

Abstract

We have investigated cation exchange reactions in copper selenide nanocrystals using two different divalent ions as guest cations (Zn2+ and Cd2+) and comparing the reactivity of close to stoichiometric (that is, Cu2Se) nanocrystals with that of nonstoichiometric (Cu2–xSe) nanocrystals, to gain insights into the mechanism of cation exchange at the nanoscale. We have found that the presence of a large density of copper vacancies significantly accelerated the exchange process at room temperature and corroborated vacancy diffusion as one of the main drivers in these reactions. Partially exchanged samples exhibited Janus-like heterostructures made of immiscible domains sharing epitaxial interfaces. No alloy or core–shell structures were observed. The role of phosphines, like tri-n-octylphosphine, in these reactions, is multifaceted: besides acting as selective solvating ligands for Cu+ ions exiting the nanoparticles during exchange, they also enable anion diffusion, by extracting an appreciable amount of selenium to the solution phase, which may further promote the exchange process. In reactions run at a higher temperature (150 °C), copper vacancies were quickly eliminated from the nanocrystals and major differences in Cu stoichiometries, as well as in reactivities, between the initial Cu2Se and Cu2–xSe samples were rapidly smoothed out. These experiments indicate that cation exchange, under the specific conditions of this work, is more efficient at room temperature than at higher temperature.
Introduction
During the last decades, cation exchange (CE) reactions have emerged as a new strategy for the fabrication of nanomaterials via postsynthetic chemical modification.1−4 At the nanoscale, CE reactions were applied and studied mostly on semiconductor II–VI, III–V, and IV–VI compounds. In this method, cations of a presynthesized parent nanocrystal (NC) can be partially or completely replaced by new guest cations with preservation of its size, shape, and, in some cases, even crystal structure. A major characteristic of such selective transformation is the overall preservation of the anion sublattice of NCs owing to the usually much larger size of anions, relative to cations, and thus their lower mobility in the lattice. Depending on the extent of CE, doped,5,6 alloyed7−12 or heterostructured NCs5,11,13−16 and completely exchanged NCs17−22 can be prepared by varying the ratio between host and guest cations. It was also shown that CE enables the synthesis of metastable NC structures,20,22 as well as specific architectures that are hardly accessible via a direct synthesis route, such as dot-in-rods ZnSe/ZnS21 and PbSe/PbS18 NCs. Moreover, CE reactions had already been successfully employed in bioassays.23 Despite CE at the nanoscale having been studied for more than one decade,19 only a few works have addressed the fundamental mechanisms of this process,13,15,24−29 as CE reactions have been used mainly as a means for synthesizing nanomaterials.
One of the most exploited classes of materials toward CE is represented by copper chalcogenide NCs, one reason being the large number of copper vacancies that these compounds can sustain, which translates in an efficient ion exchange process mediated by vacancy diffusion.1,30 In this work, we have carefully investigated room temperature CE in copper selenide (Cu2–xSe) NCs involving two divalent cations (Zn2+ and Cd2+) in the presence of trioctylphosphine (TOP) as a promoter of the exchange, with the aim of elucidating the effect of the density of copper vacancies in the starting Cu2–xSe NCs on the rate of exchange. Note that, in both cases, the entering cations have stable oxidation states (+2), and additionally, they form phases (ZnSe, CdSe) that are in principle immiscible with Cu2–xSe. This simplifies the analysis as we do not expect the occurrence of redox reactions or the pervasive formation of ternary alloys. The only remarkable difference between the two cations is that Zn2+ has an ionic radius (0.6 Å) comparable to that of Cu+ (0.6 Å), while Cd2+ is larger (0.78 Å);31 however, both Zn2+ and Cd2+ adopt a tetrahedral coordination with the Se anion sublattice. In both cases discussed here, partial CE led essentially to Janus-like NC heterostructures represented by a ZnSe (or CdSe) domain sharing a close-to-flat interface with the remaining Cu2Se portion, while core–shell geometries were never observed. Such mutual arrangement of domains in the heterostructures helps minimizing the interfacial energy and is supportive of an exchange mechanism in which ions are mobile enough to attain this stable configuration. A commonly observed trend was that the exchange was faster and could easily reach completion when performed on heavily substoichiometric Cu2–xSe NCs, that is, NCs that initially presented a high density of Cu vacancies, which points to vacancy diffusion as one of the main drivers of exchange. Our strategy of starting from NCs with a high density of Cu vacancies as templates should give therefore access to a wide range of NC materials under mild conditions (room temperature).
We found that Raman spectroscopy can easily discriminate between a sample of heavily substoichiometric NCs and a sample of closer to stoichiometric NCs, as the former presents a peak ascribable to a Se–Se vibrational mode, which is instead absent in the latter. This allowed us to monitor the stoichiometry of any remaining Cu2–xSe domains/NCs in partially exchanged samples. In all reactions tested, we always found that the stoichiometry of the nonexchanged domains was closer to Cu2Se, even when starting from NC samples with many Cu vacancies. Nevertheless, heavily substoichiometric NCs remained more reactive than the closer to stoichiometric NCs even at longer reaction times (several hours). The re-establishment of the Cu2Se stoichiometry should be mainly due to the diffusion of Cu+ ions from the exchanged domains. However, if this was the only possible mechanism operative, we would expect a quick equilibration in reactivity between samples which initially differed in number of Cu vacancies. We actually found that an important side reaction was the preferential extraction of Se atoms from Cu2–xSe NCs to the solution phase, operated by TOP, which was more efficient in heavily Cu-deficient Cu2–xSe NCs. While, over time, this extraction tended to reduce the density of Cu vacancies in the nonexchanged Cu2–xSe domains, it also involved the possibility for Se ions to diffuse through the Cu2–xSe lattice (in addition to Cu ions). Consequently, the overall lattice dynamics was likely to further facilitate cation exchange.
Another notable finding was that, at higher temperatures (150 °C here), the heavily substoichiometric Cu2–xSe NCs were quickly converted to close to stoichiometric Cu2Se, mainly by fast extraction of Se atoms by TOP. Therefore, both Cu2–xSe and Cu2Se NCs behaved similarly with respect to cation exchange, and the overall exchange efficiency was considerably lower than at room temperature.
Experimenal Section
Materials
Copper(II) acetylacetonate (Cu(acac)2, 97%), zinc nitrate hexahydrate (Zn(NO3)2·6H2O, ≥99%), zinc acetate (Zn(OAc)2, 99.99%), cadmium nitrate tetrahydrate (Cd(NO3)2·4H2O, ≥99%), 1-dodecanethiol (DDT, ≥98%), oleylamine (OlAm, 70%), octylamine (OctAm, 99%), oleic acid (OlAc, technical grade, 90%), tetrachloroethylene (TCE, ≥99%), anhydrous methanol, toluene, and tetrahydrofuran were purchased from Sigma-Aldrich. Cadmium oxide (CdO, 99.999%) and tri-n-octylphosphine (TOP, 97%) were purchased from STREM. Chemicals were used without any further purification.
Synthesis of Cu2–xSe Nanocrystals
The synthesis of Cu2–xSe NCs was performed using a standard Schlenk line technique, similarly to the synthesis of Cu2–xSeyS1–y nanoplatelets already reported by us.9 First, a Se-precursor was prepared according to the previously reported method.32 Se powder (10 mM; 0.79 g) was mixed with 5 mL of DDT and 5 mL of OlAm, and the mixture was subsequently degassed under vacuum at 50 °C for 1 h. Se powder completely dissolved through the reduction by DDT, forming a brown stock solution of alkylammonium selenide, which was cooled to room temperature and stored in a N2 filled glovebox. In a typical NC synthesis, 524 mg of Cu(acac)2 (2 mmol) was mixed with 6 mL of DDT and 19 mL of OlAm in a 100 mL three-neck round-bottom flask, and the mixture was degassed under vacuum (pressure ∼10–2 Torr) and vigorous stirring at 60 °C for 1 h. The flask was then filled with nitrogen and quickly heated to 220 °C (4–5 min to reach the temperature). Cu(acac)2 completely dissolved forming a clear yellow-orange solution. At this temperature, a mixture of 2 mL of the Se-precursor (2 mM of Se), prepared as described above, with 3 mL of DDT was swiftly injected from a syringe into the flask leading to a sudden color change from orange to greenish-brown. The reaction mixture was kept at 220 °C for 4 min and then cooled to room temperature. To purify the Cu2–xSe NCs, half of the prepared crude solution (15 mL) was centrifuged under inert gas atmosphere. The precipitate was washed twice by dissolution in 3 mL of toluene with subsequent addition of 1 mL of methanol (as a nonsolvent) and centrifugation. The Cu2–xSe NCs were dissolved in 3 mL of toluene and stored in a glovebox.
Reduction of Cu2–xSe Nanocrystals
The other half (15 mL) of the reaction mixture obtained as described above was used for the reduction of Cu2–xSe NCs toward close-to-stoichiometric Cu2Se NCs. For this, 524 mg of Cu(acac)2 (2 mmol) was mixed with 3 mL of DDT, 6 mL of OlAm, and 9 mL of ODE in a three-neck round-bottom flask, and the resulting solution was degassed under vacuum (pressure ∼10–2 Torr) and vigorous stirring at 60 °C for 1 h. Then, the flask was filled with nitrogen and the temperature was raised to 120 °C to dissolve Cu(acac)2. Thereafter, the temperature was lowered to 100 °C and 5 mL of TOP was injected followed by subsequent addition of 15 mL of the crude reaction Cu2–xSe NC mixture. The resulting mixture was maintained 20 min at 100 °C with subsequent cooling to room temperature. The purification of the reduced NCs was performed in the same way as for the Cu2–xSe NC sample.
Cation Exchange at Room Temperature
In a typical CE reaction, a certain amount of a 0.1 M methanol stock solution of a guest cation precursor (Zn(NO3)2·6H2O, or Cd(NO3)2·4H2O) was diluted with 0.5–1 mL of methanol and mixed with 1 mL of tetrahydrofuran, 0.5 mL of TOP, and 0.2 mL of Cu2–xSe (Cu2Se) NCs in toluene (Cu content = 0.05–0.06 mM). The mixture was stirred overnight under inert gas atmosphere at room temperature. Thereafter, the NCs were precipitated by centrifugation of the reaction mixture and washed by addition of 1–2 mL of toluene with subsequent sonication and centrifugation in order to remove organic residues. The precipitate obtained was washed twice in a similar way (with 1–2 mL of methanol) to remove the excess of cation salts. Finally, the NCs were dispersed in 0.5–1 mL of toluene and stored in a glovebox. In the case of TOP treatment, the same procedure, except for the addition of the guest cation precursor, was followed. For quantitative experiments, the supernatants were carefully collected after each washing step.
Cation Exchange at 150 °C
CE reactions (Cu+ → Cd2+ and Cu+ → Zn2+) were also performed at 150 °C using a standard Schlenk line technique. In the case of Cu+ → Cd2+ exchange, first, the Cd-precursor was prepared by degassing a mixture of 38.4 mg of CdO with 0.4 mL of OlAc and 12 mL of ODE in a three-neck round-bottom flask under vacuum (pressure of ∼10–2 Torr) and under vigorous stirring at 60 °C for 1 h. Afterward, the flask was filled with nitrogen, heated to 250 °C and kept at this temperature until complete dissolution of CdO (15–20 min). Then, the temperature was lowered to 150 °C and, a mixture of 1 mL of the NC suspension in toluene (νCu = 0.3 mM) with 2 mL of TOP was injected. Some 1.5–2 mL samples of the reaction mixture were collected at 1, 2, 5, and 10 min after the injection, while keeping the mixture at 150 °C. For Cu+ → Zn2+ exchange, a mixture of 1 mL of the NCs in toluene (νCu = 0.3 mM), 0.3 mL of the Zn-precursor (prepared by dissolving 10 mM of Zn(OAc)2 in 5 mL of OlAm and 5 mL of OctAm previously degassed), corresponding to a Cu/Zn ratio of 1, and 2 mL of TOP was injected in previously degassed ODE (12 mL) at 150 °C under inert gas. The mixture was kept stirring at 150 °C with sampling at 1, 2, 5, and 10 min after the injection. The samples of the reaction mixtures were centrifuged, and the NC precipitates were washed 2–3 times by redispersion in 1 mL of toluene, addition of approximately 0.5 mL of methanol and subsequent centrifugation. The TOP treatment of the NCs was performed following the same procedure, but without addition of the guest cation precursor.
Transmission Electron Microscopy (TEM)
Samples were prepared by dropping diluted NC suspensions onto carbon coated 200 mesh copper grids for conventional TEM analyses, with subsequent evaporation of the solvent. Conventional TEM imaging was done on a JEOL JEM-1011 microscope equipped with a thermionic gun (W filament) operating at 100 kV accelerating voltage. High-resolution TEM (HRTEM), energy-filtered TEM (EFTEM) and energy-dispersive X-ray spectroscopy (EDS) analyses were performed on a JEOL JEM-2200FS microscope equipped with a Schottky emitter working at 200 kV, a CEOS spherical aberration corrector of the objective lens allowing for a spatial resolution of 0.9 Å, and an in-column imaging filter (Ω-type). EDS mapping and compositional quantification of the NCs was determined in scanning TEM (STEM)-high angle annular dark field (HAADF) imaging mode, using a Bruker Quantax 400 system with a 60 mm2 XFlash 6T silicon drift detector. EDS quantification was carried out using the Cliff-Lorimer ratio method for the Cu Kα, Se Kα, Cd Lα and Zn Kα peaks, and elemental maps were obtained by integrating the corresponding peaks in the EDS spectra. For HRTEM and EDS chemical analyses, NC solutions were drop-cast onto ultrathin carbon-coated Au grids and the experiments were carried out using an analytical double tilt Be holder in order to minimize background and spurious signals. In case of partial Cd2+ exchange, better spatially resolved elemental maps were obtained by EFTEM (three-windows method), due to the favorable energy position of the M45 ionization edge of Cd and the L23 ionization edge of Cu (slit width: 30 eV for Cd M45, 50 eV for Cu L23). No comparably clear results were obtained by EFTEM mapping of Zn. The geometric phase analysis (GPA)33 tool for Gatan Digital Micrograph written by C. T. Koch and V. B. Özdöl34 was applied to HRTEM images for direct visualization of lattice constant variation in domains with different composition within individual NCs.
Powder X-ray Diffraction (XRD)
XRD patterns were recorded on a Rigaku SmartLab 9 kW diffractometer. The X-ray source was operated at 40 kV and 150 mA. The diffractometer was equipped with a Cu source and a Göbel mirror to obtain a parallel beam and to suppress Cu Kβ radiation (1.392 Å). To acquire data, a 2-θ/Ω scan geometry was used. The samples were prepared by drop casting concentrated NC dispersions onto a zero background silicon substrate. The PDXL software of Rigaku was used for phase identification.
UV–Vis-NIR Spectroscopy
Absorbance spectra of NCs dispersed in TCE were measured in 1 cm path length quartz cuvettes using a Varian Cary 5000 UV–vis-NIR spectrophotometer. Dilute NC suspensions were prepared inside a nitrogen filled glovebox.
Raman Measurements
These were performed under nitrogen atmosphere in a closed chamber (by Linkam) in order to avoid laser-induced oxidation phenomena. Spectra were collected using a Renishaw inVia MicroRaman system exciting the samples at 633 nm by means of a 50× objective with a nominal power ranging from 0.5 to 5 mW and acquisition time from 10 to 100 s.
Elemental Analysis
Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis, performed on an iCAP 6000 spectrometer (ThermoScientific), was used to quantify the composition of the NCs. The samples were digested in aqua regia (HCl/HNO3 = 3/1 (v/v)) prior to measurements.
Results and Discussion
Synthesis of Cu2–xSe Nanocrystals and Their Conversion to Cu2Se
In this work, we have developed a method for the synthesis of heavily p-doped Cu2–xSe NCs that does not require any additional postsynthetic oxidizing treatment. They were prepared similarly to a synthesis of Cu2–xSeyS1–y nanoplatelets,9 previously reported by us, with the only difference being the ratio between Cu- and Se-precursors of 1/1 (used here), as compared to 2/1 of our previous work. In the presence of an excess of Se, copper selectively reacts with selenium (instead of DDT) and ultimately yields copper selenide NCs with nearly spherical shapes and diameters around 15 nm (see Figure 1a). STEM-EDS analysis of individual NCs revealed a deviation of their composition from particle to particle: the Cu/Se ratio ranged from 1.28 to 1.50, giving an average Cu1.42Se0.97S0.03 composition. At the same time, the ICP analysis of several samples from different batches revealed an overall Cu1.60Se0.93S0.07 composition (that is, ICP gave a higher Cu/Se ratio than EDS). The minor inclusion of sulfur in these NCs comes primarily from the passivating DDT molecules, although we cannot exclude a partial inclusion of sulfur in the NC due to decomposition of some of the DDT molecules during the synthesis. The as-prepared NCs exhibited an intense localized surface plasmon resonance (LSPR) with absorption maximum at ∼1100 nm (see the red spectrum in Figure 1d), similar to that observed by our group on oxidized Cu2–xSe NCs,35 which is attributed to the collective oscillation of holes.36−43 This is indicative of the presence of a significant number of copper vacancies in the as-prepared NCs.
Figure 1.
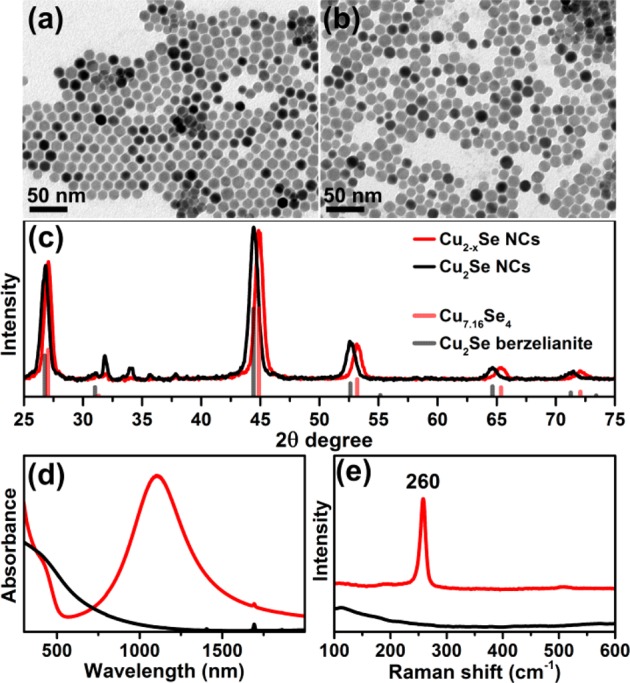
TEM images of initial (a) and reduced Cu2–xSe (b) NCs with corresponding XRD patterns (c) (red, before reduction; black, after reduction). The experimental patterns are compared to database powder diffraction files of cubic Cu7.16Se4 (PDF card #01-071-4325) and Cu2Se berzelianite (01-071-4843). Optical absorption spectra of Cu2–xSe NCs before (red) and after reduction (black)47 (d) and corresponding Raman spectra (e).
From these Cu2–xSe NCs, we could prepare closer to stoichiometric NCs by in situ incorporation of Cu+ ions. As seen from Figure 1a–c, after the reduction treatment the NCs preserved their size, shape and crystal structure (cubic berzelianite). Filling of vacancies in the reduced sample with Cu+ ions led to a slight dilation of the lattice unit cell (evidenced by a shift of the diffraction peaks in XRD to smaller Bragg angles, Figure 1c) and to the complete damping of the LSPR in the NIR (Figure 1d). The increase in Cu content in the reduced NCs was also proven by Raman analysis and by compositional analysis (both EDS and ICP). The Raman spectrum of the as-synthesized (that is, “oxidized” or vacant) Cu2–xSe NCs (x = 0.58) evidenced a feature at 260 cm–1 that can be attributed to a Se–Se vibrational mode,44−46 whereas the spectrum of the reduced Cu2Se NC sample did not exhibit any remarkable feature, as already reported in the literature and as also observed by us in Cu2–xSe11 and Cu2–xSeyS1–y NCs with x < 0.2.9 The presence/absence of this Raman-active Se–Se vibrational mode was a useful tool to discriminate between stoichiometric and nonstoichiometric NCs in all the experiments that follow. According to the EDS analysis of individual reduced NCs, the Cu/Se ratio varied from 1.63 to 2.09 (1.79 on average), with an average Cu1.65Se0.92S0.08 composition (see Supporting Information Table SI1 for details). Again, ICP analysis indicated instead a higher Cu/Se ratio (2.1, overall composition Cu1.68Se0.8S0.2). Despite these differences in chemical quantification between ICP and EDS that affected all samples, both techniques estimated a 10–20% increase in Cu content going from the oxidized to the reduced sample.
Stability of Cu2–xSe and Cu2Se Nanocrystals in Trioctylphosphine
For the sake of simplicity, in the following we will refer to the initial, oxidized NCs as “Cu2–xSe”, and to the reduced NCs as “Cu2Se” NCs. Both samples were tested in CE reactions. All these reactions involve the use of TOP as a necessary chemical favoring the CE reaction,5 since practically no CE was observed without it. The common justification is that TOP, as a soft base, promotes the extraction of the soft acid Cu+. On the other hand, it is also known that TOP can partially extract chalcogenide atoms from metal dichalcogenide NCs and transform them to metal chalcogenides (in some cases even at temperatures as low as 65 °C).48 Therefore, before starting the various experiments, we decided to test the stability of both Cu2–xSe and Cu2Se NCs against TOP under the same conditions at which the CE reactions were carried out (and which will be discussed later). The NCs were incubated at room temperature overnight, after which they were precipitated by addition of methanol followed by centrifugation and were rinsed several times with methanol. XRD patterns were acquired on the NCs, while the presence of Cu and Se in the supernatant was quantified by ICP.
Especially for the Cu2–xSe sample, i.e., the one with high density of Cu vacancies, the TOP treatment caused a considerable dilation of the unit cell, as can be seen in the XRD patterns of Figure 2a, which corroborates a variation in the composition toward Cu2Se (that is, filling of the vacancies with Cu). Also, additional peaks, compatible with Cu2Se bellidoite (see bulk patterns in Figure 2c), appeared in the TOP treated sample. This is a tetragonal phase in which the unit cell can be viewed as built from a stacking of 2 × 2 × 2 berzelianite cells along the three crystallographic directions, and slightly stretched (a bit more along c than along a and b).49 This expansion accommodates the larger number of Cu atoms of Cu2Se bellidoite compared to Cu2–xSe berzelianite. Indeed, whereas the lattice parameter of berzelianite is 5.69 Å, those of bellidoite are 11.52 Å (a, b) and 11.74 Å (c), both larger than 5.69 × 2 = 11.24. We conclude that a fraction of NCs remained in the berzelianite phase but with a stoichiometry closer to Cu2Se, while a fraction of NCs evolved to tetragonal bellidoite Cu2Se. Less drastic changes in XRD peak positions were seen instead for the Cu2Se sample (Figure 2b), although also in this case a fraction of NCs underwent a transition to Cu2Se bellidoite.
Figure 2.
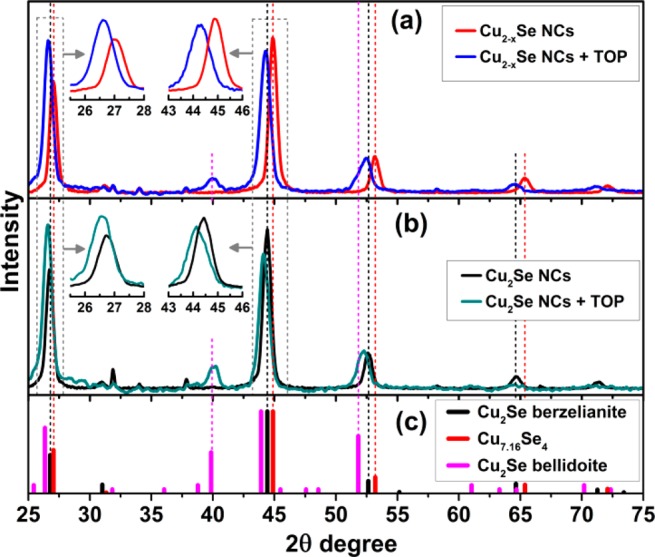
XRD patterns of oxidized (a) and reduced (b) copper selenide NCs before and after the TOP treatment. The experimental patterns are compared to database powder diffraction files of tetragonal bellidoite Cu2Se (00-029-0575), cubic Cu7.16Se4 (01-071-4325), and Cu2Se (01-071-4843) (c).
Data from XRD were then compared with the results of elemental analysis (by ICP) on the supernatant collected after precipitation of the NCs, which revealed the presence of both copper and selenium, a sign that the NCs were partially etched by TOP. Etching did not change the average size and size distribution of the particles in an appreciable way (Supporting Information Figure SI1). Taking into account 18.5% loss of Se in vacant particles and their initial Cu7.16Se4 and final Cu2Se cubic phases, we calculated their size reduction from 15 to 14.1 nm, which apparently is within the error of the NC size estimation presented in Supporting Information Figure SI1. For the Cu2Se sample, the relative amounts of Cu and Se in solution were such that their ratio was 1.85:1 (essentially the same as in the NCs, see Supporting Information Table SI2), indicating roughly equal leakage of Cu and Se from the NCs. For the Cu2–xSe sample instead many more Se atoms than Cu atoms were extracted (this time the Cu:Se ratio in solution was 0.45, that is, 1:2.2). The preferential extraction of Se by TOP from the Cu2–xSe NCs explains their evolution toward Cu2Se composition found by XRD, since the remaining unetched portions of the NCs became then enriched in Cu. The same TOP treatment, at 150 °C, also led to Cu enrichment of the NC composition, by extraction of Se by TOP, in both Cu2–xSe and Cu2Se NCs. The notable difference, with respect to the room temperature case, was that this time the Se extraction, and therefore filling of Cu vacancies, was completed already within 1 min of the reaction (see Supporting Information Table SI3), and further heating during the following 30 min did not induce any significant change in the composition of the NCs, both for the initial Cu2–xSe and Cu2Se samples. We will come back to the influence of TOP on CE later in this work.
Cu+ → Cd2+ Exchange
The as-prepared Cu2–xSe NCs and the reduced Cu2Se NCs underwent partial CE reactions. For the Cu+ → Cd2+ case, we tested various ratios of added Cd2+ ions to Cu+ ions in the NCs, at room temperature: from 1:20 (0.05) to 1:1 (see Supporting Information Table SI4). Since there will be a replacement by one Cd2+ ion every two Cu+ ions, even if all the Cd2+ ions added are taken up by the NCs, we expect that the exchange will be closer to completion only for the 1:2 (0.5) and 1:1 cases. As can be seen from the EFTEM maps of Figure 3a,b, a typical product of such reactions contained Janus-like particles of separated Cu- and Cd-containing domains, suggesting the formation of CdSe in the exchanged regions (in line with the immiscibility of Cu2Se and CdSe seen in the bulk) and indicating that CE started at one location of the particles and from there it propagated through the NC. There were additionally some unexchanged particles and some completely exchanged ones. We rationalize the formation of Janus particles by considering that cations with low coordination with the Se sublattice, such as the Cd2+ ions discussed here and the Zn2+ ions, which will be analyzed shortly, should have higher diffusivity in the CdSe and ZnSe phases, respectively, than in the Cu2–xSe phase, in analogy to the findings of Ha et al. for Cu+ → Cd2+ (Zn2+) CE reactions in Cu2S NCs.15 Therefore, ion replacement can be initiated and propagated in a way that a partially exchanged structure is the result of various steps that have eventually led to a relatively stable configuration. Such events are likely to be initial ion diffusion throughout the NC lattice, preferential exchange with the Cu+ cations in the most energetically favored locations, and subsequent growth of the CdSe and ZnSe domains by a constant supply of the guest cations through the corresponding guest phase, in a way that the overall exchanged domain (CdSe or ZnSe) of a NC can minimize its interfacial area with the remaining nonexchanged (Cu2–xSe) domain. This is certainly realized in a Janus-like type of architecture.
Figure 3.

Elastically filtered (zero-loss) TEM image of CdSe–Cu2Se NCs obtained by partial CE at room temperature from vacant Cu2–xSe NCs (a), with EFTEM mapping of Cu (red) and Cd (green) (b). Note that Se maps are not shown in the images, as no appreciable variation is observed over individual NCs. HRTEM image (c) of a CdSe–Cu2Se NC with corresponding mean dilation map as obtained by GPA (d) and FFT (e). XRD patterns in (f) display the evolution of the CdSe–Cu2Se NCs crystal structure with increasing Cd content shown by an arrow. The experimental patterns are compared to database powder diffraction files of tetragonal bellidoite and cubic alpha Cu2Se (PDF cards 00-029-0575 and 00-101-0581, respectively), Cu7.16Se4 (01-071-4325), and CdSe (01-088-2346). Raman spectra of CdSe–Cu2Se NCs obtained from Cu2–xSe and Cu2Se NC samples (g). The red vertical dotted line represents the position at which the Se–Se vibrational mode in Cu2–xSe should be observed. Diagram displaying the evolution of the Cd/Cu ratio in the heavily substoichiometric (Cu2–xSe, red) and close to stoichiometric (Cu2Se, black) NCs over the time of the Cu+ → Cd2+ CE reaction (h).
Since the lattice parameter of cubic CdSe is larger than that of Cu2Se (see Supporting Information Table SI5), we could monitor the evolution of the reactions both by HRTEM and XRD. A typical HRTEM image of a Janus particle (Figure 3c) revealed matching of lattice parameters with cubic CdSe (a = 6.1 Å) and Cu2Se (a = 5.8 Å) in the respective domains, confirmed by 4.5(±0.9)% mean dilation in the CdSe domain relative to the Cu2Se one as obtained by GPA (see Figure 3d). The fast Fourier transform (FFT) pattern presented in Figure 3e evidences the epitaxial orientation between the two phases, where red and green arrows point at spots corresponding to Cu2Se and CdSe phases, respectively. Figure 3f reports XRD patterns of partially exchanged NCs, starting from Cu2–xSe NCs, and characterized by different exchange yields. Already by incorporation of a small amount of Cd, the structure of the unexchanged Cu2–xSe NCs transformed to tetragonal Cu2Se bellidoite phase. Two concurrent effects, both causing the decrease in the density of Cu vacancies in the nonexchanged domains, may be responsible for this transformation to bellidoite: the preferential Se etching operated by TOP on NCs (as discussed earlier), and the likely diffusion of the Cu+ ions expelled from the exchanged domains toward the not yet exchanged Cu2–xSe NCs. Further exchange led to the increase of the cubic CdSe peaks and to the fading of Cu2Se ones.
Raman spectroscopy allowed us to make additional conclusions on structure and composition. A typical Raman spectrum of a sample of CdSe–Cu2Se Janus NCs obtained from Cu2–xSe NCs (Figure 3g, red spectrum) exhibited a sharp peak at 207 cm–1, which can be assigned to the longitudinal optical phonon mode of CdSe and its overtone at 414 cm–1.50 No other peaks were present, so that we could exclude the presence of alloyed phases, as well as any nonstoichiometric Cu2–xSe (as we saw no peak at 260 cm–1), which suggests that the unexchanged copper selenide domains/NCs had a low density of Cu vacancies.11 It is noteworthy that basically the same type of spectrum was recorded on partially exchanged samples starting from Cu2Se NCs (Figure 3g, black spectrum).
It appears that both partial exchange reactions, the one on Cu2–xSe NCs and the one on Cu2Se NCs, converged essentially to the same type of heterostructures. On the other hand, an important outcome of these experiments was that CE was more efficient when done on Cu2–xSe NCs than on Cu2Se ones. We compared, for example, the kinetics of the Cu+ → Cd2+ exchange on Cu2–xSe and Cu2Se NCs by monitoring two parallel reactions starting from a ratio of Cd2+ ions to Cu+ ions (feed ratio) equal to 0.5 (1:2). The evolution of the actual Cd:Cu ratio in the NCs over the reaction time is summarized in Figure 3h (additional data are reported in Supporting Information Table SI6 and Figure SI2). As follows from the results, the Cu2–xSe NCs underwent CE much faster than the Cu2Se ones: a roughly 10-fold difference between the Cd:Cu ratios in the two samples (histograms of Figure 3h) at the beginning of the reaction developed to a 30-fold difference after 3 h. After 7 h, the Cd/Cu ratio in the initial Cu2–xSe NCs had reached 0.45, while in the sample prepared from the initial Cu2Se NCs it was only 0.015.
These experiments demonstrate the impact of copper vacancies on the kinetics of CE. In fully stoichiometric compounds, cations occupying regular sites in the crystal lattice can move to an interstitial site leaving a vacancy behind. Such interstitial-vacancy pairs (known as Frenkel pairs) can move through the solid by hopping from site to site. This process is limited by solid-state diffusion within a NC, as shown by Groeneveld et al. for Zn2+ → Cd2+ CE.27 For NCs with a large number of vacancies acting as carriers of both host and guest cations, this diffusion should proceed much faster. On the other hand, we verified that, even for the Cu2–xSe NCs, the exchange rate does slow down over time. This can be seen from Figure SI2 of the Supporting Information, which reports the experimental Cd/Cu ratio in Cu2–xSe NCs over several hours (red markers). The Cd/Cu ratio followed a linear trend, corresponding to a growth rate of the volume fraction of CdSe that follows a c/(1 + ct)2 dependence over time t (with c equal to a constant, see discussion in the Supporting Information), that is, the exchange rate slowed down over time, despite our experiments still having a considerable amount of Cd2+ ions and of available TOP in solution. A linear growth of the volume fraction of CdSe, on the other hand, would correspond to a time evolution of the Cd/Cu ratio that is proportional to t/(1 – kt) with k equal to a constant (see Supporting Information), which is steeper than linear, especially at later times. One reason for such a slowdown in the growth rate can be the filling of Cu vacancies, although other parameters might play an important role and will require further scrutiny. However, a general consideration that should hold is that the ease of formation of Cu vacancies in copper selenide will make the exchange rate in this material always higher than a rate determined by Frenkel defect diffusion or by an interface-controlled reaction.
Further increase of the initial Cd:Cu ratio up to 1, i.e., by adding double excess of Cd precursor relative to Cu (considering this reaction as 2Cu+ → Cd2+), led to practically complete exchange of copper ions in the initial substoichiometric Cu2–xSe NCs, with a resulting Cd:Cu ratio of 373, whereas in the initial close to stoichiometric Cu2Se particles, this ratio was only 22 (as a reminder, the reactions were run overnight, see Supporting Information Table SI4). These results again clearly indicate the difference between the two samples and represent an important guideline when exploiting CE reactions as a means to prepare materials in which the amount of impurity atoms has to be minimized. Also, in analogy with the case of blank TOP treatment discussed above, we always found Se in significant amounts in supernatant solutions at the end of the reactions. The loss of Se was independent from the Cd:Cu feed ratio and was more pronounced for the Cu2–xSe NCs than for Cu2Se NCs. By quantifying the content of selenium in NCs and in the solution, we estimated that the Cu2–xSe NCs had lost around 30% of Se, while the Se loss for the Cu2Se NCs was half of that value, around 15%. This observation suggests that CE goes through at least partial etching of the NCs, which again is more pronounced in NCs with higher density of Cu vacancies. Also, in line with previous works on CE, we note that TOP is an important ingredient: without TOP, even starting from a Cd:Cu feed ratio equal to 1, basically no exchange took place (the Cd:Cu ratios were 0.013 starting from Cu2–xSe NCs and 0.003 starting from Cu2Se NCs).
Similar to the case of the room temperature Cu+ → Cd2+ partial CE discussed above, the reaction at a higher temperature (150 °C) yielded CdSe–Cu2Se Janus heterostructures (see Supporting Information Figure SI3). However, differently from the room temperature reactions, at 150 °C we did not observe a remarkable difference in the CE kinetics between the Cu2–xSe and the Cu2Se samples (see Supporting Information Table SI7 and Figure SI4), as both samples exchanged with a similar rate (which was even slightly higher for the initial close to stoichiometric Cu2Se NCs). This can be attributed to a quick filling of the Cu vacancies by rapid extraction of Se atoms by TOP. One additional potential reason for similarity in reactivities for the two samples is that, at 150 °C, the extracted Cu+ ions may start competing with the Cd2+ ions (or with the Zn2+ ions, as we shall see briefly) that are still in the solution phase for entry in the NCs (re-entry in the case of Cu), which should further contribute to a fast reduction of the density of Cu vacancies in the NCs.
Cu+ → Zn2+ Exchange
In a first series of experiments, the ratio of Zn2+ ions added to Cu+ ions in the NCs was set to 1:4 (0.25). Here, as in the Cd2+ case, reactions were run overnight. Figure 4a,b reports HAADF-STEM images and superimposed Cu and Zn STEM-EDS compositional maps over groups of NCs, after exchange on the Cu2–xSe (panel a) and Cu2Se (panel b) NCs. In both samples, all particles exhibited a Janus structure, in line with the results on Cd2+ discussed earlier. Unfortunately, for this system neither HRTEM (Figure 4c) nor XRD (Figure 4f) could confirm that the exchanged domain was pure ZnSe, that the nonexchanged domain had remained copper selenide (Cu2–xSe or Cu2Se), and that no partial ternary alloy compounds had formed (however never reported for the bulk), due to the low mismatch between cubic ZnSe and Cu2–xSe (see Supporting Information Table SI5). For example, in the HRTEM image of the Janus particle reported in Figure 4c, no variation of the lattice parameter (a = 5.8 Å) is appreciated throughout the NC. Raman spectroscopy, on the other hand, was more informative (Figure 4g). For the ZnSe–Cu2Se NCs samples prepared from Cu2Se NCs, as well as for the ZnSe–Cu2Se NCs prepared from Cu2–xSe, the Raman spectra exhibited only one band peaked at 240 cm–1, which can be interpreted as the longitudinal optical phonon mode of ZnSe.51 No other peaks were present, not even the one at 260 cm–1 of the initial Cu2–xSe NCs, which would fall in the region marked by the red dashed line in Figure 4g. This indicates that the unexchanged copper selenide domains, in both samples, had compositions close to Cu2Se. It also excludes the formation of alloys, again in line with the immiscibility of Cu2Se and ZnSe observed in the bulk.
Figure 4.

HAADF-STEM images and corresponding STEM-EDS Cu (red) and Zn (green) elemental maps of ZnSe–Cu2Se NCs obtained from oxidized Cu2–xSe (a) and reduced Cu2Se (b) NCs by partial CE at room temperature. Note that Se maps are not shown in the images, as selenium was homogeneously distributed over individual particles. HRTEM image of a ZnSe–Cu2Se Janus-type NC (c) with corresponding FFT (d) and STEM-EDS map (e). XRD patterns (f). The experimental patterns are compared to database powder diffraction files of cubic Cu2Se (PDF card 01-088-2043), and ZnSe (01-071-5978). Raman spectra of ZnSe–Cu2Se NCs derived from Cu2Se and Cu2–xSe NCs (g). Here again, the red vertical dotted line marks the position at which the Se–Se vibrational mode in Cu2–xSe should be observed. Diagrams displaying dependence of the Zn/Cu ratio in the vacant (red) and reduced (black) NCs on the Zn/C feed ratio in the Cu+ → Zn2+ CE reaction (h).
As in the Cd2+ case, the exchange with Zn2+ was more efficient on NCs that had initially a large number of Cu vacancies (see Supporting Information Table SI6). Figure 4h reports Zn:Cd ratios in the NCs (as measured by ICP) for both Cu2–xSe and Cu2Se NCs when working with a feed ratio of Zn:Cu of 1:4 and 1:1, at room temperature. Again, it is especially attractive that almost full exchange (Zn:Cu ratio of 67) at room temperature was possible for the Cu2–xSe NCs by employing only double excess of Zn2+ ions relative to Cu (we remind that one Zn2+ ion replaces two Cu+ ions), while, for the initial Cu2Se sample, the exchange under the same conditions yielded NCs that contained still a considerable fraction of Cu (Zn:Cu ratio was around 5). Overall, Cd2+ and Zn2+ ions behaved quite similarly at room temperature, in terms of exchange kinetics and of their dependence on initial density of Cu vacancies, as well as in terms of structure and composition of intermediate exchange products.
When the Cu+ → Zn2+ exchange was carried out at 150 °C, we did not observe any significant difference between Cu2–xSe and Cu2Se NC samples, similar to the high temperature Cu+ → Cd2+ CE reactions discussed above (see Supporting Information Table SI9 and Figure SI5): after 10 min of reaction, the Zn:Cu ratio had reached 0.33 in the case of Cu2–xSe and 0.28 in the case of Cu2Se NCs. Again, this implies that copper vacancies were quickly filled, since already after 1 min of the reaction the Zn2+ ions had replaced approximately 30% and 20% of Cu+ ions in Cu2–xSe and Cu2Se NCs, respectively, after which the exchange slowed down considerably (see Supporting Information Table SI9). Also, the structure of the resulting particles was similar to that of room temperature exchange products, i.e., Janus ZnSe–Cu2Se dimers, without detectable formation of ternary Cu–Zn–Se alloy phases.
Overall, by comparing the Cu+ → Zn2+ and Cu+ → Cd2+ exchange reactions at 150 °C, we can conclude that, under the same experimental conditions, Cd2+ ions were more reactive toward the NCs than were the Zn2+ ions. It appears that, under these conditions, CE will be favored thermodynamically by a higher bond strength of the newly forming phase, which in our case is CdSe, since the bond dissociation energies (enthalpy changes) of Cd–Se, Cu–Se, and Zn–Se bonds are 310, 293, and 136 kJ/mol,31 respectively. The advantage of room temperature CE that needs to be emphasized here is that even a Cd(Zn)/Cu feed ratio equal to 1 is sufficient to achieve almost complete exchange. At the same conditions, but at 150 °C, for example only 35% of copper ions are replaced by Zn2+ ions. The latter results are in line with published works, in which quantitative exchange of Cu+ ions by Zn2+ at high temperatures was made possible only by employing a large excess of Zn2+ ions.17,20
Conclusions
The major conclusion of this work is that Cu vacancies play a key role in cation exchange reactions involving copper selenide NCs, as their presence accelerates the exchange process. Therefore, the use of NCs with a high density of Cu vacancies, as done in this work, can simplify cation exchange reactions and make them more practical, for example, by significantly reducing the ratio between host and guest cations and by working under mild conditions, for example, at room temperature. Moreover, room temperature conditions were found advantageous compared to higher (150 °C) temperature conditions, owing to the preservation of copper vacancies over time, which resulted in a much more efficient exchange on substoichiometric Cu2–xSe NCs.
Also, since TOP acts as both complexing agent for Cu+ ions and for Se (in the form of Se-TOP), it is conceivable that its role as an enhancer of cation exchange is more multifaceted than previously thought. We additionally believe that partial exchange processes investigated in this work can be applied to other copper chalcogenide NCs yielding Janus-like structures. Moreover, by subjecting such synthesized Cu2X–Zn(Cd)X dimers to a further exchange, it should be possible to selectively convert the unexchanged Cu2X domain to yet another material, thus giving accessibility to a wide range of heterostructures.
Acknowledgments
We thank S. Vikulov for assistance in performing CE experiments and L. De Trizio and A. Castelli for fruitful discussions. V.L. gratefully acknowledges the support by a Marie Curie Intra European Fellowship within the 7th European Community Framework Programme (FP7/2007-2013) under the Grant Agreement No. 301100 (project “LOTOCON”). The research leading to these results has also received funding from the FP7 under Grant Agreement Nos. 614897 (ERC Consolidator Grant “TRANS-NANO”), and 284486 (project “SCALENANO”).
Supporting Information Available
Elemental compositions of individual Cu2–xSe and Cu2Se NCs. Ratios between Se and Cu contents in NCs and in solution after the TOP treatment. Size distribution histograms of Cu2–xSe and Cu2Se NCs before and after the TOP treatment, at room temperature and at 150 °C. Evolution of the Cd/Cu ratio in oxidized and reduced copper selenide NCs over reaction time. Elemental maps of CdSe–Cu2Se NCs prepared by partial CE at 150 °C. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b03868.
The authors declare no competing financial interest.
Supplementary Material
References
- Rivest J. B.; Jain P. K. Chem. Soc. Rev. 2013, 42, 89–96 10.1039/C2CS35241A. [DOI] [PubMed] [Google Scholar]
- Beberwyck B. J.; Surendranath Y.; Alivisatos A. P. J. Phys. Chem. C 2013, 117, 19759–19770 10.1021/jp405989z. [DOI] [Google Scholar]
- Gupta S.; Kershaw S. V.; Rogach A. L. Adv. Mater. 2013, 25, 6923–6944 10.1002/adma.201302400. [DOI] [PubMed] [Google Scholar]
- Moon G. D.; Ko S.; Min Y.; Zeng J.; Xia Y.; Jeong U. Nano Today 2011, 6, 186–203 10.1016/j.nantod.2011.02.006. [DOI] [Google Scholar]
- Gui J.; Ji M.; Liu J.; Xu M.; Zhang J.; Zhu H. Angew. Chem. 2015, 127, 3754–3758 10.1002/ange.201410053. [DOI] [Google Scholar]
- Buonsanti R.; Milliron D. J. Chem. Mater. 2013, 25, 1305–1317 10.1021/cm304104m. [DOI] [Google Scholar]
- van der Stam W.; Berends A. C.; Rabouw F. T.; Willhammar T.; Ke X.; Meeldijk J. D.; Bals S.; de Mello Donega C. Chem. Mater. 2015, 27, 621–628 10.1021/cm504340h. [DOI] [Google Scholar]
- Gupta S.; Zhovtiuk O.; Vaneski A.; Lin Y.-C.; Chou W.-C.; Kershaw S. V.; Rogach A. L. Part. Part. Syst. Charact. 2013, 30, 346–354 10.1002/ppsc.201200139. [DOI] [Google Scholar]
- Lesnyak V.; George C.; Genovese A.; Prato M.; Casu A.; Ayyappan S.; Scarpellini A.; Manna L. ACS Nano 2014, 8, 8407–8418 10.1021/nn502906z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. M.; Nie S. J. Am. Chem. Soc. 2011, 133, 24–26 10.1021/ja108482a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Trizio L.; Li H.; Casu A.; Genovese A.; Sathya A.; Messina G. C.; Manna L. J. Am. Chem. Soc. 2014, 136, 16277–16284 10.1021/ja508161c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkerman Q. A.; Genovese A.; George C.; Prato M.; Moreels I.; Casu A.; Marras S.; Curcio A.; Scarpellini A.; Pellegrino T.; Manna L.; Lesnyak V. ACS Nano 2015, 9, 521–531 10.1021/nn505786d. [DOI] [PubMed] [Google Scholar]
- Robinson R. D.; Sadtler B.; Demchenko D. O.; Erdonmez C. K.; Wang L.-W.; Alivisatos A. P. Science 2007, 317, 355–358 10.1126/science.1142593. [DOI] [PubMed] [Google Scholar]
- Miszta K.; Dorfs D.; Genovese A.; Kim M. R.; Manna L. ACS Nano 2011, 5, 7176–7183 10.1021/nn201988w. [DOI] [PubMed] [Google Scholar]
- Ha D.-H.; Caldwell A. H.; Ward M. J.; Honrao S.; Mathew K.; Hovden R.; Koker M. K. A.; Muller D. A.; Hennig R. G.; Robinson R. D. Nano Lett. 2014, 14, 7090–7099 10.1021/nl5035607. [DOI] [PubMed] [Google Scholar]
- Kriegel I.; Wisnet A.; Srimath Kandada A. R.; Scotognella F.; Tassone F.; Scheu C.; Zhang H.; Govorov A. O.; Rodriguez-Fernandez J.; Feldmann J. J. Mater. Chem. C 2014, 2, 3189–3198 10.1039/c3tc32049a. [DOI] [Google Scholar]
- van der Stam W.; Gantapara A. P.; Akkerman Q. A.; Soligno G.; Meeldijk J. D.; van Roij R.; Dijkstra M.; de Mello Donega C. Nano Lett. 2014, 14, 1032–1037 10.1021/nl4046069. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Amirav L.; Aloni S.; Alivisatos A. P. J. Am. Chem. Soc. 2010, 132, 9997–9999 10.1021/ja104126u. [DOI] [PubMed] [Google Scholar]
- Son D. H.; Hughes S. M.; Yin Y.; Alivisatos A. P. Science 2004, 306, 1009–1012 10.1126/science.1103755. [DOI] [PubMed] [Google Scholar]
- Li H.; Zanella M.; Genovese A.; Povia M.; Falqui A.; Giannini C.; Manna L. Nano Lett. 2011, 11, 4964–4970 10.1021/nl202927a. [DOI] [PubMed] [Google Scholar]
- Li H.; Brescia R.; Krahne R.; Bertoni G.; Alcocer M. J. P.; D’Andrea C.; Scotognella F.; Tassone F.; Zanella M.; De Giorgi M.; Manna L. ACS Nano 2012, 6, 1637–1647 10.1021/nn204601n. [DOI] [PubMed] [Google Scholar]
- Li H.; Brescia R.; Povia M.; Prato M.; Bertoni G.; Manna L.; Moreels I. J. Am. Chem. Soc. 2013, 135, 12270–12278 10.1021/ja404694k. [DOI] [PubMed] [Google Scholar]
- Yao J.; Schachermeyer S.; Yin Y.; Zhong W. Anal. Chem. 2011, 83, 402–408 10.1021/ac102688s. [DOI] [PubMed] [Google Scholar]
- White S. L.; Smith J. G.; Behl M.; Jain P. K. Nat. Commun. 2013, 4, 2933. 10.1038/ncomms3933. [DOI] [PubMed] [Google Scholar]
- Routzahn A. L.; Jain P. K. Nano Lett. 2014, 14, 987–992 10.1021/nl4044289. [DOI] [PubMed] [Google Scholar]
- Chan E. M.; Marcus M. A.; Fakra S.; ElNaggar M.; Mathies R. A.; Alivisatos A. P. J. Phys. Chem. A 2007, 111, 12210–12215 10.1021/jp073474u. [DOI] [PubMed] [Google Scholar]
- Groeneveld E.; Witteman L.; Lefferts M.; Ke X.; Bals S.; Van Tendeloo G.; de Mello Donega C. ACS Nano 2013, 7, 7913–7930 10.1021/nn402931y. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Wong A. B.; Yu Y.; Brittman S.; Sun J.; Fu A.; Beberwyck B.; Alivisatos A. P.; Yang P. J. Am. Chem. Soc. 2014, 136, 17430–17433 10.1021/ja511010q. [DOI] [PubMed] [Google Scholar]
- Ott F. D.; Spiegel L. L.; Norris D. J.; Erwin S. C. Phys. Rev. Lett. 2014, 113, 156803. 10.1103/PhysRevLett.113.156803. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Beberwyck B. J.; Fong L.-K.; Polking M. J.; Alivisatos A. P. Angew. Chem., Int. Ed. 2012, 51, 2387–2390 10.1002/anie.201107452. [DOI] [PubMed] [Google Scholar]
- Dean J. A.Lange’s Handbook of Chemistry, 15th ed.; McGraw-Hill, Inc.: New York, 1999. [Google Scholar]
- Liu Y.; Yao D.; Shen L.; Zhang H.; Zhang X.; Yang B. J. Am. Chem. Soc. 2012, 134, 7207–7210 10.1021/ja300064t. [DOI] [PubMed] [Google Scholar]
- Hÿtch M. J.; Snoeck E.; Kilaas R. Ultramicroscopy 1998, 74, 131–146 10.1016/S0304-3991(98)00035-7. [DOI] [Google Scholar]
- http://elim.physik.uni-ulm.de/.
- Most of the solvents used in this work led to a development of a weak plasmon band in reduced NCs, most likely due to their oxidation by oxygen traces as well as some impurities commonly present in all commercially available solvents.
- Dorfs D.; Härtling T.; Miszta K.; Bigall N. C.; Kim M. R.; Genovese A.; Falqui A.; Povia M.; Manna L. J. Am. Chem. Soc. 2011, 133, 11175–11180 10.1021/ja2016284. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Pan H.; Lou Y.; Qiu X.; Zhu J.; Burda C. J. Am. Chem. Soc. 2009, 131, 4253–4261 10.1021/ja805655b. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Jain P. K.; Ewers T.; Alivisatos A. P. Nat. Mater. 2011, 10, 361–366 10.1038/nmat3004. [DOI] [PubMed] [Google Scholar]
- Comin A.; Manna L. Chem. Soc. Rev. 2014, 43, 3957–3975 10.1039/c3cs60265f. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Burda C. Energy Environ. Sci. 2012, 5, 5564–5576 10.1039/C1EE02734D. [DOI] [Google Scholar]
- Liu X.; Swihart M. T. Chem. Soc. Rev. 2014, 43, 3908–3920 10.1039/c3cs60417a. [DOI] [PubMed] [Google Scholar]
- Faucheaux J. A.; Stanton A. L. D.; Jain P. K. J. Phys. Chem. Lett. 2014, 5, 976–985 10.1021/jz500037k. [DOI] [PubMed] [Google Scholar]
- Kriegel I.; Jiang C.; Rodríguez-Fernández J.; Schaller R. D.; Talapin D. V.; da Como E.; Feldmann J. J. Am. Chem. Soc. 2012, 134, 1583–1590 10.1021/ja207798q. [DOI] [PubMed] [Google Scholar]
- Hsu S.-W.; On K.; Tao A. R. J. Am. Chem. Soc. 2011, 133, 19072–19075 10.1021/ja2089876. [DOI] [PubMed] [Google Scholar]
- Izquierdo-Roca V.; Saucedo E.; Ruiz C. M.; Fontané X.; Calvo-Barrio L.; Álvarez-Garcia J.; Grand P.-P.; Jaime-Ferrer J. S.; Pérez-Rodríguez A.; Morante J. R.; Bermudez V. Phys. Status Solidi A 2009, 206, 1001–1004 10.1002/pssa.200881239. [DOI] [Google Scholar]
- Witte W.; Kniese R.; Powalla M. Thin Solid Films 2008, 517, 867–869 10.1016/j.tsf.2008.07.011. [DOI] [Google Scholar]
- Cho A.; Ahn S.; Yun J. H.; Gwak J.; Ahn S. K.; Shin K.; Yoo J.; Song H.; Yoon K. Thin Solid Films 2013, 546, 299–307 10.1016/j.tsf.2013.02.037. [DOI] [Google Scholar]
- Sines I. T.; Schaak R. E. J. Am. Chem. Soc. 2011, 133, 1294–1297 10.1021/ja110374d. [DOI] [PubMed] [Google Scholar]
- De Montreuil L. A. Econ. Geol. Bull. Soc. Econ. Geol. 1975, 70, 384–387 10.2113/gsecongeo.70.2.384. [DOI] [Google Scholar]
- Kelley A. M.; Dai Q.; Jiang Z.-j.; Baker J. A.; Kelley D. F. Chem. Phys. 2013, 422, 272–276 10.1016/j.chemphys.2012.09.029. [DOI] [Google Scholar]
- Lu G.; An H.; Chen Y.; Huang J.; Zhang H.; Xiang B.; Zhao Q.; Yu D.; Du W. J. Cryst. Growth 2005, 274, 530–535 10.1016/j.jcrysgro.2004.10.062. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.