

Abstract
The 2-thiouridine (s2U) at the wobble position of certain bacterial and eukaryotic tRNAs enhances aminoacylation kinetics, assists proper codon-anticodon base pairing at the ribosome A-site, and prevents frameshifting during translation. By mass spectrometry of affinity-purified native E. coli tRNA1GlnUUG, we show that the complete modification at the wobble position 34 is 5-carboxyaminomethyl-2-thiouridine (cmnm5s2U). The crystal structure of E. coli GlnRS bound to native tRNA1Gln and ATP demonstrates that cmnm5s2U34 improves the order of a previously unobserved 11 amino acid surface loop in the distal β-barrel domain of the enzyme, and imparts other local rearrangements of nearby amino acids that create a binding pocket for the 2-thio moiety. Together with previously solved structures, these observations explain the degenerate recognition of C34 and modified U34 by GlnRS. Comparative pre-steady state aminoacylation kinetics of native tRNA1Gln, synthetic tRNA1Gln containing s2U34 as sole modification, and unmodified wild-type and mutant tRNA1Gln and tRNA2Gln transcripts demonstrates that the exocyclic sulfur moiety improves tRNA binding affinity to GlnRS 10-fold compared with the unmodified transcript, and that an additional four-fold improvement arises from the presence of the cmnm5 moiety. Measurements of Gln-tRNAGln interactions at the ribosome A-site show that the s2U modification enhances binding affinity to the glutamine codons CAA and CAG, and increases the rate of GTP hydrolysis by E. coli EF-Tu by five-fold.
Keywords: glutaminyl-tRNA synthetase, ribosome, peptidyl transfer, pre-steady state kinetics, transfer RNA
Introduction
At least 92 of the over 150 known RNA modifications are found in tRNAs, which in bacteria contain on average about eight modified bases per molecule.1–4 Modifications can be found throughout the entire structure, although a difference in chemical complexity is evident between modifications in the globular core region, which are generally as simple as a single methyl group, and those in the anticodon loop, which are often more elaborate. Modifications in the core of tRNA often provide thermodynamic stability;5 in one striking case, the m1A9 nucleotide in human mitochondrial tRNALys is required for correct folding to the canonical tertiary structure.6 Recently, using a new assay in which tRNA folding is monitored by aminoacylation, we demonstrated that the ensemble of modifications in Escherichia coli tRNA1 GlnUUG improves the kinetics of folding without affecting the position of the equilibrium between folded and unfolded forms.7 Despite these clear effects on stability, equilibrium thermodynamics, and folding kinetics, in many cases the modifications in the tRNA core region exert only subtle influence on function after folding is completed: unmodified transcripts are very often utilized quite efficiently by aminoacyl-tRNA synthetases, and in certain cases also by the ribosome.8,9
For a variety of tRNAs, the more complex modifications found at tRNA anticodon positions 34 and 37 play roles in codon-anticodon pairing and in suppression of frameshifting during translation. For example, the E. coli and Salmonella typhimurium tRNA species for alanine, leucine, proline serine, threonine and valine, each possessing the modification uridine-5-oxyacetic acid (cmo5U34) at wobble position-34, are able to read all four codons of their four-fold degenerate codon boxes whereas the unmodified tRNAs are not.10,11 In another study, analysis of modified anticodon stem-loops from bacterial and eukaryotic tRNALys showed that 5-methylaminomethyl-2-thiouridine (mnm5s2U34) or 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U34), respectively, found in tRNAs that read A or G at the wobble position, are implicated in stabilization of codon-anticodon interactions.12,13 Interestingly, the identities of the modification enzymes that operate at position 34 are often specific to particular biological domains, and may have played essential roles in the refinement and standardization of the contemporary genetic code.14
The importance of modifications to U34 has also been studied in tRNAs specifying glutamate and glutamine. The modified U34 in E. coli tRNAGluUUC has been identified as mnm5s2U34, and this nucleotide is a positive determinant for recognition by GluRS.15 It has also been shown that the 2-thio moiety, and not the 5-methylaminomethyl group, is required for efficient glutamylation.16 In the case of glutamine coding, it is known that the 5-methylcarboxymethyl-2-thiouridine (mcm5s2U34) in yeast tRNAGlnUUG is important for base pairing with the wobble A in the CAA codon, and helps pairing with a wobble G.17 This study establishes an important in vivo function for wobble uridine modifications in a eukaryotic organism.
The E. coli genome encodes two glutamine-specific tRNAs: tRNA1GlnUUG and RNA2 GlnUUG.18 In addition to the anticodon base 34, these isoacceptors also differ at six base positions in the body of the tRNA (Figure 1). tRNA2Gln contains an unmodified C34 base, and the other modifications in this tRNA are not required for efficient aminoacylation by GlnRS.8 In contrast, tRNA1 GlnUUG harbors a hypermodified base of the form xm5s2U34.19 In tRNAGlnUUG from Salmonella enterica, the modified base at position 34 was tentatively identified as cmnm5s2U based on UV absorbance spectra and chromatographic migration compared with synthetic cmnm5s2U.20 However, in this case a small fraction of the U34 nucleoside was further modified by addition of a C10H17 side chain. Recently, the modification was identified as a geranyl group linked to the exocyclic sulfur in tRNAGluUUC, tRNALysUUU and tRNAGlnUUG from E. coli, S. enterica, and a number of other bacteria.21 The geranyl group is added by the enzyme SelU (product of the ybbB gene), which, remarkably, is also responsible for generating 2-selenouridine from 2-thiouridine at the wobble positions of tRNAGlu and tRNALys in at least a portion of these tRNAs in vivo.22,22 Geranylation and selenylation at the exocyclic 2-position of wobble uridines are mutually exclusive and involve distinct reaction chemistries even though promoted by the same enzyme.21
Fig. 1.

Secondary structures of E. coli tRNAGln isoacceptors. Left: tRNA1Gln is depicted in the fully modified state corresponding to native tRNA isolated from cells. It contains the hypermodified cmnm5s2U34 nucleotide in the anticodon wobble position. Right: tRNA2Gln contains a CUG anticodon is shown with known modified nucleotides. In this work, this tRNA was analyzed as an unmodified transcript. Modified nucleotides are in boldface. Differences between the two tRNAs are shown in red.
As in GluRS, modified U34 is also a GlnRS aminoacylation determinant, where it improves the catalytic efficiency of the steady state aminoacylation reaction by lowering Km for tRNA.15,24 The importance of modified U34 in E. coli tRNA1 GlnUUG occurs within the context of a crucial role for all anticodon loop nucleotides in tRNA discrimination by E. coli GlnRS.24–27 Interestingly, recognition of C34 by GlnRS involves direct hydrogen-bonding interactions at both exocyclic N4 and endocyclic N3 groups; such contacts could presumably not be formed with U34. Thus, the structural basis for degenerate recognition of both tRNA anticodons by GlnRS has not been understood.
To explore this and related questions, including the role of the 2-thio group in promoting glutamine incorporation into protein, we have carried out a comprehensive structural and functional analysis of the role of the modified U34 in both Gln-tRNAGln synthesis and ribosome utilization. In contrast to prior work in this area, we were able to specifically isolate the role of the 2-thio group at U34 by preparation of a specific tRNA substrate harboring only this modification. Structure-function parameters for this species were compared with both fully unmodified transcript and with native tRNA1 GlnUUG isolated from cells. This native tRNA was shown to contain both s2U and cmnm5 modifications at U34.
Results
Identification of the hypermodified nucleoside at the wobble position of E. coli tRNA1GlnUUG
A recent identification of the modified wobble uridine in E. coli tRNAGlnUUG by mass spectrometry was carried out only in the context of tRNA that is geranylated at the 2-position of the ring,21 while an earlier study of S. enterica tRNAGln was performed by UV absorbance and comparison of chromatographic migration with a synthetic standard. To definitively characterize this nucleoside, we first purified unfractionated E. coli tRNA from rapidly growing cells, and then obtained the tRNA1Gln isoacceptor by affinity purification using DNA hybridization.7 The tRNA was purified from natural abundance, since overproduction of tRNAs in vivo generates hypomodification of some modified nucleotides.28–30 The purified tRNAGln was digested to nucleosides and analyzed by LC-UV-MS. All of the expected nucleosides were identified by UV, based on the known retention times and the known m/z of the nucleoside and base. In the tRNA, 12 nucleosides were identified as labeled on the UV chromatogram (Figure 2A). Of particular interest was the identification of cmnm5s2U in the total nucleoside hydrolysate. A cmnm5s2U standard was analyzed with and without the nucleosides of tRNAGln to confirm the modification. The extracted ion chromatogram (XIC) for cmnm5s2U with a molecular mass of 347 ([M+H]+ m/z 348) shows that the retention time (15.9 min.) of the sample and standard are consistent (Figure 2B). A molecule with a similar m/z was not seen in the remainder of the HPLC analysis. The mass spectrum at this elution time depicts a single peak for the [M+H]+ ion of cmnm5s2U, which is also consistent with the standard (Figure 2C).
Fig. 2.

A: LC-MS/MS of the total nucleoside digest of tRNAGln. The UV trace (260 nm) of the nucleoside analysis is depicted with all the nucleosides identified by retention time, MS, and MS/MS labeled on the corresponding peak. B: Extracted ion chromatogram (XIC) of m/z 348 of a chemically synthesized cmnm5s2U standard, a digested sample of the purified tRNAGln with the standard added, and the purified tRNA showing nearly identical retention times. C: The MS data of the peak eluting at 15.9 min. in the tRNAGln sample reveals a peak at m/z 348.0, consistent with cmnm5s2U.
To obtain sequence information, purified tRNAGln was digested with RNase T1 and analyzed by LC-MS/MS.31 One digestion product with the sequence U[Um]U[cmnm5s2U]UGp eluted at approximately 20.2 min. as shown with the XIC of m/z 1004 (Figure 3A). The mass spectrum at this elution time is shown with a doubly charged ion (m/z 1004.25) (Figure 3B). To confirm the sequence of this digestion product, the m/z 1004.25 ion was used as a precursor ion for CID tandem mass spectrometry. The fragmentation spectra of the m/z 1004.25 ion resulted in the expected c- and y-type product ions for RNA, and these product ions are consistent with the sequence U[Um]U[cmnm5s2U]UGp (Figure 3C). The analysis of the RNase T1 digestion products resulted in the sequence of the tRNA shown in Figure 1B.
Fig. 3.

A: The total ion chromatogram (TIC) of RNase T1 digested E. coli tRNAGln and the accompanying extracted ion chromatogram (XIC) of m/z 1004, which corresponds to the expected m/z for the fragment with the sequence U[Um]U[cmnm5s2U]UGp. B: The MS spectrum of the eluent at 28.2 min. depicting the m/z 1004.25, consistent with the sequence U[Um]U[cmnm5s2U]UGp. C: The peak in B was selected for MS/MS analysis. The resulting fragmentation is consistent with U[Um]U[cmnm5s2U]UGp with all identified c- and y-type product ions labeled.
These sequencing results are consistent with the originally reported sequence with the exception of position 34, which had not been identified.19 They are also consistent with the more recent analyses in S. enterica, E. coli, and some other bacteria,20,21 in which the cmnm5 modification was identified in the context of a lower abundance tRNA population containing a geranyl group extending from the sulfur at the 2-position of the ring. The cmnm5s2U modification was also previously identified in Lys, Glu and Gln tRNAs of nematode mitochondria.32 In E. coli tRNAGlu and tRNALys, selenium is exchanged for sulfur at the 2-position of wobble uridine under laboratory growth conditions when selenium concentrations are high.22–23,33–34 Notably, Björk’s group had predicted that cmnm5s2U would be present in wild-type E. coli based on analysis of tRNA 5-methylaminomethyl-2-thiouridine methyltransferase activities.35 Neither the 2-seleno nor 2-geranyl derivatives of U34 were detected in this analysis.
Importance of modified U34 in E. coli tRNA1Gln assessed by glutaminylation kinetics
We analyzed aminoacylation kinetics for E. coli GlnRS utilizing a battery of substrates, including unmodified WT tRNA1GlnUUG and tRNA2GlnCUG, a mutated tRNA2Gln transcript with a C34U substitution, synthetic tRNA1Gln possessing s2U34 as the sole modification, and native, fully modified tRNA1Gln (Table 1). To confirm the suitability of synthetic tRNA1Glns2UUG as a substrate for GlnRS (and for the ribosome; see below), we first determined thermodynamic properties and base stacking contributions of the modified base by thermal denaturation/renaturation and circular dichroism (CD), and compared these measurements with the tRNA1GlnUUG transcript. The presence of s2U34 did not affect the melting temperature (Tm) (Table 2). However, s2U34 does provide a modest decrease in Gibbs free energy, and also increases the ellipticity of the characteristic tRNA circular dichroism peak at 265nm (Figure 4). This indicates increased base stacking, suggesting that s2U34 helps to stabilize the tRNA anticodon.
Table 1.
Glutaminylation kinetics of tRNAGln wobble base variants with WT GlnRSa
| tRNA2GlnCUG* | tRNA2GlnC34U | tRNA1GlnUUG | tRNA1Glns2U34 | tRNA1Glncmnm5s2U34¶ | |
|---|---|---|---|---|---|
| Plateau aminoacylation (%) | 75 ± 5 | 75 ± 5 | 75 ± 5 | 75 ± 5 | 80 ± 6 |
| Km (Glutamine) (mM) | 0.26 ± 0.04 | 0.5 ± 0.2 | 0.48 ± 0.01 | 0.44 ± 0.04 | 0.085 ± .005 |
| Km (tRNA) (μM) | 0.3 ± 0.1 | 5.5 ± 0.2 | 3.9±1.4 | 0.47 ± 0.04 | 0.7 ± 0.2 |
| bkcat(sec−1) | 3.2 ± 0.5 | 1.95 ± 0.45 | 5.1±0.5 | 0.46 ± 0.1 | 1.0 ± 0.03 |
| kcat/Km (tRNA) (M−1 · sec−1) | 1.0 × 107 | 3.5 × 105 | 1.3 × 106 | 1.0 × 106 | 1.4 ×106 |
| Kd (Glutamine) (mM) | 1.10 ± 0.04 | 1.2 ± 0.4 | 1.0±0.2 | 1.05 ± 0.6 | 1.4 ± 0.15 |
| Kd (tRNA) (μM) | 0.36 ± 0.10 | 1.70±0.04 | 1.84±0.01 | 1.6 ± 0.01 | 0.47±0.04 |
| kobs (sec−1) | 28 ± 2 | 15.8 ± 4.9 | 19.9± 0.8 | 9.0 ± 0.2 | 35.6 ± 1.2 |
| kobs/Kd (tRNA) (M−1·sec−1) | 7.8 × 107 | 9.3 × 106 | 1.1 × 107 | 5.6 × 106 | 7.6 × 107 |
Values for tRNA2Gln were identical within error to those previously reported (18)
Fully in vivo modified isoacceptor, purified from E. coli cells without overproduction.
tRNA substrates providing data reported in the first three columns were prepared by T7 RNA polymerase and are fully unmodified, the tRNA substrate providing the data in the fourth column was prepared by chemical and enzymatic synthesis and is unmodified except for s2U34, and the fully modified tRNA providing data reported in the fifth column was isolated from cells.
Pre-steady state burst kinetics with tRNA2GlnCUG, performed as described (32), yield 105% ± 5% functional active sites. kcat values are then calculated with 100% active enzyme.
Table 2.
Thermodynamic contributions of s2U
| Anticodon | ΔH (kcal/mol) | ΔS (cal/K-mol) | ΔG (kcal/mol) | Tm (°C) |
|---|---|---|---|---|
| UUG | −31 +/− 6 | −102 +/− 19 | −1.5 +/− 0.5 | 52 +/− 3 |
| s2UUG | −45 +/− 5 | −137 +/− 16 | −2.3 +/− 0.3 | 54 +/− 1 |
Fig. 4.
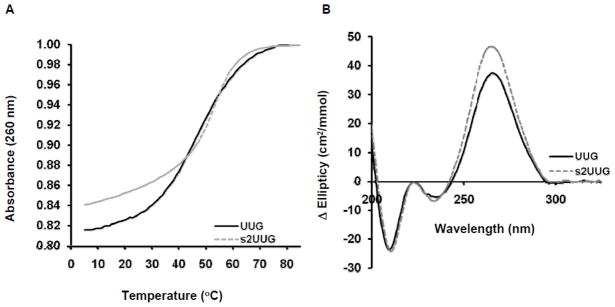
Thermal stability and base stacking contributions of s2U in tRNAGln. A) Thermal denaturation and renaturation spectra of tRNAGlnUUG transcript (black solid line, —) and tRNAGlns2UUG (grey dashed line,
 ) monitored by UV-absorbance collected at 260 nm. The profiles shown are averages of three experiments each repeated in triplicate. B) CD spectra of tRNAGlnUUG transcript (black solid line, —) and tRNAGlns2UUG (grey dashed line,
). Spectra are the averages of normalized molar circular dichroic absorption spectra (Δ Ellipticity (cm2/mmol)) collected during three independent experiments.
) monitored by UV-absorbance collected at 260 nm. The profiles shown are averages of three experiments each repeated in triplicate. B) CD spectra of tRNAGlnUUG transcript (black solid line, —) and tRNAGlns2UUG (grey dashed line,
). Spectra are the averages of normalized molar circular dichroic absorption spectra (Δ Ellipticity (cm2/mmol)) collected during three independent experiments.
Glutaminylation kinetics for the three unmodified transcripts shows that both tRNAs possessing U34 exhibit 10–20 fold elevated Km and about 5-fold elevated Kd for tRNA as compared with tRNA2GlnCUG (Table 1). Thus, as previously conjectured based on Km measurements,24 GlnRS binds tRNAGln with unmodified U34 weakly. Km for tRNA1Gln is reconstituted upon 2-thiolation of U34, with no significant further improvement in this parameter when the cmnm5U34 moiety and all other modifications are present. However, the pre-steady state analysis shows that s2U34 alone does not significantly reconstitute tRNA binding affinity; instead, Kd(tRNA) decreases by 3–4 fold from 1.6 μM when s2U34 is the sole modification, to 0.47 μM for native tRNA1Gln (Table 1). The other modified bases in native tRNA1Gln likely do not contribute to the improved affinity, since unmodified tRNA2GlnCUG binds just as strongly. Thus, it appears that tRNA1Gln is inherently deficient in tRNA binding to GlnRS if U34 is unmodified. Since the C34U tRNA2Gln mutant binds as poorly as tRNA1GlnUUG, we conclude that the tRNA binding deficit arises directly from the U34-enzyme interaction, and that tRNA binding affinity is not influenced by the six other nucleotide differences present between the two isoacceptors (Figure 1).
The kcat values for both tRNA transcripts containing unmodified U34 deviate only slightly from that measured for tRNA2GlnCUG, but kcat for both s2U34 tRNA1Gln and native tRNA1Gln are reduced 3–6 fold (Table 1). In this system kcat corresponds to the product release step, as indicated by the much higher values measured for single-turnover kobs and the observation of burst kinetics in the pre-steady state regime.36 It appears then that the presence of a modified U34 slows release of Gln-tRNAGln from the enzyme by providing additional strong interactions. In the steady-state, the decreased kcat and decreased Km for U34-modified species cancel out, so that the overall catalytic efficiency kcat/Km is similar for all three tRNA1Gln species examined (Table 1). This explains the observation that there is no difference in the in vivo aminoacylation level of tRNA1Gln regardless of whether U34 is modified.37 This congruence in the steady-state, however, masks significant differences in detailed function at the level of individual steps on the enzyme: both the tRNA substrate binding affinity and the rate of the first-order rearrangements/catalysis after binding are significantly improved by the presence of the fully modified cmnm5s2U34. The distinction shows the importance of pre-steady state analysis to understanding mechanism and providing functional correlations for the analysis of crystal structures (see below).
We also measured Km and Kd for substrate glutamine in the steady-state and pre-steady state, respectively. These data show that there are almost no differences in glutamine affinity that depend on the identity or modification status of the wobble nucleotide. However, Km(Gln) is decreased by 5-fold for fully modified native tRNA1Gln, a phenomenon related to the much faster kobs on the enzyme (Table 1). This may have significance in vivo under some conditions, since intracellular amino acid concentrations are a finely tuned aspect of cellular physiology.
Xray structures of unmodified and fully modified tRNA1GlnUUG bound to E. coli GlnRS and ATP
We obtained 250 μg of highly purified native tRNA1Gln from five liters of cells without overproduction, sufficient for cocrystallization with WT E. coli GlnRS using conditions similar to those previously reported.28 Crystals of GlnRS bound to this native tRNA and ATP exhibiting suitable diffraction properties were obtained only by successive microseeding, and application of a novel cryoprotection protocol involving stabilization in solutions containing sodium malonate (see Experimental Procedures). To enable proper comparisons, two additional structures of GlnRS bound to ATP and unmodified transcripts featuring either the CUG or UUG anticodons were also determined under identical cryostabilization conditions. Cocrystals of synthetic tRNA1Gln carrying s2U34 as the sole modification could not be obtained despite exhaustive efforts. The three structures were determined at resolutions of 2.3 – 2.5 Å and were each refined by an identical protocol with application of tight stereochemical constraints (Table 3). The structures are identical within experimental error except as described below. Details regarding the modified nucleotides s4U8, Gm18, D20, Um32, m2A37, Ψ38, T54 and Ψ55, found only in the cocrystal containing native tRNA, are provided in the supplementary material.
Table 3.
X-ray crystallography data collection and refinement statistics
| Transcript UUG | Transcript CUG | Native tRNA1Gln | |
|---|---|---|---|
| Spacegroup | C2221 | C2221 | C2221 |
| Unit cell dimensions a,b,c (Å) | 93.27 236.71 114.01 | 93.66 234.28 113.02 | 93.40 234.62 113.70 |
| α, β, γ, (°) | 90.00 90.00 90.00 | 90.00 90.00 90.00 | 90.00 90.00 90.00 |
| Wavelength (Å) | 1.0 | 1.0 | 1.0 |
| Mosaicity (°) | 1.01 | 0.89 | 0.87 |
| Resolution range | 43.37–2.3 (2.38–2.30) | 41.13–2.3 (2.38–2.30) | 47.44–2.5 (2.58–2.50) |
| Total number of reflections | 361267 | 401176 | 292670 |
| Number of unique reflections | 55306 | 55132 | 43186 |
| Average redundancy | 6.53 (6.35) | 7.28 (7.14) | 6.77 (6.62) |
| % Completeness | 98.1 (94.7) | 99.2 (97.4) | 99.0 (97.0) |
| Rmerge | 0.048 (0.266) | 0.066 (0.471) | 0.076 (0.467) |
| I/Sig I | 12.7 (3.1) | 12.0 (2.9) | 11.9 (3.0) |
| Refinement | |||
| Waters | 185 | 209 | 110 |
| No. test set reflections | 9335 (9.5%) | 5238 (9.5%) | 4103 (9.5%) |
| r.m.s.d bonds (Å)/angles (°) | 0.0067/1.158 | 0.0065/1.104 | 0.0067/1.39 |
| B-factor | 56.8 | 50.7 | 59.6 |
| Ramachandran preferred/allowed % | 92.72/6.9 | 93.05/6.2 | 91.71/7.73 |
| Rwork/Rfree | 0.2301/0.2659 | 0.2271/0.2590 | 0.2339/0.2632 |
| PDB deposition code | 4JXZ | 4JXX | 4JYZ |
The three complexes reported here differ in the local interactions at and adjacent to the position 34 wobble base, which is present as cytosine (CUG structure), uridine (UUG structure) or cmnm5s2U (native structure). First, superpositions reveal that binding of unmodified U34 by GlnRS is accompanied by small but apparently significant changes in protein tertiary structure as compared to binding of the anticodon containing C34. In the UUG structure, several peptide segments of the distal β-barrel protein domains are shifted by 1–2 Å away from the tRNA anticodon at and adjacent to the position of the wobble base (Figures 5, 6A). The precise position of the unmodified U34 base is also shifted as compared with the orientation of the cytosine base at C34 in the CUG structure (Figure 6A). While the interactions of C34 reveal a robust network of hydrogen bonds with all three discriminating Watson-Crick moieties (N4, N3 and O2),25 U34 in the UUG structure instead makes no favorable hydrogen bonds (Figures 6B, 6C). Both C34 and U34 stack on the aliphatic portion of the Arg410 side chain, but in the UUG structure only the exocyclic O4 moiety of uracil makes steric contact with GlnRS. This O4 group located 3.2 Å from the amide –NH at Ser443, but the uracil ring is oriented perpendicularly to the polypeptide at and adjacent to this amino acid, so the interaction may have little hydrogen bonding character.
Fig. 5.

Cartoon depiction of the structure of E. coli GlnRS bound to native, fully modified tRNAGln and ATP. ATP is shown in red in space-filling representation bound to the catalytic Rossmann fold domain (green). The tRNA acceptor-end binding domain is at upper right in blue, the connecting α-helical subdomain is shown in gray, and the proximal and distal β-barrels are depicted in yellow and orange, respectively. The newly ordered loop (residues 444–454) in the distal β-barrel domain is shown in space-filling representation, bound to the modified wobble nucleotide cmnm5s2U34 (shown in red, also in space-filling representation).
Fig. 6.

(A) Superposition of E. coli GlnRS structures bound to unmodified tRNA transcripts containing CUG (blue) and UUG (green) anticodons. The superposition is based on all corresponding atoms in the tRNA anticodon loops of each structure (residues 32–38 of each tRNA). Discriminatory hydrogen-bonds inferred from the structure bound to the CUG anticodon are shown as dotted black lines (see panel B for more detail). However, the protein backbone adjacent to the U34 wobble base in the UUG anticodon is shifted away, and the U34 base is rotated out, precluding any such interactions in the UUG anticodon cocrystal structure. (B) Depiction of the discriminatory hydrogen-bonding interactions made by wobble base C34 in tRNA2GlnCUG with a GlnRS peptide spanning residues 410–413 of the distal β-barrel domain. The contacts observed in this structure are very similar to those previously described, except that (i) the internal nitrogen of the Arg412 guanidinium group, and not a terminal nitrogen, donates a hydrogen bond to the O2 moiety; (ii) the hydrogen bond between N4 and the backbone amide at Leu442 was not previously observed.22 (C) Interactions of unmodified wobble base U34 with GlnRS, derived from the cocrystal structure bound to the tRNA1GlnUUG transcript (Table 3). Hydrogen bonds are shown as dashed purple lines. The U34 ring points into the polypeptide at residues 442–444, so that the exocyclic O4 moiety makes steric contacts but no favorable hydrogen bonds. The backbone at positions 412–414 is displaced too far to allow hydrogen-bonds with the O2 and N3 groups of U34 (see panel A).
These observations rationalize the substantially weaker binding of unmodified uracil as compared to cytosine, at the wobble base position of tRNAGln (Table 1). It is of interest that similar flexibility of the anticodon-binding β-barrel domains of GlnRS was previously detected based on comparing the structures of unliganded GlnRS and the tRNAGlnCUG-GlnRS complex.38 The anticodon arm and distal β-barrels are also intrinsically more mobile than the other parts of the complex, as suggested by substantially higher thermal factors throughout these domains in both the unliganded and tRNA-bound states. These new structures now indicate that this protein flexibility may have further functional significance in tRNA anticodon discrimination. The flexibility may also be generally relevant to communication of anticodon binding to the active site: mutants of anticodon nucleotides U35 and G36, and of amino acids interacting with these nucleotides, have significant effects on both active site assembly and glutamine binding affinity.39
Comparison of the native, modified tRNA cocrystal structure with the UUG complex provides new insight into the importance of cmnm5s2U34 for GlnRS recognition. The presence of the 2-thio moiety and at least a portion of the cmnm5 group was confirmed in an experimental electron density map calculated from the native data refined against the UUG structure with unmodified U34 (Figure 7A), and the presence of sulfur rather than oxygen at the exocyclic 2-position was also substantiated by the absence of residual negative electron density in a final map computed with coefficients (Fo-Fc). Further, analysis of peak areas of RNase T1 digest products by LC-MS indicates that approximately 77% of the isolated tRNA contains cmnm5s2U34, 8% contains mnm5s2U34, and 15% contains cmnm5U34 (Supplementary Figure 1). Fractional occupancies of the cmnm5 and s2 modifications in the native tRNA used for both LC-MS and crystallization experiments are thus 92% and 85%, respectively.
Fig. 7.

A. Electron density maps at 2.5 Å resolution, computed with amplitudes from cocrystals containing native, modified tRNA. The native data were first refined against the final model derived from the structure of GlnRS bound to the unmodified UUG anticodon tRNA and ATP. The gray map was then computed from amplitudes (2Fo-Fc) and is displayed at a contour level of 0.7 σ. The map shown in magenta was computed from amplitudes (Fo-Fc) and is displayed at a contour level of 3.0 σ. Peaks in the latter map adjacent to the exocyclic 2-oxygen and 5-carbon positions of U34 provide evidence for the presence of the modifications at these positions. The better-ordered surface loop spanning positions Lys444 to Gly449 appears in the (2Fo-Fc) map at bottom right. B. Interactions of cmnm5s2U34 with GlnRS. Left: primary structure of cmnm5s2U; Right: van der waals surfaces of atoms in the peptide spanning Arg410-Arg412 and at Lys444 are shown as dotted spheres, and hydrogen bonds are shown as dashed purple lines. The cmnm5 moiety lies adjacent to the backbone at Lys444 on the N-terminal end of the loop. The O2 moiety of unmodified U34 is displaced about 1.5 Å from the position occupied by the 2-thio group in cmnm5s2u34 (compare panel C). C. Superposition of the structures of GlnRS bound to the UUG anticodon transcript RNA (U34 shown in red), and bound to modified native tRNAGln containing cmnm5s2U34 (shown in blue). The loop spanning residues 444–454 is at bottom, with the salt bridge between Arg450 in the loop and adjacent Glu408 also depicted. The fully ordered loop in the native tRNA cocrystal is shown in green, and the partly disordered loop in the UUG structure is shown in yellow. The nearly identical orientations of the peptide containing Arg412 are shown at upper left. The relative orientation of the tRNA anticodon loop and protein backbone throughout the distal β-barrel domain is the same in these two structures. The side-chain of Arg410 is not shown for clarity (see panel B).
The modified uracil stacks between the side chain of Arg410 and the main and side chains of Arg412. The immediate environment of the 2-thio sulfur consists of carbon and nitrogen atoms of the Arg412 side-chain, and the backbone amide –NH group of Asn413 (Figure 7B). The exocyclic O4 group is within hydrogen-bonding distance of an outer Arg410 guanidinium nitrogen, albeit with unfavorable geometry, and also lies close to the peptide backbone at Leu442 and Ala443. Other hydrophobic atoms in the environment, at distances ranging from 4.2 Å to 4.8 Å, include the side-chains of Val455 and Arg410. It appears that the mixed polar and hydrophobic character of this environment is unsuitable for the 2-oxygen of unmodified uridine (Figure 7C). Superposition of the native and UUG structures reveals that, while there are no larger scale differences in protein conformation such as described above for the CUG structure, the unmodified uracil and cmnm5s2U bases are displaced such that the 2-oxygen and 2-thio groups are separated by 1.5 Å (Figure 7C). The specificity of this pocket for sulfur and not oxygen thus provides a structural correlate for the improved binding affinity of native tRNA possessing s2U34. We are unaware of other protein-RNA cocrystal structures in which the specific protein binding pocket for an exocyclic ring sulfur in a modified RNA base has previously been described.
Each of the three new structures exhibits increased order of a previously unobserved surface loop spanning amino acids Lys444-Gly454 in the distal anticodon-binding β-barrel of the enzyme, which is positioned adjacent to the 5′-side of the anticodon (Figure 5). The best-ordered part of this loop, observed in all three structures, spans Arg450 to Gly454. Arg450 forms a salt bridge with Glu408 elsewhere in the distal β-barrel domain, that appears to provide stability (Figures 7A, 7C). However, in the native structure but not in the CUG structure, amino acids 444–449 are also ordered (the backbone atoms for amino acids at positions 444 and 449, but not at 445–448, could be built into the UUG structure). The path of the polypeptide backbone is clear in the electron density of the native structure, although the orientations of the backbone amides within and N-terminal to this peptide are not resolved. While precise details of the interactions can thus not be described, the cmnm5 moiety does contact this loop at position Lys444 (Figures 5, 7A, 7C). This interaction may contribute to the full ordering of the surface loop, and the interactions of GlnRS with the cmnm5 moiety may help to explain the four-fold improved affinity of native tRNA as compared with tRNA containing s2U34 as sole modification (Table 1). Future mutagenesis experiments exploring the role of this surface loop in wobble base selectivity by GlnRS should be helpful in more fully defining its function.
Two other previously unobserved features of the GlnRS-tRNA complex of potential functional relevance are also found in all three structures. First, the γ phosphate of ATP makes direct rather than water-mediated contacts to the side chains of Arg260 and His43 in the active site. This conformation most closely resembles that adopted for ATP binding by tyrosyl-tRNA synthetase (TyrRS), and may have implications for catalysis of aminoacyl adenylate formation in GlnRS or in class I aaRS more generally (see Supplementary Figure 2 and legend for further discussion).25,40 Second, interactions made by G36 in the tRNA anticodon are distinctive (Supplementary Figure 3).25, 26 The side-chain of Lys401, which was previously observed to stack on one side of the guanine base and to contact an adjacent phosphate,25 is disordered in all three structures. However, electron density for Trp548, which stacks on the opposite side of the guanine base, is now observed for the first time in all structures. In the catalytic event, selective recognition of G36 may involve both Lys401 and Trp548, allowing stabilization of the guanine base by hydrophobic stacking interactions on both sides. The previously observed discriminatory hydrogen-bonds made by the 2NH2, O6 and N7 groups of G36 with Gln399 and Arg402 are maintained in all three structures.
Each of these differences with previously solved GlnRS:tRNA cocrystal structures may arise from the use of malonate as cryoprotectant, emphasizing the importance of performing detailed structural comparisons only among crystals that are treated identically.41 All interactions made by the central U35 anticodon nucleotide are identical to prior structures. The three malonate-protected crystals exhibit identical G36 interactions, suggesting that communication between binding of anticodon nucleotides 34 and 36, if any, is not reflected in the averaged conformations derived from crystal structures. Following Trp548, five further C-terminal amino acids at positions 549–553 remain disordered in all E. coli GlnRS structures.
Role of s2U34 in modulating tRNA activity on the ribosome
Both unmodified tRNAGlnUUG and tRNAGlns2UUG containing the 2-thioU34 as sole modification were tested for their ability to bind in the ribosomal A-site to cognate glutamine codons CAA and CAG, as well as to the near cognate codons CAU and CGA. While both tRNAs exhibited binding to cognate CAA, tRNAGlns2UUG exhibited three-fold higher affinity (Table 4) while also binding to a significantly greater extent to both CAA and CAG (Figure 8). In this assay neither tRNAGln could bind detectably to the near-cognate codons CAU or CGA. Together, these results suggest that s2U34 may increase tRNAGlnUUG specificity for its cognate codon CAA, and that it better accommodates wobble with glutamine codon CAG as compared with unmodified U34.
Table 4.
Binding of tRNAGln (s2UUG) and (UUG) to the ribosome A-site
|
Kd (μM)a
|
||||
|---|---|---|---|---|
| Anticodon | CAA | CAG | CAU | CGA |
|
|
||||
| UUG | 4.06 ± 0.77 | ≥5.00 | NB | NB |
| s2UUG | 1.36 ± 0.31 | 3.84 ± 2.07 | NB | NB |
Dissociation constants, Kd (μM), derived from ribosomal A-site codon binding assays. “NB” indicates no measurable binding above background. “≥5.00” indicates binding that did not saturate at 5 μM ribosome-mRNA complex. All errors are standard error of the mean.
Fig. 8.

Ribosome A-site codon binding by tRNA1GlnUUG and tRNA1Glns2UUG. A) tRNAGlnUUG transcript or B) tRNAGln s2UUG bound to glutamine codons CAA (●) and CAG (○) as well as near cognate codons CAU (▼) and CGA (△). Dissociation constants (Kd) were calculated using a one-site specific binding equation in SigmaPlot (Table 4).
Two assays were employed to measure the effects of s2U34 on ribosome kinetics. First, we determined apparent rate constants for GTP hydrolysis for Gln-tRNAGln bearing UUG and s2UUG anticodons in the presence of increasing concentrations of 70S initiation complex from 0.15–2.5 μM. At 0.75 μM, decoding as measured by the rate of GTP hydrolysis was five-fold greater with the s2UUG anticodon on both the CAA cognate and CAC noncognate codons (Table 5). Thus, the 2-thio substituent accelerates GTP hydrolysis but does not discriminate between cognate and near-cognate codons. Separate kcat and Km parameters could not be determined, since plots of kapp versus 70S initiation complex concentration were linear (Figure 9A, inset), implying that both of these tRNAs had low affinity for the ribosomal A site. The extent of GTP hydrolysis was 80–85% for tRNAGlnUUG and 25–30% for tRNAGlns2UUG (Figure 9A). The lower extent of GTP hydrolysis may be due to a specific defect in the decoding step, since ternary complex formation by tRNAGlnUUG and tRNAGlns2UUG exhibits similar stoichiometry (Figure 8). Apparent rate constants obtained in the presence of low concentrations of 70S ribosome (0.15 μM) were used to estimate catalytic efficiencies (kcat/Km) of 0.55 s−1M−1 and 2.1 s−1M−1 on the cognate CAA codon with the UUG and s2UUG anticodons, respectively.
Table 5.
Rate constants for GTP hydrolysis
|
|
|||
|---|---|---|---|
| Anticodon | CAA (s−1) | CAC (s−1) | Ratio (CAA/CAC) |
|
|
|||
| UUG | 0.30 ± 0.01 | 0.0095 ± 0.0004 | 32 |
| s2UUG | 1.57 ± 0.18 | 0.0530 ± 0.0050 | 30 |
All data were determined in the presence of 0.75 μM 70S ribosome
Fig. 9.

Comparison of tRNA1GlnUUG and tRNA1Glns2UUG on CAA cognate and CAC near-cognate codons. A: Time course of GTP hydrolysis at a ribosome concentration of 0.75 μM. Rate constants derived from single or double exponential fitting of the data sets are presented in Table 5. The inset shows a linear dependence of kobs for GTP hydrolysis on the ribosome concentration. B: Time course of dipeptide formation between fMet-tRNAfMet and Gln-tRNAGln (UUG) and (s2UUG) at an effective ribosome concentration of 0.25 μM. Each data set was fit to a single exponential. The kobs for peptide bond formation was 0.73 ± 0.06 s−1 with tRNA1Glns2UUG and 0.16 ± 0.01 s−1 with tRNA1GlnUUG.
We also measured dipeptide formation in a similar assay, which returned values of 0.73 s−1 (Gln-tRNAGlns2UUG) and 0.16 s−1 (Gln-tRNAGlnUUG). These measurements were performed by reacting 0.25 μM 70S initiation complex with 0.5 μM ternary complex consisting of EF-Tu, GTP, and Gln-tRNAGln. By comparison, nearly identical apparent rate constants of 0.77 s−1 (Gln-tRNAGlns2UUG) and 0.14 s−1 (Gln-tRNAGlnUUG) were obtained for GTP hydrolysis when 0.375 μM 70S initiation complex was reacted with a much lower concentration (8 nM) of ternary complex. Since the ratio of peptide bond formation for the two tRNAs is closely similar to the ratio for GTP hydrolysis, we infer that s2 modification of U34 exerts its effects at the level of GTP hydrolysis, and does not influence the later steps of accommodation or peptidyl transfer.
Discussion
We have carried out detailed structural and mechanistic studies that fully elaborate the key role of the modified uridine in the tRNAGln wobble position at both aminoacylation and decoding steps, and provide a structural explanation for degenerate anticodon recognition by GlnRS. It had been understood that s2U34 was essential to decrease the Km for GlnRS binding to tRNA1GlnUUG,24 but the role of the 5-position modification in conferring enhanced tRNA binding affinity and improved complementarity for glutamine (as evidenced by the reduced Km) had not been appreciated. We also demonstrate unambiguously, by comparative analysis of mutant and wild-type transcripts, and by analysis of highly purified native tRNA1Gln, that the binding deficit associated with tRNA1GlnUUG is entirely due to the presence of unmodified U34. The crystal structure of GlnRS bound to this tRNA transcript reveals that U34 makes almost no favorable interactions with protein, which provides a clear rationalization of the weak binding affinity (Figure 7).
The crystal structures presented here also provide correlates for understanding the basis of degeneracy at the wobble position by GlnRS. Modeling suggests that none of the three conformations for the wobble nucleotide can accommodate a larger purine base, by steric exclusion. Although a capacity for rearrangement of the structure is clearly present in this region, conformational changes that could provide complementarity for adenine or guanosine, that would be more favorable than interactions with solvent, may not be possible. Accommodation of both cytosine and modified uracil occurs by small rearrangements of the tRNA together with adjustments in the nearby protein groups and an improved ordering of a surface loop that appears due to interactions with the cmnm5 modification (Figures 5–7).
Perhaps the most striking observation from the structures is that the rearrangements create an apparently specific binding pocket for the 2-thio substituent of U34, that contains both polar and hydrophobic elements (Figure 7B). Such an environment may be uniquely suitable to accommodate the large and polarizable sulfur moiety. The pocket appears somewhat larger than is necessary to accommodate the sulfur atom; nonetheless, oxygen of unmodified uracil is excluded presumably on electrostatic grounds. The crystal structure of a 2-thiouracil containing tRNA fragment bound to the ribosome has been determined,42 but we are unaware of a prior structure that illustrates how protein binds to sulfur as a thio group forming a double-bond with carbon.
Genetic and structural studies have revealed that many post-transcriptional modifications in tRNA are found at positions 34 and 37 and play an important role in codon recognition and frameshift suppression.43 Yet none of these modifications have been critically studied using rapid kinetic techniques in a reconstituted in vitro translation system. Although such studies routinely use fully-modified native tRNAs, the presence of multiple modifications on each tRNA makes it difficult to attribute properties to a specific modification. Here we determined how the specific thiolation of U34 in the UUG anticodon of unmodified tRNAGln impacts two important steps in translation, decoding as measured by GTP hydrolysis and accommodation as reported by peptide bond formation. The results showed that s2U increased the decoding rate of both cognate and near-cognate triplets by 5-fold. In comparable studies a selective 5 to 10-fold increase in the decoding rate of near-cognate triplets was observed when tRNATrp contained a G24A substitution or tRNAAla contained A32-U38 to U32-A38 substitutions.44,45 Conversely, an A37G substitution in tRNACys selectively decreased the decoding rate of a near-cognate triplet by 8-fold.46 This provides an interesting contrast with s2U34 in tRNAGln, which enhances translational efficiency at cognate and near-cognate codons unselectively.
In summary, the experiments reported here demonstrate comparable quantitative effects on the ribosome to those reported previously, while offering a clear and specific view of the importance of an isolated modification in the wobble position in the context of intact tRNA.47 The methodology that we have used, employing ligation of chemically synthesized half-molecules containing site-specific modified groups,48 is generally applicable to produce intact full-length tRNA at preparative scales. It should be useful for comparable studies of other anticodon modifications.
Materials and Methods
Expression and purification of GlnRS and of the tRNAGln unmodified transcript
Wild type His-tagged and untagged E. coli GlnRS were expressed and purified as previously described.39,49 The Del 172–173 variant of T7 RNA polymerase was used during in vitro transcription reactions to synthesize tRNAGlnUUG, tRNAGlnCUG and the C34U tRNAGlnCUG mutant, as previously described.48 Oligonucleotides contained 2′ –OCH3 groups to reduce 3′ heterogeneity in transcribed tRNAs, and all tRNAs contained a catalytically neutral G1 mutation to enhance the efficiency of transcription. DNA template sequences are given in Supplemental Experimental Procedures. After transcription, the DNA template was digested with RQ1 RNase-free DNase (Promega) for one hour, then tRNA was extracted twice with phenol-chloroform and ethanol precipitated. tRNA pellets were resuspended in 200–300 μL of TE buffer, and buffer exchange was performed using centrifugal filter units with a 3K cutoff (Millipore) to remove excess nucleotides. tRNA transcripts were then loaded onto a 38 cm × 32 cm × 2mm 12% acrylamide/8M urea gel, and run at 50W for about 4–5 hours. tRNAs were visualized by UV shadowing and sliced from the gel for extraction in TE buffer overnight. Extracted transcripts were then concentrated in TE buffer. Purified tRNAs were stored in TE buffer at −20°C until required.
Synthesis of s2U34 tRNA1Gln
The modified 2-thiouridine (s2U) was synthesized using the “silyl method”.50 However, 1–O acetyl-2,3,5-O-benzene-D-ribose, SnCl4, and CH3CN were used as catalyst and solvent, instead of 1-chloro-2,3,4-tri-O-benzoyl-D-ribose, AgClO4 and benzene, respectively.51 The s2U group was then derivatized, protected and functionalized using the 5′-silyl-2′-acetoxy ethyl orthoester (ACE) chemistry to the phosphoramidite (Thermo Fisher, Lafayette, CO).52 The two tRNA half-molecules were then synthesized (Thermo Fisher Scientific). ACE-protected tRNA half-molecules were deprotected via the standard Thermo Fisher protocol, extensively dialyzed against H2O, and lyophilized for storage (−20°C).
The two lyophilized, deprotected oligoribonucleotides consisted of a 32-mer corresponding to the sequence from the 5′ tRNA end to U33, and a 43-mer initiating at the 5′-end with the s2U34 modified base. These RNAs were resuspended in 5 mM PIPES, (pH 7.5), 10 mM MgSO4 (PMS buffer) to a final concentration of 50 μM. Ligation reactions were carried out at 37°C in 500 μL volume containing 50 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM DTT, 1 mM ATP, 22.5 μM 32-mer 5′ RNA oligo, 22.5 μM 43-mer 3′ RNA oligo and 50 units of T4 RNA ligase 1 (New England Biolabs). Before adding all reagents, 3′ and 5′ oligonucleotides were mixed, heated to 80°C for 5 min on a heat block and then slowly cooled to ambient temperature. T4 RNA ligase buffer and enzyme were added, and reactions were incubated for 14 hr at 37°C. Reactions were stopped by phenol:chloroform extraction. Ligation of the two RNAs was verified by denaturing PAGE (Supplementary Figure 4). Full length tRNA was sliced from the gel and extracted as described for tRNAGln transcripts.48 The proper state of the thiouridine in the ligated tRNA was confirmed by CD and by NMR.
Isolation and affinity purification of native tRNA1Gln(UUG)
Native fully modified isoacceptor was isolated from cells without overproduction by a DNA-RNA hybridization method scaled from a previously described protocol.7,53 About 250 μg of tRNA1Gln were purified from 12 g of E. coli cells. The fraction of tRNA1Gln isolated from cells was measured by single turnover aminoacylation reactions and compared with aminoacylation of unmodified transcripts and unfractionated tRNA (Supplementary Figure 5).
Steady-state kinetics
tRNAGln was 32P-radiolabeled at the 3′-terminal internucleotide linkage using E. coli tRNA nucleotidyltransferase, as described.54 After labeling, tRNA was phenol:chloroform extracted, and purified using two P-30 columns (BioRad). To ensure tRNA homogeneity, labeled and unlabeled tRNAs were mixed and folded before aminoacylation reactions by heating to 80°C for 2–3 min, followed by addition of MgCl2 to a final concentration of 10 mM and cooling to ambient temperature for 15–20 min. Plateau levels for aminoacylation were between 70–90% for all reactions. Aminoacylation was performed in 20 μL volume in a buffer containing 50 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM DTT and substrate concentrations depending on the reaction (see below).
Km(Gln) for unmodified transcript tRNAGlnUUG was determined with 20 nM enzyme, 5 μM tRNAGlnUUG and 0.02–10 mM glutamine. Km(tRNAGlnUUG) was measured with 5–10 nM GlnRS, 0.1–10 μM tRNAGlnUUG, and 10 mM glutamine. Kinetic parameters for U34C tRNAGlnCUG were obtained in reactions containing 10–20 nM GlnRS, in the presence of 0.04–5 mM glutamine for Km(Gln), and 0.125–16 μM U34C tRNAGlnCUG for Km(tRNA). Saturating amounts of U34C tRNAGlnCUG and glutamine were 10.8 μM and 100 mM, respectively. For the chemically synthesized s2U34 tRNAGlnUUG, reactions were conducted with 20 nM enzyme. In this case, to measure Km(Gln) glutamine concentrations varied between 0.04 to 10 mM, and saturating amounts of s2U34 tRNAGlnUUG were 5 μM. The Km for tRNAGlns2UUG was determined at 5 nM and 20 nM enzyme, varying tRNA concentrations from 62.5 nM to 16 μM and keeping the glutamine concentration saturating at 50 mM. For reactions with native tRNA, 1 nM enzyme, 0.01–10 mM glutamine, and 4 μM saturating tRNA were used to determine Km(Gln). To determine Km(tRNA) we varied its concentration between 0.05–5 μM, with 10mM glutamine. ATP saturation was verified for all reactions, with 10 mM levels sufficing in every case.
Reactions were quenched by mixing 1–2 μL aliquots with 4–5 μL of 400 mM sodium acetate (pH 5.2) containing 80μg/μL P1 nuclease (Roche). Digestion proceeded for 10 min after which 1uL of ~200 CPMs digested tRNA was spotted on a pre-washed PEI cellulose TLC plate (SIGMA). TLCs were developed in a buffer containing 100 mM ammonium acetate and 5% glacial acetic acid, dried and exposed to a phosphor screen for 12–16 hours. The screen was scanned in a Typhoon variable mode imager (GE Healthcare) and unreacted reagent and product were quantified using the ImageQuant program 5.2. Data were fit to linear regressions to obtain initial velocities, which were then used to create Michaelis-Menten plots from which Km and kcat values were determined. 6–8 different substrate concentrations were used per experiment and 6–8 time points taken for each curve.
Single-turnover kinetics
All single-turnover experiments were performed with a Kin-Tek RQF-3 apparatus. 32P-labeled tRNAGln was folded as described above. Aminoacylation reactions were initiated by the addition of WT GlnRS. For all reactions the enzyme was added to a reaction mixture at 37°C containing 10–25 nM tRNAGln, and 0.01–60 mM glutamine for Kd(Gln) determination. A concentration range of 0.1–12.8 μM enzyme was used to determine Kd(tRNA). Reactions were conducted in a buffer containing 50 mM Tris (pH 7.5), 10 mM MgCl2, and 5 mM DTT. For all Kd(Gln) experiments, saturating concentrations were shown by measuring single turnover rate constants at 5 μM and 15 μM enzyme. The ATP concentration was varied between 5 mM and 10 mM for all reactions to confirm saturation. Mixing controls showed that initiation of the reaction with amino acid, tRNA, or enzyme had no effect on kobs,max.
Reactions were quenched by the addition of 400 mM sodium acetate (pH 5.2) and 0.1% SDS; a 1μL aliquot was then digested with 0.08–0.1 mg/ml P1 nuclease for 10 min and spotted on prewashed PEI cellulose TLC plates (SIGMA), developed with 100 mM ammonium acetate and 5% acetic acid. Dried TLC plates were exposed to a phosphor screen for ~12–16 hours, scanned with a Typhoon imager and quantified using ImageQuant 5.2. Nine time points were collected for each kobs determination. Time courses were fit to a single exponential equation using Prism5 and/or KaleidaGraph software. The kobs values for six different glutamine concentrations or six enzyme concentrations were taken to determine each binding curve, and these data were fit to a hyperbolic function to determine Kd(Gln) or Kd(tRNA). The reported values for kobs,max were also derived from this hyperbolic fit. No indication of a kinetic lag was found in any time course, confirming that glutamine and transcript tRNA1Gln are in rapid equilibrium with the enzyme and that no consecutive steps present similar rate constant in all cases. At least two repetitions of the single turnover glutamine-binding and tRNA-binding experiments were performed for each GlnRS mutant.
Crystallization and Xray structure determinations
Recombinant E. coli GlnRS lacking a poly-His affinity tag was dialyzed overnight in 2 L of 10 mM PIPES (pH 7.0) and 20 mM βME. After dialysis, the enzyme was concentrated to ~8mg/mL and kept on ice until ready to use. tRNAGln was folded by heating to 80°C for 3 min followed by addition of MgCl2 to a final concentration of 10 mM, and allowed to equilibrate to ambient temperature on the benchtop. The pH of a 100 mM ATP stock solution was carefully adjusted to 7.0. Enzyme, tRNA and ATP were combined to final concentrations of 78 μM, 117 μM and 5 mM, respectively for the tRNA1Gln and tRNA2Gln transcript complexes and 78 μM, 94 μM and 5 mM for the native, fully modified tRNA1Gln; these mixes were incubated on ice for 10 min before use.
All crystals were grown by the hanging drop method at 17°C in 2 μL drops. Crystals for transcript tRNA1Gln and tRNA2Gln grew in either sodium citrate or ammonium sulfate. The best diffracting crystals for the transcript tRNA1Gln:GlnRS complex grew in 2.4–2.6 M ammonium sulfate, 40 mM PIPES (pH 7.0–7.4), 20 mM MgSO4 and 20 mM βME. Crystals for the tRNA2Gln-GlnRS complex grew best in similar conditions except for the use of 0.34–0.43 w/v Na-citrate instead of ammonium sulfate. Only needle-like crystals of the in vivo tRNA1Gln complex grew under these conditions in the presence of 0.36–0.4 w/v Na-citrate. Four rounds of micro-seeding were required to obtain diffraction quality crystals in this case.
All crystals were cryo-protected by sequential washing in 30%, 40% and 49% sodium malonate (pH 7.0) (Hampton Research) containing 20 mM MgSO4, 20 mM βME and 5 mM ATP. Crystals were looped out from the drops and placed in 1 mL of 30% malonate solution, allowed to equilibrate for 5 min, then looped out and placed for 5 min in the 40% solution; finally the crystals were placed into 1 mL of the 49% sodium malonate solution, and incubated at 17°C overnight. Crystals were then frozen by submerging in liquid nitrogen, stored in a pre-cooled dewar and shipped overnight to the Advanced Light Source (ALS; Berkeley, CA) for data collection.
Data collection was performed at ALS beamline 4.2.2. Data sets were integrated and scaled using d*TREK, and data refinement and modeling were done using CNS version 1.3 and Coot version 0.6.2, respectively.55,56 Parameter and topology files for modified nucleotide bases were obtained from the HIC-Up database or modified from PRODRG.57 The Rcrane plug-in was used in Coot for regularization.58
Thermal denaturation spectroscopy
Thermal denaturation/renaturation of tRNAs in a buffer consisting of 10 mM Na2HPO4 and 10 mM KH2PO4 (final pH 6.8) was performed using a Varian Cary 3 UV–visible spectrophotometer equipped with a Peltier temperature control accessory. The temperature was increased at a rate of 1°C per minute from 5 to 85°C. Absorbance data at 260 nm were collected as a function of temperature at a rate of four data points per minute. All experiments were performed simultaneously with a control cell containing buffer only; these data were subtracted as background prior to analysis. Thermodynamic properties were determined using MeltWin v3.5 software. Data presented are the average of at least five denaturation/renaturation cycles. The error calculated is the error of the mean.
Circular dichroism (CD) spectroscopy
CD spectra of the tRNAs in a buffer consisting of 10 mM Na2HPO4 and 10 mM KH2PO4 (final pH 6.8) were collected using a Jasco J600 spectropolarimeter equipped with a Peltier temperature control accessory. Samples were analyzed at 5°C in a 0.2 cm pathlength quartz cuvette. All experiments were performed with a control containing buffer only, and these data were subtracted as background prior to analysis. Data were analyzed by normalizing the spectra to molar circular dichroic absorbance using Δε =θ/32980*(cln) where ε is molar dichroic absorbance, θ is ellipticity (mdeg), c is sample concentration (M), l is pathlength (cm), and n is the number of nucleotides. Spectra shown are the average of three independent experiments each performed in triplicate.
Ribosome binding assay
Ribosome binding assays were performed as previously described.59 Briefly, ribosomes were activated at 42°C, cooled to 37°C and programmed with the respective mRNA (see mRNAs used below). fMet-tRNAfMet was used to saturate the ribosome P-site. 5′-32P-labeled tRNA was added in increasing concentration (0–5 μM) to the readied ribosome-mRNA complex. After incubation (60 min at 37°C followed by 15 min on ice) the reactions were filtered through a nitrocellulose filter using a modified Micro-Sample Filtration Manifold (Whatman Schleicher & Schuell Minifold) dot-blot apparatus with vacuum. Filters were air-dried, exposed to a phosphor screen, and scanned using a Typhoon scanner (GE Healthcare). tRNA was quantified using ImageQuant software. 32P-labeled tRNA of known concentration was spotted on a nitrocellulose filter to create a standard curve relating picomoles of tRNA to phosphor units. Dissociation constants were determined using a one-site specific binding equation in SigmaPlot. Data presented represent at least three independent experiments each performed in triplicate. The mRNAs used are listed in Supplemental Experimental Procedures.
GTP hydrolysis and peptidyl transfer kinetics
Tight-coupled ribosomes were prepared from E. coli MRE600 cells using the methods described by Pan and colleagues.60 E. coli His-tagged EF-Tu, IF1, IF2, and IF3 were obtained by affinity purification. The templating mRNA for translation was prepared by in vitro transcription from a synthetic double-stranded DNA template using T7 RNA polymerase, and purified by electrophoresis in a 7 M urea / 12% polyacrylamide gel. This RNA contained a standard Shine-Dalgarno sequence and coded for fMQVLYKTH5. [Sequence = 5′-GGGAAG-GAGGUAAAAAUGCAAGUUCUAUACAAGACUCACCACCACCACCAC-3′.]
For the GTP hydrolysis assay, reactions were carried out in 70 mM NH4Cl, 30 mM KCl, 3.5 mM MgCl2, 1 mM dithiothreitol, 0.5 mM spermidine, and 50 mM Tris-HCl pH 7.5 (Buffer A). Ternary complex was formed using a two-step protocol in which EF-Tu was first incubated with limiting γ-32P-GTP (6000 Ci/mmole) for 15 min at 37 °C and then with excess Gln-tRNA1Gln for 15 min in an ice bath. Free radiolabeled GTP was removed from ternary complex by centrifugation through a spin cartridge (Centrispin-20; Princeton Separations). The flow-through was diluted with Buffer A to give a X2 stock of ternary complex that contained approximately 1.5 μM EF-Tu, 1.0 μM Gln-tRNA1Gln, and 16 nM γ-32P-GTP. In parallel, a X2 stock of 70S initiation complex was formed by incubating 70S ribosome (0.3–5.0 μM) with 1.33-fold molar excess IF1, IF2, IF3, fMet-tRNAiMet, and mRNA in Buffer A supplemented with 1 mM cold GTP for 25 min at 37 °C. The X2 solutions were stored in an ice bath until same day use in a Kintek quench flow apparatus. GTP hydrolysis reactions were conducted at 20 °C. Reaction aliquots were quenched with 40% formic acid. GTP was separated from Pi by thin layer chromatography on PEI-cellulose in the presence of 0.5 M NaHPO4 (pH 3.5). Spots were visualized by phosphorimaging and quantified using ImageQuant.
To measure dipeptide (fMQ) formation, 70S initiation complex was formed by incubating 0.75 μM 70S ribosome with 0.5 μM 35S-fMet-tRNAiMet and 1.0 μM each IF1, IF2, IF3, and mRNA in Buffer A with 1 mM GTP for 25 min at 37 °C. Ternary complex was formed by incubating 1.5 μM EF-Tu with 1 mM GTP in Buffer A for 15 min at 37 °C, after which Gln-tRNA1Gln was added to 1.0 μM and incubation continued for 15 min in an ice bath. The X2 stocks were rapidly mixed in a Kintek quench flow apparatus at 20 °C. Reaction aliquots were stopped in 0.8 M KOH. After 1–2 hr at 37 C, 35S-fMet was resolved from 35S-fMet-Gln by electrophoresis on a cellulose TLC plastic sheet wetted with 3.48 M HOAc / 62 mM pyridine (PYRAC; pH 2.7). Bands were visualized by phosphorimaging and quantified using ImageQuant.
LC-MS/MS analysis
For nucleoside analysis, purified tRNA1Gln from E. coli was digested to nucleosides using nuclease P1 (Sigma), snake venom phosphodiesterase I (Worthington Biochemical Corporation) and Antarctic phosphatase (New England Biolabs). The nucleosides were separated using a Hitachi D-7000 HPLC with a Hitachi L-7400 UV detector at 0.3 mL/min at room temperature on a Synergi Hydro-RP 250 × 2.00 mm column (Phenomenex), using a gradient of 5 mM ammonium acetate (pH 5.3) and acetonitrile:water (40:60 v/v).61 The eluent from the column was split, with 2/3 flowing into a UV detector and 1/3 flowing into a LTQ-XL (Thermo Scientific). The cmnm5s2U nucleoside standard was a generous gift from Prof. James McCloskey (University of Utah).
For sequence analysis, purified tRNA1Gln from E. coli was digested with 50 U/μg of RNase T1 (Worthington Biochemical Corporation) in 20 mM ammonium acetate for 2 h at 37 °C. Digestion products from 1 μg of RNA were separated using a Thermo Surveyor HPLC system with an Xbridge C18 1.0 × 150 mm column (Waters) at room temperature with a flow rate of 40 μL/min. Before each run, the column was equilibrated for 15 min at 95% Buffer A (200 mM hexafluoroisopropanol (HFIP), 8.15 mM triethylamine (TEA)) and Buffer B (200 mM HFIP, 8.15 mM TEA:methanol 50:50 v/v). A gradient of 5% B held for 5 min followed by a ramp to 30% B at 7 min and a ramp to 95% B at 50 min and held at 95% B for 5 min. The eluent was directed into a Thermo LTQ-XL linear ion trap for mass spectral analysis with a capillary temperature of 275 °C and a spray voltage of 4.5 kV. The sheath gas was set to 25 arbitrary units, auxiliary gas to 14 and sweep gas to 10. Collision induced dissociation (CID) tandem mass spectrometry set to 35% was used to obtain sequence information of the digestion products in data-dependent mode.62
Supplementary Material
Highlights.
We synthesize a tRNA containing 2-thioU34 as the only modification
The structure of tRNA with 2-thioU reveals how protein recognizes the 2-thio group
2-thioU plays a role in the efficiency of glutamine incorporation on the ribosome
The identity of the 5-position modification in E. coli glutamine tRNA is cmnm5s2U
The 5-position modification at U34 in glutamine tRNA is important to GlnRS binding
Acknowledgments
We thank Mark Rould for discussions and computer programs. We are also grateful to Jay Nix for assistance with Xray data collection at ALS beamline 4.2.2. This work was supported by the National Institute of Health grants GM63713 (to J.J.P.), GM081601 (to Y.M.H.), HGR005753 (to H.G.), and GM23037-25 (to P.F.A), and by National Science Foundation grants MCB1101859 (to PFA), CHE0910751 (to P.A.L.) and CHE1212625 (to P.A.L.).
The abbreviations used are
- aa-AMP
aminoacyl-adenylate
- aaRS
aminoacyl-tRNA synthetase
- aa-tRNA
aminoacyl-tRNA
- Ec
Escherichia coli
- GlnRS
glutaminyl-tRNA synthetase
- IPPase
inorganic pyrophosphatase
- WT
wild-type
- fmet
formylmethionine
- HFIP
hexafluoroisopropanol
- TEA
triethylamine
Footnotes
Accession Numbers
Crystal structures have been assigned the following accession numbers in the PDB; CUG structure – 4JXX; UUG structure – 4JXZ; native structure – 4JYZ.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Motorin Y, Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49:4934–4944. doi: 10.1021/bi100408z. [DOI] [PubMed] [Google Scholar]
- 3.Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39(Database issue):D195–201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, Helm M, Bujnicki JM, Grosjean H. MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res. 2013;41(Database issue):D262–7. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davanloo P, Sprinzl M, Watanabe K, Albani M, Kersten H. Role of ribothymidine in the thermal stability of transfer RNA as monitored by proton magnetic resonance. Nucleic Acids Res. 1979;6:1571–1581. doi: 10.1093/nar/6.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helm M, Giegé R, Florentz C. A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry. 1999;38:13338–13346. doi: 10.1021/bi991061g. [DOI] [PubMed] [Google Scholar]
- 7.Bhaskaran H, Rodriguez-Hernandez A, Perona JJ. Kinetics of tRNA folding monitored by aminoacylation. RNA. 2012;18:569–580. doi: 10.1261/rna.030080.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibba M, Hong KW, Sherman JM, Sever S, Söll D. Interactions between tRNA identity nucleotides and their recognition sites in glutaminyl-tRNA synthetase determine the cognate amino acid affinity of the enzyme. Proc Natl Acad Sci USA. 1996;93:6953–6958. doi: 10.1073/pnas.93.14.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrington KM, Nazarenko IA, Dix DB, Thompson RC, Uhlenbeck OC. In vitro analysis of translational rate and accuracy with an unmodified tRNA. Biochemistry. 1993;32:7617–7622. doi: 10.1021/bi00081a003. [DOI] [PubMed] [Google Scholar]
- 10.Ashraf SS, Sochacka E, Cain R, Guenther R, Malkiewicz A, Agris PF. Single atom modification (O-->S) of tRNA confers ribosome binding. RNA. 1999;5:188–194. doi: 10.1017/s1355838299981529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weixlbaumer A, Murphy FV, 4th, Dziergowska A, Malkiewicz A, Vendeix FA, Agris PF, Ramakrishnan V. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat Struct Mol Biol. 2007;14:498–502. doi: 10.1038/nsmb1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vendeix FA, Dziergowska A, Gustilo EM, Graham WD, Sproat B, Malkiewicz A, Agris PF. Anticodon domain modifications contribute order to tRNA for ribosome-mediated codon binding. Biochemistry. 2008;47:6117–6129. doi: 10.1021/bi702356j. [DOI] [PubMed] [Google Scholar]
- 13.Yarian C, Marszalek M, Sochacka E, Malkiewicz A, Guenther R, Miskiewicz A, Agris PF. Modified nucleoside dependent Watson-Crick and wobble codon binding by tRNALysUUU species. Biochemistry. 2000;39:13390–13395. doi: 10.1021/bi001302g. [DOI] [PubMed] [Google Scholar]
- 14.Grosjean H, de Crecy-Lagard V, Marck C. Deciphering synonymous codons in the three domains of life: coevolutino with specific tRNA modification enzymes. FEBS Lett. 2010;584:252–264. doi: 10.1016/j.febslet.2009.11.052. [DOI] [PubMed] [Google Scholar]
- 15.Sylvers LA, Rogers KC, Shimizu M, Ohtsuka E, Söll D. A 2-thiouridine derivative is a positive determinant for aminoacylation by Escherichia coli glutamyl-tRNA synthetase. Biochemistry. 1993;32:3836–3841. doi: 10.1021/bi00066a002. [DOI] [PubMed] [Google Scholar]
- 16.Madore E, Florentz C, Giegé R, Sekine S, Yokoyama S, Lapointe J. Effect of modified nucleotides on Escherichia coli tRNAGlu structure and on its aminoacylation by glutamyl-tRNA synthetase. Predominant and distinct roles of the mnm5 and s2 modifications of U34. Eur J Biochem. 1999;266:1128–1135. doi: 10.1046/j.1432-1327.1999.00965.x. [DOI] [PubMed] [Google Scholar]
- 17.Johansson MJ, Esberg A, Huang B, Björk GR, Byström AS. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol Cell Biol. 2008;28:3301–3312. doi: 10.1128/MCB.01542-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakajima N, Ozeki H, Shimura Y. Organization and structure of an E. coli tRNA operon containing seven tRNA genes. Cell. 1981;23:239–249. doi: 10.1016/0092-8674(81)90288-9. [DOI] [PubMed] [Google Scholar]
- 19.Yaniv M, Folk WR. The nucleotide sequences of the two glutamine transfer ribonucleic acids from Escherichia coli. J Biol Chem. 1975;250:3243–3253. [PubMed] [Google Scholar]
- 20.Chen P, Crain PF, Nasvall J, Pomerantz SC, Bjork GR. A ‘gain of function’ mutation in a protein mediates productino of novel modified nucleosides. EMBO J. 2005;24:1842–1851. doi: 10.1038/sj.emboj.7600666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dumelin CE, Chen Y, Leconte AM, Chen YG, Liu DR. Discovery and characterization of geranylated RNA in bacteria. Nat Chem Biol. 2012;8:913–919. doi: 10.1038/nchembio.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stadtman TC. Specific occurrence of selenium in enzymes and amino acid tRNAs. FASEB J. 1987;1:375–379. doi: 10.1096/fasebj.1.5.2445614. [DOI] [PubMed] [Google Scholar]
- 23.Wolfe MD, Ahmed F, Lacourciere GM, Lauhon CT, Stadtman TC, Larson TJ. Functional diversity of the rhodanese homology domain: the Escherichia coli ybbB gene encodes a selenophosphate-dependent tRNA 2-selenouridine synthase. J Biol Chem. 2004;279:1801–1809. doi: 10.1074/jbc.M310442200. [DOI] [PubMed] [Google Scholar]
- 24.Rogers KC, Söll D. Discrimination among tRNAs intermediate in glutamate and glutamine acceptor identity. Biochemistry. 1993;32:14210–14219. doi: 10.1021/bi00214a021. [DOI] [PubMed] [Google Scholar]
- 25.Rould MA, Perona JJ, Steitz TA. Structural basis of anticodon loop recognition by glutaminyl-tRNA synthetase. Nature. 1991;352:213–218. doi: 10.1038/352213a0. [DOI] [PubMed] [Google Scholar]
- 26.Jahn M, Rogers MJ, Söll D. Anticodon and acceptor stem nucleotides in tRNA(Gln) are major recognition elements for E. coli glutaminyl-tRNA synthetase. Nature. 1991;352:258–260. doi: 10.1038/352258a0. [DOI] [PubMed] [Google Scholar]
- 27.Frugier M, Söll D, Giegé R, Florentz C. Identity switches between tRNAs aminoacylated by class I glutaminyl- and class II aspartyl-tRNA synthetases. Biochemistry. 1994;33:9912–9921. doi: 10.1021/bi00199a013. [DOI] [PubMed] [Google Scholar]
- 28.Perona JJ, Swanson R, Steitz TA, Söll D. Overproduction and purification of E. coli tRNA2Gln and its use in the crystallization of the glutaminyl-tRNA synthetase:tRNAGln complex. J Mol Biol. 1988;202:121–126. doi: 10.1016/0022-2836(88)90524-4. [DOI] [PubMed] [Google Scholar]
- 29.Arnez JG, Steitz TA. Crystal structure of unmodified tRNA(Gln) complexed with glutaminyl-tRNA synthetase and ATP suggests a possible role for pseudo-uridines in stabilization of RNA structure. Biochemistry. 1994;33:7560–7567. doi: 10.1021/bi00190a008. [DOI] [PubMed] [Google Scholar]
- 30.Addepalli B, Limbach PA. Mass spectrometry-based quantification of pseudouridine in RNA. J Am Soc Mass Spectrom. 2011;22:1363–1372. doi: 10.1007/s13361-011-0137-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandal D, Köhrer C, Su D, Russell SP, Krivos K, Castleberry CM, Blum P, Limbach PA, Söll D, RajBhandary UL. Agmatidine, a modified cytidine in the anticodon of archaeal tRNA(Ile), base pairs with adenosine but not with guanosine. Proc Natl Acad Sci USA. 2010;107:2872–2877. doi: 10.1073/pnas.0914869107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakurai M, Ohtsuki T, Suzuki T, Watanabe K. Unusual usage of wobble modifications in mitochondrial tRNAs of the nematode Ascaris suum. FEBS Lett. 2005;579:2767–2772. doi: 10.1016/j.febslet.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Wittwer AJ. Specific incorporation of selenium into lysine- and glutamate- accepting tRNAs from Escherichia coli. J Biol Chem. 1983;258:8637–8641. [PubMed] [Google Scholar]
- 34.Mihara H, Kato S, Lacourciere GM, Stadtman TC, Kennedy RA, Kurihara T, Tokumoto U, Takahashi Y, Esaki N. The iscS gene is essential for the biosynthesis of 2-selenouridine in tRNA and the selenocysteine-containing formate dehydrogenase H. Proc Natl Acad Sci USA. 2002;99:6679–6683. doi: 10.1073/pnas.102176099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hagervall TJ, Edmonds CG, McCloskey JA, Björk GR. Transfer RNA (5-methylaminomethyl-2-thiouridine)-methyltransferase from Escherichia coli K-12 has two enzyme activities. J Biol Chem. 1987;262:8488–8495. [PubMed] [Google Scholar]
- 36.Uter NT, Perona JJ. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady state kinetics. Proc Natl Acad Sci USA. 2004;101:14396–14401. doi: 10.1073/pnas.0404017101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krüger MK, Sørensen MA. Aminoacylation of hypomodified tRNAGlu in vivo. J Mol Biol. 1998;284:609–620. doi: 10.1006/jmbi.1998.2197. [DOI] [PubMed] [Google Scholar]
- 38.Sherlin LD, Perona JJ. tRNA-dependent active-site assembly in a class I aminoacyl-tRNA synthetase. Structure. 2003;11:591–603. doi: 10.1016/s0969-2126(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez-Hernandez A, Perona JJ. Heat maps for intramolecular communication in an RNP enzyme encoding glutamine. Structure. 2011;19:386–396. doi: 10.1016/j.str.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 40.Rould MA, Perona JJ, Söll D, Steitz TA. Structure of glutaminyl-tRNA synthetase complexed with tRNAGln and ATP at 2.8 Å resolution. Science. 1989;246:1135–1142. doi: 10.1126/science.2479982. [DOI] [PubMed] [Google Scholar]
- 41.Bujacz B, Wresniewska B, Bujacz A. Cryoprotection properties of salts of organic acids: a case study for a tetragonal crystal of HEW lysozyme. Acta Cryst. 2010;D66:789–796. doi: 10.1107/S0907444910015416. [DOI] [PubMed] [Google Scholar]
- 42.Vendeix FA, Murphy FV, 4th, Cantara WA, Leszczynska G, Gustilo EM, Sproat B, Malkiewicz A, Agris PF. Human tRNA(Lys3)(UUU) is pre-structured by natural modifications for cognate and wobble codon binding through ketoenol tautomerism. J Mol Biol. 2012;416:467–485. doi: 10.1016/j.jmb.2011.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gustilo EM, Vendeix FAP, Agris PF. tRNA’s modifications bring order to gene expression. Curr Opin Microbiol. 2008;11:135–140. doi: 10.1016/j.mib.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cochella L, Green R. An active role for tRNA in decoding beyond codon:anticodon pairing. Science. 2005;308:1178–1180. doi: 10.1126/science.1111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ledoux S, Olejniczak M, Uhlenbeck OC. A sequence element that tunes Escherichia coli tRNAAlaGGC to ensure accurate decoding. Nature Struct Mol Biol. 2009;16:359–364. doi: 10.1038/nsmb.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu C, Gamper H, Liu H, Cooperman BS, Hou YM. Potential for interdependent development of tRNA determinants for aminoacylation and ribosome decoding. Nature Comm. 2011;2:329. doi: 10.1038/ncomms1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Agris PF. Wobble position modified nucleosides evolved to select transfer RNA codon recognition: A modified-wobble hypothesis. Biochimie. 1991;73:1345–1349. doi: 10.1016/0300-9084(91)90163-u. [DOI] [PubMed] [Google Scholar]
- 48.Sherlin LD, Bullock TL, Nissan TA, Perona JJ, LaRiviere F, Uhlenbeck OC, Scaringe S. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA. 2001;7:1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 49.Hoben P, Royal N, Cheung A, Yamao F, Biømann K, Söl1 D. Escherichia coli glutaminyl-tRNA synthetase. II Characterization of the glnS gene product. J Biol Chem. 1984;257:11644–11650. [PubMed] [Google Scholar]
- 50.Vorbruggen H, Strehlke P. Nucleosidsynthesen, VII. Eine einfache Synthese von 2 Thiopyrimidin nucleosiden. Chem Berichte. 1973;106:3039–3061. [Google Scholar]
- 51.Sierzputowska-Gracz H, Sochacka E, Malkiewicz A, Kuo K, Gehrke CW, Agris PF. Chemistry and structure of modified uridines in the anticodon, wobble position of transfer RNA are determined by thiolation. J Amer Chem Soc. 1987;109:7171–7177. [Google Scholar]
- 52.Scaringe SA. RNA oligonucleotide synthesis via 5′-silyl-2′-orthoester chemistry. Methods. 2001;23:206–217. doi: 10.1006/meth.2000.1132. [DOI] [PubMed] [Google Scholar]
- 53.Yokogawa T, Kitamura Y, Nakamura D, Ohno S, Nishikawa K. Optimization of the hybridization-based method for purification of thermostable tRNAs in the presence of tetraalkylammonium salts. Nucleic Acids Res. 2010;238:e89. doi: 10.1093/nar/gkp1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci USA. 2002;99:5965–5970. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nature Protocols. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schüttelkopf AW, van Aalten DMF. PRODRG - a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004;D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 58.Kevin S, Keating KS, Pyle AM. RCrane: semi-automated RNA model building. Acta Crystallogr D Biol Crystallogr. 2012;68:985–995. doi: 10.1107/S0907444912018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cantara WA, Bilbille Y, Kim J, Kaiser R, Leszczyska G, Malkiewicz A, Agris PF. Modifications modulate anticodon loop dynamics and codon recognition of E. coli tRNA(Arg1,2) J Mol Biol. 2012;416:5795–5797. doi: 10.1016/j.jmb.2011.12.054. [DOI] [PubMed] [Google Scholar]
- 60.Pan D, Kirillov S, Zhang CM, Hou YM, Cooperman BS. Rapid ribosomal translocation depends on the conserved 18–55 base pair in P-site transfer RNA. Nat Struct Mol Biol. 2006;13:354–359. doi: 10.1038/nsmb1074. [DOI] [PubMed] [Google Scholar]
- 61.Pomerantz SC, McCloskey JA. Analysis of RNA hydrolyzates by liquid chromatography-mass spectrometry. Methods Enzymol. 1990;193:796–824. doi: 10.1016/0076-6879(90)93452-q. [DOI] [PubMed] [Google Scholar]
- 62.Krivos KL, Addepalli B, Limbach PA. Removal of 3′-phosphate group by bacterial alkaline phosphatase improves oligonucleotide sequence coverage of RNase digestion products analyzed by collision-induced dissociation mass spectrometry. Rapid Commun Mass Spectrom. 2011;25:3609–3616. doi: 10.1002/rcm.5266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.