

Abstract
Human gnathostomiasis is an emerging food-borne parasitic disease caused by nematodes in the genus Gnathostoma. In spite of their significance as pathogens, these parasites remain poorly understood at the molecular level. In the present study, we sequenced the mitochondrial (mt) genome of G. spinigerum, which infects a range of definitive hosts including dogs, cats, tigers, leopards and humans. The mt genome of G. spinigerum is 14,079 bp in size and shows substantial changes in gene order compared to other nematodes studied to date. Phylogenetic analyses of mt genome sequences by Bayesian inference (BI) revealed that the infraorder Gnathostomatomorpha (represented by G. spinigerum) is closely related to the infraorder Ascaridomorpha. G. spinigerum is the first species from the infraorder Gnathostomatomorpha for which a complete mt genome has been sequenced. The new data will help understand the evolution, population genetics and systematics of this medically important group of parasites.
Human gnathostomiasis caused by Gnathostoma spp. is a highly endemic disease in some under-developed communities in Asia, particularly in China1. Recently, it has become an emerging disease among travelers from Europe and other continents who are coming into contact with endemic areas2. Nematodes of the genus Gnathostoma (Nematoda: Gnathostomatidae) are the etiological agents of human gnathostomiasis and may also infest dogs, cats, tigers and leopards3. There are 12 species in this genus with four species recorded in humans: G. spinigerum, commonly found in India, China, Japan and southeast Asia; G. hispidum, found in Europe, Asia and Australia; G. doloresi, found in southeast Asia; and G. nipponicum, found in Japan3. G. spinigerum is frequently reported in Asia as being responsible for human gnathostomiasis, but G. hispidum, G. doloresi, and G. nipponicum have also occasionally been reported4,5,6. Humans are infected when consuming raw or uncooked infected meats of intermediate hosts (fish) or other paratenic hosts (snake, pig and poultry). The majority of gnathostomiasis patients present with cutaneous lesions; involvement of eyes and central nervous system has also been sporadically reported7,8,9.
The genus Gnathostoma is in the infraorder Gnathostomatomorpha of the suborder Spirurina. There are four other infraorders in this suborder: Ascaridomorpha, Spiruromorpha, Rhigonematomorpha and Oxyuridomorpha10. The phylogenetic relationships among the infraorders of the Spirurina have been assessed using nuclear small subunit (SSU) rRNA (five infraorders) gene and mitochondrial (mt) gene/genome sequences (four infraorders) and there are inconsistencies between the nuclear phylogeny and the mt phylogeny11,12,13,14,15,16,17,18. In the phylogeny inferred from SSU rRNA gene sequences, the Ascaridomorpha is sister to the Rhigonematomorpha, and the Spiruromorpha is sister to the Ascaridomorpha + Rhigonematomorpha, the Oxyuridomorpha is sister to the Spiruromorpha + Ascaridomorpha + Rhigonematomorpha, and the Gnathostomatomorpha is sister to the Oxyuridomorpha + Spiruromorpha + Ascaridomorpha + Rhigonematomorpha12,19. In the mt gene/genome phylogeny, however, the Ascaridomorpha is sister to the Rhabditomorpha + Diplogasteromorpha in most analyses, and the Spiruromorpha is sister to the Rhabditomorpha + Diplogasteromorpha + Ascaridomorpha + Rhigonematomorpha + Panagrolaimomorpha + Tylenchomorpha + Oxyuridomorpha15,16,17. More recently, Kim et al.20 inferred the phylogeny with mt genome sequences and showed that the Rhigonematomorpha is sister to the Ascaridomorpha. Taxon sampling was limited in both the nuclear SSU rRNA and the mt gene/genome phylogenetic analysis; furthermore, no species from the Gnathostomatomorpha was included in any of these mt analyses.
Animal mt genomes are typically a circular DNA, 15–20 kb in size, containing 36–37 genes: 12–13 protein-coding genes (PCGs), 22 transfer RNAs (tRNA) and two ribosomal RNA (rRNA) genes21,22. Mt genome sequences are commonly used for phylogentic, population genetic and taxonomic investigations of animals23,24. To understand the phylogenetic relationship of the infraorder Gnathostomatomorpha with other infraorders of the class Chromadorea, we sequenced the mt genome of G. spinigerum.
Results and Discussion
General features of the mt genome of G. spinigerum
The complete mt genome of G. spinigerum (GenBank accession no. KP410547) was 14,079 bp in size (Fig. 1). This genome contains 12 PCGs (cox1-3, nad1-6, nad4L, atp6 and cytb), 22 tRNA genes, two rRNA genes (rrnL and rrnS) and two non-coding (AT-rich) regions. All genes are transcribed in the same direction. As in most other nematodes of the class Chromadorea, atp8 gene is not present in the mt genome of G. spinigerum (Table 1). The mt genome sequence of G. spinigerum is biased toward A and T (71.1%), similar to that of other nematodes in the suborder Spirurina25,26,27,28. This nucleotide composition of the 12 PCGs of G. spinigerum was strongly skewed away from A, in favour of T (AT skew between −0.58 and –0.27), and the GC skew was between 0.41 and 0.77 (Table 2). Codons composed of A and T were more frequently used in PCGs, reflecting the high A + T content in the mt genome of G. spinigerum. The most frequently used amino acid was TTT (Phe; 13.14%), followed by TTG (Leu; 8.79%), ATT (IIe; 5.96%) and GTT (Val; 5.46%) (Table 3). ATA, TTG and ATT were used as initiation codons and TAA and TAG as termination codons; incomplete termination codons (T or TA) were also identified (Table 1), which is consistent with the arrangement in the mt genomes of other nematodes29,30,31. Twenty-two tRNA genes were identified in the mt genome of G. spinigerum, which range from 54 to 68 bp in size. The secondary structures inferred for the 22 tRNAs of G. spinigerum are similar to those of other nematodes29,30,31 (Fig. 2). rrnL is located between trnH and nad3 in the G. spinigerum mt genome; rrnS is between trnE and trnS2 (Table 1). The two non-coding regions in the mt genome of G. spinigerum were located between trnI and trnN (designated NCL, 750 bp), and between nad1 and atp6 (designated NCR, 77 bp) respectively. No repetitive sequences were detected in the non-coding regions of G. spinigerum, as in other Spirurina nematodes26,27.
Figure 1. The mitochondrial genome of Gnathostoma spinigerum.
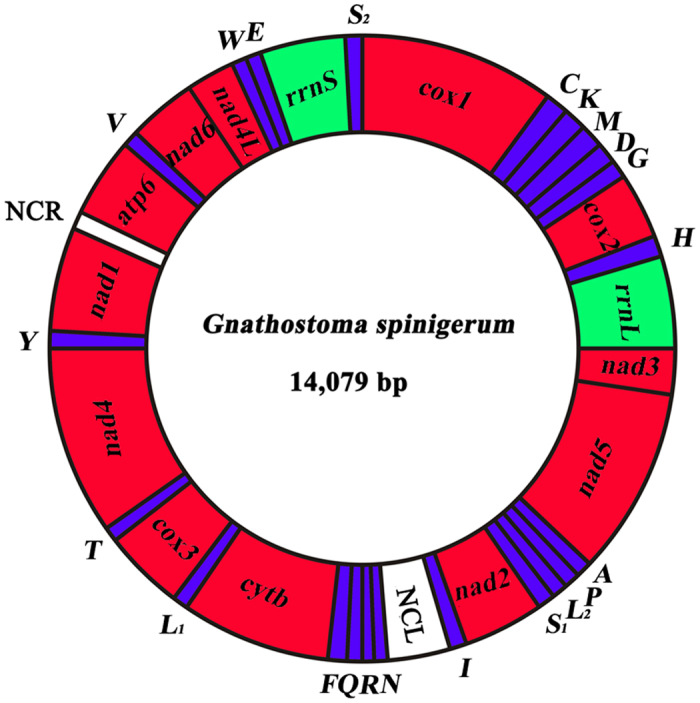
All genes are on the same DNA strand and are transcribed clockwise. Protein-coding and rRNA genes are indicated with the standard nomenclature. tRNA genes are indicated with the one-letter code of their corresponding amino acids. There are two tRNA genes for leucine: L1 for codons CUN and L2 for UUR; and two tRNA genes for serine: S1 for codons AGN and S2 for UCN. “NCL” refers to the large non-coding region. “NCR” refers to a small non-coding region.
Table 1. The organization of the mitochondrial genome of Gnathostoma spinigerum.
| Genes | Positions | Lengths (bp) | Start codons | Stop codons | Anticodons |
|---|---|---|---|---|---|
| cox1 | 1–1572 | 1572 | ATA | TAG | |
| tRNA-Cys (C) | 1573–1629 | 57 | GCA | ||
| tRNA-Lys (K) | 1629–1691 | 63 | TTT | ||
| tRNA-Met (M) | 1699–1752 | 53 | CAT | ||
| tRNA-Asp (D) | 1753–1808 | 56 | GTC | ||
| tRNA-Gly (G) | 1813–1868 | 56 | TCC | ||
| cox2 | 1869–2556 | 688 | TTG | T | |
| tRNA-His (H) | 2557–2612 | 56 | GTG | ||
| rrnL | 2613–3554 | 942 | |||
| nad3 | 3555–3890 | 336 | TTG | TAA | |
| nad5 | 3894–5478 | 1585 | TTG | T | |
| tRNA-Ala (A) | 5479–5533 | 55 | TGC | ||
| tRNA-Pro (P) | 5535–5592 | 58 | TGG | ||
| tRNA-Leu UUR (L2) | 5598–5652 | 55 | TAA | ||
| tRNA-Ser AGN (S1) | 5651–5710 | 60 | TCT | ||
| nad2 | 5721–6553 | 833 | ATA | TA | |
| tRNA-Ile (I) | 6554–6609 | 56 | GAT | ||
| Non-coding region | 6610–7359 | 750 | |||
| tRNA-Asn (N) | 7360–7416 | 57 | GTT | ||
| tRNA-Arg (R) | 7448–7504 | 57 | TCG | ||
| tRNA-Gln (Q) | 7504–7557 | 54 | TTG | ||
| tRNA-Phe (F) | 7557–7624 | 68 | GAA | ||
| cytb | 7648–8721 | 1074 | ATA | TAG | |
| tRNA-Leu CUN (L1) | 8722–8776 | 55 | TAG | ||
| cox3 | 8777–9544 | 768 | TTG | TAG | |
| tRNA-Thr (T) | 9547–9601 | 55 | TGT | ||
| nad4 | 9602–10826 | 1225 | TTG | T | |
| tRNA-Tyr (Y) | 10827–10880 | 54 | GTA | ||
| nad1 Non-coding region | 10881–11753 11754–11830 | 873 77 | TTG | TAA | |
| atp6 | 11831–12409 | 579 | ATT | TAG | |
| tRNA-Val (V) | 12463–12517 | 55 | TAC | ||
| nad6 | 12518–12955 | 438 | TTG | TAG | |
| nad4L | 12963–13190 | 228 | TTG | TAG | |
| tRNA-Trp (W) | 13191–13247 | 57 | TCA | ||
| tRNA-Glu (E) | 13255–13311 | 57 | TTC | ||
| rrnS | 13312–13985 | 674 | |||
| tRNA-Ser UCN (S2) | 13986–14039 | 54 | TGA |
Table 2. Nucleotide composition and skews of the Gnathostoma spinigerum mitochondrial protein-coding genes.
| Gene | Proportion of nucleotides |
A + T (%) | AT Skew | GC Skew | |||
|---|---|---|---|---|---|---|---|
| A | T | G | C | ||||
| cox1 | 20.6 | 46.4 | 23.4 | 9.7 | 67.0 | −0.39 | 0.41 |
| cox2 | 22.7 | 45.9 | 24.1 | 7.3 | 68.6 | −0.34 | 0.54 |
| cox3 | 19.8 | 50.9 | 21.2 | 8.1 | 70.7 | −0.44 | 0.45 |
| nad1 | 21.2 | 47.8 | 22.2 | 8.8 | 69.0 | −0.39 | 0.43 |
| nad2 | 25.0 | 51.0 | 17.6 | 6.4 | 76.0 | −0.34 | 0.47 |
| nad3 | 21.1 | 52.7 | 23.2 | 3.0 | 73.8 | −0.43 | 0.77 |
| nad4 | 20.2 | 51.2 | 21.1 | 7.5 | 71.4 | −0.43 | 0.48 |
| nad4L | 15.8 | 57.0 | 22.4 | 4.8 | 72.8 | −0.57 | 0.65 |
| nad5 | 21.0 | 50.1 | 22.3 | 6.6 | 71.1 | −0.41 | 0.54 |
| nad6 | 15.3 | 57.1 | 22.4 | 5.3 | 72.4 | −0.58 | 0.62 |
| cytb | 20.8 | 49.2 | 21.9 | 8.2 | 70.0 | −0.41 | 0.46 |
| atp6 | 25.9 | 44.7 | 22.1 | 7.3 | 70.6 | −0.27 | 0.50 |
Table 3. Codon usage of Gnathostoma spinigerum mitochondrial protein-coding genes.
| Amino acid | Codon | Number | Frequency (%) | Amino acid | Codon | Number | Frequency (%) |
|---|---|---|---|---|---|---|---|
| Phe | TTT | 447 | 13.14 | Met | ATA | 92 | 2.70 |
| Phe | TTC | 13 | 0.38 | Met | ATG | 114 | 3.35 |
| Leu | TTA | 183 | 5.38 | Thr | ACT | 78 | 2.29 |
| Leu | TTG | 299 | 8.79 | Thr | ACC | 2 | 0.05 |
| Ser | TCT | 114 | 3.35 | Thr | ACA | 7 | 0.20 |
| Ser | TCC | 6 | 0.17 | Thr | ACG | 11 | 0.32 |
| Ser | TCA | 16 | 0.47 | Asn | AAT | 92 | 2.70 |
| Ser | TCG | 9 | 0.26 | Asn | AAC | 4 | 0.11 |
| Tyr | TAT | 177 | 5.20 | Lys | AAA | 43 | 1.26 |
| Tyr | TAC | 4 | 0.11 | Lys | AAG | 48 | 1.41 |
| Stop | TAA | 2 | 0.05 | Ser | AGT | 98 | 2.88 |
| Stop | TAG | 6 | 0.17 | Ser | AGC | 7 | 0.20 |
| Cys | TGT | 59 | 1.73 | Ser | AGA | 60 | 1.76 |
| Cys | TGC | 2 | 0.05 | Ser | AGG | 57 | 1.67 |
| Trp | TGA | 27 | 0.79 | Val | GTT | 186 | 5.46 |
| Trp | TGG | 46 | 1.35 | Val | GTC | 9 | 0.26 |
| Leu | CTT | 32 | 0.94 | Val | GTA | 62 | 1.82 |
| Leu | CTC | 1 | 0.02 | Val | GTG | 80 | 2.35 |
| Leu | CTA | 7 | 0.20 | Ala | GCT | 81 | 2.38 |
| Leu | CTG | 18 | 0.52 | Ala | GCC | 5 | 0.14 |
| Pro | CCT | 66 | 1.94 | Ala | GCA | 13 | 0.38 |
| Pro | CCC | 1 | 0.02 | Ala | GCG | 5 | 0.14 |
| Pro | CCA | 3 | 0.08 | Asp | GAT | 74 | 2.17 |
| Pro | CCG | 8 | 0.23 | Asp | GAC | 4 | 0.11 |
| His | CAT | 50 | 1.47 | Glu | GAA | 37 | 1.08 |
| His | CAC | 3 | 0.08 | Glu | GAG | 48 | 1.41 |
| Gln | CAA | 16 | 0.47 | Gly | GGT | 103 | 3.02 |
| Gln | CAG | 23 | 0.67 | Gly | GGC | 9 | 0.26 |
| Arg | CGT | 23 | 0.67 | Gly | GGA | 29 | 0.85 |
| Arg | CGC | 3 | 0.08 | Gly | GGG | 63 | 1.85 |
| Arg | CGA | 3 | 0.08 | IIe | ATT | 203 | 5.96 |
| Arg | CGG | 5 | 0.14 | IIe | ATC | 5 | 0.14 |
Figure 2. The predicted secondary structures of representing tRNAs of the Gnathostoma spinigerum mitochondrial DNA determined in this study.

Gene arrangement in the mt genome of G. spinigerum
The mt genome of G. spinigerum shows a different gene arrangement pattern from the other 29 patterns seen in nematodes revealed by previous studies (pattern GA26 hereafter; see17 for GA1-25; GA27 for Rhigonema thysanophora20; GA28-30 for Meloidogyne chitwoodi, M. graminicola and M. incognita, respectively32,33). In this study, we compared the 30 gene arrangement patterns in nematodes, and found that the gene order for all of the protein-coding and rRNA genes were principally conserved, but tRNA genes/clusters were not conserved in nematodes. Furthermore, mt gene arrangement events among the 30 patterns observed in nematodes were analyzed with CREx34; numerous transposition and tandem-duplication-random-loss (TDRL) events could be inferred. Our results support the view that the evolution of mt gene order in nematodes was mostly driven by transposition and TDRL14; inversion and reverse-transposition played a minor role relatively.
Compared to the most common pattern of mt gene arrangement seen in nematodes (i.e. GA3)17, a block of 12 genes (from trnV to trnK, Fig. 3) in G. spinigerum has been broken and moved to four locations. Seven genes (trnV, nad6, nad4L, trnW, trnE, rrnS and trnS2) between trnP and trnN in GA3 pattern, was found between apt6 and cox1 in GA26 pattern in G. spinigerum. trnN was located between trnS2 and trnY in GA3 pattern but was between trnI and trnR in GA26 pattern. Three genes (trnY, nad1 and atp6) between trnN and trnK in GA3 pattern were located between nad4 and trnV in GA26 pattern. Additionally, trnK, which was between atp6 and trnL2 in GA3 pattern, was located between trnC and trnM in GA26 pattern. CREx analysis modeled that one transposition and two TDRL events were required to convert GA3 to GA26. Two rearrangement events involved PCGs while the other two events involved only tRNA genes.
Figure 3. Rearrangement of mitochondrial genes in Gnathostoma spinigerum (pattern GA26) relative to the most common pattern of mitochondrial gene arrangement observed in nematodes (GA3).

Phylogenetic analyses
We inferred the phylogenetic relationship between G. spinigerum and other 57 species of Chromadorea nematodes with concatenated amino acid sequences of the 12 mt PCGs (Fig. 4). Phylogenies of the Chromadorea nematodes were inferred with mt genome sequences in previous studies14,15,16,17,18; however, several major lineages including the infraorder Gnathostomatomorpha were not represented.
Figure 4. Phylogenetic relationships among 58 species of Chromadorea nematodes inferred from Bayesian inference of deduced amino acid sequences of 12 mitochondrial protein-coding genes.

Trichuris suis (GenBank accession number GU070737) was used as the outgroup. Bayesian posterior probabilities (Bpp) values were indicated at nodes.
Our Bayesian analysis showed that G. spinigerum was most closely related to Cucullanus robustus with moderate support [Bayesian posterior probabilities (Bpp) = 0.88, Fig. 4]. Our maximum likelihood (ML) and maximum parsimony (MP) analyses also recovered this relationship but the bootstrapping frequency (Bf) was weak (not shown). This grouping was inconsistent with those from morphological and molecular studies12,19,35,36,37. The infraorder Ascaridomorpha was monophyletic in the present study (Bpp = 0.67, Fig. 4). The two species from the two families, Cucullanidae and Ascaridiidae, of the infraorder Ascaridomorpha were more closely related to G. spinigerum (infraorder Gnathostomatomorpha) and R. thysanophora (infraorder Rhigonematomorpha) than they were to other six species from the infraorder Ascaridomorpha. R. thysanophora was most closely related to Ascaridia galli (Bpp = 0.67). A recent study based on mt genome sequences also indicated that R. thysanophora was most closely related to an Ascaridia species20. Using nuclear SSU rRNA gene sequences, Meldal et al.12 showed that the infraorder Ascaridomorpha was sister to the group that included the infraorders Ascaridomorpha, Rhabditomorpha, Spiruromorpha, Oxyuridomorpha, and Gnathostomatomorpha. These controversial results surrounding phylogenetic placement of members in Spirurina may reflect the different evolutionary rates of the nuclear and mt genomes38,39.
Our analysis also showed that the infraorder Rhabditomorpha was paraphyletic with respect to the Diplogasteromorpha. Two species from the families Rhabditidae and Heterorhabditidae of the Rhabditomorpha were more closely related to Pristionchus pacificus (Neodiplogasteridae) than they were to the other 22 species from the Rhabditomorpha. The close relationship between the species of the families Rhabditidae and Heterorhabditidae and P. pacificus was strongly supported in BI (Bpp = 1, Fig. 4). The results were consistent with that of a previous study using nuclear SSU rRNA gene dataset11. The Oxyuridomorpha (2 species) and the Spiruromorpha (12 species) were both monophyletic with strong support in the present analysis (Bpp = 1, Fig. 4). The nine species of the suborder Tylenchina included in this study were from two infraorders: Panagrolaimorpha (2 species), and Tylenchomorpha (7 species). Both of these infraorders were paraphyletic in the present analysis (Bpp ≥ 0.67, Fig. 4). The Oxyuridomorpha and the Spiruromorpha were both monophyletic with strong support in the current analyses (Bpp = 1 for Oxyuridomorpha and Bpp ≥ 0.79 for Spiruromorpha, Fig. 4).
For decades, there have been controversies surrounding the systematics of the suborder Spirurina (infraorders Ascaridomorpha, Spiruromorpha, Rhigonematomorpha, Gnathostomatomorpha and Oxyuridomorpha)10. Given the demonstrated utility of mt datasets, there is now an opportunity to test the phylogenetic relationships of a wide range of Spirurina nematodes using expanded mt datasets. Analyses of mt genome sequences in the current study and several previous studies14,15,16,17,18 have provided insights into the phylogenetic relationships among major lineages of the Spirurina nematodes. However, some lineages of the suborder Spirurina are underrepresented or not represented in these analyses. So, more mt genome data from the suborder Spirurina would be required in future analyses to understand the phylogeny of the suborder Spirurina.
In summary, this is the first determination of a complete mt genome of a parasite belonging to the infraorder Gnathostomatomorpha. Although the length, gene and AT content are similar to other nematode mt genomes, the mt genome of G. spinigerum exhibits some interesting features. The gene order of G. spinigerum is distinct from that of other nematodes. Phylogenetic analysis shows that G. spinigerum was most closely related to Cucullanus robustus with moderate support, which is inconsistent with that from morphological and molecular studies. Our results provided insights into the phylogenetic relationships among several major lineages of nematodes.
Methods
Ethics statement
Specimens of G. spinigerum were collected from an Asian swamp eel, in accordance with the animal ethics procedures and guidelines China. All experimental protocols were approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences.
Collection of G. spinigerum and DNA isolation
Larval specimens of G. spinigerum were collected from an infected Asian swamp eel, Monopterus albus, imported from Indonesia, and were identified to species morphologically40. The specimens were fixed in ethanol and stored at –20 °C until use. Total genomic DNA was isolated from individual worms using small-scale sodium dodecyl-sulphate (SDS)/proteinase K digestion and spin-column purification (Wizard® SV Genomic DNA Purification System, Promega). The identity of G. spinigerum specimens (coded GS5) was also verified by sequencing regions of ITS-2 and cox1 genes41; both regions had 100% similarity with those of G. spinigerum from Thailand and Indonesia (GenBank accession Nos. AB181155 and JN408304).
Long-PCR amplification and sequencing
Fragments of cox1 and nad1 genes of G. spinigerum were amplified by PCR with primer pairs JB3-JB4.542 and JB11-JB1243 (Table 4). After we obtained partial cox1 and nad1 sequences for G. spinigerum, we designed specific primers from these fragments for long PCR amplification. The complete mt genome of G. spinigerum (coded GS5) was amplified by long-PCR as two segments (~10 kb and ~4 kb) using genomic DNA extracted from a single specimen; the gaps between the two segments were filled by the short cox1 and nad1 fragments amplified initially. PCR was conducted in 25 μl using 2 mM MgCl2, 0.2 mM each of dNTPs, 2.5 μl 10 × Taq buffer, 2.5 μM of each primer and 0.5 μl LA Taq DNA polymerase (5 U/μl, Takara) in a thermocycler (Biometra). The cycling conditions were: 92 °C for 2 min (initial denaturation), then 92 °C for 10 s (denaturation), 56 °C (10 kb) or 54 °C (4 kb) for 30 s (annealing) and 60 °C for 10 min (extension) for 10 cycles, followed by 92 °C for 10 s, 56 °C (~10 kb) or 54 °C (~4 kb) for 30 s (annealing), and 60 °C for 10 min for 20 cycles, with a cycle elongation of 10 s for each cycle and a final extension at 60 °C for 10 min. PCR products were sequenced at Sangon Company (Shanghai, China) using a primer walking strategy44.
Table 4. Sequences of primers used to amplify PCR fragments from Gnathostoma spinigerum.
| Name of primer | Sequence (5’ to 3’) | Reference |
|---|---|---|
| Short PCR | ||
| JB3 | TTTTTTGGGCATCCTGAGGTTTAT | 42 |
| JB4.5 | TAAAGAAAGAACATAATGAAAATG | |
| JB11 | AGATTCGTAAGGGGCCTAATA | 43 |
| JB12 | ACCACTAACTAATTCACTTTC | |
| Long PCR | ||
| GSCF | GGTTTTCGTATGATGTTTTCTCCTT | This study |
| GSNR | CCACCATTCCATACTTAGACTTCCT | |
| GSNF | CCTGGAGTCGCTTTTGTAACTATGT | This study |
| GSCR | GTAGCCAACCATCTAAAAACCTTCA | |
Sequence analyses
Sequences obtained from the PCR amplicons of G. spinigerum were assembled manually and aligned with the mt genome sequences of roundworm and pinworm (GenBank accession numbers: NC_016128 and NC_011300) using the program MAFFT 7.12245 to identify gene boundaries. The sequence of each protein-coding gene was translated into amino acid sequence using the invertebrate mt genetic code in MEGA 546; the amino acid sequences were aligned using default settings. tRNAscan-SE47 and ARWEN48 were used to identify all of the tRNA genes except trnS2, which was identified manually by sequence comparison with trnS2 of other nematodes reported previously49. The two rRNA genes were identified by BLAST searches and were verified by sequence comparison with these two genes of other nematodes reported previously14. Tandem repeats in the non-coding regions were found using Tandem Repeat Finder program (http://tandem. bu.edu/trf/trf.html)50. The rearrangement events in the mt genomes were modelled with CREx (http://pacosy.informatik.uni-leipzig.de/crex)34.
Phylogenetic analyses
We combined the mt genome sequence of G. spinigerum with those of selected 57 other Chromadorea nematodes (Table S1) retrieved from the GenBank for phylogenetic analysis; Trichuris suis (GenBank accession number GU070737) was used as the outgroup51. Amino acid sequences inferred from the sequences of 12 mt PCGs were aligned individually first using MAFFT 7.122 and were then concatenated to form a single dataset; ambiguously aligned regions were excluded using Gblocks 0.91b (doc)52 with the default parameters (allow smaller final blocks, allow gap positions within the final blocks and allow less strict flanking positions). Phylogenetic analyses were conducted using Bayesian inference (BI). The MtArt + I + G + F model of amino acid evolution was selected as the most suitable model of evolution by ProtTest 2.453 based on the Akaike information criterion (AIC). As MtArt model is not implemented in the current version of MrBayes, an alternative model, CpREV, was used in BI and four chains (three heated and one cold) were run simultaneously for the Monte Carlo Markov Chain. Two independent runs for 2,000,000 metropolis-coupled MCMC generations, sampling a tree every 100 generations in MrBayes 3.1.154; the first 5,000 trees represented burn-in and the remaining trees were used to calculate Bpp. Bayesian analysis was run until the potential scale reduction factor approached 1 and the average standard deviation of split frequencies was less than 0.01. All sites were coded as unordered and equally weighted characters. The topology was reconstructed using the 50% majority rule and the support values were assessed by 1000 bootstrap replicates. Phylograms were drawn using the program FigTree v.1.4 (http://tree.bio.ed.ac.uk/software/figtree).
Additional Information
How to cite this article: Liu, G.-H. et al. Gnathostoma spinigerum Mitochondrial Genome Sequence: a Novel Gene Arrangement and its Phylogenetic Position within the Class Chromadorea. Sci. Rep. 5, 12691; doi: 10.1038/srep12691 (2015).
Supplementary Material
Acknowledgments
This work was supported, in part, by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840) and the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006) to XQZ. GHL acknowledges the funding support from Hunan Province Graduate Research and Innovation Project (Grant No. CX2013B285). RS acknowledges the funding support from the Australian Research Council (Discovery Project DP120100240) and the Australian and Chinese Governments (Australia-China Science & Research Fund, ACSRF00980).
Footnotes
The authors declare no competing financial interests.
Author Contributions X.Q.Z., R.S. and G.H.L. designed the research. G.H.L. performed the research. X.Q.C. and W.W.L. contributed reagents/materials/analyses. G.H.L., R.S. and X.Q.Z. analyzed the data. G.H.L., R.S. and X.Q.Z. wrote the manuscript.
References
- Ma A. & Gan X. X. Diagnosis and treatment of human gnathostomiasis. J Pathol Biol. 22, 385–388 (2010). In Chinese. [Google Scholar]
- Cole R. A., Choudhury A., Nico L. G. & Griffin K. M. Gnathostoma spinigerum in live Asian swamp eels (Monopterus spp.) from food markets and wild populations, United States. Emerg Infect Dis. 20, 634–642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman J. S. & Chiodini P. L. Gnathostomiasis, another emerging imported disease. Clin Microbiol Rev. 22, 484–492 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K., Tanaka H., Taniguchi Y., Shimizu M. & Kondo K. Two human cases of gnathostomiasis and discovery of a second intermediate host of Gnathostoma nipponicum in Japan. J Parasitol. 74, 623–627 (1988). [PubMed] [Google Scholar]
- Taniguchi Y., Ando K., Isoda K., Shimizu M. & Sonobe K. Human gnathostomiasis: successful removal of Gnathostoma hispidum. Int J Dermatol. 31, 175–177 (1992). [DOI] [PubMed] [Google Scholar]
- Yang Q. S., Chen K. G. & Lu X. S. Stomach perforation due to infection of Gnathostoma doloresi. Chin J Parasitol Parasit Dis. 23, 427 (2005). In Chinese. [PubMed] [Google Scholar]
- Katchanov J., Sawanyawisuth K., Chotmongkoi V. & Nawa Y. Neurognathostomiasis, a neglected parasitosis of the central nervous system. Emerg Infect Dis. 17, 1174–1180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J. & Auer H. Parasitoses of the human central nervous system. J Helminthol. 87, 257–270 (2013). [DOI] [PubMed] [Google Scholar]
- Sujata D. N. & Renu B. S. Intraocular gnathostomiasis from coastal part of Maharashtra. Trop Parasitol. 3, 82–84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ley P. & Blaxter M. Systematic position and phylogeny. In The Biology of Nematodes. Edited by Lee D. L.London and New York, Taylor & Francis, 1–30 (2002). [Google Scholar]
- Blaxter M. L. et al. A molecular evolutionary framework for the phylum Nematoda. Nature. 392, 71–75 (1998). [DOI] [PubMed] [Google Scholar]
- Meldal B. H. et al. An improved molecular phylogeny of the Nematoda with special emphasis on marine taxa. Mol Phylogenet Evol. 42, 622–636 (2007). [DOI] [PubMed] [Google Scholar]
- Nadler S. A. et al. Molecular phylogeny of clade III nematodes reveals multiple origins of tissue parasitism. Parasitology. 134, 1421–1442 (2007). [DOI] [PubMed] [Google Scholar]
- Kang S. et al. The mitochondrial genome sequence of Enterobius vermicularis (Nematoda: Oxyurida)–an idiosyncratic gene order and phylogenetic information for chromadorean nematodes. Gene. 429, 87–97 (2009). [DOI] [PubMed] [Google Scholar]
- Park J. K. et al. Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genomics. 12, 392 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana T. et al. Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol Biol. 13, 12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. H. et al. The complete mitochondrial genomes of three parasitic nematodes of birds: a unique gene order and insights into nematode phylogeny. BMC Genomics. 14, 414 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J. F. et al. Comparative analyses of the complete mitochondrial genomes of the two ruminant hookworms Bunostomum trigonocephalum and Bunostomum phlebotomum. Gene. 541, 92–100 (2014). [DOI] [PubMed] [Google Scholar]
- Holterman M. et al. Phylum-wide analysis of SSU rDNA reveals deep phylogenetic relationships among nematodes and accelerated evolution toward crown Clades. Mol Biol Evol. 23, 1792–800 (2006). [DOI] [PubMed] [Google Scholar]
- Kim T. et al. Phylogeny of Rhigonematomorpha based on the complete mitochondrial genome of Rhigonema thysanophora (Nematoda: Chromadorea). Zool Scr. 43, 289–303 (2014). [Google Scholar]
- Wolstenholme D. R. Animal mitochondrial DNA, structure and evolution. Int Rev Cytol. 141, 173–216 (1992). [DOI] [PubMed] [Google Scholar]
- Boore J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M. & Gasser R. B. Mitochondrial genomes of parasitic nematodes-progress and perspectives. Trends Parasitol. 22, 78–84 (2006). [DOI] [PubMed] [Google Scholar]
- Shao R. & Barker S. C. Mitochondrial genomes of parasitic arthropods: implications for studies of population genetics and evolution. Parasitology. 134, 153–167 (2007). [DOI] [PubMed] [Google Scholar]
- Liu G. H. et al. The complete mitochondrial genome of Toxascaris leonina: comparison with other closely related species and phylogenetic implications. Infect Genet Evol. 21, 329–333 (2014). [DOI] [PubMed] [Google Scholar]
- Liu G. H. et al. Mitochondrial genome of the eyeworm, Thelazia callipaeda (nematoda: spirurida), as the first representative from the family Thelaziidae. PLoS Negl Trop Dis. 7, e2029 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. H. et al. Characterization of the complete mitochondrial genome of Spirocerca lupi: sequence, gene organization and phylogenetic implications. Parasit Vectors. 6, 45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. H. et al. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene. 492, 110–116 (2012). [DOI] [PubMed] [Google Scholar]
- Kim K. H., Eom K. S. & Park J. K. The complete mitochondrial genome of Anisakis simplex (Ascaridida: Nematoda) and phylogenetic implications. Int J Parasitol. 36, 319–328 (2006). [DOI] [PubMed] [Google Scholar]
- Li M. W., Lin R. Q., Song H. Q., Wu X. Y. & Zhu X. Q. The complete mitochondrial genomes for three Toxocara species of human and animal health significance. BMC Genomics. 9, 224 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto R., Macfarlane J. L., Clary D. O. & Wolstenholme D. R. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 130, 471–498 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys-Pereira D. A. & Elling A. A. Mitochondrial genomes of Meloidogyne chitwoodi and M. incognita (Nematoda: Tylenchina): comparative analysis, gene order and phylogenetic relationships with other nematodes. Mol Biochem Parasitol. 194, 20–32 (2014). [DOI] [PubMed] [Google Scholar]
- Besnard G. et al. Fast assembly of the mitochondrial genome of a plant parasitic nematode (Meloidogyne graminicola) using next generation sequencing. C R Biol. 337, 295–301 (2014). [DOI] [PubMed] [Google Scholar]
- Bernt M. et al. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics. 23, 2957–2958 (2007). [DOI] [PubMed] [Google Scholar]
- Wijová M., Moravec F., Horák A. & Lukes J. Evolutionary relationships of Spirurina (Nematoda: Chromadorea: Rhabditida) with special emphasis on dracunculoid nematodes inferred from SSU rRNA gene sequences. Int J Parasitol. 36, 1067–1075 (2006). [DOI] [PubMed] [Google Scholar]
- Schneider A. Monoden. Berlin, Georg Reimer. 357, (1866). [Google Scholar]
- Cobb N. A. The orders and classes of nemas. Contrib Sci Nematol. 8, 213–216 (1919). [Google Scholar]
- Yang Z. On the best evolutionary rate for phylogenetic analysis. Syst Biol. 47, 125–133 (1998). [DOI] [PubMed] [Google Scholar]
- Lin R. et al. Analysis of the complete mitochondrial genome of Pochonia chlamydosporia suggests a close relationship to the invertebrate-pathogenic fungi in Hypocreales. BMC Microbiol. 15, 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. W. et al. Morphological and molecular identification of larval gnathostomes isolates from Heilongjiang and Guangzhou. Chin J Prev Vet Med. 34, 104–107 (2012). In Chinese. [Google Scholar]
- Ando K. et al. Comparative study on DNA sequences of ribosomal DNA and cytochrome c oxidase subunit 1 of mitochondrial DNA among five species of gnathostomes. J Helminthol. 80, 7–13 (2006). [DOI] [PubMed] [Google Scholar]
- Bowles J., Blair D. & McManus D. P. Genetic variants within the genus Echinococcus identified by mitochondrial DNA sequencing. Mol Biochem Parasitol. 54, 165–174 (1992). [DOI] [PubMed] [Google Scholar]
- Bowles J. & McManus D. P. NADH dehydrogenase 1 gene sequences compared for species and strains of the genus Echinococcus. Int J Parasitol. 23, 969–972 (1993). [DOI] [PubMed] [Google Scholar]
- Hu M., Jex A. R., Campbell B. E. & Gasser R. B. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nature Protoc. 2, 2339–2344 (2007). [DOI] [PubMed] [Google Scholar]
- Katoh K. & Standley D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T. M. & Eddy S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D. & Canback B. Arwen: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 24, 172–175 (2008). [DOI] [PubMed] [Google Scholar]
- Hu M., Chilton N. B. & Gasser R. B. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int J Parasitol. 32, 145–158 (2002). [DOI] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. H. et al. Clear genetic distinctiveness between human- and pig-derived Trichuris based on analyses of mitochondrial datasets. PLoS Negl Trop Dis. 6, e1539 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G. & Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56, 564–577 (2007). [DOI] [PubMed] [Google Scholar]
- Abascal F., Zardoya R. & Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 21, 2104–2105 (2005). [DOI] [PubMed] [Google Scholar]
- Ronquist F. & Huelsenbeck J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 19, 1572–1574 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.