

Abstract
The biofilm state is an integral part of the lifecycle of many bacterial pathogens. Identifying inhibitors as molecular probes against bacterial biofilms has numerous potential biomedical applications. Here we report quinoline amino alcohol 20 as a highly potent disruptor of V. cholerae biofilms. Additionally, 20 was able to disperse preformed biofilms, an activity exhibited by few compounds with biofilm inhibiting activity.
Cholera is a disease that disproportionately affects those in countries with inadequate access to clean drinking water. The absence of strong sanitation infrastructure is particularly challenging in this regard, as this can lead to the contamination of water supplies, which in turn increases the chances of an outbreak.1 Normally, standard care includes fluid replacement and administration of antibiotics, making cholera a treatable disease provided that treatment is initiated quickly. Unfortunately natural disasters and the growing problem of bacterial drug resistance can affect accessibility to and efficacy of both these items, as was observed after the Haiti earthquake in 2010.2
These challenges are exacerbated by V. cholerae’s ability to exist in both planktonic and biofilm states. Bacteria that inhabit the biofilm state are an estimated 10–1000 times more resistant to traditional antibiotics than planktonic cells.3 Bacterial biofilm formation is a stochastic process driven by a complex and dynamic regulatory network.4 Once formed, bacterial biofilms become communities of sessile cells packaged in an extracellular polymeric substance (EPS) matrix comprised of polysaccharides, proteins, and nucleic acids5 that is less susceptible to the effects of antibiotics. Bacterial biofilms have been the focal point of significant attention because of this role in antibiotic resistance as well as their impacts on the progression and persistence of nosocomial infections. This is particularly true in the case of cholera, where biofilm formation is a key element of the lifecycle and transmission mechanism.6 Therefore the discovery of new inhibitors, antibiotics, and chemical genetic probes for V. cholerae biofilms is of high value for the development of next-generation therapeutics against this global health pathogen.
Our laboratory has recently developed an image-based high content screening platform for the discovery of biofilm inhibitors and disruptors against both V. cholerae and P. aeruginosa.7,8 Application of these screens to both natural products and synthetic screening libraries has led to the discovery of a number of new lead structures that inhibit biofilm formation and progression.9,10 A previous report by our group described the synthetic optimization of one of these lead series, and demonstrated a clear pharmacophore model for this compound class.11 This model requires a quinoline core with an essential amino alcohol at the 4-position of the heterocycle, as well as an aryl substituent at the 2-position. This led to the development of a synthetically tractable lead compound with a simpler structure and equal potency to the original lead compounds (Figure 1). Motivated by the fact that our synthetic strategy can rapidly diversify the 2 and 4-positions, the goal of this new study was to identify more potent analogs with a particular interest in compounds with differential biofilm and antibiotic activities. This is relevant because elimination of biofilm persister cells through non-antibiotic Mechanisms of Action (MOA) gives less evolutionary pressure for resistance.12
Figure 1.
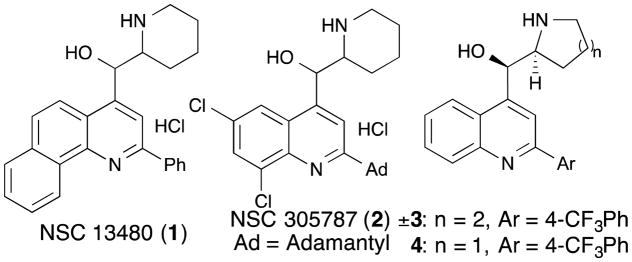
Initial lead compounds (1 and 2) and first generation library leads (3 and 4).
Our first generation library explored the effects of systematic variation of functional groups at both the 2 and 4-positions of the quinoline core (Scheme 1). This work identified the L-proline-derived 5-membered ring as the optimal substructure at the 4-position. This is an improvement over the original racemic piperidinyl scaffold because L-proline is a cheap, commercial starting material and racemic compounds are now difficult to advance as drug leads due to increased regulatory requirements for clinical testing laid down by the FDA.13 Compound 4 was therefore selected as a starting point for this new study, whose goal was to prepare a second-generation library that extended the diversity of functionalization on the aryl ring of the 2-position, and to examine whether the substitution pattern and degree of electron withdrawing effect on this aryl ring could improve biofilm disruption and identify new dose response phenotypes. These studies began by taking compound 5 and via a regiospecific Grignard reaction, prepared 4.15 g of 7 using our previously reported protocol. This intermediate was then diversified via Suzuki cross-coupling reactions to introduce 14 different boronic acids at the 2-position (Scheme 2). These boronic acids varied in size, lipophilicity, and degree of electron withdrawing capacity and were selected to probe the substitution and electronic requirements for biofilm disruption. Finally, the crude material from each Suzuki cross-coupling reaction was separately treated with HCl to remove the trityl protecting group, and the final compounds purified using a combination of silica gel chromatography and HPLC separation to give compounds 8 – 21.
Scheme 1.

Retrosynthetic approach to second-generation library.
Scheme 2.

Second-generation library analogs.
Currently there are no FDA-approved antibiotics that specifically target bacteria in the biofilm state. From an applications perspective, compounds that modulate biofilm dynamics are of value in two different arenas: 1) compounds that can inhibit the initial attachment of bacterial cells to native and non-native substrates, and 2) compounds that are capable of dispersing preformed biofilm colonies.14 In the first case, inhibitors of initial attachment have potential applications as prophylactics during implant surgery, or as additives to coatings for medical implant devices, while in the second case compounds that disperse mature biofilms could be used as co-dose therapies for the treatment of established biofilm-mediated infections. Our current image-based high content screening platform is capable of identifying compounds with either of these activities, depending upon the configuration of the assay platform (biofilm inhibition model (BIM), or biofilm dispersal model (BDM)). In the BIM, test wells are treated with compound at the initial point of inoculation to model inhibition of initial attachment of bacterial cells, while in the BDM biofilms are allowed to preform for two hours prior to compound addition, to model the effect of test compounds on existing biofilm colonies. In both cases, incubation of the screening plates is then followed by imaging (% biofilm coverage) and optical density (OD) readings to quantify both biofilm coverage and cell viability in each well.
We subjected compounds 8 to 21 to evaluation in both the biofilm inhibition and the biofilm dispersal models to examine whether any of these molecules were capable of disrupting biofilm progression in V. cholerae. Consideration of these screening results (Table 1) revealed several interesting SAR features for this new suite of compounds. As expected, compound 8, which contains a chlorine atom in place of the aryl substituent at the 2-position, was inactive while compound 9 (2-phenyl derivative) possessed only moderate activity in the inhibition screen. Our previous exploration of this compound class revealed para-CF3 derivative 4 as a potent biofilm inhibitor. Using the underivatized phenyl derivative 9 as a baseline, we therefore first performed a trifluoromethylation scan to examine the effect of varying the substitution of the –CF3 group around the aryl ring. Interestingly, the position of the –CF3 group had a pronounced impact on activity, with ortho-derivative (10) displaying no activity in either the inhibition or dispersal assays, while the meta- and para- derivatives (4 and 11) were both more active than the parent phenyl derivative in the inhibition model with BIC50 activities of 25.3 and 56.5 μM respectively. In line with our previous work in this area, both of these compounds displayed weaker activities in the dispersal assay, With BDC50 activities in the mid- to high-micromolar range.
Table 1.
Biological activity of second-generation library (μM)
| Compound | Substitution | BIC50 | BDC50 |
|---|---|---|---|
| 4 | 4-CF3 | 25.3 | 62.6 |
| 8 | Cl | >625 | >625 |
| 9 | Ph | 77.8 | >250 |
| 10 | Ortho-CF3 | >250 | >250 |
| 11 | Meta-CF3 | 56.5 | 151.5 |
| 12 | 4-NO2 | >625 | >625 |
| 13 | 4-Me | 75.8 | 148.3 |
| 14 | 4-F | 123.9 | >250 |
| 15 | 4-SF5 | 12.9 | 15.6 |
| 16 | 4-Cl | 79.2 | 119.3 |
| 17 | 2,4-CF3 | 29.4 | 49.1 |
| 18 | 2,4-Me | 169.5 | 175.9 |
| 19 | 2,4-F | >250 | >250 |
| 20 | 3-SF5 | 4.4 | 7.4 |
| 21 | 2,4-Cl | 52.5 | 153.3 |
We next explored the impact of varying substituents at the para-position of the phenyl ring, including -CF3 (4), -H (9), -NO2 (12), -CH3 (13), -F (14), -SF5 (15), and -Cl (16). The comparable activity of 9 and 14 suggests that F is not sufficiently electron withdrawing to impart improved activity. The activity of 12 on the other hand suggests that robust electron withdrawing groups like –NO2 that change other properties (H-bond acceptors, and polarity) also do not improve activity. Analogs 4 and 13 are isosteres of one another (CH3 vs CF3) and their activities show the importance of electron withdrawing groups on the activity, with compound 4 showing improved potency over 13. Consistent with this we observed strong activity for the –SF5 derivative 15, which was significantly more active (BIC50 = 12.9 μM, BDC50 = 15.6 μM) than any of the other derivatives in this set.
In an effort to further interrogate the importance of substitution patterns on activity, disubstituted derivatives 17, 18, 19, and 21 were prepared from the corresponding boronic acids (2,4-RPhB(OH)2: R=CF3, CH3, F, Cl). The activity trend for these analogs (17>21>18>19) is consistent with the monosubstituted analogs (4, 13–16). However what was unexpected was the comparable activity of the 4-substituted CF3 and Cl derivatives (4 and 16) and their corresponding 2,4-disubstituted derivatives (17 and 21). The inclusion of an additional -CF3 (17) or -Cl (21) motif greatly increased the AlogP values, however the change in activity in each case was negligible, suggesting that the activity of these compounds is not driven by their lipophilic properties.
Up to this point, compound 15 was the most active in this new series with improved activity over first-generation lead 4. The improvement in activity observed by inclusion of the increasingly popular -SF5 group encouraged us to further explore the use of this motif in our lead development efforts.15 Gratifyingly when the meta-SF5 derivative 20 was synthesized and screened it was five-fold more potent (BIC50 = 4.4 μM) than 15 at disrupting biofilm formation. This result was unexpected because in the trifluoromethylation scan of the phenyl ring it was observed that para- substitution was optimal, whereas in the -SF5 series it is the meta-substituted derivative that possesses the most potent activity. Most interesting is that OD (optical density) measurements in the inhibition assay show a window of efficacy between the BIC50 and the concentration at which the compound became bactericidal (minimum inhibitory concentration (MIC) = 78.1 μM). This result is significant because up to this point almost all derivatives examined in this series have BIC50 values close to the corresponding MICs. The differentiation between inhibitory and antibacterial activities is therefore intriguing, and suggests that this compound may have a mode of action for biofilm inhibition and dispersal that is distinct from its antibiotic activity.
This compound has the most potent inhibition and dispersal activities observed to date for this structural class, and is one of the most active biofilm disruptors known for V. cholerae. We further examined the value of this new lead by comparing its activity to the activity of the FDA-approved antibiotic doxycycline, which is part of the standard of care for treatment of patients suffering from cholera. In the initial attachment assay, where test wells are treated with compound at the point of inoculation, doxycycline was capable of inhibiting both bacterial growth and biofilm formation (Figure 2). However, in line with clinical observations, doxycycline was not able to reduce biofilm coverage up to very high testing concentrations in the dispersal model, where biofilms are allowed to preform prior to compound addition. By contrast, 20 was capable of reducing both planktonic cell density (as measured by OD600) and biofilm coverage in both the initial attachment and preformed biofilm assay models. This activity profile is consistent with the application of these compounds as part of a co-dose therapy to treat established biofilm mediated infections by clearing established biofilm colonies from native and non-native surfaces.
Figure 2.

Biofilm Inhibitory Concentration (BIC50), and Biofilm Dispersal Concentration (BDC50) curves and in-well images (●% Biofilm Coverage,
 Optical Density)
Optical Density)
Finally, to explore the therapeutic potential of this new lead series we also examined the cytotoxicity of this compound series in our image-based cytological profiling assay in HeLa cells.16 Results from this screen (Supplementary Information, Table S1) indicated that many of these compounds possessed measurable cytotoxic activities, in line with other recent reports that have examined the cytotoxic activities of members of this compound class.17 In most cases in our assay system the SAR of this cytotoxic activity paralleled the anti-biofilm activities with similar absolute potencies. With this data in mind the preferred application of these materials is likely as the bioactive components of coatings for implant medical devices, where local concentrations can be high, but systemic concentrations would remain low. Given that the best treatment for biofilm-mediated infections on medical implant devices is removal of the device.4 There is clearly a strong need for such materials, providing an incentive for the development of compounds of this type.
Conclusions
In conclusion, we report the creation and biological evaluation of a library of quinoline amino alcohol derivatives and their evaluation as biofilm inhibitors against the Gram-negative pathogen V. cholerae. This synthetic route is amenable to large-scale production of an enantiopure intermediate derived from L-proline, and subsequent late-stage diversification using palladium cross coupling chemistry. Biological examination of this new library has led to the discovery of a new lead compound (20) with improved BIC50 and BDC50 activities over the initial lead molecule which possesses a unique activity profile compared to other members of this class, providing encouragement for further development in the area of bioactive coatings for medical implant devices.
Supplementary Material
Acknowledgments
Financial support for this work was provided by NIH grant 1R21AI098836 (RGL) and an NIH IMSD Fellowship 3R25GM058903-15S1 (BL). We thank F. Yildiz and A. Cheng for assistance with bacterial cultures.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Aibana O, Franke M, Teng J, Hilaire J, Raymond M, Ivers LC. PLoS Negl Trop Dis. 2013;7:e2576. doi: 10.1371/journal.pntd.0002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barzilay EJ, Schaad N, Magloire R, Mung KS, Boncy J, Dahourou GA, Mintz ED, Steenland MW, Vertefeuille JF, Tappero JW. N Engl J Med. 2013;368:599–609. doi: 10.1056/NEJMoa1204927. [DOI] [PubMed] [Google Scholar]
- 3.Jefferson KK, Goldmann DA, Pier GB. Antimicrob Agents Chemother. 2005;49:2467–2473. doi: 10.1128/AAC.49.6.2467-2473.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostakioti M, Hadjifrangiskou M, Hultgren SJ. Cold Spring Harb Perspect Med. 2013;3:a010306. doi: 10.1101/cshperspect.a010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berk V, Fong JCN, Dempsey GT, Develioglu ON, Zhuang X, Liphardt J, Yildiz FH, Chu S. Science. 2012;337:236–239. doi: 10.1126/science.1222981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Clin Infect Dis. 2010;50:1081–1083. doi: 10.1086/652237. [DOI] [PubMed] [Google Scholar]; (b) Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, Lynfield R, Maloney M, McAllister-Hollod L, Nadle J, Ray SM, Thompson DL, Wilson LE, Fridkin SK. N Engl J Med. 2014;370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peach KC, Bray WM, Shikuma NJ, Gassner NC, Lokey RS, Yildiz FH, Linington RG. Mol Biosyst. 2011;7:1176. doi: 10.1039/c0mb00276c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Navarro G, Cheng AT, Peach KC, Bray WM, Bernan VS, Yildiz FH, Linington RG. Antimicrob Agents Chemother. 2014;58:1092–1099. doi: 10.1128/AAC.01781-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warner CJA, Cheng AT, Yildiz FH, Linington RG. Chem Commun. 2014;51:1305–1308. doi: 10.1039/c4cc07003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peach KC, Cheng AT, Oliver AG, Yildiz FH, Linington RG. ChemBioChem. 2013;14:2209–2215. doi: 10.1002/cbic.201300131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.León B, Fong JCN, Peach KC, Wong WR, Yildiz FH, Linington RG. Org Lett. 2013;5:2011–2014. doi: 10.1021/ol400150z. [DOI] [PubMed] [Google Scholar]
- 12.Worthington RJ, Richards JJ, Melander C. Org Biomol Chem. 2012;10:7457–7474. doi: 10.1039/c2ob25835h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Rouf A, Taneja SC. Chirality. 2014;26:63–78. doi: 10.1002/chir.22268. [DOI] [PubMed] [Google Scholar]; (b) Murakami H. Top Curr Chem. 2007;269:273–299. doi: 10.1007/128_2006_072. [DOI] [PubMed] [Google Scholar]
- 14.de la Fuente-Núñez C, Reffuveille F, Haney EF, Straus SK, Hancock REW. PLoS Pathog. 2014;10:e1004152. doi: 10.1371/journal.ppat.1004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jiménez-Díaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. J Med Chem. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wipf P, Mo T, Geib SJ, Caridha D, Dow GS, Gerena L, Roncal N, Milner EE. Org Biomol Chem. 2009;7:4163–4165. doi: 10.1039/b911483a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Matsuzaki K, Okuyama K, Tokunaga E, Saito N, Shiro M, Shibata N. Org Lett. 2015;17:3038–3041. doi: 10.1021/acs.orglett.5b01323. [DOI] [PubMed] [Google Scholar]
- 16.Schulze CJ, Bray WM, Woerhmann MH, Stuart J, Lokey RS, Linington RG. Chem Biol. 2013;20:285–295. doi: 10.1016/j.chembiol.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitambi SS, Toledo EM, Usoskin D, Wee S, Harisankar A, Svensson R, Sigmundsson K, Kalderén C, Niklasson M, Kundu S, Aranda S, Westermark B, Uhrbom L, Andäng M, Damberg P, Nelander S, Arenas E, Artursson P, Walfridsson J, Forsberg Nilsson K, Hammarström LGJ, Ernfors P. Cell. 2014;157:313–328. doi: 10.1016/j.cell.2014.02.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.