

Abstract
We report an unusual combination of features comprising delayed tooth eruption and closure of the anterior fontanel as the sole presenting features in a child with cleidocranial dysplasia (CCD). Radiological survey revealed the presence of wormian bones in the skull, pseudoepiphysis at the base of the bilateral second metacarpal, and midline ossification defects at pubic symphysis in the presence of essentially normal clavicles. DNA sequencing of the RUNX2 gene detected a novel nonsense mutation in exon1 (c.166C>T; p.Q56X) in its glutamine-alanine (Q/A) repeat domain. The genotypes of all published cases of CCD with normal clavicles were reviewed. Additional dental and otolaryngological features were enlisted. Three cases with a milder dental phenotype and normal clavicles were associated with a mutation in the Q/A domain. Collectively, we found a novel CCD-causing nonsense mutation p.Q56X in the Q/A domain of the RUNX2 gene.
Key Words: Clavicles, Cleidocranial dysplasia, RUNX2
Cleidocranial dysplasia (CCD; OMIM 119600), inherited in an autosomal dominant manner, is a generalized skeletal dysplasia. Individuals with this disorder present with persistently open sutures with bulging calvaria, hypoplasia or aplasia of clavicles, dental anomalies, wide pubic symphysis, short middle phalanx of the fifth finger, and associated vertebral anomalies. Its incidence is estimated to be 1:1,000,000.
Familial clustering of cases is known, but most cases are reported to be sporadic. CCD is caused by mutations in the RUNX2 gene in the small arm of chromosome 6 at 6p21.1 [Mundlos, 1999; Lin et al., 2011]. The condition was initially designated as cleidocranial dysostosis by Marie and Sainton [1898]. Because this condition is now recognized as a generalized disorder of bone development, it has been renamed as cleidocranial dysplasia [Rimoin, 1978].
Case Report
An 11-year-old boy presented to the pediatric dentistry clinic with a history of failure to shed the primary dentition and a delay in appearance of the secondary teeth. The boy was born by normal vaginal delivery after an uneventful pregnancy. Three-generation pedigree analysis was uninformative. The mother also had a history of delayed closure of the anterior fontanel compared to her siblings. All deciduous teeth appeared by 2 years of age. The patient presented with 35 kg weight (∼50th percentile), 145 cm height (∼50th percentile), a head circumference of 51 cm (∼50th percentile), and US/LS ratio of 0.9 which were normal for his age. There was no obvious facial dysmorphology (fig. 1). Oral examination showed retention of primary dentition with nonappearance of secondary dentition; an oral pantogram revealved the latter being normally developed lying beneath the primary teeth (fig. 2). All fontanels and sutures were closed. Both shoulders could not be approximated to midline either actively or passively, suggesting the presence of normally developed clavicles. Assessment of IQ was performed using the Binet Kulshreshtha test and suggested normal intelligence [Jain et al., 2013].
Fig. 1.

Facial appearance of the patient showing a minimal overbite due to abnormal dentition.
Fig. 2.
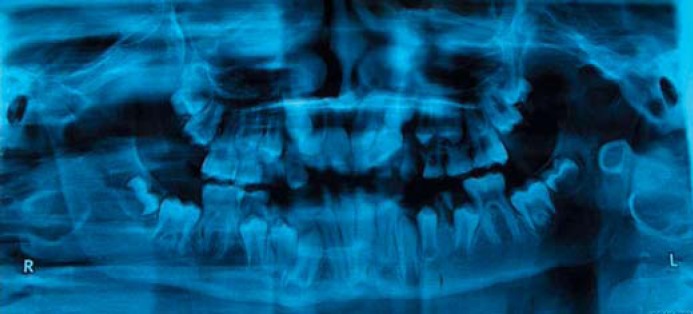
Orthopantomogram showing retained primary and appearing secondary dentition.
Radiographs of the skull revealed wormian bones in the occipital region and perisutural sclerosis. Chest radiographs demonstrated normally developed clavicles, ribs, and thoracic vertebral bodies (fig. 3). Radiographs of the hands depicted tapered distal phalanges of both thumbs, absence of tufting of distal phalanges of index fingers with second metacarpals showing accessory ossification centers at the bases (fig. 4). The bone age was between 8 and 10 years as assessed by the Greulich and Pyle atlas [Heyworth et al., 2013]. Radiographs of the pelvis showed delayed ossification of pubic symphysis; spine and long bones were essentially normal. Blood calcium, phosphate, alkaline phosphatase and thyroid profiles were normal. Sonographic examination of the abdomen and renal region revealed no abnormal findings. Sequencing of the RUNX gene identified a sporadic novel nonsense mutation (c.166C>T; p.Q56X) in the glutamine-alanine (Q/A) repeat domain of the RUNX2 transcription factor (fig. 5). This mutation could not be identified in his parents, siblings, or 100 ethnic controls.
Fig. 3.
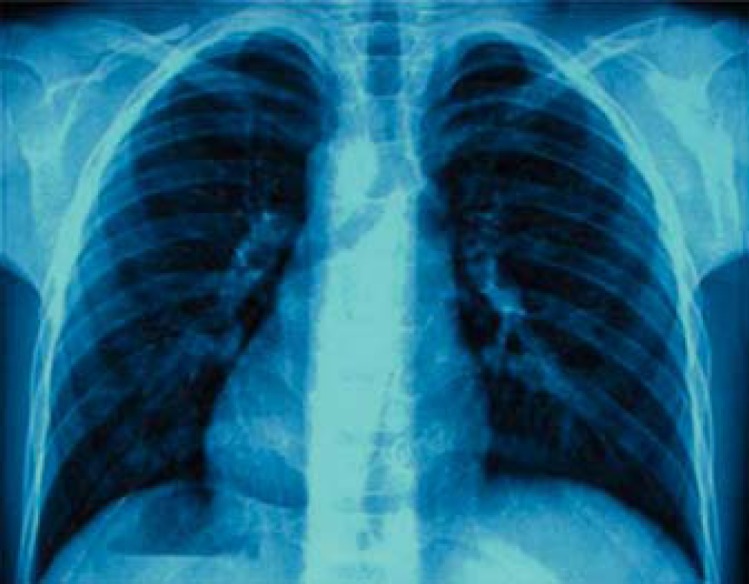
Chest radiograph showing the presence of normal bilateral clavicles with no rib or vertebral anomaly.
Fig. 4.
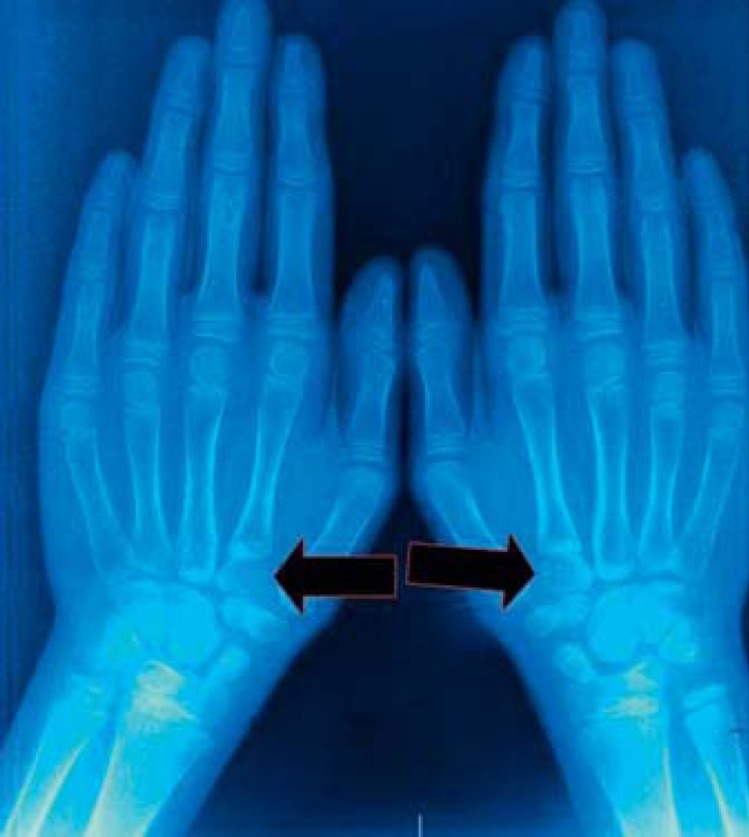
Psuedoepiphysis at the base of the second metacarpal (arrows).
Fig. 5.

Novel nonsense mutation (c.166C> T; p.Q56X) in the proband.
Discussion
CCD is a generalized skeletal dysplasia affecting bones of intermembranous and enchondral ossification [Jensen, 1990]. The phenotypic spectrum of the condition varies from mild cases presenting supernumerary teeth only to the phenotypic features characteristic for CCD. A high index of clinical signs and symptoms together with molecular confirmation are required to make a definitive diagnosis.
Our patient presented dental abnormalities, as mentioned above, delayed closure of fontanels, presence of wormian bones, an accessory epiphyseal ossification center at the base of the bilateral second metacarpal, delayed ossification of pubic symphysis, and normally developed bilateral clavicles. Accessory epiphysis and delayed ossification of pubic symphysis have been previously reported in the CCD literature [Jensen, 1990].
The term cleidocranial implies defective bone modeling or formation of the clavicles as a mandatory feature; however, there are 8 case reports mentioning the presence of normal clavicles in CCD. Three cases were published prior to identification of the causative gene, rendering correlation difficult. Two cases presented characteristic skull changes with normal clavicles [Stocks and Barrington, 1925]. The third case was the brother of a typical case of CCD and had other features in form of delayed ossification of pubic symphysis, pseudoepiphyses at the base of second metacarpals, and dysostosis [Cole and Levine, 1951]. We have attempted to tabulate the phenotypic features of the 5 patients (including our patient) presenting with normal clavicles. Baumert et al. [2005] proposed a classification system for CCD which we found useful for eliciting the features and assigning a score. We scored the patients for each variable to reach the cumulative score for genotype-phenotype correlation (table 1). This analysis provided 2 interesting clinical observations. We noticed a correlation between dental abnormalities and normal clavicles with mutations of the Q/A domain. Nonsense mutations of the Q/A domain presented milder dental scores, more severe otorhinolarygological scores, and normal clavicles (table 1). On the other hand, a severe dental score and a mild otolaryngological score with normal clavicles were associated with missense mutations in the RUNT domain (table 1).
Table 1.
Phenotype and genotype of 5 male patients with normal clavicles
| Patient | Age, years | Affected family members | Otorhinolaryngological scorea | Dental scorea | Craniofacial scoreb | Nucleotide change | Spontaneous mutation | Predicted protein change | Mutation type | Domain | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 23 | 1 | 5 | 1 | 6 | c.169C>T | no | p.Q57X | nonsense | Q/A | Baumert et al. [2005] |
| 2 | 50 | 1 | 4 | 2 | 4 | c.169C>T | no | p.Q57X | nonsense | Q/A | Baumert et al. [2005] |
| 3 | 11 | 1 | 1 | 6 | 4 | c.436G>A | no | p.G146R | missense | RHD | Baumert et al. [2005] |
| 4 | 37 | 1 | 2 | 6 | 8 | c.436G>A | no | p.G146R | missense | RHD | Baumert et al. [2005] |
| 5c | 11 | 0 | 3 | 3 | 2 | c.166C>T | yes | p.Q56X | nonsense | Q/A | present study |
Score: mild = 1 – 3; severe = 4 – 6.
Score: mild = 1 – 5; severe = 6 – 10. These scores are cumulative.
Patient 5 refers to the present study. Classification scheme given by Baumert et al. [2005] was used in patients 1 – 4. In all cases, shoulder mobility was zero (clavicles could not be approximated to midline). RHD = Runt homology domain.
Diagnosis of CCD is straight forward in cases with craniofacial, dental and clavicular involvement. Cases with milder phenotypes and normal clavicles require analysis of the RUNX2 gene to confirm the diagnosis in the absence of positive family history. We found a novel nonsense mutation (c.166C>T; p.Q56X) in exon1 of the Q/A domain of RUNX2 gene (fig. 5). This mutation results in a truncated protein which cannot bind to DNA and the core-binding factor beta. Therefore, the phenotype is due to haploinsufficiency of the RUNX2 gene [Kim et al., 2006]. The parents, siblings of the proband and ethnic controls were negative for this mutation. This further suggests that the mutation was de novo, although we could not perform gene expression studies. Pathogenicity was predicted with the help of bioinformatics software (in silico analysis) using Polyphen and SIFT.
Our observations are limited to only 5 patients, and thus, further studies are needed to confirm this genotype-phenotype correlation. We could not find any genotype-phenotype correlation between craniofacial score and mutation type. Therefore, if our results are validated by larger studies, the location of mutations in the domains may also help in predicting the phenotypic severity.
Acknowledgements
This study was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health and Welfare (A1111487), and the NRF grant by the Korea government (No. 2010-0026741).
References
- 1.Baumert U, Golan I, Redlich M, Aknin JJ, Muessig D. Cleidocranial dysplasia: molecular genetic analysis and phenotypic-based description of a Middle European patient group. Am J Med Genet A. 2005;139A:78–85. doi: 10.1002/ajmg.a.30927. [DOI] [PubMed] [Google Scholar]
- 2.Cole WR, Levine S. Cleidocranial dysostosis. Br J Radiol. 1951;24:549–555. doi: 10.1259/0007-1285-24-286-549. [DOI] [PubMed] [Google Scholar]
- 3.Heyworth BE, Osei DA, Fabricant PD, Schneider R, Doyle SM, et al. The shorthand bone age assessment: a simpler alternative to current methods. J Pediatr Orthop. 2013;33:569–574. doi: 10.1097/BPO.0b013e318293e5f2. [DOI] [PubMed] [Google Scholar]
- 4.Jain S, Chowdhury V, Juneja M, Kabra M, Pandey S, et al. Intellectual disability in Indian children: experience with a stratified approach for etiological diagnosis. Indian Pediatr. 2013;50:1125–1130. doi: 10.1007/s13312-013-0306-7. [DOI] [PubMed] [Google Scholar]
- 5.Jensen BL. Somatic development in cleidocranial dysplasia. Am J Med Genet. 1990;35:69–74. doi: 10.1002/ajmg.1320350113. [DOI] [PubMed] [Google Scholar]
- 6.Kim HJ, Nam SH, Kim HJ, Park HS, Ryoo HM, et al. Four novel RUNX2 mutations including a splice donor site result in the cleidocranial dysplasia phenotype. J Cell Physiol. 2006;207:114–122. doi: 10.1002/jcp.20552. [DOI] [PubMed] [Google Scholar]
- 7.Lin WD, Lin SP, Wang CH, Tsai Y, Chen CP, et al. RUNX2 mutations in Taiwanese patients with cleidocranial dysplasia. Genet Mol Biol. 2011;34:201–204. doi: 10.1590/S1415-47572011005000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marie P, Sainton P. On hereditary cleido-cranial dysostosis. Rev Neurol. 1898;6:835. [Google Scholar]
- 9.Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet. 1999;36:177–182. [PMC free article] [PubMed] [Google Scholar]
- 10.Rimoin DL. International nomenclature of constitutional diseases of bone. J Pediatr. 1978;93:614–618. doi: 10.1016/s0022-3476(78)80897-x. [DOI] [PubMed] [Google Scholar]
- 11.Stocks P, Barrington A. Treasury of Human Inheritance. Cambridge University Press: London; 1925. [Google Scholar]