

Abstract
Epidemiological studies have demonstrated an association between malaria and invasive non-typhoid Salmonella (NTS) infections, especially in children. We explore the role of iron as a possible co-factor in this association. Malarial disease, among others, is associated with enhanced erythrophagocytosis and inflammation, which increases the iron content of macrophages and thereby also the survival of Salmonellae spp within macrophages. Whether iron supplementation programs augment the risk of invasive NTS infections in malaria endemic regions is an important global health issue that still needs to be determined.
Keywords: malaria, Plasmodium falciparum, Salmonella, iron, co-infection
An introduction to the association between malaria, Salmonella and iron
Malaria and invasive non-typhoidal Salmonella (NTS) infections are both prominent causes of severe illness and death in sub-Saharan Africa, particularly in children. While infections with NTS usually cause a self-limiting diarrheal illness in high-income countries, these micro-organisms have emerged as a leading cause of bloodstream infections with subsequent increased mortality in sub-Saharan Africa [1, 2]. In these areas, malaria is predominantly caused by Plasmodium falciparum, and many people are estimated to be at high transmission risk [3].
Iron deficiency is very common in areas which suffer from a high burden of malaria and invasive NTS disease [4]. Iron supplementation was a common intervention until a large clinical trial in preschool children on the Tanzanian island Pemba, which is an area of holo-endemic malaria transmission, showed that routine iron and folic acid supplementation was associated with an increased risk for hospital admission and death [5]. In a sub-analysis of this trial in children whose baseline iron status was assessed, adverse events of iron and folic acid supplementation were more common in children without iron deficiency. In response, the World Health Organization (WHO) recommended to restrict iron supplementation to those children with proven iron deficiency [6]. The debate on benefits and possible harm of iron supplementation and fortification in malaria endemic areas continues [7]. For example, recent Cochrane reviews concluded that iron supplementation and fortification in malaria endemic areas are probably safe, in the case that access to health care facilities are present [8-10]. The mechanisms underlying the possible adverse outcome of iron interventions remain poorly understood. One of the ways that iron may exert its harmful effects in malaria endemic areas is through predisposing individuals to malaria and NTS co-infection. In this review, we first focus on the epidemiologic evidence of the co-occurrence of Salmonella bacteremia and P. falciparum malaria, followed by a discussion of how malaria influences macrophageal iron homeostasis. Subsequently, we address the role of iron in the individual pathogenesis of Salmonella and malaria infections and, finally, explore the iron-related pathways that may be involved in the co-occurrence of malaria and invasive NTS disease.
Epidemiologic evidence for malaria and Salmonella co-infections
The association between malaria and Salmonella dates back to the 19th century, when ‘typhomalarial fever’ was a common diagnosis of army surgeons [11]. In addition, a physician reported in 1929: “The epidemiological relation of paratyphoid C to malaria in British Guiana is interesting. Not only does the disease become much more prevalent in coincidence with malarial outbreaks, but its virulence increases tenfold” [12]. Many decades later, in 1987, Mabey and coworkers renewed attention to this topic. They described the co-occurrence of NTS bacteremia and malaria parasitemia in Gambian children [13].
Since then, several prospective studies have reported similar findings. A study in 166 Kenyan children with NTS bacteremia reported that around three quarters of them had concurrent malaria parasitemia or recent malaria [14]. There was a clear seasonal trend in NTS bacteremia with the highest incidence during the rainy season when malaria rates peaked. In a study in children in Malawi with severe malaria, nearly 5% had positive blood cultures, of which NTS was the most common isolate [15]. Children with severe malarial anemia had the highest risk for NTS bacteremia. A similar outcome was found in two Kenyan studies and one Tanzanian study, in which NTS was the most common bloodstream isolate in children with parasitemia [16, 17], while Streptococcus pneumoniae and Haemophilus influenza were common isolates in aparasitemic children [17, 18]. In Kenya, NTS bacteremia was also significantly more common in rural areas with intense malaria transmission, in contrast to urban sites with less malaria burden where Salmonella typhi and S. pneumoniae were more prevalent [19].
More evidence came from a systematic review and meta-analysis of community acquired bloodstream infections in Africa [20]. This meta-analysis included 58 296 patients with febrile illness of whom almost three quarters were children. A total number of 5578 (9.6%) bacterial or fungal bloodstream infections were diagnosed, of which NTS was the most common isolate, accounting for 29.1% of the isolates recovered overall and 42.3% of pathogenic isolates in adults. Malaria parasitemia was documented in 11 814 of the cases, and 769 (6.5%) of these cases had concurrent bacterial or fungal bloodstream infection. Additional support for the existence of co-infection was derived from an observation in the Gambia and Kenya where the reduction in malaria infections was associated with a concurrent decline in NTS, while the incidence of pneumococcal bacteremia remained stable [21, 22].
Together, these studies show that invasive NTS disease and malaria are among the most common causes of fever in sub-Saharan Africa and, even though blood cultures are often not available or can be falsely negative, the data presented above suggest that co-infection of malaria and invasive NTS are common. This is in contrast to findings from Northern Africa and Asia, where invasive NTS disease appears to be relatively rare, and enteric fever caused by S. typhi and S. paratyphi are more common causes of fever [23]. The lower infection burden of both malaria and HIV in these areas and the fact that parasite densities are considerably lower in vivax malaria most likely contribute to these regional differences in NTS epidemiology.
How can this apparent association of malaria and invasive NTS disease in sub-Saharan Africa be explained? While there is no doubt that environmental factors (e.g., rainy season, humidity [1]) are important, several observations also support the importance of host factors. For example, prevalence rates of Salmonella-positive stools remain relatively stable year round, while NTS-bacteremia has a seasonal variation with a peak in the number of cases when malaria rates are also highest [13, 14]. The well-described link between invasive NTS disease, HIV infection, malnutrition, and (severe) malarial anemia also suggests host specific modulations [14-16]. These modulations involve several immune and non-immune mechanisms, and we suggest to include malaria-induced changes in the host iron homeostasis as one of the possible factors that predispose to invasive NTS, as described below.
Malaria affects macrophage iron homeostasis
In humans, the Plasmodium parasite first grows and multiplies in hepatocytes, followed by a blood stage characterized by a cycle of red blood cell (RBC) invasion, intra-erythrocytic parasite multiplication, and RBC burst, which is responsible for the characteristic inflammatory storm that is a feature of clinical malaria. Both infected and uninfected RBCs are cleared from the bloodstream by macrophages of the reticuloendothelial system taken up in phagosomes (Figure 1a). Iron is liberated from heme by the enzyme heme oxygenase (HO-1) and exported to the cytosol via the divalent metal transporter-1 (DMT-1) and natural resistance-associated macrophage protein-1 (Nramp-1), which are localized predominantly to early and late phagosomes, respectively [24, 25]. In the cytosol, the excess iron is incorporated in ferritin molecules, which is the main iron storage protein, or exported back to the plasma via the sole cellular iron exporter ferroportin. To deal with the excessive supply of heme and free iron in conditions with increased erythrophagocytosis, these proteins are all upregulated to maximize iron recycling by macrophages (Figure 1,Table 1) [26]. Malaria is also associated with intra- and extravascular hemolysis [27]. Free circulating hemoglobin and heme are bound by haptoglobin and hemopexin, respectively, taken up by macrophages via specific receptors (CD163 and CD91, respectively) and then processed as described above via the HO-1 system [28].
Figure 1. Interaction of malaria and Salmonella with macrophage iron.
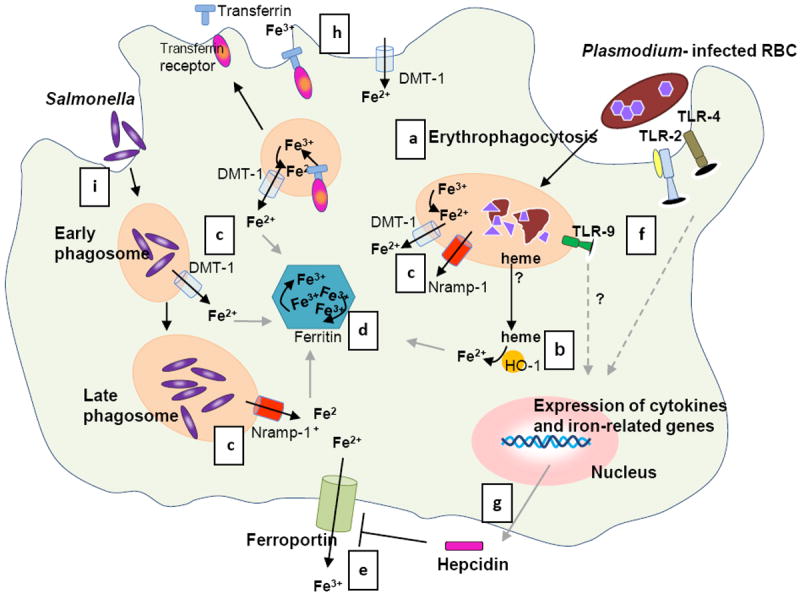
Phagocytosis of both uninfected and infected RBCs (a) is increased during the blood stage malaria infection [83], which results in degradation of the RBCs by proteolytic enzymes into heme. (b) HO-1 converts heme into iron (and carbon monoxide and biliverdin). (c) Excess iron is transported to the cytosol via phagosomal transporters DMT-1 and Nramp-1 [24] and further processed: (i) stored in ferritin (d), and (ii) used in metabolic processes or released from the cell via ferroportin (e) [26, 79]. (f) Meanwhile, parasite products activate the innate immune system via Toll Like receptors (TLR) 2, 4 and 9 [84]. This systemic response during malaria induces hepatic hepcidin production; (e) hepcidin functions by blocking ferroportin [29, 78]. In addition, monocytes and macrophages also express hepcidin upon stimulation with various pro-inflammatory cytokines and parasitized RBCs [36-38], (g) which may result in autocrine ferroportin blocking. (h) In addition, inflammatory stimuli inhibit ferroportin and modulate cellular iron uptake by DMT-1 and transferrin receptor [36, 39]. As a consequence of these processes iron is sequestered in macrophages. (i) Salmonella enters the cell via endocytosis and proliferates in phagosomes. Nramp-1 expression is required to control Salmonella growth by depleting the phagosome of iron (c) [46]. In a co-infection, Salmonellae spp. may benefit from the increased cellular iron induced by a malaria infection and establish an infection. Whether both pathogens reside in the same macrophage during invasive NTS infection and malaria as depicted in the figure is unknown. Illustration by A. Kartikasari.
Table 1.
Regulation of iron regulatory proteins in macrophagesa
| stimuli | iron | heme | inflammation | pRBCs | Salmonella | Refs | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| hepcidin | no effect | [36] | n.a. | ↑ transcriptional e.g., IL-6, LPS | [36, 37] | ↑ transcriptional | [38, 76] | ↑ transcriptional | [47] | |
|
| ||||||||||
| ferroportin | ↑ post-transcriptional | [78] | ↑ transcriptional | [26, 79] | ↓posttranslational by hepcidin | [36, 39] | n.a. | ↑ transcriptional /no effect | [44, 47] | |
| ↑ transcriptional | [36] | ↓ transcriptional LPS, IFNγ, TNFα | ||||||||
|
| ||||||||||
| H- ferritin | ↑ post-transcriptional | [78] | ↑ transcriptional | [26] | ↑transcriptional? | n.a. | ↑ transcriptional /no effect | [44, 47] | ||
|
| ||||||||||
| HO-1 | n.a. | ↑ transcriptional | [26, 79] | ↑ IL- 10, not LPS | [80] | ↑ transcriptional | [63] | ↑ transcriptional | [44, 47] | |
|
| ||||||||||
| DMT-1 | ↓posttranslational | [78] | no effect | [81] | ↑ transcriptional LPS, IFNγ, TNFα | [39] | n.a. | ↑ transcriptional | [44, 47] | |
| ↑ transcriptional? | [36] | |||||||||
|
| ||||||||||
| Nramp-1 | n.a. | ↑ transcriptional | [81] | ↑ transcriptional LPS | [82] | n.a. | n.a. | |||
Overview of how iron regulatory proteins of macrophages (vertical column) are affected by various components related to malaria infection (iron, heme, inflammatory stimuli, pRBCs) as well as by Salmonella. The consequence of these alterations is discussed in Figure 1.
Abbreviations: LPS, lipopolysaccharide; TNFα, tumor necrosis factor α; IFNγ, interferon γ; n.a., no reports available.
Besides erythrophagocytosis and hemolysis, malaria is also associated with a pronounced inflammatory response. Inflammation is well known to have a major impact on iron homeostasis, predominantly mediated by the iron regulatory hormone hepcidin. Human iron homeostasis is tightly regulated via hepcidin, produced in the liver by pro-inflammatory cytokines like interleukin (IL)-6 and suppressed in conditions of iron deficiency or increased erythropoietic activity [29]. Hepcidin acts by binding and subsequent degradation of the sole cellular iron exporter ferroportin, mainly situated on duodenal enterocytes and macrophages [29]. As a consequence, hepcidin simultaneously prevents iron absorption from the diet and the recycling of iron after erythrophagocytosis in macrophages. This hepcidin-mediated, macrophage iron withholding is nowadays regarded as an ancient host defense mechanism against extracellular pathogens by depriving these pathogens of iron [30]. Previous work has shown that febrile malaria is associated with a strong increase in systemic hepcidin levels and pronounced disturbances in iron distribution, characterized by hypoferremia and increased ferritin levels [31-33]. Studies in Indonesian schoolchildren [34] and Beninese women [35] with asymptomatic P. falciparum parasitemia and a study in volunteers participating in an experimental malaria infection [33] showed that disturbances in iron homeostasis already occur with mild inflammation.
Macrophages also express hepcidin upon stimulation with an array of infectious and inflammatory stimuli including parasitized RBCs [36-38], which may lead to autocrine ferroportin degradation and iron withholding. Besides the effects of hepcidin, inflammation also leads to increased uptake of both transferrin-bound and non-transferrin-bound iron via modulation of transferrin receptors and DMT-1 [39, 40]. A schematic description of systemic iron metabolism during malaria infections is given in Figure 2. The role of iron in malaria infections is discussed in Box 1.
Figure 2. Systemic effects of malaria on body iron stores.
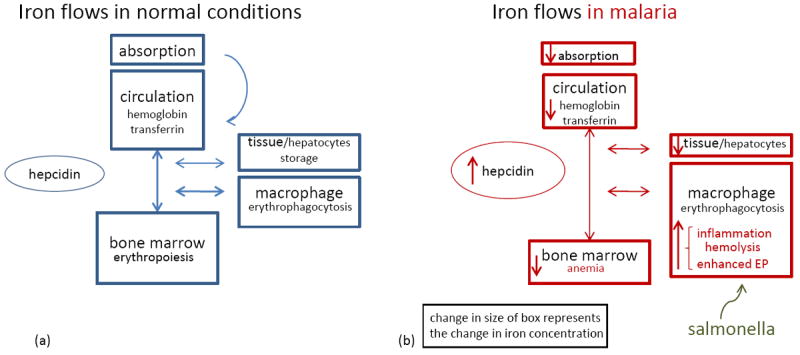
(a) A schematic presentation of iron flows in normal circumstances. Hepcidin controls the amount of iron absorbed from the diet and the release of iron from macrophages from the reticulo-endothelial system [29]. There is a steady state of iron recycling from senescent RBCs that are degraded in macrophages into iron. This iron is transported via transferrin in the circulation towards the bone marrow where iron is essential for erythropoiesis. Body iron losses are minimal and not regulated. (b) Iron flows in malaria. During malaria infection the body iron homeostasis changes, but total amount of body iron remains similar as is visualized by the change in the size of boxes. Inflammatory factors increase hepcidin release [30, 33, 76, 77]. As a consequence, absorption of iron from the diet is impaired, iron is redistributed to macrophages, less iron is bound to transferrin and iron stores become depleted. Finally, the erythropoiesis is impaired, due to hepcidin-mediated iron restriction, in addition to malaria-specific inhibitory factors (e.g., cytokines, hemozoin) [71]. Also, a blood stage malaria infection is hallmarked by hemolysis and increased phagocytosis of parasitized and non infected RBCs, which also augments the macrophage iron content. As suggested, the increased iron availability in malaria could facilitate the growth and replication of Salmonellae spp. Abbreviation: EP, erythrophagocytosis.
Box 1. The importance of iron in malaria infections.
The Plasmodium parasite is dependent on iron and other nutrients for its development. However, it is currently unclear what the iron source for the parasite is, since neither activity of HO-1 [68], nor functional iron transport or active iron acquisition systems have been detected in P. falciparum [69]. Possible sources are residual intra-erythrocytic ferritin, hemeiron or extracellular (transferrin) iron [69].
Several explanations for the possible harmful effects of iron supplementation on clinical outcome in malaria have been postulated [8, 9]. The increased availability of serum non-transferrin bound iron after iron supplementation could facilitate hepatic parasite growth, or result in an increased expression of endothelial adhesion molecules and, as such, influence malaria morbidity [70, 71]. However, evidence is scarce. In line with a harmful interaction between iron and malaria are observations of community studies in Tanzania and Malawi that young infants with iron deficiency were protected against P. falciparum infection [72, 73].
The hypothesis that this harmful effect is caused by the iron need of Plasmodium spp may be oversimplified as the host iron status influences immune effector functions as well. For instance, mice fed an iron deficient diet had lower levels of parasitemia, which related to increased clearance of RBCs and not to lower proliferation of erythrocytic parasites [74]. On the contrary, iron chelators were reported to inhibit the intra-erythrocytic parasite growth in association with a decrease in the intracellular iron pool of the infected erythrocytes and reticulocytes [71]. This observation could argue for iron-dependency, or to the generation of nitric oxide after iron chelation, an important component of the host immune response against malaria [75].
Interestingly, recent studies in mice showed that hepatic hepcidin gene expression was increased during an infection with the plasmodial mouse strain Plasmodium berghei and resulted in impaired hepatic proliferation of the parasite and lower rates of parasitemia [76], while treatment of these mice with anti-hepcidin antibodies deteriorated their survival upon malaria infection [77]. These studies suggest that increased hepcidin levels redistribute iron away from the hepatocyte, resulting in impaired growth of secondary liver stage malaria parasites during bloodstream malaria infection and protection against malarial superinfections [76, 77]. However, the question remains as to what the consequence is of this redistribution of iron toward macrophages and whether these findings can be translated to humans.
Nevertheless, it seems likely that iron-deficiency impairs malaria infection, and both nutritional and immunological aspects should be considered to play a role in which hepcidin might function as an arm of immunity linking malaria with iron.
In summary, during malaria infections macrophageal iron contents increase as a result of these putative effects of enhanced erythrophagocytosis, cytokine activation and hepcidin upregulation.
Iron is an essential micronutrient for Salmonella
To establish a systemic infection, Salmonellae spp have to invade the epithelial wall of the intestine, after which the bacteria are ingested by immune effector cells and transported to lymph nodes, the spleen, and other organs [41, 42]. Salmonellae spp reside within modified phagosomes in macrophages where replication is promoted and killing evaded [41, 42]. Iron is an essential micronutrient for replication, and Salmonellae spp (like other intracellular pathogens) harbor various iron acquisition systems, such as the siderophores enterobactin and salmochelin [43]. Salmonellae spp can also upregulate HO-1 expression in macrophages, resulting in increased iron supply from heme [44]. Several studies have directly related macrophage iron content to Salmonella pathogenesis. These studies showed that iron loading is associated with increased survival of Salmonellae spp within macrophages, while cellular iron deprivation results in the opposite [44, 45]. In response to a Salmonella infection, macrophages try to reduce the availability of intracellular iron both by increasing the incorporation of free iron into ferritin and cellular iron release via ferroportin (Table 1) [44, 46, 47]. Another iron transporter that appears crucial in the control of Salmonella is Nramp-1. As mentioned above, Nramp-1 is localized to late phagosomes in which Salmonellae spp reside, where it depletes the phagosomes of iron (Figure 1c). This limits growth of intracellular pathogens [25], and its expression was required to control Salmonella growth [46]. Apart from Nramp-1, ferroportin and DMT-1 are also located in phagosomal and endosomal membranes and to mycobacterium-containing phagosomes [48]. Although little is known about the functional role of ferroportin and DMT-1 in these endosomal and bacteria-containing phagosomes, it is conceivable that they are important in modulating endosomal iron flows. Hepcidin peptide was also detected intracellularly in mycobacterium-containing phagosomes [49], where it may control local ferroportin expression [36], and in high concentrations exert direct antibacterial activity [50].
In addition, the interaction with iron also occurs in the gut lumen, the main starting point of invasive NTS disease. For example, oral iron supplementation influenced the colonization of Salmonellae spp in the intestines of African children [51]. The number of fecal enterobacteriae, predominantly Salmonellae spp, significantly increased at the cost of non-iron dependent, non-pathogenic lactobacilli, and this correlated with a fecal marker (calprotectin) of gut inflammation, although no overt clinical disease was observed [51]. Recent in vitro data also demonstrated that iron increased the virulence of prevalent enteric pathogens, including Salmonellae [52]. Indeed, gut microbiota differed in iron-depleted versus iron replete rats, and iron supplementation increased neutrophilic infiltration of the colonic mucosa [53]. This agreed with observations suggesting that the development and function of the gut immune system is iron-dependent [54].
Taken together, the interaction between the host and Salmonellae spp includes the modulation of intracellular iron availability. On the one hand, Salmonellae spp utilize various iron acquisition systems to access iron; on the other hand, the host response aims to deprive phagosomes of iron, via the endosomal iron exporter Nramp-1, as well as the cytosol by way of upregulation of ferroportin. It is conceivable, but speculative, that P. falciparum, or Salmonellae in phagosomal compartments, modulate these iron regulatory proteins to increase their iron availability, while the host aims to deplete the phagosome of iron.
How malaria leads to more Salmonella infections: the link with iron
Despite the strong epidemiological evidence for a link between malaria and invasive NTS disease, the biological mechanisms underlying this link are still incompletely understood. Several mechanisms have been implicated, including malaria-associated impairments of immune function, antigen presentation, cytokine dysregulation, bacterial seeding of the bloodstream because of microvascular parasite sequestration in gut mucosa, and the effects of iron, heme, and the malaria pigment hemozoin [22]. The relative importance of iron, heme and hemozoin and the exact pathways are unknown. In any case, the following partly overlapping pathways are potentially involved.
Macrophage iron retention in malaria favors NTS multiplication
As described above, malaria leads to intramacrophageal iron sequestration, which is of nutritional benefit to the intracellularly growing Salmonellae. Apart from the systemic effects of hepcidin and cytokines on macrophageal iron homeostasis, the redistribution to specific locations of macrophages also seems important [30]. The observation that malaria parasites and Salmonellae spp frequently reside in close proximity is therefore of special interest: malaria-infected RBCs are retained in the slow microcirculation of the red pulp zone in the spleen [55], while Salmonellae spp reside predominantly in phagocytic macrophages of the red pulp and marginal zone of the spleen [56]. Another study in which mice were co-infected with malaria and NTS also found a marked increase in macrophages containing hemozoin and neutrophil infiltrates of Salmonellae in the red pulp of the spleen [57]. The latter is also exemplified by the preferential presence of Salmonellae spp in (iron-rich) hemophagocytic macrophages [41].
Modulation of the host immune response by iron
Besides a direct influence on Salmonella growth, iron status also influences the immune effector functions of macrophages, and disturbances in the iron load of the body have been linked to increased susceptibility to infections [30, 58]. An interesting example is the finding that iron loading of macrophages inhibits their interferon gamma (IFNγ) expression [43]. IFNγ is an essential part of the immune response against intracellular pathogens and crucially involved in protection against both malaria [59] and Salmonella [41]. The suggestion that hemophagocytic cells provide a place for Salmonellae spp to establish a chronic infection as a consequence of iron overload that interferes with immune function is in line with this hypothesis [41]. Conversely, iron deficiency may negatively affect innate and cellular immunity [60], both processes important for Salmonella host defense [30].
Iron-dependent effects of erythrophagocytosis, hemozoin and hemolysis on macrophage function
The vast proportion of iron in the human body is incorporated in hemoglobin. During the intra-erythrocytic stage, Plasmodium digests large amounts of hemoglobin into heme, which is highly deleterious for both the parasite and the host and therefore polymerized by the parasite into inert hemozoin [61]. Macrophages clear both infected and uninfected RBCs and hemozoin, which impairs macrophage function [57, 61, 62]. Hemolysis is another component that may explain the increased susceptibility to Salmonellae spp during malaria infection [57, 63]. Host defense strategies to prevent heme toxicity include the binding of heme to hemopexin and albumin, the internalization of these heme protein complexes by macrophages and, ultimately, the degradation of heme into iron via upregulation of HO-1 [28]. A recent study showed that malaria NTS co-infection of mice causes acute, fatal bacteremia with increased bacterial load; features reproduced by phenylhydrazine hemolysis or hemin administration [63]. In this study, heme and upregulation of HO-1 were associated with dysfunctional maturation and mobilization of granulocytes upon NTS infection, which resulted in an impairment of their oxidative burst and bacterial killing ability. In addition, pharmacotherapeutic inhibition of HO-1 prolonged survival of NTS and malaria co-infected mice. However, HO-1 was also reported to confer protection against non-cerebral forms of severe malaria [64] and may mediate the well known protection of sickle cell anemia against malaria [65]. This suggests a dual role for heme and HO-1 and suggests that chronic hemolysis facilitates NTS infections as a result of modulation of host immune responses in combination with the increased iron availability induced by HO-1 activity [63].
The three mechanisms described above for the connection between malarial infection and invasive NTS may also explain the reported link between severe malarial anemia and invasive NTS. The pathogenesis of malarial anemia is multifactorial and includes hemolysis, erythrophagocytosis of infected and uninfected erythrocytes, and iron delocalization due to the effects of hepcidin, all processes that favor invasive NTS, as delineated above and illustrated in the Figures 1 and 2.
Concluding remarks and future directions
The interplay between iron, malaria and invasive NTS infections is intriguing and complex to unravel. The epidemiological evidence supporting an association between these infections is convincing. Also, the specific features of malaria on one hand, whereby parasites reside in erythrocytes and hemolysis, erythrophagocytosis and inflammation are important, and the widely present and intracellular growing NTS on the other hand, makes iron a logical candidate to explain this association. Moreover, NTS, more than other bacteria, especially benefits in this scenario from its sophisticated iron acquisition systems. However, the host iron status also influences the risk for other intracellular infections such as Mycobacterium tuberculosis [66, 67].
Can these observations also explain the debated findings of an increased risk for malaria-associated morbidity and mortality after iron supplementation [5]? An increased incidence of invasive NTS may contribute to the increased morbidity and mortality after iron supplementation in malaria-endemic regions. Diagnostic facilities for invasive NTS disease are not widely available in many malaria-endemic countries, and, especially in areas where asymptomatic parasitemia is common, severe febrile disease may wrongly be attributed to malaria. Therefore, we suggest that future trials assessing the safety of iron supplementation should specifically include diagnostics for invasive NTS. Moreover, molecular studies exploring the interaction between P. falciparum and macrophage iron homeostasis are needed to understand the role of iron in the interplay with host immune responses. Modulation of macrophage iron regulatory proteins and iron distribution may also provide an explanation for NTS bacteremia during malaria infections that warrant further studies. These insights may have important implications in nutritional public health programs in developing countries.
Highlights.
Malaria and non-typhoid Salmonella infections frequently co-occur
We speculate that iron may play a central role in this association
Malaria leads to iron sequestration in macrophages
Iron sequestration in macrophages increases survival of Salmonellae spp
This might also contribute to the adverse effects of iron therapy in malaria areas
Acknowledgments
We thank April Kartikasari for her drawing of Figure 1. This work is supported by a grant from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the Office of Dietary Supplements (ODS) (grant number: 5UO1HD061246).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morpeth SC, et al. Invasive non-typhi Salmonella disease in Africa. Clin Infect Dis. 2009;49:606–611. doi: 10.1086/603553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feasey NA, et al. Invasive non-typhoidal Salmonella disease: an emerging and neglected tropical disease in Africa. Lancet. 2012;379:2489–2499. doi: 10.1016/S0140-6736(11)61752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hay SI, et al. A world malaria map: Plasmodium falciparum endemicity in 2007. PLoS Med. 2007;6:e1000048. doi: 10.1371/journal.pmed.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLean E, et al. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993-2005. Public Health Nutr. 2009;12:444–454. doi: 10.1017/S1368980008002401. [DOI] [PubMed] [Google Scholar]
- 5.Sazawal S, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006;367:133–143. doi: 10.1016/S0140-6736(06)67962-2. [DOI] [PubMed] [Google Scholar]
- 6.WHO. Conclusions and recommendations of the WHO consultation on prevention and control of iron deficiency in infants and young children in malaria-endemic areas. Food Nutr Bull. 2007;28:S621–627. doi: 10.1177/15648265070284s414. [DOI] [PubMed] [Google Scholar]
- 7.Prentice AM, Cox SE. Iron and malaria interactions: research needs from basic science to global policy. Adv Nutr. 2012;3:583–591. doi: 10.3945/an.111.001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ojukwu JU, et al. Oral iron supplementation for preventing or treating anaemia among children in malaria-endemic areas. Cochrane Database Syst Rev. 2009;9 doi: 10.1002/14651858.CD006589.pub2. CD006589. [DOI] [PubMed] [Google Scholar]
- 9.Okebe JU, et al. Oral iron supplements for children in malaria-endemic areas. Cochrane Database Syst Rev. 2011;10 doi: 10.1002/14651858.CD006589.pub3. CD006589. [DOI] [PubMed] [Google Scholar]
- 10.De-Regil LM, et al. Home fortification of foods with multiple micronutrient powders for health and nutrition in children under two years of age. Cochrane Database Syst Rev. 2011;9 doi: 10.1002/14651858.CD008959.pub2. CD008959. [DOI] [PubMed] [Google Scholar]
- 11.Smith DC. The Rise and Fall of Typhomalarial Fever: II. Decline and Fall. J Hist Med All Sc. 1982;37:287–321. doi: 10.1093/jhmas/xxxvii.3.287. [DOI] [PubMed] [Google Scholar]
- 12.Giglioli G. Paratyphoid C an endemic disease of British Guiana: A clinical and pathological outline. B. paratyphosum C as a pyogenic organism. Proc R Soc Med. 1929;23:165–177. doi: 10.1177/003591572902300247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mabey DC, et al. Plasmodium falciparum malaria and Salmonella infections in Gambian children. J Infect Dis. 1987;155:1319–1321. doi: 10.1093/infdis/155.6.1319. [DOI] [PubMed] [Google Scholar]
- 14.Brent AJ, et al. Salmonella bacteremia in Kenyan children. Pediatr Infect Dis. 2006;25:230–236. doi: 10.1097/01.inf.0000202066.02212.ff. [DOI] [PubMed] [Google Scholar]
- 15.Bronzan RN, et al. Bacteremia in Malawian children with severe malaria: prevalence, etiology, HIV coinfection, and outcome. J Infect Dis. 2007;195:895–904. doi: 10.1086/511437. [DOI] [PubMed] [Google Scholar]
- 16.Were T, et al. Bacteremia in Kenyan children presenting with malaria. J Clin Microbiol. 2011;49:671–676. doi: 10.1128/JCM.01864-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berkley JA, et al. HIV Infection, malnutrition, and invasive bacterial infection among children with severe malaria. Clin Infect Dis. 2009;49:336–343. doi: 10.1086/600299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nadjm B, et al. WHO guidelines for antimicrobial treatment in children admitted to hospital in an area of intense Plasmodium falciparum transmission: prospective study. B M J. 2010;340:c1350. doi: 10.1136/bmj.c1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tabu C, et al. Differing burden and epidemiology of non-typhi Salmonella bacteremia in rural and urban Kenya, 2006-2009. PLoS One. 2012;7:e31237. doi: 10.1371/journal.pone.0031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy EA, et al. Community-acquired bloodstream infections in Africa: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:417–432. doi: 10.1016/S1473-3099(10)70072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackenzie G, et al. A decline in the incidence of invasive non-typhoidal Salmonella infection in the Gambia temporally associated with a decline in malaria infection. PLoS ONE. 2010;5:e10568. doi: 10.1371/journal.pone.0010568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott JA, et al. Relation between falciparum malaria and bacteraemia in Kenyan children: a population-based, case-control study and a longitudinal study. Lancet. 2011;378:1316–1323. doi: 10.1016/S0140-6736(11)60888-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khan MI, et al. Non-typhoidal Salmonella rates in febrile children at sites in five Asian countries. Trop Med Int Health. 2010;15:960–963. doi: 10.1111/j.1365-3156.2010.02553.x. [DOI] [PubMed] [Google Scholar]
- 24.Soe-Lin S, et al. Both Nramp1 and DMT1 are necessary for efficient macrophage iron recycling. Exp Hematol. 2010;38:609–617. doi: 10.1016/j.exphem.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Cellier MF, et al. Nramp1 phagocyte intracellular metal withdrawal defense. Microbes Infect. 2007;9:1662–1670. doi: 10.1016/j.micinf.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Delaby C, et al. Sequential regulation of ferroportin expression after erythrophagocytosis in murine macrophages: early mRNA induction by haem, followed by iron-dependent protein expression. Biochem J. 2008;411:123–131. doi: 10.1042/BJ20071474. [DOI] [PubMed] [Google Scholar]
- 27.Fendel R, et al. Hemolysis is associated with low reticulocyte production index and predicts blood transfusion in severe malarial anemia. PLoS One. 2010;5:e10038. doi: 10.1371/journal.pone.0010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira A, et al. A central role for free heme in the pathogenesis of severe malaria: the missing link? J Mol Med. 2008;86:1097–1111. doi: 10.1007/s00109-008-0368-5. [DOI] [PubMed] [Google Scholar]
- 29.Kroot JJ, et al. Hepcidin in human iron disorders: diagnostic implications. Clin Chem. 2011;57:1650–1669. doi: 10.1373/clinchem.2009.140053. [DOI] [PubMed] [Google Scholar]
- 30.Drakesmith H, Prentice AM. Hepcidin and the iron-infection axis. Science. 2012;338:768–72. doi: 10.1126/science.1224577. [DOI] [PubMed] [Google Scholar]
- 31.de Mast Q, et al. Assessment of urinary concentrations of hepcidin provides novel insight into disturbances in iron homeostasis during malarial infection. J Infect Dis. 2009;199:253–262. doi: 10.1086/595790. [DOI] [PubMed] [Google Scholar]
- 32.Howard CT, et al. Relationship of hepcidin with parasitemia and anemia among patients with uncomplicated Plasmodium falciparum malaria in Ghana. Am J Trop Med Hyg. 2007;77:623–626. [PubMed] [Google Scholar]
- 33.de Mast Q, et al. Mild increases in serum hepcidin and interleukin-6 concentrations impair iron incorporation in haemoglobin during an experimental human malaria infection. Br J Haematol. 2009;145:657–664. doi: 10.1111/j.1365-2141.2009.07664.x. [DOI] [PubMed] [Google Scholar]
- 34.de Mast Q, et al. Increased serum hepcidin and alterations in blood iron parameters associated with asymptomatic P. falciparum and P. vivax malaria. Haematol. 2010;95:1068–1074. doi: 10.3324/haematol.2009.019331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cercamondi CI, et al. Afebrile Plasmodium falciparum parasitemia decreases absorption of fortification iron but does not affect systemic iron utilization: a double stable-isotope study in young Beninese women. Am J Clin Nutr. 2010;92:1385–1392. doi: 10.3945/ajcn.2010.30051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen NB, et al. Hepcidin expression and iron transport in alveolar macrophages. Am J Physiol Lung Cell Mol. 2006;291:L417–L425. doi: 10.1152/ajplung.00484.2005. [DOI] [PubMed] [Google Scholar]
- 37.Armitage AE, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood. 2011;118:4129–4139. doi: 10.1182/blood-2011-04-351957. [DOI] [PubMed] [Google Scholar]
- 38.Armitage AE, et al. Plasmodium falciparum infected erythrocytes induce hepcidin (HAMP) mRNA synthesis by peripheral blood mononuclear cells. Br J Haematol. 2009;147:769–771. doi: 10.1111/j.1365-2141.2009.07880.x. [DOI] [PubMed] [Google Scholar]
- 39.Ludwiczek S, et al. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003;101:4148–4154. doi: 10.1182/blood-2002-08-2459. [DOI] [PubMed] [Google Scholar]
- 40.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352:1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 41.Silva-Herzog E, Detweiler CS. Intracellular microbes and haemophagocytosis. Cell Microbiol. 2008;10:2151–2158. doi: 10.1111/j.1462-5822.2008.01192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibarra JA, Steele-Mortimer O. Salmonella-the ultimate insider. Salmonella virulence factors that modulate intracellular survival. Cell Microbiol. 2009;11:1579–1586. doi: 10.1111/j.1462-5822.2009.01368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nairz M, et al. The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol. 2010;12:1691–702. doi: 10.1111/j.1462-5822.2010.01529.x. [DOI] [PubMed] [Google Scholar]
- 44.Nairz M, et al. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007;9:2126–2140. doi: 10.1111/j.1462-5822.2007.00942.x. [DOI] [PubMed] [Google Scholar]
- 45.Paradkar PN, et al. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112:866–874. doi: 10.1182/blood-2007-12-126854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nairz M, et al. Slc11a1 limits intracellular growth of Salmonella enterica sv. Typhimurium by promoting macrophage immune effector functions and impairing bacterial iron acquisition. Cell Microbiol. 2009;11:1365–1381. doi: 10.1111/j.1462-5822.2009.01337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan X, et al. Modulation of iron homeostasis in macrophages by bacterial intracellular pathogens. BMC Microbiol. 2010;10:64–77. doi: 10.1186/1471-2180-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Zandt KE, et al. The iron export protein ferroportin 1 is differentially expressed in mouse macrophage populations and is present in the mycobacterial-containing phagosome. J Leukoc Biol. 2008;84:689–700. doi: 10.1189/jlb.1107781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sow FB, et al. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol. 2007;82:934–945. doi: 10.1189/jlb.0407216. [DOI] [PubMed] [Google Scholar]
- 50.Park CH, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 51.Zimmermann MB, et al. The effects of iron fortification on the gut microbiota in African children: a randomized controlled trial in Cote d’Ivoire. Am J Clin Nutr. 2010;92:1406–1415. doi: 10.3945/ajcn.110.004564. [DOI] [PubMed] [Google Scholar]
- 52.Kortman GA, et al. Iron availability increases the pathogenic potential of Salmonella typhimurium and other enteric pathogens at the intestinal epithelial interface. PLoS One. 2012;7:e29968. doi: 10.1371/journal.pone.0029968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dostal A, et al. Iron depletion and repletion with ferrous sulfate or electrolytic iron modifies the composition and metabolic activity of the gut microbiota in rats. J Nutr. 2012;142:271–277. doi: 10.3945/jn.111.148643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cherayil BJ, et al. Iron and intestinal immunity. Curr Opin Gastroenterol. 2011;27:523–528. doi: 10.1097/MOG.0b013e32834a4cd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Safeukui I, et al. Retention of Plasmodium falciparum ring-infected erythrocytes in the slow, open microcirculation of the human spleen. Blood. 2008;112:2520–2528. doi: 10.1182/blood-2008-03-146779. [DOI] [PubMed] [Google Scholar]
- 56.Salcedo SP, et al. Intracellular replication of Salmonella typhimurium strains in specific subsets of splenic macrophages in- vivo. Cell Microb. 2001;3:587–597. doi: 10.1046/j.1462-5822.2001.00137.x. [DOI] [PubMed] [Google Scholar]
- 57.Roux CM, et al. Both hemolytic anemia and malaria parasite-specific factors increase susceptibility to nontyphoidal Salmonella enterica Serovar Typhimurium infection in mice. Infect Immun. 2010;78:1520–1527. doi: 10.1128/IAI.00887-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson EE, Wessling-Resnick M. Iron metabolism and the innate immune response to infection. Microb Infect. 2012;14:207–216. doi: 10.1016/j.micinf.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCall MB, Sauerwein RW. Interferon y- central mediator of protective immune responses against the pre-erythrocytic and blood stage of malaria. J Leukoc Biol. 2010;88:1131–1143. doi: 10.1189/jlb.0310137. [DOI] [PubMed] [Google Scholar]
- 60.Oppenheimer SJ. Iron and Its Relation to Immunity and Infectious Disease. J Nutr. 2001;131:616S–635S. doi: 10.1093/jn/131.2.616S. [DOI] [PubMed] [Google Scholar]
- 61.Hanscheid T, et al. Haemozoin: from melatonin pigment to drug target, diagnostic tool, and immune modulator. Lancet Infect Dis. 2007;7:675–685. doi: 10.1016/S1473-3099(07)70238-4. [DOI] [PubMed] [Google Scholar]
- 62.Cambos M, Scorza T. Robust erythrophagocytosis leads to macrophage apoptosis via a hemin-mediated redox imbalance: role in hemolytic disorders. J Leukoc Biol. 2011;89:159–171. doi: 10.1189/jlb.0510249. [DOI] [PubMed] [Google Scholar]
- 63.Cunnington AJ, et al. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat Med. 2012;18:120–127. doi: 10.1038/nm.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seixas E, et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A. 2009;106:15837–15842. doi: 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferreira A, et al. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. 2011;145:398–409. doi: 10.1016/j.cell.2011.03.049. [DOI] [PubMed] [Google Scholar]
- 66.Gangaidzo IT, et al. Association of pulmonary tuberculosis with increased dietary iron. J Infect Dis. 2001;184:936–939. doi: 10.1086/323203. [DOI] [PubMed] [Google Scholar]
- 67.McDermid JM, et al. Host iron redistribution as a risk factor for incident tuberculosis in HIV infection: an 11-year retrospective cohort study. BMC Infect Dis. 2013;13:48. doi: 10.1186/1471-2334-13-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sigala PA, et al. Direct tests of enzymatic heme degradation by the malaria parasite Plasmodium falciparum. J Biol Chem. 2012;287:37793–37807. doi: 10.1074/jbc.M112.414078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scholl PF, et al. Bioavailable iron and heme metabolism in Plasmodium falciparum malaria. Curr Top Microbiol Immunol. 2005;295:293–324. doi: 10.1007/3-540-29088-5_12. [DOI] [PubMed] [Google Scholar]
- 70.Raiten DJ, et al. Considerations for the safe and effective use of iron interventions in areas of malaria burden - executive summary. Int J Vitam Nutr Res. 2011;81:57–71. doi: 10.1024/0300-9831/a000051. [DOI] [PubMed] [Google Scholar]
- 71.Portugal S, et al. Superinfection in malaria: Plasmodium shows its iron will. EMBO Rep. 2011;12:1233–1242. doi: 10.1038/embor.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gwamaka M, et al. Iron deficiency protects against severe Plasmodium falciparum malaria and death in young children. Clin Infect Dis. 2012;37:1137–1144. doi: 10.1093/cid/cis010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonker FAM, et al. Iron Status Predicts Malaria Risk in Malawian Preschool Children. PLoS ONE. 2012;7:e42670. doi: 10.1371/journal.pone.0042670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matsuzaki-Moriya C, et al. A critical role for phagocytosis in resistance to malaria in iron-deficient mice. Eur J Immunol. 2011;41:1365–1375. doi: 10.1002/eji.201040942. [DOI] [PubMed] [Google Scholar]
- 75.Fritsche G, et al. Regulatory interactions between iron and nitric oxide metabolism for immune defense against Plasmodium falciparum infection. J Infect Dis. 2001;183:1388–1394. doi: 10.1086/319860. [DOI] [PubMed] [Google Scholar]
- 76.Portugal S, et al. Host-mediated regulation of superinfection in malaria. Nat Med. 2011;17:732–737. doi: 10.1038/nm.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang HZ, et al. Hepcidin is regulated during blood-stage malaria and plays a protective role in malaria infection. J Immunol. 2011;187:6410–6416. doi: 10.4049/jimmunol.1101436. [DOI] [PubMed] [Google Scholar]
- 78.Muckenthaler MU, et al. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Ann Rev Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 79.Marro S, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematol. 2010;95:1261–1268. doi: 10.3324/haematol.2009.020123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 81.Knutson MD, et al. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood. 2003;102:4191–4197. doi: 10.1182/blood-2003-04-1250. [DOI] [PubMed] [Google Scholar]
- 82.Govoni G, et al. Cell-specific and inducible Nramp1 gene expression in mouse macrophages in vitro and in vivo. J Leukoc Biol. 1997;62:277–286. doi: 10.1002/jlb.62.2.277. [DOI] [PubMed] [Google Scholar]
- 83.Waitumbi JN, et al. Red cell surface changes and erythrophagocytosis in children with severe Plasmodium falciparum anemia. Blood. 2000;95:1481–1486. [PubMed] [Google Scholar]
- 84.Gowda DC. TLR-mediated cell signaling by malaria GPIs. Trends Parasitol. 2007;23:596–604. doi: 10.1016/j.pt.2007.09.003. [DOI] [PubMed] [Google Scholar]