VCAM-1 has been shown to decrease atherosclerotic plaque in low-density lipoprotein receptor-deficient (LDLR−/−) mice. In contrast, ICAM-1 is constitutively expressed on endothelial cells. Although its expression increases in the atherosclerotic plaque, there are conflicting results of ICAM-1 in atherosclerosis using knock-out mouse model (35, 36).
IL-35 shares the subunit IL-12A with IL-12 and EBI3 with IL-27. IL-35 receptor shares the subunits IL-12Rβ2 with IL-12 receptor, and shares gp130 with the receptors for IL-6 family cytokines including IL-6, IL-11, leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF), and cardiotrophin-1 (CT-1) (37). The single gene deficiency approach for studying IL-35 is difficult because knock-out of either IL-35 subunit IL-12p35 or EBI3, or IL-35 receptor subunit gp130 or IL-12Rβ2, could lead to the deficiencies of IL-12, IL-27, or six cytokines in the IL-6 cytokine family. Our results suggest that IL-35 inhibition of EC activation is not dependent on IL-27 and that EBI3−/− mice have no deficiency in IL-35 receptor signaling. Therefore, our new IL-35 therapy model in the EBI3−/− mice provides a better solution to examine the pathophysiological functions of IL-35 in various disease models.
Previous studies from us and other laboratories have demonstrated that CD4+CD25highFoxp3+ Tregs play a critical role in suppressing vascular inflammation and atherosclerosis (4, 5). However, the role of Tregs in acute sepsis and endotoxemia remained open (38) until it was demonstrated that adaptive immune cells including Tregs regulate initial innate responses (30). One of the potential mechanisms underlying Treg inhibition of acute sepsis is that Tregs may secrete immunosuppressive/anti-inflammatory cytokines. Future study is needed to clarify whether Tregs or any other cell type(s) contribute to the induction of plasma IL-35 after LPS injection in mice. Taken together, by integrating our data, we propose a new working model (Fig. 8). LPS stimulation may trigger Tregs and potentially other cell types to secrete IL-35 alongside with proinflammatory cytokines/chemokines into plasma. IL-35 acts on IL-35 dimeric receptor(s) on ECs and inhibits LPS-induced EC activation and leukocyte adhesion to ECs via suppressing VCAM-1 up-regulation in a MAPK-AP-1-dependent manner. Our findings provide a novel insight on the roles of this responsive, inflammation scale-controlling cytokine IL-35 on acute inflammation, which may have important functions in facilitating inflammation resolution.
FIGURE 8.
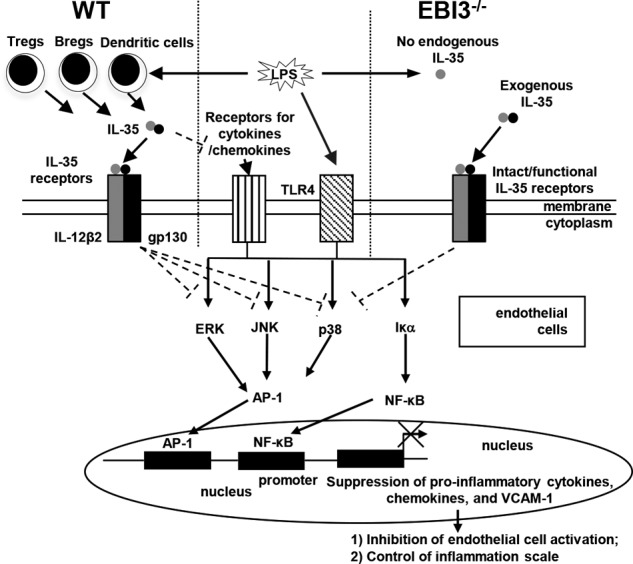
A new working model. LPS can induce inflammatory response in endothelial cells by engaging Toll-like receptor 4 (TLR4) and activating the MAPK-AP-1 and NF-κB pathways. IL-35 is induced by LPS presumably in regulatory T cells, B cells, and dendritic cells at the same time, which suppresses LPS-induced endothelial cell activation by inhibiting expressions of VCAM-1 in endothelial cells, proinflammatory cytokines, and chemokines in plasma and monocyte adhesion to endothelium, thereby controlling inflammation scale in sepsis. In IL-35 subunit EBI3 gene-deficient (EBI3−/−) mice, endogenous IL-35 could not be induced by LPS, but exogenous IL-35 could still suppress LPS-induced inflammatory response in endothelial cells because of intact and functional IL-35 receptors and its signaling pathway(s) in EBI3−/− mice.
Author Contributions
X. S., S. M., X. L., H. X., H. W., and X. F. Y designed research; X. S., S. M., X. L., H. X., and H. S. performed research; X. J., M. M., and D. W. P provided reagents and critically read the manuscript; X. S., S. M., X. L., H. X., H. W., and X. F. Y analyzed data; and X. S. and X. F. Y wrote the paper.
Supplementary Material
Acknowledgments
We are very grateful to Dr. Barrie Ashby and Dr. Anthony Virtue for the critical reading and English editing. We are also very grateful to Dr. Xiaoxuan Fan from Temple University School of Medicine Flow Cytometry Core Facility for technical assistance for the flow cytometry study.
This work was partially supported by National Institutes of Health Grants 5R01HL108910-05; 5R01HL116917-03; 5R01HL077288-11; 5R01HL110764-05; and 5R01HL117654-03 (to X. F. Y and H. W). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movies 1–3.
Y. Iida, H. Tanaka, X. Hu, X. Xiong, H. Zhao, B. Xu, and R. L. Dalman, poster presented at Vascular Research Initiatives Conference (April 30, 2014).
- Treg
- regulatory T cell
- EC
- endothelial cell
- HAEC
- human aortic EC
- PBMC
- peripheral blood mononuclear cell
- BALF
- bronchoalveolar lavage fluid
- rIL
- recombinant IL
- MPO
- myeloperoxidase.
References
- 1. Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R. (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 2. Wood K. A., Angus D. C. (2004) Pharmacoeconomic implications of new therapies in sepsis. PharmacoEconomics 22, 895–906 [DOI] [PubMed] [Google Scholar]
- 3. Stoll L. L., Denning G. M., Weintraub N. L. (2004) Potential role of endotoxin as a proinflammatory mediator of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 2227–2236 [DOI] [PubMed] [Google Scholar]
- 4. Xiong Z., Yan Y., Song J., Fang P., Yin Y., Yang Y., Cowan A., Wang H., Yang X. F. (2009) Expression of TCTP antisense in CD25(high) regulatory T cells aggravates cuff-injured vascular inflammation. Atherosclerosis 203, 401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ait-Oufella H., Salomon B. L., Potteaux S., Robertson A. K., Gourdy P., Zoll J., Merval R., Esposito B., Cohen J. L., Fisson S., Flavell R. A., Hansson G. K., Klatzmann D., Tedgui A., Mallat Z. (2006) Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180 [DOI] [PubMed] [Google Scholar]
- 6. Yang W. Y., Shao Y., Lopez-Pastrana J., Mai J., Wang H., Yang X. F. (2015) Pathological conditions re-shape physiological Tregs into pathological Tregs. Burns Trauma 3, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Libby P., Ridker P. M., Hansson G. K. (2011) Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 [DOI] [PubMed] [Google Scholar]
- 8. Chowienczyk P. J., Watts G. F., Cockcroft J. R., Ritter J. M. (1992) Impaired endothelium-dependent vasodilation of forearm resistance vessels in hypercholesterolaemia. Lancet 340, 1430–1432 [DOI] [PubMed] [Google Scholar]
- 9. He S., Li M., Ma X., Lin J., Li D. (2010) CD4+CD25+Foxp3+ regulatory T cells protect the proinflammatory activation of human umbilical vein endothelial cells. Arterioscler. Thromb. Vasc. Biol. 30, 2621–2630 [DOI] [PubMed] [Google Scholar]
- 10. Wang R. X., Yu C. R., Mahdi R. M., Egwuagu C. E. (2012) Novel IL27p28/IL12p40 cytokine suppressed experimental autoimmune uveitis by inhibiting autoreactive Th1/Th17 cells and promoting expansion of regulatory T cells. J. Biol. Chem. 287, 36012–36021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen P., Roch T., Lampropoulou V., O'Connor R. A., Stervbo U., Hilgenberg E., Ries S., Dang V. D., Jaimes Y., Daridon C., Li R., Jouneau L., Boudinot P., Wilantri S., Sakwa I., Miyazaki Y., Leech M. D., McPherson R. C., Wirtz S., Neurath M., Hoehlig K., Meinl E., Grützkau A., Grün J. R., Horn K., Kühl A. A., Dörner T., Bar-Or A., Kaufmann S. H., Anderton S. M., Fillatreau S. (2014) IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dixon K. O., van der Kooij S. W., Vignali D. A., van Kooten C. (2015) Human tolerogenic dendritic cells produce IL-35 in the absence of other IL-12 family members. Eur. J. Immunol. 45, 1736–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi J., Leung P. S., Bowlus C., Gershwin M. E. (2015) IL-35 and autoimmunity: a comprehensive perspective. Clin. Rev. Allergy Immunol. 10.1007/s12016-015-8468-9 [DOI] [PubMed] [Google Scholar]
- 14. Whitehead G. S., Wilson R. H., Nakano K., Burch L. H., Nakano H., Cook D. N. (2012) IL-35 production by inducible costimulator (ICOS)-positive regulatory T cells reverses established IL-17-dependent allergic airways disease. J. Allergy Clin. Immunol. 129, 207–215.e1–5, 10.1016/j.jaci.2011.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kochetkova I., Golden S., Holderness K., Callis G., Pascual D. W. (2010) IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J. Immunol. 184, 7144–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niedbala W., Wei X. Q., Cai B., Hueber A. J., Leung B. P., McInnes I. B., Liew F. Y. (2007) IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur. J. Immunol. 37, 3021–3029 [DOI] [PubMed] [Google Scholar]
- 17. Li X., Mai J., Virtue A., Yin Y., Gong R., Sha X., Gutchigian S., Frisch A., Hodge I., Jiang X., Wang H., Yang X. F. (2012) IL-35 is a novel responsive anti-inflammatory cytokine: a new system of categorizing anti-inflammatory cytokines. PLoS One 7, e33628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vignali D. A., Kuchroo V. K. (2012) IL-12 family cytokines: immunological playmakers. Nat. Immunol. 13, 722–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collison L. W., Delgoffe G. M., Guy C. S., Vignali K. M., Chaturvedi V., Fairweather D., Satoskar A. R., Garcia K. C., Hunter C. A., Drake C. G., Murray P. J., Vignali D. A. (2012) The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 13, 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Howard M., Muchamuel T., Andrade S., Menon S. (1993) Interleukin 10 protects mice from lethal endotoxemia. J. Exp. Med. 177, 1205–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang X. F., Yin Y., Wang H. (2008) Vascular inflammation and atherogenesis are activated via receptors for PAMPs and suppressed by regulatory T cells. Drug Discov. Today Ther. Strateg. 5, 125–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mannam P., Zhang X., Shan P., Zhang Y., Shinn A. S., Zhang Y., Lee P. J. (2013) Endothelial MKK3 is a critical mediator of lethal murine endotoxemia and acute lung injury. J. Immunol. 190, 1264–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shaulian E., Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–136 [DOI] [PubMed] [Google Scholar]
- 24. Iademarco M. F., McQuillan J. J., Rosen G. D., Dean D. C. (1992) Characterization of the promoter for vascular cell adhesion molecule-1 (VCAM-1). J. Biol. Chem. 267, 16323–16329 [PubMed] [Google Scholar]
- 25. Jersmann H. P., Hii C. S., Ferrante J. V., Ferrante A. (2001) Bacterial lipopolysaccharide and tumor necrosis factor α synergistically increase expression of human endothelial adhesion molecules through activation of NF-κB and p38 mitogen-activated protein kinase signaling pathways. Infect. Immun. 69, 1273–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rawlings J. S., Rosler K. M., Harrison D. A. (2004) The JAK/STAT signaling pathway. J. Cell Sci. 117, 1281–1283 [DOI] [PubMed] [Google Scholar]
- 27. Wirtz S., Tubbe I., Galle P. R., Schild H. J., Birkenbach M., Blumberg R. S., Neurath M. F. (2006) Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. J. Exp. Med. 203, 1875–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koltsova E. K., Kim G., Lloyd K. M., Saris C. J., von Vietinghoff S., Kronenberg M., Ley K. (2012) Interleukin-27 receptor limits atherosclerosis in Ldlr−/− mice. Circ. Res. 111, 1274–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guzzo C., Ayer A., Basta S., Banfield B. W., Gee K. (2012) IL-27 enhances LPS-induced proinflammatory cytokine production via upregulation of TLR4 expression and signaling in human monocytes. J. Immunol. 188, 864–873 [DOI] [PubMed] [Google Scholar]
- 30. Kim K. D., Zhao J., Auh S., Yang X., Du P., Tang H., Fu Y. X. (2007) Adaptive immune cells temper initial innate responses. Nat. Med. 13, 1248–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tedgui A., Mallat Z. (2001) Anti-inflammatory mechanisms in the vascular wall. Circ. Res. 88, 877–887 [DOI] [PubMed] [Google Scholar]
- 32. Henke P. K., DeBrunye L. A., Strieter R. M., Bromberg J. S., Prince M., Kadell A. M., Sarkar M., Londy F., Wakefield T. W. (2000) Viral IL-10 gene transfer decreases inflammation and cell adhesion molecule expression in a rat model of venous thrombosis. J. Immunol. 164, 2131–2141 [DOI] [PubMed] [Google Scholar]
- 33. Jackson S. M., Parhami F., Xi X. P., Berliner J. A., Hsueh W. A., Law R. E., Demer L. L. (1999) Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler. Thromb. Vasc. Biol. 19, 2094–2104 [DOI] [PubMed] [Google Scholar]
- 34. Tsoyi K., Kim W. S., Kim Y. M., Kim H. J., Seo H. G., Lee J. H., Yun-Choi H. S., Chang K. C. (2009) Upregulation of PTEN by CKD712, a synthetic tetrahydroisoquinoline alkaloid, selectively inhibits lipopolysaccharide-induced VCAM-1 but not ICAM-1 expression in human endothelial cells. Atherosclerosis 207, 412–419 [DOI] [PubMed] [Google Scholar]
- 35. Cybulsky M. I., Iiyama K., Li H., Zhu S., Chen M., Iiyama M., Davis V., Gutierrez-Ramos J. C., Connelly P. W., Milstone D. S. (2001) A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest. 107, 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Collins R. G., Velji R., Guevara N. V., Hicks M. J., Chan L., Beaudet A. L. (2000) P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J. Exp. Med. 191, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Taga T., Kishimoto T. (1997) Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 15, 797–819 [DOI] [PubMed] [Google Scholar]
- 38. Wisnoski N., Chung C. S., Chen Y., Huang X., Ayala A. (2007) The contribution of CD4+ CD25+ T-regulatory-cells to immune suppression in sepsis. Shock 27, 251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.