

Abstract
Vascular oxidative stress and inflammation play an important role in angiotensin II–induced hypertension, and mitogen-activated protein kinases participate in these processes. We questioned whether mitogen-activated protein kinase–activated protein kinase 2 (MK2), a downstream target of p38 mitogen–activated protein kinase, is involved in angiotensin II–induced vascular responses. In vivo experiments were performed in wild-type and Mk2 knockout mice infused intravenously with angiotensin II. Angiotensin II induced a 30 mm Hg increase in mean blood pressure in wild-type that was delayed in Mk2 knockout mice. Angiotensin II increased superoxide production and vascular cell adhesion molecule-1 in blood vessels of wild-type but not in Mk2 knockout mice. Mk2 knockdown by small interfering RNA in mouse mesenteric vascular smooth muscle cells caused a 42% reduction in MK2 protein and blunted the angiotensin II–induced 40% increase of MK2 expression. Mk2 knockdown blunted angiotensin II–induced doubling of intracellular adhesion molecule-1 expression, 2.4-fold increase of nuclear p65, and 1.4-fold increase in Ets-1. Mk2 knockdown abrogated the angiotensin II–induced 4.7-fold and 1.3-fold increase of monocyte chemoattractant protein-1 mRNA and protein. Angiotensin II enhanced reactive oxygen species levels (by 29%) and nicotinamide adenine dinucleotide phosphate oxidase activity (by 48%), both abolished by Mk2 knockdown. Reduction of MK2 blocked angiotensin II–induced p47phox translocation to the membrane, associated with a 53% enhanced catalase expression. Angiotensin II–induced increase of MK2 was prevented by the nicotinamide adenine dinucleotide phosphate oxidase inhibitor Nox2ds-tat. Mk2 small interfering RNA prevented the angiotensin II–induced 30% increase of proliferation. In conclusion, MK2 plays a critical role in angiotensin II signaling, leading to hypertension, oxidative stress via activation of p47phox and inhibition of antioxidants, and vascular inflammation and proliferation.
Keywords: MK2, angiotensin II, oxidative stress, vascular inflammation, hypertension, arterial
Vascular wall inflammation plays a key role in the pathogenesis of hypertension, atherosclerosis, and other forms of cardiovascular disease. Angiotensin II (Ang II) induces vascular injury by modulating release of inflammatory chemokines such as monocyte chemoattractant protein-1 (MCP-1)1 and the nuclear action of proinflammatory transcription factors such as nuclear factor-κB (NF-κB). The latter, in turn, regulates expression of vascular cell adhesion molecule-1 (VCAM-1)2 and intracellular adhesion molecule-1 (ICAM-1). These events induce inflammation within the vascular wall, deposition of extracellular matrix and hypertrophy or hyperplasia of vascular smooth muscle cells (VSMCs). Ang II actions in the vascular wall are mediated in large measure through increase in the generation of reactive oxygen species (ROS).3 The major source of ROS in the vessel wall is reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.4 Upon stimulation by Ang II or other agents, the cytosolic subunits are translocated to the membrane and assemble with the membrane subunits to form the active NADPH oxidase capable of producing superoxide anion (·O2−). Redox-sensitive pathways are important in Ang II–stimulated growth and differentiation of cells, probably through activation of mitogen-activated protein kinases (MAPKs).5
p38 MAPK is markedly activated in cardiovascular cells by a variety of stimuli, including Ang II, and typically mediates apoptosis and inflammatory responses. Ang II rapidly stimulates the phosphorylation of p38 MAPK in cultured VSMCs6 as well as in cardiac and vascular tissues.7 Some in vitro studies suggest that Ang II–induced activation of NADPH oxidase results in upregulation of p38 MAPK.8,9 In addition, p38 MAPK plays an important role in endothelial inflammation and dysfunction associated with salt-sensitive hypertension.10 Treatment with SB202190, a p38 MAPK inhibitor, resulted in marked reductions in blood pressure in a diabetic rat model, which underlines the role of p38 MAPK in hemodynamic control.11 Upregulation of p38 MAPK induces early response gene expression and cell growth.12,13 Finally, in VSMCs from spontaneously hypertensive rat, p38 MAPK has been shown to influence Ang II–induced collagen production.14
p38 MAPK activates many protein kinases, such as MAPK-activated protein kinase 2 (MK2),15 MK3, MAPK-interacting kinases 116 and 2, and p38-regulated/activated protein.17 Phosphorylation of MK2 by p38 MAPK unmasks a nuclear export signal in the C-terminal part of the molecule.18,19 Several substrates have been described for MK2, including mRNA-binding proteins such as tristetraproline, transcription factors such as heat shock protein-1, proteins interacting with the cytoskeleton such as heat shock protein-25, as well as regulators of the cell cycle and apoptosis such as Cdc25B/C.20–24 Interaction of p38 MAPK with the preexisting complex of heat shock protein-27 and Akt is mediated by MK2.25 MK3, an isoform of MK2, was shown by Ronkina et al using a gene targeting approach to be coexpressed with MK2 in cells and tissues and to share similar physiological functions.26 More recently, Mk2 knockout (KO) mice were demonstrated to be more resistant to lipopolysaccharide-induced endotoxic shock as a consequence of a reduction of tumor necrosis factor-a production.27
In the present study, we investigated in vivo and in vitro whether MK2, one of the downstream targets of p38 MAPK, is involved in the pathway that mediates Ang II–induced inflammation, oxidative stress, and proliferation in VSMCs from mesenteric arteries, and in Ang II–induced hypertension. In vivo experiments were performed examining the effects of Ang II infusion into Mk2 KO mice. In vitro, Ang II action was tested on VSMCs in which Mk2 knockdown was achieved using small interfering RNA (siRNA).
Methods
An expanded Methods section can be found in an online data supplement available at http://hyper.ahajournals.org.
Animals and Blood Pressure Determination
Ten-week-old male C57BL/6 mice (Harlan Laboratories; Indianapolis, IN) were killed humanely by exsanguination under isoflurane anesthesia. Mesenteric arteries were dissected and used for the isolation of VSMCs. Fourteen- to 17-week-old male Mk2 KO and wild-type (WT) littermates were anesthetized with isoflurane, surgically instrumented with PA-C10 radiotelemetry transmitters as recommended by the manufacturer (Data Sciences International; St. Paul, MN), allowed to recover for 10 days, and then blood pressure was determined every 5 minutes for 10 s for 3 consecutive days. Thereafter, mice were infused with Ang II (400 ng/kg/min; Calbiochem; EMD Biosciences Inc.) or vehicle (Veh) using Alzet osmotic mini-pumps (DURECT Corp.) for 14 days. Blood pressure was determined as above during the 2-week treatment successfully in 5 WT+Veh, 3 WT+Ang II, 3 Mk2+Veh, and 4 Mk2+Ang II. At the end of the experiment, mice were killed as above, and the mesenteric vascular bed and aorta were dissected, frozen, and stored at −80°C for VCAM-1 and superoxide determination.
Cell Culture and Stimulation
VSMCs were isolated from mesenteric arteries as described previously28 and were used after 3 to 6 passages. They were transfected with siRNA (5 nmol/L; Qiagen; Mississauga, ON, Canada) targeting Mk2 (siMK2) or luciferase used as a negative control for 30 hours, or with a control fluorescein isothiocyanate (FITC)– coupled siRNA (10 µmol/L; Santa Cruz Biotechnology; Santa Cruz, CA) used to determine the efficiency of transfection. VSMCs were serum starved for 18 hours and treated with vehicle or Ang II (100 nmol/L) for 10 minutes to 24 hours, depending on the experiments. In some experiments, cells were also pre-exposed for 30 minutes to 10 µmol/L Nox2ds-tat (NADPH oxidase inhibitor) or to scrambled peptide used as negative control before stimulation with Ang II.29 For the efficiency of transfection after being transfected with FITC-coupled siRNA, VSMCs were stained with 4′,6-diamidino-2-phenylindole (Sigma Chemicals). Fluorescence was visualized with a DM2000 microscope with an FITC filter (488 nm; Leica Microsystems).
Nuclear Extract Preparation
VSMC nuclear extracts were prepared as described previously30 (details in the online supplement).
Western Blotting
Total or fractioned proteins (15 to 20 µg) were extracted from VSMCs, separated by SDS-PAGE, transferred to nitrocellulose membranes, and incubated overnight at 4°C with antibodies (1:1000) against the following: MK2, p38 MAPK, and NF-κB p65 subunit (from Cell Signaling Technology), and ICAM-1, VCAM-1, Ets-1, and p47phox (from Santa Cruz Biotechnology). After incubation with secondary antibodies, signals were revealed by chemiluminescence (SuperSignal West Pico chemiluminescent signal; Thermo Scientific), with the Molecular Imager Chemidoc XRS system (Bio-Rad), and quantified by densitometry using Quantity One software (Bio-Rad). Membranes were subsequently stripped and reprobed with anti–ß-actin antibody (Sigma Chemicals) to verify equal loading. VCAM-1 was also determined in protein extracts of mesenteric arteries from Mk2 KO and WT mice.
Quantitative RT-PCR
MCP-1 mRNA expression levels were determined by quantitative real-time PCR. RNA was extracted from VSMCs using the TRIzol reagent (Invitrogen). One microgram of total RNA was reversed transcribed with a Quantitect RT kit (Qiagen). Quantitative real-time PCR was performed using a QuantiTect SYBR Green PCR Kit (Qiagen) with the Mx3005P real-time PCR cycler (Stratagene). Quantitative real-time PCR results were normalized with ribosomal protein S16 and expressed as fold change of negative-control siRNA-treated samples. Primers were designed using Primer331 and are presented in the online data supplement.
MCP-1 Assay
MCP-1 levels were measured in the supernatant of VSMCs using a simplex microbead-based immunoassay kit on a Bio-Plex 200 (Bio-Rad) according to the manufacturer protocol.
ROS Levels
Intracellular ROS levels were evaluated using 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl– ester mixed isomers (CM-H2DCFDA; Invitrogen), a chloromethyl derivative of dichlorodihydrofluorescein diacetate that becomes fluorescent when oxidized. Dihydroethidium (DHE) was used to evaluate in situ production of superoxide (·O2−)32 (online supplement).
NADPH Oxidase Activity
The lucigenin-enhanced chemiluminescence assay was used to determine NADPH oxidase activity in VSMC homogenates as described previously.33 VSMCs were transfected with siMK2 or siRNA targeting luciferase, serum starved, and stimulated or not with Ang II for 5 minutes as above (online supplement).
Proliferation Assay
Ang II–stimulated DNA synthesis was determined by measuring incorporation of [3H]thymidine into DNA34 (online supplement).
Statistical Analysis
Experiments were repeated 3 to 6 times in duplicate. Results are presented as means±SEM and were compared by analysis of variance followed by a Student Newman–Keuls post hoc test. P<0.05 was considered significant.
Results
Mk2 Deletion Delays and Blunts the Ang II–Induced Rise in Mean Blood Pressure
To establish whether MK2 participates in Ang II–induced blood pressure rise in vivo, we measured mean blood pressure (MBP) by radiotelemetry in WT and Mk2 KO mice infused or not with Ang II for 14 days. No significant difference in day and night MBP was observed between Mk2 KO and WT mice at baseline and during the 14-day infusion with the vehicle (Figure 1). Ang II caused a 30- to 35-mm Hg increase in day and night MBP from day 2, which was maintained up to the end of the protocol in WT mice. Mk2 deletion delayed and blunted the rise in day and night MBP, respectively. Day MBP of Ang II–infused Mk2 KO mice increased significantly only at day 11 to day 14, whereas night MBP only tended to increase during the last few days of the protocol.
Figure 1.

Mk2 deletion delays and blunts, respectively, the Ang II–induced day and night blood pressure rise in mice. MBP was determined by radiotelemetry in WT and Mk2 KO mice at baseline and during the infusion with vehicle (Veh) or Ang II (400 ng/kg/min) for 14 days. The day (6 am to 8 pm) and night (8 pm to 6 am) average MBPs are presented. Results are means±SEM; *P<0.05 and **P<0.01 vs respective Veh, with n=5 for WT+Veh, n=3 for WT+Ang II, n=3 for Mk2+Veh, and n=4 for Mk2+Ang II.
Mk2 KO Reduces Ang II–Induced Vascular Superoxide Generation
·O2− production was evaluated by DHE fluorescence in aorta sections of WT and Mk2 KO mice infused or not with Ang II for 14 days. Basal production of ·O2− was significantly enhanced by Mk2 deletion (Figure 2A). Ang II increased ·O2− production in WT mice, whereas this change was blunted in Mk2 KO mice.
Figure 2.

Absence of MK2 prevents Ang II–induced superoxide and proinflammatory VCAM-1 production. In situ detection of superoxide generation in the aorta using DHE staining (A) and mesenteric artery protein level of VCAM-1 using Western blot (B) were determined in WT and Mk2 KO mice infused with vehicle (Veh) or Ang II (400 ng/kg/min) for 14 days. Representative Western blots of VCAM-1 and bar graphs representing VCAM-1 expression in different conditions are presented. Results are means±SEM; n=3 to 4; *P<0.05 vs respective WT+Veh.
Mk2 Deletion Blunts Ang II–Induced Inflammatory Responses
The levels of the proinflammatory molecule VCAM-1 were determined in mesenteric arteries of WT and Mk2 KO mice infused or not with Ang II for 14 days. Basal levels of VCAM-1 were increased 1.7-fold in Mk2 KO mice compared with WT mice (Figure 2B). Ang II infusion increased 1.4-fold the levels of VCAM-1 in WT mice and had no effect in Mk2 KO mice.
Ang II Upregulates p38 MAPK and MK2
Ang II increased p38 MAPK phosphorylation by 45% (Figure 3A) as well as MK2 phosphorylation and total expression in VSMCs by 29% and 39%, respectively (Figure 3B and 3D). For gene silencing experiments, the efficiency of VSMCs siRNA transfection was determined using a control FITC-coupled siRNA. We observed an efficiency of 56% (Figure 3C). siMK2 decreased MK2 protein by 42% and blunted the Ang II–induced increase of MK2 expression (Figure 3D). The expression of p38 MAPK was not significantly affected by MK2 knockdown (data not shown).
Figure 3.
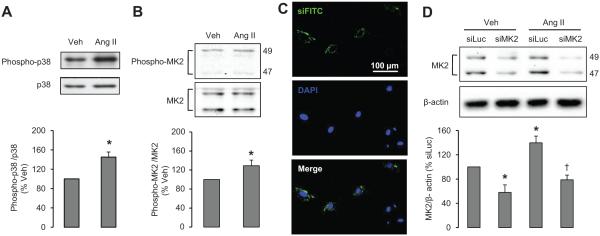
A and B, Ang II increases p38 MAPK and MK2 phosphorylation levels. VSMCs were stimulated with vehicle (Veh) or Ang II for 10 minutes. Representative Western blots of phosphorylated and total p38 MAPK (A) and MK2 (B) and corresponding bar graphs are represented. C, VSMCs were transfected with a nontargeting control FITC-siRNA (siFITC) for 24 hours. Representative siFITC and 4′,6-diamidino-2-phenylindole (DAPI) images are presented. D, MK2 siRNA transfection blunts Ang II–induced MK2 expression. VSMCs were transfected with siMK2 or negative control siRNA targeting luciferase (siLuc) and treated with Veh or Ang II for 24 hours. Representative Western blots of MK2 and bar graphs representing MK2 expression in different conditions are depicted. Results are means±SEM; n=4 to 5; *P<0.05 vs Veh for A and B and vs siLuc+Veh for D, and †P<0.01 vs siLuc+Ang II for D.
MK2 Is Involved in Ang II–Induced Increase of ICAM-1, NF-κB, Ets-1, and MCP-1
VSMC Mk2 knockdown did not significantly alter protein levels of ICAM-1, VCAM-1, and Ets-1, but resulted in a1.5-fold increase of the nuclear level of the p65 subunit of NF-κB (Figure 4). Ang II increased ICAM-1 and nuclear p65 2-fold and Ets-1 1.4-fold without altering the levels of VCAM-1. siMK2 blunted these Ang II responses. siMK2 did not alter the basal expression of MCP-1, but abrogated the Ang II–induced 4.7- and 1.3-fold increase of MCP-1 at mRNA and protein levels, respectively (Figure 5). These results demonstrate the involvement of MK2 activation in some of Ang II–induced inflammatory pathways.
Figure 4.

Downregulation of MK2 prevents Ang II–induced proinflammatory molecules. The protein levels of ICAM-1 (A), VCAM-1 (B), NF-κB p65 subunit (C), and Ets-1 (D) were determined in VSMCs transfected with siMK2 or with the luciferase siRNA (siLuc) used as a negative control and treated with vehicle (Veh) or Ang II for 24 hours. Representative Western blots of proinflammatory mediators and bar graphs representing proinflammatory molecule expression in different conditions are shown. Results are means±SEM; n=4 to 5; *P<0.05 vs siLuc+Veh and †P<0.05 vs siLuc+Ang II.
Figure 5.

Downregulation of MK2 prevents Ang II–induced increase of MCP-1. The mRNA (A) and protein (B) levels of MCP-1 were determined in VSMCs transfected with siMK2 or with the luciferase siRNA (siLuc) used as a negative control and treated with vehicle (Veh) or Ang II for 24 hours. Results are means±SEM; n=4; *P<0.05 vs siLuc+Veh and †P<0.05 vs siLuc+Ang II.
MK2 Mediates Ang II–Induced ROS Generation Via NADPH Oxidase
Intracellular ROS levels and in situ production of ·O2− were evaluated respectively in VSMCs by dichlorodihydrofluorescein diacetate and by DHE fluorescence. Mk2 knockdown did not affect the basal levels of intracellular ROS or generation of ·O2− in VSMCs (Figure 6A and 6B). Ang II increased intracellular ROS levels (by 29%) and production of ·O2− revealed by DHE staining in VSMCs, both abrogated by MK2 knockdown. Ang II increased NADPH oxidase activity (48%), the main vascular source of ·O2− (Figure 6C). Mk2 knockdown abolished the increase in NADPH oxidase activity induced by Ang II without altering basal levels. These results suggest that Ang II–induced NADPH oxidase activation involves MK2.
Figure 6.
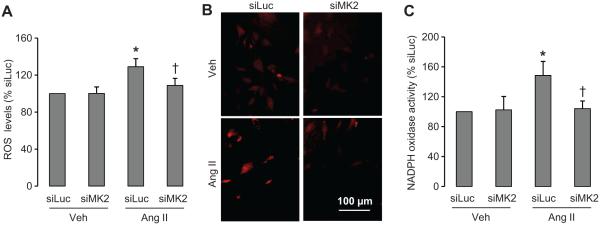
Downregulation of MK2 abolished Ang II–induced increase of oxidative stress. ROS were measured by DCF-DA fluorescence (A), superoxide generation was detected in situ with DHE staining (B), and NADPH oxidase activity was determined by lucigenin chemiluminescence (C) in VSMCs transfected with siMK2 or with the luciferase siRNA (siLuc) used as a negative control and treated with vehicle (Veh) or Ang II for 10 minutes. Results are means±SEM; n=5; *P<0.05 vs siLuc+Veh and †P<0.05 vs siLuc+Ang II.
Ang II/MK2–Induced NADPH Oxidase Involves p47phox Translocation
To further understand the molecular pathways of activation of NADPH oxidase by MK2, we examined the translocation of p47phox, one of the main cytosolic subunits of NADPH oxidase, the translocation of which from the cytosol to the membrane is essential for the assembling of the active NADPH oxidase in the cell membrane. Ang II increased p47phox translocation to the VSMC membrane 2-fold, an effect that was prevented by Mk2 knockdown (Figure 7A). Basal p47phox membrane/cytosolic ratio was not altered by Mk2 knockdown.
Figure 7.

MK2 is involved in an Ang II–induced NADPH oxidase activation positive feedback loop. A, p47phox translocation from cytoplasm to the membrane was demonstrated in VSMCs transfected with siMK2 or with the luciferase siRNA (siLuc) used as a negative control and treated with vehicle (Veh) or Ang II for 5 minutes. B, MK2 protein level was determined in VSMCs exposed or not to the NADPH oxidase inhibitor Nox2ds-tat (Nox2) or a scrambled peptide and treated with vehicle (Veh) or Ang II for 24 hours. Representative Western blots of p47phox, MK2, and bar graphs representing membrane and cytoplasmic p47phox and MK2 expression in different conditions are depicted. Results are means±SEM; n=4, *P<0.01 vs siLuc+Veh and †P<0.05 vs siLuc+Ang II for A and n=5, *P<0.01 vs Veh for B.
Ang II–Induced NADPH Oxidase Upregulates MK2 Expression
To determine whether activation of NADPH oxidase affects the expression of MK2, VSMCs were pretreated with the NADPH oxidase inhibitor Nox2ds-tat and then stimulated or not with Ang II. Nox2ds-tat is a small peptide that was designed to inhibit assembly of p47phox with gp91-phox (Nox2) and thus inhibit NADPH oxidase (Nox2) activation. An Ang II–induced increase of MK2 protein levels was prevented by ds-tat but not by the ds-tat scrambled peptide (Figure 7B). This result suggests that Ang II–induced NADPH oxidase-driven superoxide generation induces a positive feedback loop by increasing the expression of MK2.
Downregulation of MK2 Caused Ang II to Upregulate Catalase in VSMCs
We then evaluated the effects of modulation of MK2 on expression of the antioxidant catalase in VSMCs. Neither Ang II nor MK2 knockdown had any effect on the basal levels of catalase (Figure 8). However, in presence of siMK2, Ang II increased catalase expression by 53%. These results suggest that MK2 has an inhibitory effect on catalase in oxidative conditions, which further increases ROS production.
Figure 8.

MK2 knockdown causes Ang II to upregulate catalase in VSMCs. Catalase protein levels were determined in VSMCs transfected with siMK2 or with the luciferase siRNA (siLuc) used as a negative control and treated with vehicle (Veh) or Ang II for 24 hours. Representative Western blots of catalase and bar graphs representing catalase expression in different conditions are presented. Results are means±SEM; n=4; *P<0.05 vs siLuc+Veh.
Downregulation of MK2 Prevents Ang II–Induced VSMC Proliferation
To determine whether MK2 mediates Ang II–induced vascular remodeling, VSMC proliferation was evaluated using [3H]thymidine incorporation. MK2 knockdown decreased by 50% [3H]thymidine VSMC incorporation under basal conditions (Figure 9). Ang II increased [3H]thymidine incorporation into VSMCs by 30%, which was abolished by MK2 knockdown, suggesting that MK2 is a mediator of Ang II–induced proliferation.
Figure 9.

Downregulation of MK2 blocks Ang II–induced VSMC proliferation. [3H]thymidine incorporation was determined in VSMCs transfected with siMK2 or with the luciferase siRNA (siLuc) used as a negative control and treated with vehicle (Veh) or Ang II for 24 hours. Results are means±SEM; n=5; *P<0.05 vs siLuc+Veh and †P<0.05 vs siLuc+Ang II.
Discussion
The present study demonstrates the following novel findings. In vivo, Mk2 plays a key role in Ang II–induced rise in blood pressure and vascular superoxide production and inflammation. Similarly, in vitro MK2, one of the direct downstream targets of p38 MAPK, mediates Ang II–induced increase of inflammatory mediators as well as superoxide production from NADPH oxidase, through regulation of the p47phox subunit of NADPH oxidase and the antioxidant enzyme catalase, and VSMC proliferation.
To investigate the role of MK2 in Ang II–induced hypertension; we performed in vivo experiments using Mk2 KO mice. There are contradictory results on the effect of p38 MAPK pharmacological inhibitors on hypertension in different experimental models.10,35,36 However, our results showed for the first time that the absence of MK2, which is downstream of p38 MAPK, delayed Ang II–induced hypertension. In addition, our in vivo data showed a decrease of Ang II–induced oxidative stress and inflammation in Mk2 KO mice, demonstrating a crucial role for MK2 in Ang II signaling, leading to increases in blood pressure. To determine the molecular mechanisms involved, we performed in vitro experiments by knocking down MK2 in mesenteric VSMCs. Vascular MK2 in mice in our study was upregulated by Ang II. Previous in vitro studies reported that Ang II activates p38 MAPK in VSMCs from resistance and conduit arteries.37 In addition, acute and chronic infusion of Ang II upregulated p38 MAPK in hypertensive models.38 Indeed, activated MK2 is cotransported with active p38 MAPK to the cytoplasm.18 siMK2 blunted the Ang II–induced increase of MK2 but did not affect p38 MAPK expression. This result is in agreement with a previous study on MK2 KO mice embryonic fibroblasts that showed that p38 MAPK activity was unchanged by Ang II.27 p38 MAPK is involved in Ang II–induced inflammatory responses.39 Accordingly, we examined the effect of MK2 downregulation on Ang II–induced increase of vascular inflammatory mediators. MK2 is involved in Ang II–induced increase of inflammatory adhesion molecule such as ICAM-1, as well as the proinflammatory chemokine MCP-1, and the proinflammatory transcription factor NF-κB. Previous experiments have shown that SB203580, an inhibitor of p38 MAPK, blocks the synthesis of proinflammatory cytokines at the transcriptional level.40 Moreover, mice lacking MK2 gene showed a defect in lipopolysaccharide-induced biosynthesis of inflammatory cytokines.27 Recently, it was reported that Ang II stimulates MCP-1 expression via distinct p38 MAPK- and NF-κB–dependent pathways in VSMCs.41 Thus, it appears that Ang II–induced upregulation of MCP-1 occurs through a coactivation of MK2 and NF-κB, with MK2 influencing as well the NF-κB pathway. Further, it has been reported that p38 MAPK is involved in monocyte/macrophage survival regulated by the macrophage colony-stimulating factor.42 Recently, a novel role for transcription factor Ets-1 has been described as a transcriptional mediator of vascular inflammation and remodeling.43 The induction of MCP-1 by Ang II is largely dependent on Ets-1. We have shown here that MK2 downregulation led to a marked reduction in the induction of Ets-1 by Ang II. Further experiments are needed to determine which inflammatory factor is initially upregulated by Ang II–activated MK2.
A central mechanism by which Ang II promotes inflammation is via the generation of ROS.44 In fact, p38 MAPK is a redox-sensitive kinase.6,45 Ang II–induced MK2 upregulation was significantly reduced by NADPH oxidase inhibition, suggesting that ROS produced by NADPH oxidase act as second messengers in Ang II–triggered MK2 upregulation (Figure 10). Some in vitro studies suggest an important influence of NADPH oxidase in p38 MAPK activation.9,33 We found that downregulation of MK2 abolished Ang II–induced increase of ROS production and NADPH oxidase activation. This suggests that MK2 plays an important role in NADPH oxidase activation by Ang II and is in agreement with our in vivo data as well as with what has been reported previously for p38 MAPK. p38 MAPK activates NADPH oxidase in human endothelial cells by enhancing phosphorylation and assembly of its subunits.8 SB239063, a p38 MAPK inhibitor, blunted Ang II–induced increase of NADPH oxidase subunits and restored endothelium-dependent relaxation in stroke-prone spontaneously hypertensive rats fed a high-salt/high-fat diet.38 Our results suggest that MK2 is one of the most important downstream targets of p38 MAPK and is involved directly in Ang II–induced redox signaling. This suggests that upregulation of MK2 by Ang II activates proinflammatory molecules that could regulate NADPH oxidase activation and ROS generation. Our data also appear to suggest the involvement of Nox2 NADPH oxidase (also known as, Nox2 oxidase). Activation of Nox2 oxidase, which has been shown to be present in resistance artery smooth muscle,46 is a multistep process that is initiated by serine phosphorylation of the cytosolic regulatory subunit p47phox. In the canonical Nox2 oxidase system, the activated p47phox subunit translocates to the membrane along with other essential cytosolic subunits and associates with the membrane-bound Nox2 (also known as gp91-phox) and p22phox subunits to produce active NADPH oxidase. We were able to demonstrate that MK2 plays a critical role in Ang II–induced p47phox translocation. Further, we demonstrated that the Nox2 oxidase inhibitor Nox2ds-tat, which targets p47phox and interferes with subunit assembly, blunted Ang II–induced MK2, indicating that MK2 is in fact part of a positive-feedback loop. Ang II increased NADPH oxidase activity through MK2, and NADPH oxidase activation induced MK2. Interestingly, a recent study reported that Ets-1 is a critical transcriptional mediator of ROS generation by Ang II by regulating the expression of p47phox subunit.47 Because we have shown that MK2 plays a role in Ang II–induced Ets-1 upregulation, we can hypothesize that one of the mediators involved in the regulation of p47phox by MK2 could be Ets-1. Moreover, we found that only in the absence of MK2, Ang II induced a slight increase of catalase, a major antioxidant enzyme expressed in the vasculature.48 In hypertensive animal models, H2O2 levels are higher compared with normotensive animals.49 In addition, H2O2 is specifically involved in the blood pressure–induced enhancement of responses to Ang II.50 The generation of H2O2 after Ang II stimulation is a critical step in arterial wall hypertrophy.51 This could explain in part the regulation of catalase by MK2 in hypertensive models and suggests that active MK2 regulates cellular ROS levels in part by inhibiting antioxidant enzymes.
Figure 10.
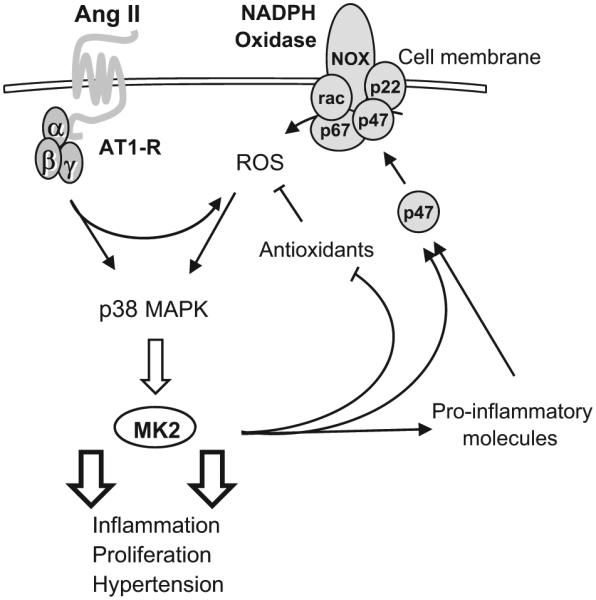
Proposed role for MK2 in Ang II signaling in mesenteric VSMCs. Ang II activates the NADPH oxidase through angiotensin receptor type 1 (AT1R). The increased production of ROS activates p38 MAPK, which leads to the upregulation of MK2. Activated MK2 in turn activates proinflammatory molecules, inhibits antioxidants, and potentiates NADPH oxidase activation by regulating the translocation of the p47 phox (p47) subunit to the membrane. These effects lead to vascular inflammation, cell proliferation, and hypertension. NOX, rac, p67, p47, and p22 are NADPH oxidase subunits.
Vascular inflammation is closely related to vascular cell proliferation. Thus, to further understand the functional effect of MK2 activation by Ang II, we investigated VSMC proliferation and demonstrated that MK2 plays an important role in Ang II–induced cell growth. p38 MAPK is activated by G-protein– coupled receptors, such as Ang II type 1 receptors, and plays an important role in the induction of proliferative responses in susceptible cells.12 However, in rat VSMCs, Ang II–induced growth may be p38 MAPK independent.52 It was reported recently that endothelin-1 drives proliferation of VSMCs through stimulation of extracellular signal-regulated kinase and p38 MAPK.53 Moreover, very recently, using a cardiomyocyte-specific and inducible transgenic approach, MK2 activity was shown to play an important role in p38 MAPK–mediated cardiac hypertrophy and heart failure.54 Our findings suggest that MK2 activation by Ang II may contribute to vascular remodeling by promoting cell growth.
Our in vivo data on blood pressure support clearly the implication of MK2 in Ang II– induced hypertension, and the potential molecular mechanism was demonstrated in VSMCs. However, additional in vivo experiments will be performed to confirm the proposed signaling pathway, and experiments with shorter time of Ang II infusion may be needed to determine whether the observed Ang II effects involving MK2 are related to blood pressure increase.
In summary, the data reported here demonstrate the novel findings that MK2 is involved in Ang II–induced hypertension and is upregulated by Ang II in VSMCs through generation of ROS via NADPH oxidase, likely Nox2 oxidase. Activation of MK2 upregulates proinflammatory molecules that further activate ROS generation, via effects on the p47phox subunit of NADPH oxidase as well as on antioxidant enzymes. Upregulation of MK2 appears to be part of a vicious circle that potentiates inflammatory responses to Ang II, leading as well to an increase in VSMC proliferation (Figure 10).
Perspectives
Clinical trials with p38 MAPK pharmacological inhibitors as anti-inflammatory therapies have been shown to be ineffective, attributable in part to the influence of p38 MAPK on diverse cellular pathways. The present results show for the first time that MK2 is an essential component of Ang II–induced hypertension and vascular oxidative stress and inflammation and could be a promising target for specific cardiovascular anti-inflammatory therapy.
Supplementary Material
Acknowledgments
We thank Dr Bruce G. Allen of the Montreal Heart Institute for the Mk2 KO breeder pair mice. We are also grateful to André Turgeon and Marie-Éve Deschênes for excellent technical support and animal care.
Sources of Funding
This work was funded by Canadian Institutes of Health Research (CIHR) grant MOP82790 to E.L.S., by Canada Research Chairs (CRC) from the CRC Government of Canada/CIHR Program and the Canada Fund for Innovation (CFI) to E.L.S. and S.W. T.E. was supported by a fellowship from the Heart and Stroke Foundation of Canada.
Footnotes
Disclosures
None.
References
- 1.Chen XL, Tummala PE, Olbrych MT, Alexander RW, Medford RM. Angiotensin II induces monocyte chemoattractant protein-1 gene expression in rat vascular smooth muscle cells. Circ Res. 1998;83:952–959. doi: 10.1161/01.res.83.9.952. [DOI] [PubMed] [Google Scholar]
- 2.Tummala PE, Chen XL, Sundell CL, Laursen JB, Hammes CP, Alexander RW, Harrison DG, Medford RM. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: a potential link between the renin-angiotensin system and atherosclerosis. Circulation. 1999;100:1223–1229. doi: 10.1161/01.cir.100.11.1223. [DOI] [PubMed] [Google Scholar]
- 3.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept. 2000;91:21–27. doi: 10.1016/s0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 4.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 5.Touyz RM, Cruzado M, Tabet F, Yao G, Salomon S, Schiffrin EL. Redox-dependent MAP kinase signaling by Ang II in vascular smooth muscle cells: role of receptor tyrosine kinase transactivation. Can J Physiol Pharmacol. 2003;81:159–167. doi: 10.1139/y02-164. [DOI] [PubMed] [Google Scholar]
- 6.Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem. 1998;273:15022–15029. doi: 10.1074/jbc.273.24.15022. [DOI] [PubMed] [Google Scholar]
- 7.Zhang GY, Li X, Yi CG, Pan H, He GD, Yu Q, Jiang LF, Xu WH, Li ZJ, Ding J, Lin DS, Gao WY. Angiotensin II activates connective tissue growth factor and induces extracellular matrix changes involving Smad/activation and p38 mitogen-activated protein kinase signaling pathways in human dermal fibroblasts. Exp Dermatol. 2009;18:947–953. doi: 10.1111/j.1600-0625.2009.00880.x. [DOI] [PubMed] [Google Scholar]
- 8.Lal AS, Clifton AD, Rouse J, Segal AW, Cohen P. Activation of the neutrophil NADPH oxidase is inhibited by SB 203580, a specific inhibitor of SAPK2/p38. Biochem Biophys Res Commun. 1999;259:465–470. doi: 10.1006/bbrc.1999.0759. [DOI] [PubMed] [Google Scholar]
- 9.Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97:772–780. doi: 10.1161/01.RES.0000185804.79157.C0. [DOI] [PubMed] [Google Scholar]
- 10.Ju H, Behm DJ, Nerurkar S, Eybye ME, Haimbach RE, Olzinski AR, Douglas SA, Willette RN. p38 MAPK inhibitors ameliorate target organ damage in hypertension: part 1. p38 MAPK-dependent endothelial dysfunction and hypertension. J Pharmacol Exp Ther. 2003;307:932–938. doi: 10.1124/jpet.103.057422. [DOI] [PubMed] [Google Scholar]
- 11.Komers R, Schutzer W, Xue H, Oyama TT, Lindsley JN, Anderson S. Effects of p38 mitogen-activated protein kinase inhibition on blood pressure, renal hemodynamics, and renal vascular reactivity in normal and diabetic rats. Transl Res. 2007;150:343–349. doi: 10.1016/j.trsl.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Clerk A, Michael A, Sugden PH. Stimulation of multiple mitogen-activated protein kinase sub-families by oxidative stress and phosphorylation of the small heat shock protein, HSP25/27, in neonatal ventricular myocytes. Biochem J. 1998;333:581–589. doi: 10.1042/bj3330581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.New L, Jiang Y, Zhao M, Liu K, Zhu W, Flood LJ, Kato Y, Parry GC, Han J. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998;17:3372–3384. doi: 10.1093/emboj/17.12.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touyz RM, He G, El Mabrouk M, Schiffrin EL. p38 Map kinase regulates vascular smooth muscle cell collagen synthesis by angiotensin II in SHR but not in WKY. Hypertension. 2001;37:574–580. doi: 10.1161/01.hyp.37.2.574. [DOI] [PubMed] [Google Scholar]
- 15.Stokoe D, Campbell DG, Nakielny S, Hidaka H, Leevers SJ, Marshall C, Cohen P. MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO J. 1992;11:3985–3994. doi: 10.1002/j.1460-2075.1992.tb05492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukunaga R, Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16:1921–1933. doi: 10.1093/emboj/16.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol. 1998;8:1049–1057. doi: 10.1016/s0960-9822(98)70442-7. [DOI] [PubMed] [Google Scholar]
- 19.Adams RH, Porras A, Alonso G, Jones M, Vintersten K, Panelli S, Valladares A, Perez L, Klein R, Nebreda AR. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol Cell. 2000;6:109–116. [PubMed] [Google Scholar]
- 20.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14–3-3 binding. J Biol Chem. 2004;279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 21.Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 22.Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. MK2-induced tristetraprolin:14–3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M. Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett. 1992;313:307–313. doi: 10.1016/0014-5793(92)81216-9. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Khaleque MA, Zhao MJ, Zhong R, Gaestel M, Calderwood SK. Phosphorylation of HSF1 by MAPK-activated protein kinase 2 on serine 121, inhibits transcriptional activity and promotes HSP90 binding. J Biol Chem. 2006;281:782–791. doi: 10.1074/jbc.M505822200. [DOI] [PubMed] [Google Scholar]
- 25.Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J Biol Chem. 2006;281:37215–37226. doi: 10.1074/jbc.M603622200. [DOI] [PubMed] [Google Scholar]
- 26.Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27:170–181. doi: 10.1128/MCB.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez JB, Pitman D, Lin LL, Gaestel M. Distinct cellular functions of MK2. Mol Cell Biol. 2002;22:4827–4835. doi: 10.1128/MCB.22.13.4827-4835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Touyz RM, Tolloczko B, Schiffrin EL. Mesenteric vascular smooth muscle cells from spontaneously hypertensive rats display increased calcium responses to angiotensin II but not to endothelin-1. J Hypertens. 1994;12:663–673. [PubMed] [Google Scholar]
- 29.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 30.Morin S, Paradis P, Aries A, Nemer M. Serum response factor-GATA ternary complex required for nuclear signaling by a G-protein-coupled receptor. Mol Cell Biol. 2001;21:1036–1044. doi: 10.1128/MCB.21.4.1036-1044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, Chen AF. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation. 2003;107:1053–1058. doi: 10.1161/01.cir.0000051459.74466.46. [DOI] [PubMed] [Google Scholar]
- 33.Touyz RM, Yao G, Viel E, Amiri F, Schiffrin EL. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J Hypertens. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. [DOI] [PubMed] [Google Scholar]
- 34.Bunkenburg B, van AT, Rogg H, Wood JM. Receptor-mediated effects of angiotensin II on growth of vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 1992;20:746–754. doi: 10.1161/01.hyp.20.6.746. [DOI] [PubMed] [Google Scholar]
- 35.Behr TM, Nerurkar SS, Nelson AH, Coatney RW, Woods TN, Sulpizio A, Chandra S, Brooks DP, Kumar S, Lee JC, Ohlstein EH, Angermann CE, Adams JL, Sisko J, Sackner-Bernstein JD, Willette RN. Hypertensive end-organ damage and premature mortality are p38 mitogen-activated protein kinase-dependent in a rat model of cardiac hypertrophy and dysfunction. Circulation. 2001;104:1292–1298. doi: 10.1161/hc3601.094275. [DOI] [PubMed] [Google Scholar]
- 36.Widder J, Behr T, Fraccarollo D, Hu K, Galuppo P, Tas P, Angermann CE, Ertl G, Bauersachs J. Vascular endothelial dysfunction and superoxide anion production in heart failure are p38 MAP kinase-dependent. Cardiovasc Res. 2004;63:161–167. doi: 10.1016/j.cardiores.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Sugden PH, Clerk A. “Stress-responsive” mitogen-activated protein kinases (c-Jun N-terminal kinases and p38 mitogen-activated protein kinases) in the myocardium. Circ Res. 1998;83:345–352. doi: 10.1161/01.res.83.4.345. [DOI] [PubMed] [Google Scholar]
- 38.Bao W, Behm DJ, Nerurkar SS, Ao Z, Bentley R, Mirabile RC, Johns DG, Woods TN, Doe CP, Coatney RW, Ohlstein JF, Douglas SA, Willette RN, Yue TL. Effects of p38 MAPK Inhibitor on angiotensin II-dependent hypertension, organ damage, and superoxide anion production. J Cardiovasc Pharmacol. 2007;49:362–368. doi: 10.1097/FJC.0b013e318046f34a. [DOI] [PubMed] [Google Scholar]
- 39.Hippenstiel S, Soeth S, Kellas B, Fuhrmann O, Seybold J, Krull M, Eichel-Streiber C, Goebeler M, Ludwig S, Suttorp N. Rho proteins and the p38-MAPK pathway are important mediators for LPS-induced interleukin-8 expression in human endothelial cells. Blood. 2000;95:3044–3051. [PubMed] [Google Scholar]
- 40.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi M, Suzuki E, Takeda R, Oba S, Nishimatsu H, Kimura K, Nagano T, Nagai R, Hirata Y. Angiotensin II and tumor necrosis factor-alpha synergistically promote monocyte chemoattractant protein-1 expression: roles of NF-kappaB, p38, and reactive oxygen species. Am J Physiol Heart Circ Physiol. 2008;294:H2879–H2888. doi: 10.1152/ajpheart.91406.2007. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Zeigler MM, Lam GK, Hunter MG, Eubank TD, Khramtsov VV, Tridandapani S, Sen CK, Marsh CB. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte/macrophage survival. Am J Respir Cell Mol Biol. 2007;36:68–77. doi: 10.1165/rcmb.2006-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, Ho IC, Oettgen P. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest. 2005;115:2508–2516. doi: 10.1172/JCI24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taniyama Y, Hitomi H, Shah A, Alexander RW, Griendling KK. Mechanisms of reactive oxygen species-dependent downregulation of insulin receptor substrate-1 by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:1142–1147. doi: 10.1161/01.ATV.0000164313.17167.df. [DOI] [PubMed] [Google Scholar]
- 45.Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kubler W, Kreuzer J. Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol. 2000;20:940–948. doi: 10.1161/01.atv.20.4.940. [DOI] [PubMed] [Google Scholar]
- 46.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 47.Ni W, Zhan Y, He H, Maynard E, Balschi JA, Oettgen P. Ets-1 is a critical transcriptional regulator of reactive oxygen species and p47(phox) gene expression in response to angiotensin II. Circ Res. 2007;101:985–994. doi: 10.1161/CIRCRESAHA.107.152439. [DOI] [PubMed] [Google Scholar]
- 48.Jeney V, Itoh S, Wendt M, Gradek Q, Ushio-Fukai M, Harrison DG, Fukai T. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ Res. 2005;96:723–729. doi: 10.1161/01.RES.0000162001.57896.66. [DOI] [PubMed] [Google Scholar]
- 49.Zhou X, Bohlen HG, Miller SJ, Unthank JL. NAD(P)H oxidase-derived peroxide mediates elevated basal and impaired flow-induced NO production in SHR mesenteric arteries in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H1008–H1016. doi: 10.1152/ajpheart.00114.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bagi Z, Erdei N, Koller A. High intraluminal pressure via H2O2 upregulates arteriolar constrictions to angiotensin II by increasing the functional availability of AT1 receptors. Am J Physiol Heart Circ Physiol. 2008;295:H835–H841. doi: 10.1152/ajpheart.00205.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension. 2005;46:732–737. doi: 10.1161/01.HYP.0000182660.74266.6d. [DOI] [PubMed] [Google Scholar]
- 52.Touyz RM, He G, El MM, Diep Q, Mardigyan V, Schiffrin EL. Differential activation of extracellular signal-regulated protein kinase 1/2 and p38 mitogen activated-protein kinase by AT1 receptors in vascular smooth muscle cells from Wistar-Kyoto rats and spontaneously hypertensive rats. J Hypertens. 2001;19:553–559. doi: 10.1097/00004872-200103001-00006. [DOI] [PubMed] [Google Scholar]
- 53.Chen S, Qiong Y, Gardner DG. A role for p38 mitogen-activated protein kinase and c-myc in endothelin-dependent rat aortic smooth muscle cell proliferation. Hypertension. 2006;47:252–258. doi: 10.1161/01.HYP.0000198424.93598.6b. [DOI] [PubMed] [Google Scholar]
- 54.Streicher JM, Ren S, Herschman H, Wang Y. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ Res. 2010;106:1434–1443. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.