

Background: Assimilatory NADPH-sulfite reductase (SiR) is an essential metalloenzyme for sulfur metabolism made from two subunits.
Results: We defined how the subunits of SiR assemble, with or without cofactors.
Conclusion: One region of the metalloenzyme interacted either with its reductase partner when cofactors were formed or with itself when they were not.
Significance: We propose a novel mechanism to regulate SiR assembly.
Keywords: electron transfer, hydrogen-deuterium exchange, iron-sulfur protein, metalloenzyme, small angle x-ray scattering (SAXS), electrospray ionization, Fourier transform mass spectrometry, ion cyclotron resonance, iron-sulfur cluster biogenesis, sulfite reductase
Abstract
Assimilatory NADPH-sulfite reductase (SiR) from Escherichia coli is a structurally complex oxidoreductase that catalyzes the six-electron reduction of sulfite to sulfide. Two subunits, one a flavin-binding flavoprotein (SiRFP, the α subunit) and the other an iron-containing hemoprotein (SiRHP, the β subunit), assemble to make a holoenzyme of about 800 kDa. How the two subunits assemble is not known. The iron-rich cofactors in SiRHP are unique because they are a covalent arrangement of a Fe4S4 cluster attached through a cysteine ligand to an iron-containing porphyrinoid called siroheme. The link between cofactor biogenesis and SiR stability is also ill-defined. By use of hydrogen/deuterium exchange and biochemical analysis, we show that the α8β4 SiR holoenzyme assembles through the N terminus of SiRHP and the NADPH binding domain of SiRFP. By use of small angle x-ray scattering, we explore the structure of the SiRHP N-terminal oligomerization domain. We also report a novel form of the hemoprotein that occurs in the absence of its cofactors. Apo-SiRHP forms a homotetramer, also dependent on its N terminus, that is unable to assemble with SiRFP. From these results, we propose that homotetramerization of apo-SiRHP serves as a quality control mechanism to prevent formation of inactive holoenzyme in the case of limiting cellular siroheme.
Introduction
Assimilatory NADPH-sulfite reductase (SiR)3 (NADPH-sulfite reductases other than EC 1.8.1.2 are indicated with other abbreviations throughout) is an oligomeric oxidoreductase that reduces sulfur to the biologically available form, SH−. In Escherichia coli, the SiR holoenzyme is composed of two polypeptides (1). One is an α subunit that is a multimeric flavoprotein (SiRFP) when it is dissociated from the holoenzyme. The other is a β subunit that is a monomeric hemoprotein (SiRHP) when it is dissociated from the holoenzyme. SiRFP is a homolog of NADPH-cytochrome p450 reductase (CYPOR) and delivers electrons to SiRHP through coupled NADPH, FAD, and FMN cofactors (2). SiRHP channels those electrons through an Fe4S4 cluster that is covalently attached through one of its four cysteine ligands to the iron of an iron-containing porphyrinoid called siroheme (3). As assembled, SiR is believed to be an α8β4 oligomer on the basis of amino acid analysis mass measurements from the early 1970s (1, 4). Recombinant α1β1 generated through N-terminal truncation of SiRFP has reduced but measurable activity and similar flavin content, bringing the 2:1 SiRFP/SiRHP stoichiometry into question (5).
SiR catalyzes the six-electron reduction of sulfite to sulfide. The only other known six-electron reduction reaction is that of nitrite to ammonia, which is catalyzed either by a homologous siroheme-dependent, ferredoxin-nitrite reductase (NiR) or by a multiheme NiR (6). In this way, SiR is a powerful enzyme that performs chemistry that is difficult to replicate synthetically. Biologically, SiR is essential for preparing sulfur for incorporation into sulfur-containing amino acids and cofactors. SiRs are found in diverse organisms from bacteria through plants and use either SiRFP or ferredoxin as the source of their electrons (6). SiR is not found in humans, in part explaining the indispensable nature of methionine in human diets (7).
Monomeric SiRHP is closely related to the heterodimeric dissimilatory sulfite reductase (DSR) from sulfate-reducing prokaryotes that relies on a ferredoxin domain for its reducing equivalents (6). The two polypeptides of DSR are homologs, suggesting that the heterodimer evolved through an early gene duplication event (8). SiRHP has pseudo-2-fold symmetry defined by two tandem sulfite/nitrite reductase repeats (S/NiRRs (9)) that recalls the pseudosymmetry of the dimeric DSR. SiRHP probably evolved through a gene fusion event of the duplicated reading frames, potentially explaining the mismatch in stoichiometry between the oxidase and reductase subunits (9). Each DSR subunit contains two siroheme active sites. In contrast, SiRHP contains only one, but side chains from a linker mimic siroheme, filling the vestigial active site (9).
The multimeric nature of SiRFP suggests that it is distinct from other CYPOR homologs that do not form such a large complex. CYPOR itself is a monomer (10). The nitric-oxide synthase reductase homolog is a dimer (11). Methionine synthase reductase is also a monomer (12). Further, SiR also seems to be unique, because if the disputed α8β4 stoichiometry is correct, then the SiRFP subunits are in 2-fold excess over the SiRHP subunits. In other words, we cannot easily extrapolate details about the mechanism by which these subunits work together. Within SiRHP, the covalently coupled siroheme and Fe4S4 cluster are essential to the catalytic power of its six-electron reduction. The covalently bound cofactors work together to push electrons while three positively charged amino acids pull the charge onto the evolving substrate (13). A fourth basic amino acid, Arg83, is not required for catalysis but is essential for siroheme binding (13). Despite the intricate and essential nature of the metal sites, we know very little about how the active site is assembled in the apoprotein.
We have undertaken biochemical analysis of the complex to understand the mechanism by which SiRFP and SiRHP assemble to facilitate electron transfer. We present the first evidence for how SiR assembles, based on interaction motifs identified by means of hydrogen/deuterium exchange (HDX). Further, we have characterized an apo-form of SiRHP that assembles into a homotetramer but is unable to form a holoenzyme-like complex with SiRFP. Finally, we propose a novel mechanism by which this tetrameric apo-SiRHP serves a quality control function by blocking inactive holoenzyme complex formation in the absence of its metalloenzyme cofactors.
Experimental Procedures
Recombinant Protein Production
SiRHP
Full-length wild-type or R83S E. coli SiRHP was expressed as an N-terminal six-histidine (His6) fusion from a bicistronic construct with the SiRHP open reading frame (cysI) and the siroheme synthase open reading frame (cysG) cloned into the pBAD/myc/his A plasmid (Life Technologies, Inc.) in LMG194 E. coli cells, as described previously (13). N-terminally His6-tagged apo-full-length SiRHP (apo-SiRHP) or C-terminally His6-tagged apo-Δ60 and -Δ80 truncations were expressed without cysG as a single cistron cloned into pBAD/myc/his A or pBAD/myc A and in a cysG− strain (Keio collection strain JW3331 (14)), following the same induction/expression procedure. All His6 SiRHP variants were purified by use of nickel affinity and size exclusion chromatography (SEC) as described previously (13). Untagged SiRHP or untagged SiRHP Δ60 and Δ80 truncations were also independently overexpressed in LMG194 E. coli from cysG-containing bicistronic pBAD/myc A plasmids and purified as described previously for untagged SiRHP (15).
Throughout, cofactor state was monitored with UV-visible spectroscopy, and the oligomeric state of the protein was monitored with native gel and SEC analysis, described below. Protein concentration was determined for each sample by use of the bicinchoninic acid (BCA) assay (Life Technologies), per the manufacturer's protocol. Table 1 summarizes all expression details and protein oligomeric states.
TABLE 1.
SiR proteins and their expression plasmids
| Protein | Expression plasmid | Strain | Tag | Oligomeric state |
|---|---|---|---|---|
| His6-SiRHP | pBAD/myc/his A | LMG194 | N-terminal His6 | Monomer |
| SiRHP | pBAD/myc A | LMG194 | N-terminal His6 | Monomer |
| Δ60-SiRHP | pBAD/myc A | LMG194 | N-terminal His6 | Monomer |
| Δ80-SiRHP | pBAD/myc A | LMG194 | N-terminal His6 | Monomer |
| Apo-wild type SiRHP | pBAD/myc/his A | JW3331 (cVsG−) | N-terminal His6 | Tetramer |
| Apo-Δ60-SiRHP | pBAD/myc/his A | JW3331 (cVsG−) | C-terminal His6 | Monomer |
| Apo-Δ80-SiRHP | pBAD/myc/his A | JW3331 (cVsG−) | C-terminal His6 | Monomer |
| SiRFP | pCDFDuet-1 | BL21(DE3) | Octomer | |
| SiRFPFMN | pCDFDuet-1 | BL21(DE3) | N-terminal His6 | Octomer |
| SiRFP43 | pET-HTG | BL21(DE3) | N-terminal His6-GST/cleaved | Monomer |
| SiR | pBAD/myc/his A | LMG194 | N-terminal His6 (SiRFP) | Octomer/Tetramer |
SiRFP
Wild-type SiRFP (cysJ) was recombinantly expressed as an untagged construct in the pCDFDuet1 plasmid (EMD Millipore, Billerica, MA) in BL21(DE3) E. coli cells induced with 1 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) and grown at 25 °C for 4 h. The purification protocol was modified from Ref. 16. The pellet from 3 liters of cells was resuspended in 30 ml of 50 mm KH2PO4/K2HPO4 (KPi), pH 7.8, 200 mm NaCl, 1 mm EDTA and lysed by sonication. Cell lysate was clarified by centrifugation at 16,000 × g for 35 min. All centrifugation steps were performed in an Eppendorf AG (Hamburg, Germany) F-34-6-38 fixed angle rotor in a 5810R centrifuge. The supernatant was stirred with 0.1% polyethyleneimine (PEI) for 20 min and then centrifuged at 10,000 × g for 25 min. The supernatant was recovered and precipitated with 27% ammonium sulfate, stirred for 1 h, and centrifuged for 30 min at 16,000 × g. This second supernatant was recovered and brought to 45% ammonium sulfate final concentration, stirred for 30 min, and centrifuged for 40 min at 10,000 × g. The pellet was dissolved in a small volume of 50 mm KPi, pH 7.8, and then passed through a 60-ml G-25 column (GE Healthcare) equilibrated with the same buffer (the top 10 ml of the column was equilibrated in 50 mm KPi, 200 mm NaCl, pH 7.8) to eliminate excess PEI. The protein was then incubated for 1 h with 10 ml of hydroxyapatite Bio-Gel HT gel (Bio-Rad) equilibrated in 10 mm KPi, pH 7.8. The resin was washed twice with 20 volumes of 100 mm KPi, pH 7.8, before the protein was eluted with 60 volumes of 200 mm KPi, pH 7.8. Immediately following elution, EDTA was added to 1 mm final concentration. The elution fraction was concentrated to 30 ml; dialyzed overnight in 10 mm KPi, pH 7.8, 1 mm EDTA; loaded onto a 5-ml HiTrap Q-Sepharose Fast Flow column (GE Healthcare); and eluted with a gradient of KCl from 100 to 1,000 mm. SiRFP-containing fractions were concentrated, loaded onto a Superose 6 size exclusion column (GE Healthcare), and eluted with 65 mm KPi, 200 mm KCl, 1 mm EDTA, pH 7.8. All steps were performed at 4 °C.
SiRFPFMN
A gene fragment encoding SiRFPFMN (amino acids 1–208) was subcloned into the pCDFDuet1 plasmid. SiRFPFMN was recombinantly expressed as an N-terminal His6 fusion in BL21(DE3) E. coli cells induced with 1 mm IPTG and grown at 25 °C for 4 h. The pellet from a 1-liter culture of SiRFPFMN-expressing cells was resuspended in 20 ml of 50 mm KPi, pH 7.8, 200 mm NaCl and lysed by sonication. Cell lysate was clarified by centrifugation at 16,000 × g for 35 min, and SiRFPFMN was purified by nickel affinity chromatography as described previously (13). SiRFPFMN-containing fractions were concentrated and loaded onto a Superose 6 column and eluted with 65 mm KPi, 200 mm KCl, 1 mm EDTA, pH 7.8. Peak fractions were pooled and concentrated. All steps were performed at 4 °C.
SiRFP43
A gene fragment encoding SiRFP43 (amino acids 209–599, named for its 43-kDa theoretical molecular mass) was subcloned into the pET-HTG plasmid (17) for recombinant expression as an N-terminal His6/glutathione S-transferase (GST) fusion in BL21(DE3) E. coli cells, induced with 1 mm IPTG. The pellet from a 1-liter culture of cells expressing His6-GST-SiRFP43 was resuspended in 20 ml of 50 mm KPi, pH 7.8, 200 mm NaCl and lysed by sonication. Cell lysate was clarified by centrifugation at 16,000 × g for 35 min. The supernatant was purified by glutathione affinity chromatography over a 5-ml GSTrap affinity column (GE Healthcare) as described by the manufacturer. Fractions containing His6-GST-SiRFP43 were concentrated and dialyzed in 65 mm KPi, pH 7.8, 200 mm NaCl, before the His6-GST tag was cleaved with His6-tagged tobacco etch virus protease (Life Technologies) for 2 h at 30 °C. To remove the His6-GST tag and residual tobacco etch virus, the reaction mix was loaded onto a 5-ml nickel-nitrilotriacetic acid (Ni-NTA) column (GE Healthcare). Unbound protein in the flow-through and subsequent 30-ml wash with 65 mm KPi, pH 7.8, 200 mm NaCl was collected, concentrated, loaded onto a Superose 6 column, and eluted with 65 mm KPi, 200 mm KCl, 1 mm EDTA, pH 7.8. All steps were performed at 4 °C unless otherwise mentioned.
SiR
Recombinant SiR holoenzyme was generated from a pBAD/myc/his A tricistronic cysJIG construct as an N-terminal His6 SiRFP fusion. A pellet from a 3-liter culture of SiR-expressing LMG194 E. coli cells was resuspended in 30 ml of 50 mm KPi, pH 7.8, 200 mm NaCl and lysed by sonication. Cell lysate was clarified by centrifugation at 16,000 × g for 35 min. The supernatant was stirred with 0.1% PEI for 20 min and then centrifuged at 10,000 × g for 25 min. The supernatant was purified by use of nickel-affinity chromatography as described previously (13). Upon elution, 1 mm EDTA was immediately added to the SiR-containing fractions, which were then concentrated and loaded onto a Superose 6 column and eluted with 65 mm KPi, 200 mm KCl, 1 mm EDTA, pH 7.8. All steps were performed at 4 °C.
SiR, SiRFP43, and Δ60/Δ80-SiRHP Activity Assays
SiR holoenzyme activity was measured by spectrophotometrically following the oxidation of the electron donor NADPH at 340 nm, according to methods described previously (18). To measure the activity of recombinant SiR, an anaerobic reaction mix containing 100 mm KPi, pH 7.7, 0.5 mm NaHSO3, 0.2 mm of NADPH, 10 units glucose oxidase (Sigma-Aldrich), 10 mm glucose, and 8 μg of enzyme, in 1-ml quartz cuvettes with a 1-cm path length, was followed with an Agilent 8453 UV-visible spectrophotometer. We then tested SiRFP43 for activity by mixing it with SiRHP at 1:1 and 2:1 ratios in the same assay. Additionally, we measured the activity of Δ60- and Δ80-SiRHP by chemical reduction with methyl viologen/Cr(II)-EDTA activity assays, performed as described previously with 0.5 mm NH2OH substrate (13). All reactions were performed in triplicate.
Free Cysteine Analysis
To determine the number of solvent-accessible thiols in SiRHP, apo-SiRHP, and R83S SiRHP, 500 μl of 1 mg/ml protein was reduced for 30 min with 5 mm tris(2-carboxyethyl)phosphine (per the Ellman's reagent manufacturer's recommendation) in 100 mm KPi, pH 7.8, 1 mm EDTA at 4 °C. Excess reductant was removed by passing the sample through two successive PD MiniTrap G-25 columns (GE Healthcare). SiRHP was then concentrated to 15 μm with a 30,000 molecular weight cut-off spin concentrator before being mixed with a final concentration of 200 μm 5-(3-carboxy-4-nitrophenyl)disulfanyl-2-nitrobenzoic acid according to according to the manufacturer's recommendations (Ellman's reagent; Sigma-Aldrich). After a 15-min reaction time, the absorbance at 412 nm was recorded on a Cary 300 UV-Vis Spectrophotometer. To account for the excess of reagent to reactive protein, the samples were blanked against 200 μm 5-(3-carboxy-4-nitrophenyl)disulfanyl-2-nitrobenzoic acid and background-corrected for absorbance of the SiRHP protein (the siroheme cofactor in the wild-type enzyme absorbs strongly at 386 nm; Fig. 1A). All reactions were performed in triplicate.
FIGURE 1.
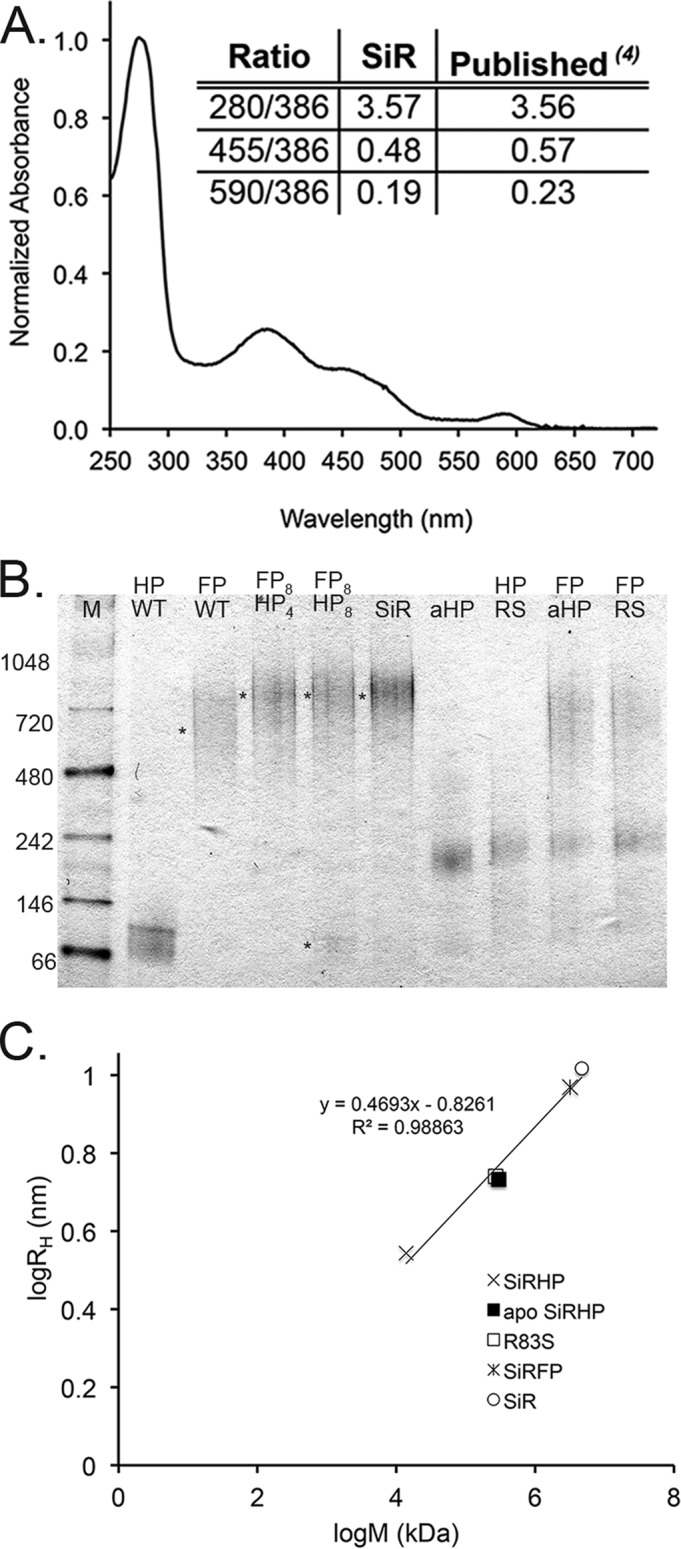
UV-visible spectroscopy, native PAGE, and DLS analysis of SiR. A, the UV-visible spectrum of recombinant SiR shows the expected peaks at 386, 455, and 590 nm at the expected ratios, characteristic of the overlapping siroheme and flavin cofactors of the SiR holoenzyme (4) (inset). B, 4–16% BisTris NativePAGE gel. Lane 1, NativeMark size standards (in kDa; Life Technologies). SiRHP (HP) and SiRFP (FP) controls are shown in lanes 2 and 3, respectively. Mixtures of SiRFP and SiRHP in a ratio of 8:4 subunits, (lane 4) shift the band to correspond to recombinant SiR (lane 6). Mixing SiRFP and SiRHP in a ratio of 8:8 subunits results in an excess of unbound SiRHP (lane 5). Apo-SiRHP (lane 7) and R83S (lane 8) are significantly larger than WT SiRHP. Neither apo-SiRHP nor R83S binds to SiRFP (lanes 9 and 10). C, log/log plot of hydrodynamic radius of SiR, SiRFP, SiRHP, apo-SiRHP, and R83S determined by DLS versus molecular weight (69) as determined by blue native PAGE.
Blue Native PAGE
Blue native gel analysis was performed according to the manufacturer's recommendations on 4–16% NativePAGE gels with NativeMark size standards (Life Technologies). Briefly, 2 μg of protein was added along with the Coomassie dye and electrophoresed for 3 h at 150 V. Gels were fixed and destained overnight in a 40% methanol, 10% acetic acid solution. Masses of SiRHP, apo-SiRHP, SiRFP, and SiR were determined from a calibration curve generated from the known sizes of the NativeMark protein standards versus migration distance from the top of the gel. Gel electrophoresis was performed in triplicate, and the masses reported are an average of three independent measurements.
Dynamic Light Scattering (DLS)
DLS experiments were carried out with a DynaPro-99 instrument attached to a DynaPro-MSXTC temperature-controlled microsampler (Wyatt/Protein Solutions, Santa Barbara, CA). Each protein sample was centrifuged (14,000 × g for 30 min at 5 °C) and then placed into a 45-μl DynaPro cuvette. The mean hydrodynamic radius (RH) and percentage of polydispersity were derived from the autocorrelation curve of the scattered light intensity based on the diffusion coefficient (Dt) from Stokes-Einstein's equation,
in which k is the Boltzmann constant, T is temperature in Kelvin, and η is the viscosity of water. Each 0.5 mg/ml protein sample was measured at 5 °C and presented as an average of 10 measurements. All data were analyzed at optimal resolution with Dynamics version 7.0.0.94 software.
Far UV Circular Dichroism Analysis
Circular dichroism (CD) experiments were carried out with an AVIV, model 410, spectrometer (AVIV Biomedical, Lakewood, NJ) attached to a CFT-33 circulating chiller (NESLAB, Portsmouth, NH). All samples were dialyzed overnight into 50 mm KPi, pH 7.8, and adjusted to 0.25 mg/ml. CD spectra were collected from 260–195 nm in a 1-mm path length quartz cuvette with a 1-nm bandwidth and a 3-s averaging time. An average of three scans was baseline-corrected, smoothed, and converted to mean residue ellipticity for comparison.
In Vivo Complex Formation
BL21(DE3) E. coli cells were co-transformed with SiRFP pcCDFDuet1 plasmid and His6 R83S pBAD plasmid; four independent colonies were grown under ampicillin/streptomycin selection. Expression of proteins was induced with 1 mm IPTG and 0.05% l-arabinose. Cells were lysed by repetitive freeze-thaw, and the His6-tagged SiRHP, along with bound SiRFP, was captured on Ni-NTA affinity agarose. Recovered protein was analyzed by SDS-PAGE and stained with colloidal Coomassie stain (19).
HDX and Liquid Chromatography (LC)-Electrospray Ionization, Fourier Transform Ion Cyclotron Resonance (FT-ICR) Mass Spectrometry Analysis
HDX experiments were optimized and automated with an HTC PAL autosampler (Eksigent Technologies, Dublin, CA). HDX samples were prepared in a 5-μl volume at 20 μm (in monomer SiRHP, octomer SiRFP, or SiR oligomer) in 65 mm KPi, 200 mm NaCl, and 1 mm EDTA at pH 7.8. HDX was initiated when this stock was diluted with 50 μl of HEPES buffer in D2O at pH 7.5. Triplicate HDX incubation periods were performed for 0, 0.5, 1, 2, 4, 8, 15, 30, 60, 120, and 140 min. Quenching by the addition of 25 μl of 200 mm tris(2-carboxyethyl)phosphine and 6 m urea in 1.0% formic acid was followed by the addition of 25 μl of saturated protease type XIII (Sigma-Aldrich) in 1.0% formic acid, yielding a final pH of 2.3. Digestion proceeded for 3 min at 1 °C before injection for LC-MS analysis.
Peptide separation and desalting were performed over a Pro-Zap Expedite MS C18 column (1.5-μm particle size, 500-Å pore size, 2.1 × 10 mm2; Grace Davidson, Deerfield, IL) with a Jasco high performance liquid chromatography/supercritical fluid chromatography system controlled by an HTC PAL autosampler. Peptides were eluted at a flow rate of 300 μl/min over 2.5 min with a gradient from 2 to 95% Buffer B (Buffer A: acetonitrile/H2O/formic acid (4.5:95:0.5); Buffer B: acetonitrile/H2O/formic acid (95:4.5:0.5)). A post-column splitter reduced the flow rate by 1:1,000 for efficient electrospray ionization.
After ionization, the sample was introduced into a custom-built hybrid LTQ 14.5 T FT-ICR mass spectrometer (Thermo Fisher Scientific) (20). Over a period of 6.5 min, 319 mass spectra were collected from m/z 400 to 1,300, at high mass resolving power (m/Δm50% = 200,000 at m/z 400, in which Δm50% is the peak full width at half-maximum peak height). External ion accumulation (21) was performed in the linear ion trap with a target ion population of 3 million charges for each FT-ICR measurement. LTQ-accumulated ions were transferred (∼1-ms transfer period) (22) through three octopole ion guides (2.2 MHz, 250 Vp-p) to a capacitively coupled (23) closed cylindrical ICR cell (55 mm inner diameter) (24) for analysis. The ion accumulation period was typically less than 100 ms during peptide elution, and the FT-ICR time domain signal acquisition period was 767 ms (leading to an overall duty cycle of 1 Hz/acquisition). Automatic gain control (25) and high magnetic field (26) provided excellent external calibration (27, 28) for mass accuracy (typically less than 500 ppb root mean square mass error).
After peptide masses were measured for the free and complexed proteins, the measured deuterium uptake (D) at each time point was calculated by dividing the measured deuteration level at each time point by the calculated maximum uptake (Dmax; an n-amino acid-long peptide can take up n − 1 deuteriums in the absence of prolines).
Deuterium uptake profiles were fitted by a well characterized maximum entropy method algorithm (29). Although the maximum entropy method generally fits the experimental data well, it is limited to three rate constants and can deviate for very rapid exchange if the initial time points are not accurate due to incomplete mixing.
Small Angle X-ray Scattering (SAXS)
SAXS measurements on the full-length, monomeric SiRHP were made at the BioCAT/18ID beamline at the Advanced Photon Source, Argonne National Laboratory (Chicago, IL) (30). A photon-counting PILATUS 3 1M detector was used to record the scattered x-rays at a wavelength of 1.03 Å. The 3.5-m sample-to-detector distance yielded a range of 0.005–0.33 Å−1 for the momentum transfer (q = 4π sinθ/λ, where 2θ is the angle of scatter between the incident and scattered beam and λ is the x-ray wavelength). The SiRHP sample was loaded onto a Superdex 200 column in 65 mm KPi, 200 mm KCl, 1 mm EDTA, pH 7.8, through a chromatography system (ÄKTA pure, GE Healthcare). The standard data reduction procedure for biological SAXS was performed with the programs in the ATSAS package (31). The program CRYSOL was used to calculate the theoretical x-ray scattering profile with the atomic model from the crystal structure of spinach NiR (Protein Data Bank accession code 2AKJ (9)) (32). Missing N-terminal amino acids were represented by a chain of dummy residues and added to the crystal structure with the program BUNCH to best fit the experimental data from the full-length SiRHP (33). The ensemble optimization method (34) was used to investigate the flexibility of the 80 missing residues.
SiR Complementation Assays
SiRHP-deficient E. coli (cysI−, Keio strain JW2733 (14)) cells were transformed with one of the following: empty pBAD plasmid, Δ60/untagged SiRHP-expressing pBAD plasmid, Δ80/untagged SiRHP-expressing pBAD plasmid, or full-length/untagged wild-type SiRHP-expressing pBAD plasmid as a control. All SiRHP-expressing plasmids were bicistronic cysI-cysG constructs. Cells were grown overnight in Luria-Bertani (LB) medium (35) selected with 50 μg/ml kanamycin and 100 μg/ml ampicillin. All cultures were then harvested, washed gently in M9 medium, and plated through serial dilution onto either M9-agar plates containing 50 μg/ml kanamycin, 100 μg/ml ampicillin, and 0.05% l-arabinose or LB medium with the same antibiotics and induction agent. The latter was used as a positive control for cell growth. Kanamycin and ampicillin maintained the cysI− deficiency and the pBAD plasmid and l-arabinose induced SiRHP expression. M9 medium provided sulfur in the form of sulfate (SO32−) (35), so cysI− bacteria were unable to sustain growth on this medium without SiRHP activity. Cell growth was assessed after 48 h.
Analytical SEC
SiRFP and SiRHP variants were mixed in 65 mm KPi at a 1:2 ratio of subunit monomers before being applied to a Superose 6 column in 65 mm KPi, 200 mm KCl, 1 mm EDTA, pH 7.8. All UV-visible traces of the protein elutions were normalized to a maximum of 1 for the highest peak. Likewise, SiR, SiRHP, Δ80-SiRHP, apo-SiRHP, and apo-Δ80-SiRHP were applied individually to a Superose 6 column in 65 mm KPi, 200 mm KCl, 1 mm EDTA, pH 7.8, and compared to assess their oligomeric state.
Results
Recombinant SiRHP Assembles Like Native SiR
Spectroscopic analysis of recombinant SiR agreed with the defining optical spectrum that derives from overlap of the protein signature (A280), the siroheme (A386 and A590), and the flavin cofactors (A390 and A455) (4). The ratio of these signature peaks absolutely defines the stoichiometry of the cofactors, showing that our recombinant enzyme matched the native enzyme (Fig. 1A).
Wild-type SiR holoenzyme dissociates as monomeric SiRHP and octomeric SiRFP when treated with a high concentration of urea (1). Similarly, when either component is expressed recombinantly in isolation, wild-type SiRHP is monomeric, and full-length SiRFP is octomeric (36, 37). SiRHP has a calculated molecular mass of 64 kDa, and our recombinant protein, with a His6 tag, ran at a slightly higher mass in native gel analysis, corresponding to a protein of 71 ± 3 kDa (Figs. 1B (lane 2) and 2B). Recombinantly expressed, untagged SiRFP ran as a broad band, associated with a molecular mass of about 660 ± 30 kDa (Fig. 1B, lane 3).
FIGURE 2.

Analysis of apo-SiRHP. A, UV-visible spectra of wild-type SiRHP (solid line), Δ60-SiRHP (short dashed/dotted line), and Δ80-SiRHP (long dashed/dotted line) showing siroheme's characteristic absorbances at 386 and 590 nm, absent in the apo-SiRHP (dotted line) and R83S (dashed line) spectra. B, Ellman's reagent test showing the absence of the Fe4S4 cluster in apo-SiRHP and R83S via detection of solvent-accessible thiols. The Fe4S4 cluster is coordinated by four cysteine residues, which, when absent, interact with Ellman's reagent to produce a signal of four reduced sulfur equivalents. C, CD spectra of wild-type SiRHP (solid line), Δ80-SiRHP (M-dash/dot), apo-SiRHP (dotted line) and apo-Δ80-SiRHP (dashed line). Apo-SiRHP and apo-Δ80-SiRHP have a similar change in secondary structure compared with SiRHP and Δ80-SiRHP upon the loss of siroheme cofactor. D, apo-SiRHP does not bind SiRFP in vivo. PageRuler protein markers (in kDa; Thermo Scientific) are shown in lanes 1 and 4. SiRFP (*) co-elutes with wild-type SiRHP (#) (lane 1) but is significantly reduced when co-precipitated with the R83S variant (lane 2). SiRFP is expressed strongly in both, shown by residual SiRFP in the unbound fractions (lanes 3 and 4). Residual SiRFP that co-elutes in the R83S variant probably corresponds to the small amount of R83S that is able to bind siroheme. Error bars, S.D.
To address the molecular mass and stoichiometry of the holoenzyme, we showed that octomeric SiRFP shifted into SiR holoenzyme when mixed with 4 molar eq of SiRHP (Fig. 1B, lane 4). Additional SiRHP at 8 molar eq did not shift the complex to a higher molecular mass, and excess monomeric SiRHP remained (Fig. 1B, lane 5). In addition to having the correct distribution of protein, cofactors, and siroheme, purified recombinant SiR ran at the expected molecular mass for an α8β4 oligomer at 800 ± 13 kDa (Fig. 1B, lane 6) and showed sulfite reduction activity similar to that originally reported, 2.6 ± 0.1 μmol NADPH/min·mg SiR (4).
To assess the integrity of the proteins in solution, we performed DLS on the individual subunits and the holoenzyme. As expected from a sample that behaved similarly in the native gel environment and in solution, there was a linear log-log relationship between the mass determined from blue native PAGE and the RH determined by DLS (Fig. 1C).
Apo-SiRHP Binds neither Siroheme nor the Fe4S4 Cluster and Is Tetrameric
We recently showed that, despite predictions, Arg-83 is not directly involved in catalysis but is rather more important for siroheme binding (13). Further, we observed that the R83S SiRHP point variant is more α-helical than the wild-type enzyme (13). We wished to explore whether the altered CD spectrum was a consequence of the R83S amino acid alteration or the absence of siroheme. Therefore, we endeavored to make a wild-type but “apo” enzyme. Expression in a cysG− strain (Keio collection strain JW3331 (14)) resulted in fully apo-SiRHP. The absence of siroheme was clear from the UV-visible spectrum of the purified protein, which lacked the characteristic absorbances at 386 and 590 nm (Fig. 2A). Ellman's reagent did not react with reduced, wild-type SiRHP but indicated the presence of four reduced sulfur atoms for both apo-SiRHP and R83S variants; the sulfur atoms correspond to the four cysteine residues that would otherwise coordinate the Fe4S4 cluster (Fig. 2B).
As expected from CD analysis of R83S (13), apo-SiRHP also had different secondary structure than wild-type SiRHP (Fig. 2C). Further, apo-SiRHP, generated either by purification of the wild-type enzyme in the absence of siroheme synthase (CysG) or through the R83S mutation was also higher in apparent molecular mass than wild-type SiRHP (Fig. 1B, lanes 7 and 8). From its position in native gel electrophoresis, apo-SiRHP ran at about 240 kDa, consistent with a tetrameric assembly of 64-kDa monomers.
Only Wild-type SiRHP and SiRFP Assemble into the Holoenzyme Complex
Next, we wanted to know if the apo, tetrameric form of SiRHP could form a complex with SiRFP. In contrast to wild-type SiRHP, neither apo-SiRHP nor R83S SiRHP formed a complex with SiRFP when purified subunits were mixed, regardless of their stoichiometry (Fig. 1B, lanes 9 and 10). We then tested for in vivo assembly by co-expressing either wild-type or His6-tagged R83S SiRHP with untagged SiRFP in BL21(DE3) E. coli and capturing the resulting complex by Ni-NTA affinity purification. We then compared the efficiency of SiRFP capture for each SiRHP variant with SDS-PAGE analysis (Fig. 2D). When the two wild-type SiR components were expressed in the presence of CysG, SiRFP co-eluted with SiRHP at the same stoichiometry as it did for the purified holoenzyme. In contrast, SiRFP co-eluted with R83S at significantly lower efficiency. What residual SiRFP co-precipitated is probably due to the small amount of R83S that remained bound to siroheme (Fig. 2A) (13).
The N terminus of SiRHP Is Buried in the Holoenzyme
To gain insight into the nature of the holoenzyme, we measured the hydrogen/deuterium exchange rate for free SiRHP compared with that for the SiR holoenzyme. We discovered that SiRHP has a solid core composed of amino acids 81–570, corresponding to the region of the structure that is resolved in the x-ray crystallographic structure (9). The accessibility of these amino acids changed little upon complex formation, marked by low or no change in the exchange rate of these hydrogens for deuteriums for SiRHP relative to SiR (Fig. 3). In contrast, the hydrogen-deuterium exchange rates for the N-terminal 80 amino acids of SiRHP decreased upon assembly with SiRFP, indicative of protection (Figs. 3 and 4). These amino acids are not resolved in the x-ray crystallographic structure because they are either missing from the construct (amino acids 1–60) or disordered (amino acids 61–80) (9). This result was observed both with N-terminally His-tagged or untagged protein with >98% sequence coverage and >99% without the His6 tag.
FIGURE 3.

SiRHP HDX. Representative absolute deuterium uptake profiles show deuterium incorporation versus exchange time (in h) for the SiRHP monomer (blue) versus SiRHP in the holoenzyme complex (red). The peptide is indicated for each.
FIGURE 4.

Heat map for SiRHP HDX and SAXS modeling. HDX shows that the N terminus of SiRHP is buried in the SiR complex. A, sequence of SiRHP in which light-to-dark blue shows increasingly buried residues and pink-to-red shows increasingly exposed residues, measured by the absolute deuterium uptake. Leu-80 is in boldface type. B, six independent models of the N terminus in E. coli SiRHP built from a SAXS curve, superimposed, show that the compact N-terminal 80 amino acids (yellow, displayed as pseudochain models), which are missing in the x-ray crystallographic structure (9), sit over the active site, as in the NiR homolog (C). Colors are as in A, modeled from Protein Data Bank files 2AKJ (38) (yellow, N-terminal amino acids 1–80, as in B) and 2GEP (46) (gray, amino acids 80–570).
The SiRHP N Terminus Is Not Flexible and Sits near the Active Site
We next wanted to learn more about the nature of the missing or disordered SiRHP amino acids that were identified as important for complex formation. We performed SAXS on full-length, monomeric SiRHP to test the N-terminal 80 amino acids' flexibility and position relative to its core S/NiRR domains. Ensemble optimization method analysis showed a broad distribution of radius of gyration (Rg), ranging from 22 to 39 Å, for a random pool that was generated by assuming highly flexible N-terminal residues as a random chain (Fig. 5A). In contrast, after optimizing the ensemble to fit the experimental data, the Rg peak was narrow (Fig. 5A). Although the N terminus of SiRHP is easily cleaved by proteolysis (9, 13), this SAXS analysis shows that it is not disordered in solution. Further, the peak was centered at the theoretical Rg of 23.8 Å, estimated from the x-ray crystal structure of the homologous siroheme-containing spinach NiR (Protein Data Bank accession code 2AKJ (38)), suggesting that full-length SiRHP was compact, as in NiR, where the full-length protein is ordered in the crystal structure.
FIGURE 5.
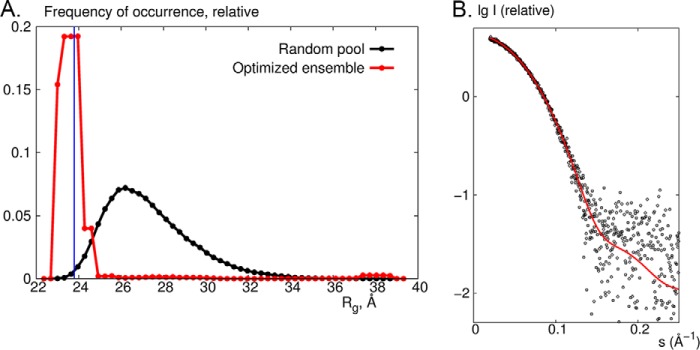
SAXS on SiRHP. A, ensemble optimization method analysis showing the Rg distributions from a pool of 10,000 models generated by adding 80 N-terminal residues as a random loop of a native-like chain (black) and for the optimized ensemble of which the averaged theoretical scattering profiles fit the experimental data (red). The blue line corresponds to Rg = 23.8 Å estimated from the x-ray crystal structure of spinach NiR (Protein Data Bank accession code 2AKJ (38)). B, comparison of experimental data (black circles) with the theoretical x-ray scattering profile calculated from the x-ray crystal structure of NiR (red) by use of the program CRYSOL (32).
To further obtain information about the position of the missing N-terminal amino acids, we added the residues to the existing structure to fit the experimental data by use of rigid body and ab initio modeling. Six independent runs yielded models that fit the data equally well (goodness of fit, χ2 ∼0.7) (Fig. 4B). Despite the non-uniqueness and low resolution (∼25 Å) of SAXS models, which precludes absolute positioning of the residues in question, the 80 N-terminal residues were clearly held tightly near the active site. Moreover, the experimental SAXS curve agreed perfectly with a theoretical curve calculated from the crystal structure of NiR (38) (Figs. 4C and 5B). These N-terminal amino acids are the most variable in what is otherwise a strongly conserved protein. Overall, the sequences are 26% identical. The C-terminal S/NiRR domains are 27% identical, whereas the first 80 amino acids are only 17% identical, and the NiR has long insertions in this region.
The N-terminal 80 Amino Acids of SiRHP Are Required for Complex Formation and in Vivo Function
HDX measures the rate at which hydrogens exchange to deuteriums in proteins. By comparing the identical protein in two different states, such as with or without a binding partner, one can infer details about how the two components change upon interaction. We performed biochemical analysis of SiRHP truncations to show that the region we identified in HDX experiments is the true interaction interface and not the result of a conformational change upon assembly that is far from the interface. We generated N-terminal truncation constructs of SiRHP missing either the first 60 amino acids or the first 80 amino acids (Fig. 6A). Next, we tested both truncated proteins for the presence of cofactor by use of UV-visible spectroscopy and for reductase activity by use of a chemical assay. Both truncations have a spectroscopic signal indicating formed siroheme-Fe4S4 clusters (Fig. 2A). Δ60-SiRHP showed 3-fold higher activity, and Δ80-SiRHP showed 6-fold higher activity than full-length SiRHP with NH2OH substrate when supplied with electrons from reduced methyl viologen. Nonetheless, neither truncated construct was able to complement SiRHP deficiency in a cysI− E. coli strain (Fig. 6B). Similarly, neither Δ60- nor Δ80-SiRHP was able to assemble with SiRFP in vitro, measured by SEC (Fig. 6, C and D).
FIGURE 6.

Functional analysis SiRHP truncations. A, schematic of the SiRHP domains showing the variable N terminus, the first S/NiRR (9), and a short linker followed by a second S/NiRR. B, SiRHP complementation assay of cysI− E. coli (14) transformed with SiRHP-expressing pBAD, empty pBAD, Δ60-SiRHP-expressing pBAD, or Δ80-expressing pBAD vectors grown on minimal M9 (35) or complete LB medium as a positive control. Only wild-type SiRHP expression can complement for growth. C, SEC analysis of SiRHP (red), SiRFP (blue), and SiR (green) show the relative sizes of the complex and its components. D, when SiRFP and excess SiRHP are mixed to ensure complete complex formation, all SiRFP shifts into a higher molecular weight complex, consistent with the size of SiR (purple, solid line). When SiRFP and excess Δ60 SiRHP are mixed, the peaks remain consistent with SiRFP and a slightly smaller SiRHP (purple, dashed lines). When SiRFP and Δ80 are mixed, the peaks remain consistent with SiRFP and an even smaller SiRHP (purple, dotted lines). mAU, milliabsorbance units.
Two Patches of SiRFP Alter Their Accessibility upon Holoenzyme Complex Formation but Only One Forms a Stable Interface with SiRHP
We also measured the hydrogen/deuterium exchange rates for SiRFP compared with SiR holoenzyme (Fig. 7), with 97% sequence coverage. In full-length SiRFP, two distinct peptides were protected upon complex formation, one within the N-terminal FMN domain (amino acids 94–112) and another within the C-terminal NADPH domain (amino acids 496–502) (Fig. 8, A and B). No other regions changed significantly upon complex assembly.
FIGURE 7.
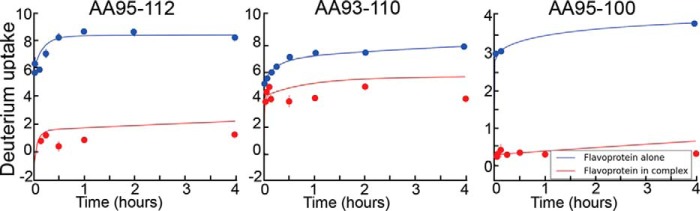
SiRFP HDX. Representative absolute deuterium uptake profiles showing deuterium incorporation versus exchange period (in h) for the SiRFP octomer (blue) and SiRFP in the holoenzyme complex (red). The peptide is indicated for each.
FIGURE 8.

HDX of SiRFP. A, sequence of SiRFP, in which light-to-dark blue shows increasingly buried residues and pink-to-red shows increasingly exposed residues. Alternating italic and Roman type denotes domains (amino acids 1–60 make up the N-terminal octomerization domain, amino acids 61–207 make up the FMN domain, amino acids 208–443 make up the FAD domain, and amino acids 444–599 make up the C-terminal NADPH domain). B, SiRFP, modeled by combining the two partial structures of its FMN (SiRFPFMN) (42) or FAD and NADPH (SiRFP43) (40) domains in the “closed” conformation defined by the human CYPOR (48). The mapped sequences that are buried upon complex formation are far from one another, colored as in A. C, charged surface of SiRFP, in which red is negatively charged and blue is positively charged, determined by the Coulomic potential defined by Chimera (70). D, charged surface of SiRHP with its modeled N terminus, represented as in C.
Given that HDX reports only on regions that change upon complex formation and not directly about specific interface regions, we generated SiRFP truncations that have previously been shown to fold independently (39–42). To ensure that each SiRFP construct presented proper cofactor binding, we measured their UV-visible spectra (Fig. 9, insets). Next, we mixed SiRFPFMN (amino acids 1–208) and SiRHP and measured formation of complex with SEC (Fig. 9A). This domain of SiRFP was unable to form a complex with SiRHP. Although the exchange rate of this peptide changes upon formation of the holoenzyme, it does not make a sufficiently strong interaction with SiRHP to persist on its own. In contrast, SiRFP43 formed a complex with SiRHP that shows spectroscopic features similar to the holoenzyme (Figs. 1A and 9B (inset)). SiRFP43 mixed with SiRHP showed NADPH-catalyzed sulfite reduction activity similar to the protein-free control, which was unmeasurable.
FIGURE 9.

SEC analysis of SiRFP truncations and Apo-SiRHP. A, SiRHP was mixed with C-terminally truncated SiRFP, an octomer of the FMN domains (SiRFPFMN). No higher-order structure was formed, indicating that the patch of amino acids 93–110 is not sufficient for forming a tight complex with SiRHP. Inset, UV-visible spectroscopy shows that SiRFPFMN binds flavin cofactor. B, SiRHP was mixed with N-terminally truncated SiRFP, a monomer of the FAD and NADPH domains (SiRFP43). A larger complex of higher molecular weight appears, indicating that the patch of amino acids 496–502 is sufficient for forming a tight complex. Inset, UV-visible spectroscopy shows that SiRFP43 binds flavin cofactor, and SiRHP/SiRFP43 shows a spectrum similar to that of SiR holoenzyme (Fig. 1A). C, wild-type SiRHP and Δ80-SiRHP run on SEC as tight peaks, corresponding to monomers of ∼64 kDa that differ slightly in size because of the N-terminal 80-amino acid truncation in Δ80-SiRHP. D, apo-SiRHP and apo-Δ80-SiRHP run at positions significantly different from those of their metallated counterparts because apo-SiRHP is a homotetramer, whereas apo-Δ80-SiRHP is a monomer. Both are broad, shouldered peaks indicative of loosely packed protein.
The N Terminus of SiRHP Also Modulates Oligomerization of the Apo-SiRHP Tetramer
We next tested whether the N-terminal 80 amino acids of SiRHP also played a role in the altered structure and assembly state of apo-SiRHP with CD and SEC. Wild-type SiRHP and Δ80-SiRHP were monomeric with largely α-β secondary structure (Figs. 1, 2C, and 9C). In contrast, apo-SiRHP formed a 240-kDa complex with a broad SEC peak and had a higher α-helical content than the metallated form (Figs. 1B, 2C, and 9D). Apo-Δ80-SiRHP shared the altered CD spectrum with apo-SiRHP (Fig. 2C). Like apo-SiRHP, apo-Δ80-SiRHP had a broad, shouldered SEC peak; unlike apo-SiRHP, the position of the N-terminally truncated peak corresponded to the position of monomeric SiRHP (Fig. 9, C and D).
Discussion
Despite 50 years of study of SiR structure and enzyme activity (4, 9, 13, 40, 42–46), we do not know how its subunits assemble and, therefore, how they work to catalyze electron transfer. Siroheme-dependent assimilatory NADPH-SiR is not a particularly efficient enzyme and does not discriminate strongly between sulfite and nitrite substrates (18). Recent discovery of a non-siroheme SiR that is both more efficient and better able to discriminate between sulfite and nitrite (47) suggests that there is perhaps something unique about the architecture of this SiR holoenzyme that constrained its evolutionary progress in becoming an ideal enzyme. Here, we report the structural elements of each subunit that are important for assimilatory NADPH-SiR assembly and propose a model that invokes four structural SiRFP molecules for each functional SiRFP-SiRHP dimer. Further, we have shown that the N terminus of SiRHP plays important roles in mediating both the SiRFP-SiRHP interaction and oligomerization of apo-form, non-functional SiRHP proteins that cannot make a complex with SiRFP. Taken together, these observations suggest a model for quality control of SiR assembly in organisms that use this combination of reductase/oxidase subunits.
SiR Asymmetry and Conformational Dynamics
SiR is unique in the octomeric state of its reductase subunit, a CYPOR homolog that uses NADPH, FAD, and FMN to transfer electrons to the active site of the accompanying SiRHP metalloenzyme. All other known examples of this type of flavoprotein exist in a conformationally dynamic, 1:1 partnership with its oxidase, either α1β1 (CYP (10) and methionine synthase (12)) or α2β2 (nitric-oxide synthase (11)). Significant modification to their protein architecture was necessary to constrain the molecules for structure determination (10, 48). Monomeric SiRFP, formed by removing its N-terminal 60 amino acids, is also dynamic, evidenced by the disordered FMN-binding domain in multiple crystal forms (40); likewise, octomeric SiRFP is conformationally complex, evidenced by its broad band in native gel analysis, even when it is part of the holoenzyme (Fig. 1B). In SiR, early cofactor and amino acid analysis pointed to an asymmetric stoichiometry between the α (SiRFP) and β (SiRHP) subunits, where the holoenzyme is an α8β4 oligomer (1) but monomeric SiRFP can form an active α1β1 oligomer (5). Binding assays reported here indicate that the asymmetric stoichiometry of SiR is real; there is an extra reductase unit per oxidase (Figs. 1 and 6C).
Importance of the SiRHP N Terminus in SiR Formation
The N-terminal 80 amino acids from SiRHP are required for complex formation with SiRFP, measured either for residual transient interactions in vivo by complementation of a SiRHP defect (Fig. 6B) or directly by SEC (Fig. 6D). N-terminally truncated SiRHP variants cannot complement the SiRHP defect, but they have intact active sites (Fig. 2A) and are active for catalysis when they are chemically reduced (see “Results”). Taken together, these results suggest that the growth deficiency in complementation assays is due to the truncations' inability to form holoenzyme in the cell, even transiently, rather than a catalytic defect. In fact, N-terminal truncations are more active than full-length SiRHP, akin to the N149W active site loop variant (13), suggesting an interaction between these functionally important structural elements. Interestingly, SiR and NiR seem to share a similar N-terminal architecture in that the N terminus of SiR is also packed tightly against its core despite those amino acids being disordered in the x-ray crystal structure (Fig. 4, B and C) (9). Spinach NiR relies on a single ferredoxin for its reducing equivalents, with which it interacts only transiently (49); clearly, further experimentation is needed to learn whether the N terminus of NiR plays a similar role in electron transfer from the different oxidase.
Apo-SiRHP Is a Tetramer
The SiRHP N-terminal 80 amino acids are also implicated in oligomerization of the apo-form of SiRHP, which has significant structural differences, beyond the absence of the cofactors, from the metallated form. Other apo-metalloenzymes, like ferredoxin (50–52) or carbonic anhydrase (53, 54), take on altered structures that are consistent with a molten globule state, marked by disruption of tertiary structure, maintenance of secondary structure, and a slight (∼20%) increase in size (55). Still other apo-metalloenzymes like copper-zinc superoxide dismutase take on amyloid structure (56).
Apo-SiRHP has some properties consistent with a molten globule state but others that are somewhat unique. As in a molten globule, apo-SiRHP maintains secondary structure, measured by CD, although it is different from (more α-helical than) the wild-type form (Fig. 2C). The apparent molecular mass of apo-SiRHP shifts from 64 to 240 kDa (Fig. 1), a larger increase than would be expected simply from loss of tertiary structure but consistent with formation of a homotetramer. The increase in apparent molecular mass disappears when the N terminus is removed (Fig. 9D), supporting the idea that oligomerization, not only loss of tertiary structure, is the cause. Nonetheless, apo-Δ80-SiRHP is slightly larger than its tightly packed metallated counterparts, judging by SEC (Fig. 9, C and D), as would be expected in a molten globule state.
The altered protein secondary structure seen in the absence of cofactors could arise from two non-exclusive sources. First, it might come from structural elements that make up the active site itself, suggesting that the active site is not preformed but reorganizes upon assembly with its cofactors. Second, altered secondary structure might directly come from changes to the N terminus, which modulates oligomerization with the SiRFP subunit (Figs. 3, 4, and 6) and into the apo-SiRHP homotetramer (Fig. 9D). Apo-Δ80-SiRHP shares its CD spectrum with apo-SiRHP (Fig. 2C), suggesting that the conformation of those 80 amino acids does not explain the altered secondary structure. Rather, altered secondary structure at the active site may serve as a signal to attract the bacterial chaperone and co-chaperone HscA and HscB that play a role in ISC-mediated iron-sulfur cluster biogenesis (57–59). Perhaps these chaperones additionally help to refold the active site for cofactor assembly and/or the SiRHP N terminus for SiRFP assembly.
Apo-SiRHP Is SiR Assembly-deficient
We propose that formation of an apo-SiRHP homotetramer through the same N-terminal SiRHP interaction domain that is required for SiRFP assembly blocks inactive apo-SiRHP from joining with SiRFP in the case of insufficient cellular siroheme (Fig. 10). SiRFP and SiRHP are encoded by cysJ and cysI as part of the cysteine regulon. Transcription of the cysJIH region of the operon is enhanced in response to N-acetyl-l-serine, the cysteine precursor, by the LysR-like transcription factor CysB (60). Despite its name, siroheme synthase (CysG) is part of the nitrate regulon, expressed constitutively at low levels but not overexpressed in response to sulfite starvation (61, 62). Transcription of the nirBDCG-cysG region of the operon is enhanced in response to NarL and NarP, which sense nitrite/nitrate, or the Fnr transcription factor, which senses oxygen (63). We propose that the apo-SiRHP homotetramer is a quality control check so that the cell does not expend energy in assembling inactive complexes that might arise because of differential transcriptional regulation of SiRHP and the enzyme that makes its cofactor. Perhaps there is a further mechanism in cofactor biogenesis by which siroheme insertion is regulated, either to prevent tetramerization of apo-SiRHP or reform active, monomeric SiRHP that can go on to assemble with SiRFP. Precedence for this type of metalloenzyme-chaperone partnering exists, for example in the role of MeaB in cofactor insertion into the coenzyme B12-dependent methylmalonyl-CoA mutase (64), in the role of NarJ in assembling the molybdenum cofactor in respiratory nitrate reductase (65), and in chaperoning copper to Cu-Zn superoxide dismutase (66).
FIGURE 10.

Oligomerization of Apo-SiRHP plays a role in quality control of SiR assembly. A proposed model for the role of the N-terminal 80 amino acids of SiRHP (blue) in blocking apo-SiRHP (gray) from assembling with the octomeric SiRFP and in modulating the interaction with four structural SiRFP molecules (yellow). Four functional SiRFP molecules interact transiently through their FMN domains to pass electrons to SiRHP.
SiR Holoenzyme Stoichiometry
Why is SiR composed of an asymmetric assembly of subunits? At first glance, the answer could lie in the apparent gene duplication-gene fusion event that ultimately resulted in the monomeric, rather than heterodimeric, SiRHP found in the assimilatory SiR enzyme. Most of the progenitors of this enzyme, the hetero-oligomeric, respiratory DSR, depend on a membrane-associated cytochrome c3 hydrogenase and a fused ferredoxin rather than the CYPOR homolog (67). Assimilatory plant SiRs and NiRs use a single copy of NADPH ferredoxin (6). If the asymmetry stems from a phylogenetic one, then the ancestor used an uncommon combination of hemoprotein/flavoprotein subunits. Another possibility is that the excess of reductase to oxidase subunits has a functional role in making SiR an efficient electron transfer enzyme, perhaps by allowing two different SiRFPs to provide electrons to a single SiRHP. Alternatively, four of the SiRFP molecules could serve as a scaffold for four active SiRFP-SiRHP heterodimers.
We show that two different interfaces on SiRFP become less accessible when it is bound to SiRHP. One surface is part of the N-terminal FMN-binding domain, and the other is part of the C-terminal NADPH-binding domain, far from the final FMN cofactor that ultimately passes electrons to SiRHP (18) (Fig. 8). Given this, we anticipated that there would be a strong interaction between the FMN-binding domain and SiRHP that would be the contact point for electron transfer; however, SiRFPFMN does not bind SiRHP (Fig. 9A). In contrast, SiRFP43, which contains the NADPH-binding domain but is missing the FMN-binding domain, does bind SiRHP (Fig. 9B). The NADPH binding domain is largely negatively charged, and the N terminus of SiRHP is predicted to be predominantly positively charged (Fig. 8, C and D), supporting the feasibility of their interaction.
A strong electrostatic interaction between SiRFP and SiRHP could be mediated by SiRFP amino acids Glu-495, Asp-496, and Glu-501 and SiRHP amino acids Arg-66, Arg-69, Gln-72, and Lys-73. This interaction would form a structural, rather than functional, interface, demonstrated by the catalytically inactive SiRFP43-SiRHP complex (see “Results”). We propose that only four of the eight SiRFP molecules bind to the four SiRHP molecules at this position. The N-terminal SiRFP octomerization domain would then scaffold the remaining four functional SiRFP molecules, akin to the multimeric metabolic enzyme pyruvate dehydrogenase, which has a solid core that tethers flexible enzymes at its periphery (68). Our model does not exclude the possibility that the structural SiRFP molecules could also serve as electron donors, but if so, there would be additional steric constraints to an already highly complex assembly. Further, all other known CYPOR homologs work as 1:1 complexes with their oxidase subunits. Likewise, all other known SiR/NiR hemoproteins work as 1:1 complexes with their reductase subunits, so a functional asymmetry would be inconsistent with what we know about the evolutionary history of SiR.
The second SiRFP-SiRHP contact point, on the FMN domain, is not sufficient to reconstitute a minimal subcomplex and is not strongly negatively charged (Figs. 8C and 9A). Nonetheless, this interface could form transiently on the functional SiRFP molecules and modulate electron transfer between the subunits. This model suggests that the active SiRFP molecules are highly dynamic, held in contact with the other SiRFPs through their N-terminal octomerization domain but free to come on and off SiRHP as NADPH/NAD+ cycle on and off the protein, passing electrons two at a time to the FAD and FMN cofactors (Fig. 10).
Author Contributions
M. E. S. conceived and coordinated the study and wrote the paper with contributions from I. A., J. M. P., A. G. M., and W. S., M. E. S., I. A., and J. P. G. designed, performed, and analyzed the experiments shown in Figs. 1, 2, 6, and 9. M. E. S., I. A., Y. T., N. L. Y., and A. G. M. designed, performed, and analyzed the experiments shown in Figs. 3, 4 (A and C), 7, and 8 (A and B). M. E. S., I. A., and W. S. designed, performed, and analyzed the experiments shown in Fig. 5. M. E. S. conceived of the model illustrated in Fig. 10. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. CD and DLS were performed in Florida State University Physical Biochemistry Facility, managed by Dr. Claudius Mundoma. HDX experiments were performed at the National High Magnetic Field Laboratory in Tallahassee, FL. We kindly thank Drs. Christopher Stroupe, Scott Stagg, and Kathryn Jones for helpful conversations.
This work was supported, in whole or in part, by National Institutes of Health, NIGMS, Grants 9 P41 GM103622 (for access to the Biophysics Collaborative Access Team (BioCAT) beamline) and 1S10OD018090-01 (for use of the PILATUS 3 1M detector). This work was also supported by National Science Foundation award MCB1149763 (to M. E. S.) and National Science Foundation award DMR-11–57490. The authors declare that they have no conflicts of interest with the contents of this article.
- SiR
- sulfite reductase
- SiRHP
- sulfite reductase hemoprotein
- SiRFP
- sulfite reductase flavoprotein
- CYPOR
- NADPH-cytochrome p450 reductase
- NiR
- nitrite reductase
- DSR
- dissimilatory sulfite reductase
- S/NiRR
- sulfite/nitrite reductase repeat
- HDX
- hydrogen/deuterium exchange
- BCA
- bicinchoninic acid
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- SEC
- size-exclusion chromatography
- SiRFPFMN
- SiRFP FMN domain (residues 1–208)
- SiRFP43
- SiRFP FAD/NADPH domains (residues 209–599)
- Ni-NTA
- nickel-nitrilotriacetic acid
- DLS
- dynamic light scattering
- FT
- Fourier transform
- ICR
- ion cyclotron resonance
- SAXS
- small angle x-ray scattering
- CysG
- siroheme synthase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- RH
- mean hydrodynamic radius
- Rg
- radius of gyration.
References
- 1. Siegel L. M., Davis P. S. (1974) Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. IV. The Escherichia coli hemoflavoprotein: subunit structure and dissociation into hemoprotein and flavoprotein components. J. Biol. Chem. 249, 1587–1598 [PubMed] [Google Scholar]
- 2. Faeder E. J., Davis P. S., Siegel L. M. (1974) Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. V. Studies with the Escherichia coli hemoflavoprotein depleted of flavin mononucleotide: distinct roles for the flavin adenine dinucleotide and flavin mononucleotide prosthetic groups in catalysis. J. Biol. Chem. 249, 1599–1609 [PubMed] [Google Scholar]
- 3. Siegel L. M., Rueger D. C., Barber M. J., Krueger R. J., Orme-Johnson N. R., Orme-Johnson W. H. (1982) Escherichia coli sulfite reductase hemoprotein subunit. Prosthetic groups, catalytic parameters, and ligand complexes. J. Biol. Chem. 257, 6343–6350 [PubMed] [Google Scholar]
- 4. Siegel L. M., Murphy M. J., Kamin H. (1973) Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. I. The Escherichia coli hemoflavoprotein: molecular parameters and prosthetic groups. J. Biol. Chem. 248, 251–264 [PubMed] [Google Scholar]
- 5. Zeghouf M., Fontecave M., Coves J. (2000) A simplifed functional version of the Escherichia coli sulfite reductase. J. Biol. Chem. 275, 37651–37656 [DOI] [PubMed] [Google Scholar]
- 6. Crane B. R., Getzoff E. D. (1996) The relationship between structure and function for the sulfite reductases. Curr. Opin. Struct. Biol. 6, 744–756 [DOI] [PubMed] [Google Scholar]
- 7. Rose W. C., Johnson J. E., Haines W. J. (1950) The amino acid requirements of man. 1. The role of valine and methionine. J. Biol. Chem. 182, 541–556 [Google Scholar]
- 8. Wagner M., Roger A. J., Flax J. L., Brusseau G. A., Stahl D. A. (1998) Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J. Bacteriol. 180, 2975–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crane B. R., Siegel L. M., Getzoff E. D. (1995) Sulfite reductase structure at 1.6 Å: evolution and catalysis for reduction of inorganic anions. Science 270, 59–67 [DOI] [PubMed] [Google Scholar]
- 10. Wang M., Roberts D. L., Paschke R., Shea T. M., Masters B. S., Kim J. J. (1997) Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. U.S.A. 94, 8411–8416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J., Martàsek P., Paschke R., Shea T., Siler Masters B. S., Kim J. J. (2001) Crystal structure of the FAD/NADPH-binding domain of rat neuronal nitric-oxide synthase: comparisons with NADPH-cytochrome P450 oxidoreductase. J. Biol. Chem. 276, 37506–37513 [DOI] [PubMed] [Google Scholar]
- 12. Olteanu H., Banerjee R. (2001) Human methionine synthase reductase, a soluble P-450 reductase-like dual flavoprotein, is sufficient for NADPH-dependent methionine synthase activation. J. Biol. Chem. 276, 35558–35563 [DOI] [PubMed] [Google Scholar]
- 13. Smith K. W., Stroupe M. E. (2012) Mutational analysis of sulfite reductase hemoprotein reveals the mechanism for coordinated electron and proton transfer. Biochemistry 51, 9857–9868 [DOI] [PubMed] [Google Scholar]
- 14. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ostrowski J., Wu J. Y., Rueger D. C., Miller B. E., Siegel L. M., Kredich N. M. (1989) Characterization of the cysJIH regions of Salmonella typhimurium and Escherichia coli B. DNA sequences of cysI and cysH and a model for the siroheme-Fe4S4 active center of sulfite reductase hemoprotein based on amino acid homology with spinach nitrite reductase. J. Biol. Chem. 264, 15726–15737 [PubMed] [Google Scholar]
- 16. Ostrowski J., Barber M. J., Rueger D. C., Miller B. E., Siegel L. M., Kredich N. M. (1989) Characterization of the flavoprotein moieties of NADPH-sulfite reductase from Salmonella typhimurium and Escherichia coli: physicochemical and catalytic properties, amino acid sequence deduced from DNA sequence of cysJ, and comparison with NADPH-cytochrome P-450 reductase. J. Biol. Chem. 264, 15796–15808 [PubMed] [Google Scholar]
- 17. Shibuya T., Tange T. Ø., Stroupe M. E., Moore M. J. (2006) Mutational analysis of human eIF4AIII identifies regions necessary for exon junction complex formation and nonsense-mediated mRNA decay. RNA 12, 360–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siegel L. M., Davis P. S., Kamin H. (1974) Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. 3. The Escherichia coli hemoflavoprotein: catalytic parameters and the sequence of electron flow. J. Biol. Chem. 249, 1572–1586 [PubMed] [Google Scholar]
- 19. Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L., Righetti P. G. (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333 [DOI] [PubMed] [Google Scholar]
- 20. Schaub T. M., Hendrickson C. L., Horning S., Quinn J. P., Senko M. W., Marshall A. G. (2008) High-performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 tesla. Anal. Chem. 80, 3985–3990 [DOI] [PubMed] [Google Scholar]
- 21. Senko M. W., Hendrickson C. L., Emmett M. R., Shi S. D. H., Marshall A. G. (1997) External accumulation of ions for enhanced electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 8, 970–976 [Google Scholar]
- 22. Wilcox B. E., Hendrickson C. L., Marshall A. G. (2002) Improved ion extraction from a linear octopole ion trap: SIMION analysis and experimental demonstration. J. Am. Soc. Mass Spectrom. 13, 1304–1312 [DOI] [PubMed] [Google Scholar]
- 23. Beu S. C., Laude D. A. (1992) Elimination of axial ejection during excitation with a capacitively coupled open trapped-ion cell for Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 64, 177–180 [Google Scholar]
- 24. Kaiser N. K., Savory J. J., McKenna A. M., Quinn J. P., Hendrickson C. L., Marshall A. G. (2011) Electrically compensated FT-ICR cell for complex mixture analysis. Anal. Chem. 83, 6907–6910 [DOI] [PubMed] [Google Scholar]
- 25. Schwartz J. C., Senko M. W., Syka J. E. (2002) A two-dimensional quadrupole ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 13, 659–669 [DOI] [PubMed] [Google Scholar]
- 26. Marshall A. G., Guan S. (1996) Advantages of high magnetic field for Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 10, 1819–1823 [DOI] [PubMed] [Google Scholar]
- 27. Ledford E. B., Jr., Rempel D. L., Gross M. L. (1984) Space charge effects in Fourier transform mass spectrometry mass calibration. Anal. Chem. 56, 2744–2748 [DOI] [PubMed] [Google Scholar]
- 28. Shi S. D. H., Drader J. J., Freitas M. A., Hendrickson C. L., Marshall A. G. (2000) Comparison and interconversion of the two most common frequency-to-mass calibration functions for Fourier transform ion cyclotron resonance mass spectrometry. Int. J. Mass Spectrom. 196, 591–598 [Google Scholar]
- 29. Zhang Z., Li W., Logan T. M., Li M., Marshall A. G. (1997) Human recombinant [C22A] FK506-binding protein amide hydrogen exchange rates from mass spectrometry match and extend those from NMR. Protein Sci. 6, 2203–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fischetti R., Stepanov S., Rosenbaum G., Barrea R., Black E., Gore D., Heurich R., Kondrashkina E., Kropf A. J., Wang S., Zhang K., Irving T. C., Bunker G. B. (2004) The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and x-ray absorption spectroscopy at the Advanced Photon Source. J. Synchrotron Radiat. 11, 399–405 [DOI] [PubMed] [Google Scholar]
- 31. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., Svergun D. I. (2012) New developments in the program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Svergun D., Barberato C., Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 28, 768–773 [Google Scholar]
- 33. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tria G., Mertens H. D., Kachala M., Svergun D. I. (2015) Advanced ensemble modelling of flexible macromolecules using x-ray solution scattering. IUCr J. 2, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., p. A.3, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 36. Wu J. Y., Siegel L. M., Kredich N. M. (1991) High-level expression of Escherichia coli NADPH-sulfite reductase: requirement for a cloned cysG plasmid to overcome limiting siroheme cofactor. J. Bacteriol. 173, 325–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeghouf M., Fontecave M., Macherel D., Covès J. (1998) The flavoprotein component of the Escherichia coli sulfite reductase: expression, purification, and spectral and catalytic properties of a monomeric form containing both the flavin adenine dinucleotide and the flavin mononucleotide cofactors. Biochemistry 37, 6114–6123 [DOI] [PubMed] [Google Scholar]
- 38. Swamy U., Wang M., Tripathy J. N., Kim S. K., Hirasawa M., Knaff D. B., Allen J. P. (2005) Structure of spinach nitrite reductase: implications for multi-electron reactions by the iron-sulfur:siroheme cofactor. Biochemistry 44, 16054–16063 [DOI] [PubMed] [Google Scholar]
- 39. Coves J., Zeghouf M., Macherel D., Guigliarelli B., Asso M., Fontecave M. (1997) Flavin mononucleotide-binding domain of the flavoprotein component of the sulfite reductase from Escherichia coli. Biochemistry 36, 5921–5928 [DOI] [PubMed] [Google Scholar]
- 40. Gruez A., Pignol D., Zeghouf M., Covès J., Fontecave M., Ferrer J. L., Fontecilla-Camps J. C. (2000) Four crystal structures of the 60 kDa flavoprotein monomer of the sulfite reductase indicate a disordered flavodoxin-like module. J. Mol. Biol. 299, 199–212 [DOI] [PubMed] [Google Scholar]
- 41. Gruez A., Zeghouf M., Bertrand J., Eschenbrenner M., Covès J., Fontecave M., Pignol D., Fontecilla-Camps J. C. (1998) The FNR-like domain of the Escherichia coli sulfite reductase flavoprotein component: crystallization and preliminary x-ray analysis. Acta Crystallogr. D Biol. Crystallogr. 54, 135–136 [DOI] [PubMed] [Google Scholar]
- 42. Sibille N., Blackledge M., Brutscher B., Covès J., Bersch B. (2005) Solution structure of the sulfite reductase flavodoxin-like domain from Escherichia coli. Biochemistry 44, 9086–9095 [DOI] [PubMed] [Google Scholar]
- 43. Siegel L. M., Monty K. J. (1966) Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation: application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta 112, 346–362 [DOI] [PubMed] [Google Scholar]
- 44. Murphy M. J., Siegel L. M., Kamin H., Rosenthal D. (1973) Reduced nicotinamide adenine dinucleotide phosphate-sulfite reductase of enterobacteria. II. Identification of a new class of heme prosthetic group: an iron-tetrahydroporphyrin (isobacteriochlorin type) with eight carboxylic acid groups. J. Biol. Chem. 248, 2801–2814 [PubMed] [Google Scholar]
- 45. Crane B. R., Siegel L. M., Getzoff E. D. (1997) Structures of the siroheme- and Fe4S4-containing active center of sulfite reductase in different states of oxidation: heme activation via reduction-gated exogenous ligand exchange. Biochemistry 36, 12101–12119 [DOI] [PubMed] [Google Scholar]
- 46. Crane B. R., Siegel L. M., Getzoff E. D. (1997) Probing the catalytic mechanism of sulfite reductase by x-ray crystallography: structures of the Escherichia coli hemoprotein in complex with substrates, inhibitors, intermediates, and products. Biochemistry 36, 12120–12137 [DOI] [PubMed] [Google Scholar]
- 47. Hermann B., Kern M., La Pietra L., Simon J., Einsle O. (2015) The octahaem MccA is a haem c-copper sulfite reductase. Nature 520, 706–709 [DOI] [PubMed] [Google Scholar]
- 48. Xia C., Hamdane D., Shen A. L., Choi V., Kasper C. B., Pearl N. M., Zhang H., Im S. C., Waskell L., Kim J. J. (2011) Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem. 286, 16246–16260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krueger R. J., Siegel L. M. (1982) Spinach siroheme enzymes: isolation and characterization of ferredoxin-sulfite reductase and comparison of properties with ferredoxin-nitrite reductase. Biochemistry 21, 2892–2904 [DOI] [PubMed] [Google Scholar]
- 50. Abdel-Ghany S. E., Ye H., Garifullina G. F., Zhang L., Pilon-Smits E. A., Pilon M. (2005) Iron-sulfur cluster biogenesis in chloroplasts: involvement of the scaffold protein CpIscA. Plant Physiol. 138, 161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leal S. S., Gomes C. M. (2007) Studies of the molten globule state of ferredoxin: structural characterization and implications on protein folding and iron-sulfur center assembly. Proteins 68, 606–616 [DOI] [PubMed] [Google Scholar]
- 52. Pilon M., Rietveld A. G., Weisbeek P. J., de Kruijff B. (1992) Secondary structure and folding of a functional chloroplast precursor protein. J. Biol. Chem. 267, 19907–19913 [PubMed] [Google Scholar]
- 53. Uversky V. N., Ptitsyn O. B. (1996) Further evidence on the equilibrium “pre-molten globule state”: four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. J. Mol. Biol. 255, 215–228 [DOI] [PubMed] [Google Scholar]
- 54. Andersson D., Hammarström P., Carlsson U. (2001) Cofactor-induced refolding: refolding of molten globule carbonic anhydrase induced by Zn(II) and Co(II). Biochemistry 40, 2653–2661 [DOI] [PubMed] [Google Scholar]
- 55. Ptitsyn O. B. (1995) Molten globule and protein folding. Adv. Protein Chem. 47, 83–229 [DOI] [PubMed] [Google Scholar]
- 56. Strange R. W., Antonyuk S., Hough M. A., Doucette P. A., Rodriguez J. A., Hart P. J., Hayward L. J., Valentine J. S., Hasnain S. S. (2003) The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J. Mol. Biol. 328, 877–891 [DOI] [PubMed] [Google Scholar]
- 57. Rouault T. A. (2012) Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech. 5, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Silberg J. J., Tapley T. L., Hoff K. G., Vickery L. E. (2004) Regulation of the HscA ATPase reaction cycle by the co-chaperone HscB and the iron-sulfur cluster assembly protein IscU. J. Biol. Chem. 279, 53924–53931 [DOI] [PubMed] [Google Scholar]
- 59. Bandyopadhyay S., Chandramouli K., Johnson M. K. (2008) Iron-sulfur cluster biosynthesis. Biochem. Soc. Trans. 36, 1112–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ostrowski J., Kredich N. M. (1989) Molecular characterization of the cysJIH promoters of Salmonella typhimurium and Escherichia coli: regulation by cysB protein and N-acetyl-l-serine. J. Bacteriol. 171, 130–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Belyaeva T., Griffiths L., Minchin S., Cole J., Busby S. (1993) The Escherichia coli cysG promoter belongs to the “extended −10” class of bacterial promoters. Biochem. J. 296, 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Peakman T., Busby S., Cole J. (1990) Transcriptional control of the cysG gene of Escherichia coli K-12 during aerobic and anaerobic growth. Eur. J. Biochem. 191, 325–331 [DOI] [PubMed] [Google Scholar]
- 63. Wu H., Tyson K. L., Cole J. A., Busby S. J. (1998) Regulation of transcription initiation at the Escherichia coli nir operon promoter: a new mechanism to account for co-dependence on two transcription factors. Mol. Microbiol. 27, 493–505 [DOI] [PubMed] [Google Scholar]
- 64. Lofgren M., Padovani D., Koutmos M., Banerjee R. (2013) A switch III motif relays signaling between a B12 enzyme and its G-protein chaperone. Nat. Chem. Biol. 9, 535–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Blasco F., Dos Santos J. P., Magalon A., Frixon C., Guigliarelli B., Santini C. L., Giordano G. (1998) NarJ is a specific chaperone required for molybdenum cofactor assembly in nitrate reductase A of Escherichia coli. Mol. Microbiol. 28, 435–447 [DOI] [PubMed] [Google Scholar]
- 66. Casareno R. L., Waggoner D., Gitlin J. D. (1998) The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J. Biol. Chem. 273, 23625–23628 [DOI] [PubMed] [Google Scholar]
- 67. Steuber J., Cypionka H., Kroneck P. M. (1994) Mechanism of dissimilatory sulfite reduction by Desulfovibrio desulfuricans: purification of a membrane-bound sulfite reductase and coupling iwth cytochrome c3 and hydrogenase. Arch. Microbiol. 162, 255–260 [Google Scholar]
- 68. Yu X., Hiromasa Y., Tsen H., Stoops J. K., Roche T. E., Zhou Z. H. (2008) Structures of the human pyruvate dehydrogenase complex cores: a highly conserved catalytic center with flexible N-terminal domains. Structure 16, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rollings J. E. (1992) in Laser Light Scattering in Biochemistry (Harding S. E., Satelle D. B., Bloomfield V. A., eds) pp. 275–293, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 70. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]