

Abstract
The virally encoded protease is an important drug target for AIDS therapy. Despite the potency of the current drugs, infections with resistant viral strains limit the long-term effectiveness of therapy. Highly resistant variants of HIV protease from clinical isolates have different combinations of about 20 mutations and several orders of magnitude worse binding affinity for clinical inhibitors. Strategies are being explored to inhibit these highly resistant mutants. The existing inhibitors can be modified by introducing groups with the potential to form new interactions with conserved protease residues, and the flexible flaps. Alternative strategies are discussed, including designing inhibitors to bind to the open conformation of the protease dimer, and inhibition of the protease-catalyzed processing of the Gag-Pol precursor.
Successful treatment of the HIV/AIDS epidemic is due to a combination of medical, scientific and pharmaceutical efforts in the last three decades [1]. In lieu of an effective vaccine, highly active antiretroviral therapy (HAART) with a cocktail of different drugs remains the standard regimen for improved clinical outcomes [2]. The drugs target several essential steps in the viral replication, including virus entry and fusion with the host cell, and the viral enzymes, protease, reverse transcriptase, and integrase. Complications for HAART stem from immune dysfunction, increased risk for non-AIDS related disorders, and side effects from long term large pharmaceutical exposure [3]. The selection of viral strains that are resistant to current inhibitors remains a prominent challenge to successful long-term treatment of HIV infected individuals.
The HIV protease is an important drug target for HIV/AIDS therapy, and its structure and function have been reviewed in [4]. HIV protease performs an essential role in viral maturation by processing specific cleavage sites in the Gag and Gag-Pol precursor polyproteins to release the mature proteins (Figure 1). The protease-catalyzed processing of viral precursors is essential for formation of infectious virus particles. The 99 amino acid aspartic protease forms a homodimer, which is characterized by a central active site cavity capped by two flexible flap regions. The dimer presents a closed conformation when bound to substrate or inhibitor, and the flaps open for entry or release of the ligands. The structure-guided strategy for drug design has been extremely successful, resulting in competitive inhibitors that tightly bind in the active site with high affinity by mimicking the transition state of the natural protein substrate. Clinical protease inhibitors (PIs) have significantly improved patient outcomes since their introduction in 1995. Nine PIs have been approved for clinical therapy to date (Table 1). Resistance to inhibitors arises from mutations in HIV protease that alter inhibitor binding site or the subunit-subunit interface of the protease dimer, while still permitting viable levels of hydrolysis of the substrate allowing formation of infectious virus particles. Accumulation of drug resistant mutations can result in highly resistant variants. The two newest PIs, tipranavir and darunavir (DRV), were specifically designed for increased efficacy against resistant mutants. However, no new PIs have been introduced in the clinic since DRV was approved in 2006. The identification of clinically isolated proteases that are highly resistant to DRV and other PIs has sparked renewed interest in developing strategies for inhibition of resistant HIV protease.
Figure 1. Gag and Gag-Pol precursors.
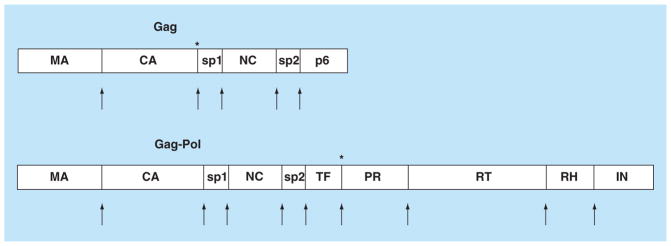
HIV PR processes the Gag and Gag-Pol precursor proteins during the maturation stage of viral replication. PR releases itself and the individual proteins: MA, CA, NC, RT, IN and RH, as well as small, probably unstructured, spacer peptides sp1, sp2 and p6. The arrows indicate the cleavage sites. The maturation inhibitor, Bevirimat, targets the CA-sp1 cleavage site and cleavage of TF-PR at the N-terminus of PR is another potential target for novel inhibitors, as indicated by asterisks in Gag and Gag-Pol.
CA: Capsid; IN: Integrase; MA: Matrix; NC: Nucleocapsid; PR: Protease; RH: RNaseH; RT: Reverse transcriptase.
Table 1.
Clinical inhibitors and resistance mutations in HIV-1 protease.
| Amino acid | L | V | G | K | L | V | D | L | E | M | K | M | I | G | I | F | I | Q | D | I | L | I | H | A | G | T | L | V | V | N | I | I | N | L | L | I |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||||||||||||||||||||||||
| Location | 10 | 11 | 16 | 20 | 24 | 32 | 30 | 33 | 34 | 36 | 43 | 46 | 47 | 48 | 50 | 53 | 54 | 58 | 60 | 62 | 63 | 64 | 69 | 71 | 73 | 74 | 76 | 77 | 82 | 83 | 84 | 85 | 88 | 89 | 90 | 93 |
| Inhibitor | ||||||||||||||||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Saquinavir 1995 | I | I | V | V | V | V | S | I | A | V | M | |||||||||||||||||||||||||
| R | L | T | F | |||||||||||||||||||||||||||||||||
| V | T | |||||||||||||||||||||||||||||||||||
| S | ||||||||||||||||||||||||||||||||||||
| 10 | 20 | 24 | 32 | 36 | 46 | 54 | 71 | 73 | 76 | 77 | 82 | 84 | 90 | |||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Indinavir 1996 | I | M | I | I | I | I | V | V | S | V | I | A | V | M | ||||||||||||||||||||||
| R | R | L | T | A | F | |||||||||||||||||||||||||||||||
| V | T | |||||||||||||||||||||||||||||||||||
| 10 | 30 | 36 | 46 | 71 | 77 | 82 | 84 | 88 | 90 | |||||||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Nelfinavir 1997 * w/o RTV | F | N | I | I | V | I | A | V | D | M | ||||||||||||||||||||||||||
| I | L | T | F | S | ||||||||||||||||||||||||||||||||
| T | ||||||||||||||||||||||||||||||||||||
| S | ||||||||||||||||||||||||||||||||||||
| 10 | 32 | 46 | 47 | 50 | 54 | 73 | 76 | 82 | 84 | 90 | ||||||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Fosamprenavir 1999, 2003 | F | I | I | V | V | L | S | V | A | V | M | |||||||||||||||||||||||||
| I | L | V | F | |||||||||||||||||||||||||||||||||
| R | M | S | ||||||||||||||||||||||||||||||||||
| V | T | |||||||||||||||||||||||||||||||||||
| 10 | 20 | 24 | 32 | 34 | 46 | 47 | 50 | 53 | 54 | 63 | 71 | 73 | 76 | 82 | 84 | 90 | ||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Lopinavir 2000 | F | M | I | I | F | I | V | V | L | V | P | V | S | V | A | V | M | |||||||||||||||||||
| I | R | L | A | L | T | F | ||||||||||||||||||||||||||||||
| R | A | T | ||||||||||||||||||||||||||||||||||
| V | M | S | ||||||||||||||||||||||||||||||||||
| T | ||||||||||||||||||||||||||||||||||||
| S | ||||||||||||||||||||||||||||||||||||
| 10 | 16 | 20 | 24 | 32 | 34 | 34 | 36 | 46 | 48 | 50 | 53 | 54 | 60 | 62 | 64 | 71 | 73 | 82 | 84 | 85 | 88 | 90 | 93 | |||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Atazanavir 2003 *w/ and w/o RTV | I | E | R | I | I | I | Q | I | I | V | V | L | L | E | V | L | V | C | A | V | V | S | M | L | ||||||||||||
| F | M | F | L | L | Y | V | M | I | S | T | M | |||||||||||||||||||||||||
| V | I | V | V | M | V | T | T | F | ||||||||||||||||||||||||||||
| C | T | T | L | A | I | |||||||||||||||||||||||||||||||
| V | A | |||||||||||||||||||||||||||||||||||
| 10 | 34 | 36 | 43 | 46 | 47 | 54 | 58 | 69 | 74 | 82 | 83 | 84 | 89 | |||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Tipranavir 2005 | V | F | I | T | L | V | A | E | K | P | L | D | V | I | ||||||||||||||||||||||
| L | M | R | E | M | ||||||||||||||||||||||||||||||||
| V | V | V | ||||||||||||||||||||||||||||||||||
| 11 | 32 | 33 | 47 | 50 | 54 | 74 | 76 | 84 | 89 | |||||||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||
| Darunavir 2006 | I | I | F | V | V | M | P | V | V | V | ||||||||||||||||||||||||||
| L | ||||||||||||||||||||||||||||||||||||
Clinical inhibitors are shown with the year of approval. Major mutations are in bold and underlined.
Clinical inhibitors (PIs) and the sites of their drug resistant mutations in HIV protease. PIs are used with ritonavir unless stated.
Adapted from [21].
Causes of resistance to protease inhibitors
Since the successful debut of the clinical inhibitor saquinavir (SQV) in 1995, viral resistance to protease inhibitors has arisen as an urgent challenge to HAART [5]. Evolution of HIV to acquire resistance to PIs emerges from a combination of several factors. The viral reverse transcriptase lacks a proofreading function; low fidelity transcription drives an increased rate of spontaneous mutagenesis [6]. Selection of resistant strains is increased by loss of adherence to the drug program due to the necessity for long-term use of HAART [7,8]. Patient noncompliance due to drug side-effects, pill-burden, and toxicity has increased viral selection for resistance [9,10] Primary transmission of resistant strains from HAART treated patients to antiretroviral-naive patients is also common in middle to low income countries [11,12].
Viral resistance to protease inhibitors evolves initially by mutations in the protease gene. The majority of these mutations associated with resistance are single amino acid substitutions, as illustrated in Table 1. Rarely, insertions have been observed in the protease gene and may play a role in PI resistance [13–15]. Multiple mutations accumulate, and high level resistance may require a combination of 10–20 mutations in the protease. Many different combinations are possible. As resistance evolves, additional mutations occur in the cleavage sites of the polyproteins.
Virus containing resistant protease mutants must still be capable of reproducing in vivo by successful processing of the Gag and Gag-Pol precursors in the maturation stage. Several mutations in the precursor cleavage sites have been shown to improve the activity of resistant protease [16,17]. Mutations in the gag cleavage sites of NC-p2 and p2-p6 are commonly associated with drug resistance [18]. Structural modeling suggests that the mutations in the cleavage site act to improve interactions between the substrate and the mutated protease and increase the efficiency of cleavage. Hence, mutations in the Gag and Gag-Pol substrates can compensate for the lower catalytic efficiency of resistant protease mutants [18,19]. This mechanism of resistance can be difficult to detect as the protease genotype from the patient would not necessarily match the expected PI-resistance profile [20]. Initial mutations in the protease gene co-evolve with compensatory mutations in the cleavage sites of the Gag and Gag-Pol substrates. The result of this network of influences is specific combinations of mutations that confer resistance to PIs but still provide adequate proteolytic activity, viral maturation and production of infectious virus particles.
Mutated residues in HIV PR are classified as either major or minor resistance mutations by their effect in HAART, as summarized in Wensing et al. [21]. Minor resistance mutations are assumed to have ancillary roles such as compensation for lower efficiency of proteolysis caused by major mutations. Major resistance mutations tend to confer high levels of resistance to one or multiple inhibitors and develop early in patient treatment. When possible, testing for resistance is performed following diagnosis of HIV infection [22]. For example, specific mutations I47A/V and V32I confer high resistance to lopinavir (LPV) [23–25]. The L76V mutation significantly increases LPV resistance when combined with common partner substitutions M46I, I54V, V82A and L90M [26]. One mutation, I84V, is associated with resistance to all nine currently approved PIs [21].
The 15 sites described for major mutations are distributed between the active site, flap and distal regions of the protease dimer (Figure 2). Patients exhibiting pan-PI-resistance were shown to have convergent evolved patterns of accumulated protease mutations [27]. Different multiple genotypic mutation patterns frequently yield similar phenotypic resistance profiles [28]. A library of laboratory designed recombinant molecular clones with common PI-resistant substitutions is publicly available as representative sequences for studying novel therapeutics for resistant strains [29]. The effects of individual mutations and their combinations are described in the next section.
Figure 2. Protease dimer showing sites of resistance mutations.
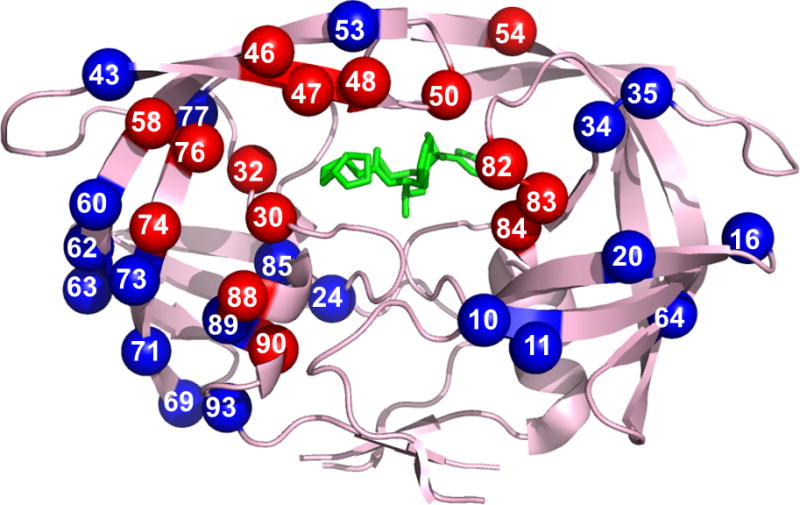
Locations of resistance associated mutations mapped on the HIV protease dimer bound with DRV (PDB: 2IEN). The protease dimer is shown in pink ribbons with DRV in green sticks. Major and minor mutations are distributed among both monomers and shown in red and blue spheres, respectively.
Adapted with permission from [30] (2009); using updated resistance mutations from [21].
For color images please see online www.future-science.com/doi/full/10.4155/FMC.15.44
Diverse mechanisms of resistance
Distinct mechanisms of resistance to PIs have been described. Earlier studies focused on mutants with single substitutions, and the molecular mechanisms contributing to resistance are reviewed in Weber and Agniswamy 2009 [30]. Resistant mutants often demonstrate reduced but viable catalytic activity and defective processing of the polyproteins. Analysis of the changes in structure and activity of the mutants suggested three categories of mutations:
Single substitutions in active site cavity residues can result in altered drug interactions. This mechanism is a straightforward way of reducing the binding affinity of drugs yet still maintaining catalytic activity. Known mutations using this mechanism include D30N, V32I, I47V, G48V, I50V, Val82 and I84V. These mutations produce small changes in the size and shape of the amino acid side chains that interact with inhibitors. A recent example of this mechanism is a double mutant, G48T/L89M, that displays resistance to SQV via an expanded active site cavity from a defective hydrophobic sliding mechanism [31];
Mutations can lower dimer stability by interrupting the interactions at the interface between the two subunits in the dimer. The result will be increased dimer dissociation and release of bound inhibitor. This mechanism has been described for mutants with L24I, I50V and F53L by kinetic and structural studies [30]. HIV protease dimerization is thought to occur in two sequential steps, initial intermolecular interactions at the active site lead to more interactions along the dimer interface [32]. Mutations of Ile50 alter both dimer stability and interactions with PIs [30];
Distal mutations can cause PI resistance in unique ways. The major mutation of L76V is associated with resistance to DRV, fosamprenavir (APV), indinavir (IDV) and LPV, while conferring susceptibility to the other PIs. Protease bearing the L76V mutation showed low dimer stability and defects in autoprocessing [33]. Analysis of crystal structures showed significant loss of interactions with DRV, while protease interactions were maintained with SQV. Molecular dynamic studies combined with crystal structures suggested L76V contributed to resistance by altering the overall enzyme dynamics [34]. Many distal mutations are classified as minor mutations that modulate the effects of major mutations. One example is G73S, which showed an altered network of interactions leading to the inhibitor binding site [30]. Another indirect effect was observed for the distal mutation N88D, which had altered interactions with water in a conserved region of the protease structure [35]. Changes due to a single distal mutation may be small and difficult to analyze in the absence of major mutations. However, the cooperative influences of distal mutations with active site mutations may be required for high level resistance to multiple drugs [36].
High level resistance from multiple resistance mutations
Multidrug and highly resistant variants often employ a combination of mechanisms that act synergistically to evade inhibition. Several multidrug and highly resistant HIV protease mutants have been identified and characterized in order to elucidate how the mutations affect the structure and function of the enzyme. Several mutants containing different sets of 10–22 substitutions have been investigated by x-ray crystallography and isothermal titration calorimetry, and selected examples are listed in Table 2. The mutants contain one or more major mutations combined with a variety of minor mutations. The binding affinity for DRV decreases by nearly four orders of magnitude in the worst cases, as shown in Table 2. The drastically lower binding affinity for inhibitor suggests that viral strains with these protease variants have lost susceptibility to DRV. Commonly observed structural features of these highly resistant variants include an expanded PI binding site and altered flap dynamics. The structures of the highly resistant mutant PR20 and wild type protease are compared in Figure 3. The different flap conformations highlight the variation observed in the crystal structures. The two closed conformation dimers are complexed with DRV and are similar in overall structure. The open conformation of the flaps is observed in the absence of bound inhibitor. In the absence of inhibitor, PR20 exhibits extremely open conformations of the flaps, and the two flaps in the dimer can exist in asymmetrical conformations, which is atypical for wild type enzyme.
Table 2.
Selected highly resistant protease mutants and Kd for Darunavir.
| Protease | Kd DRV (nM) | Relative Kd | Amino acid substitutions |
|---|---|---|---|
| Wild type | 0.005 | 1.0 | |
| PR20† | 41.000 | 8200 | Q7K, L10F, I13V, I15V, D30N, V32I, L33F, E35D, M36I, S37N, I47V, I54L, Q58E, I62V, L63P, A71V, I84V, N88D, L89T, L90M |
| P51†‡ | 37.000 | 7400 | L10I, I15V, K20R, L24I, V32I, I33F, M36I, M46L, I54M, I63P, K70Q, V82I, I84V, L89M |
| PRdrv1§ | 15.000 | 3000 | L10I, M36V, M46L, I54V, I62V, L63P, A71V, V82A, I84V, L90M |
| PRdrv2§ | 0.750 | 150 | T12V, I13V, I15V, K20M, V32I, L33F, K43T, I54L, K55N, I62V, L63P, A71V, I72V, G73S, V77I, V82L, I84V, L89V, L90M |
| PRdrv3§ | 0.004 | 0.82 | I13V, K20R, V32I, L33F, E35D, M36I, R41K, K43T, I47V, I54M, I62V, L63V, A71V, I72T, G73S, T74P, V82L, L89V, I93L |
| PRdrv4§ | 35.000 | 7000 | L10F, I13V, D30N, K45I, I50L, L63P, A71I, V77I, N88D |
| PRdrv5§ | 0.370 | 74 | L10I, I13V, K14R, V32I, L33F, K43T, M46I, I47V, I54L, I62V, L63P, A71T, I72T, G73T, V77I, P79S, I84V, L90M |
| PRdrv6§ | 1.800 | 360 | L10I, I13V, G16E, L33F, M36L, N37T, P39S, K45R, M46L, I54V, K55R, I62V, L63P, A71V, G73D, V82T, I84V, L89V, L90M, I93L |
Comparison of drug resistant HIV protease variants bearing multiple substitutions and the DRV binding affinity measured by isothermal titration calorimetry. Assays used different conditions, and may not be directly comparable to each other. Major mutations from Table 1 are shown in bold and underlined.
Data taken from [42].
Selected in vitro. Other mutants are from clinical isolates.
Data taken from [41].
Figure 3. Closed and open conformations of the dimer of wild type protease and highly resistant mutant PR20.
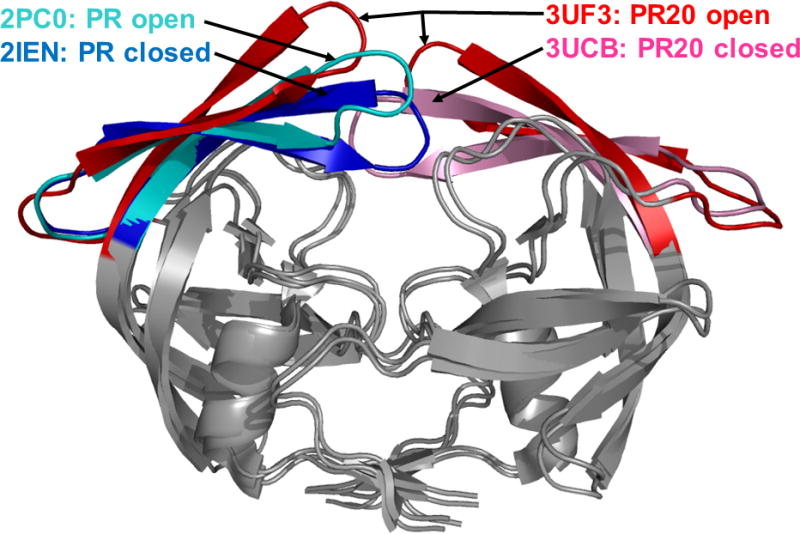
Superposition of the crystal structures of PR20 and wild type HIV protease. The dimers are shown in gray, except for the flaps, which are in different colors. The PDB ID is indicated for each structure. The different flap conformations highlight the variation observed in the crystal structures. The two closed conformation dimers are complexed with DRV and are similar in overall structure. The open conformation dimers contain no inhibitor. Open PR20 is shown with the two flaps in the dimer to indicate its atypical flap asymmetry, and the other structures are represented as single flaps.
For color images please see online www.future-science.com/doi/full/10.4155/FMC.15.44
An inactive (D25N) variant of a multidrug resistant clinical isolate deemed MDR769 carrying 10 mutations L10I, M36V, M46L, I54V, I62V, L63P, A71V, V82A, I84V, and L90M has been studied by x-ray crystallography. A variant of MDR769 with V82T displays 11–2600-fold resistance to all nine PIs [37]. Analysis of a 1.3Å resolution crystal structure describes three major differences between MDR769 and wild type protease [38]. First, the flaps of the MDR769 display a ‘wide open’ form as a result of structural rearrangements. Furthermore, the commonly occurring substitution L90M reshapes the drug interactions of the MDR769 active site by a domino effect of perturbations in the 80’s loop, which distorts the S1/S1′ subsites of the binding pocket [39]. The active site is further expanded by approximately 3.0Å via the dual reduction in atom volume from mutations V82A and I84V.
A clinical isolate from a pediatric patient failing HAART that harbors 22 mutations in the protease (T4S, L10V, I13A, K14R, K20I, A22V, L33I, E35D, M36I, N37D, R41K, K43S, G48A, I54V, I66F, H69K, T74S, V82A, I84V, L89I, L90M and T91S) was studied by kinetic and x-ray analysis in complex with DRV [19]. This mutant is highly resistant (>3600-fold increase from wild type) to SQV, RTV, IDV, ATV and NFV, cross-resistant to LPV and APV, and less resistant to DRV. Structural information indicates rearrangements of the flap elbow region leading to loss of direct interactions between protease and DRV. Additionally, V82A/I84V/G48A mutations contribute to the collapse of the 80’s loop by directly affecting inhibitor binding to the S1/S1′ subsites. Furthermore, distal mutations likely induce long-range structural changes altering inhibitor and substrate binding kinetics. The usual loss of catalytic efficiency of highly mutated protease variants was rescued in this case by co-evolution selecting for changes in the Gag-Pol cleavage sites [19]. The accumulated mutations of this protease enable viral replication through both decreased affinity for inhibitor and co-evolution of enzyme and its substrate.
Six clinically derived variants of HIV protease with up to 21 mutations known to be highly resistant to DRV with cross-resistance to several other clinical PIs were characterized extensively [40,41]. Isothermal titration calorimetry (ITC) analysis showed that the six DRV-resistant proteases lose inhibitor binding affinity due to less favorable enthalpy of binding when compared with the wild type enzyme. The analysis of crystal structures of three of these protease mutants complexed with DRV suggested a significant loss of protease-DRV hydrogen bond interactions as the likely explanation for the changes in binding enthalpy.
A different clinical isolate with extremely resistant HIV protease bearing 20 mutations (PR20) has been studied extensively with several different PIs and a substrate analog. This variant exhibits extreme resistance to all clinical PIs of several orders of magnitude [42], and the PR20 dimer dissociates more readily than wild type protease, while the monomer stability is enhanced [43]. Additionally, autocatalytic cleavage of PR20 precursor is unaffected by PIs, in contrast to the inhibition of the wild type precursor autoprocessing by DRV and SQV. To date, the PR20 structure has been solved in the absence of inhibitor or complexed to DRV, SQV, p2-NC peptide analog, APV and two investigational inhibitors GRL0519 and GRL02031 [44,45]. Like other resistant variants of HIV protease, the flaps observed in the different apo-structures of PR20 adopt a more open conformation as shown in Figure 3. The crystal structure of the open conformation of PR20 indicates the higher dissociation constant likely stems from loss of intersubunit contacts between the more flexible flaps. The flexibility of the flaps in PR20 is proposed to arise from structural rearrangements caused by the combination of the M36I/I15V/133F mutations. Unlike other highly resistant mutants, PR20 retains the majority of hydrogen bond interactions with clinical inhibitors DRV, SQV and APV [44,45]. Instead, the expanded binding site of PR20 results in loss of hydrophobic interaction with inhibitors. Additionally, the intersubunit interactions involving Arg8/8′ and Arg29/29′ are perturbed in PR20 by the presence of the L10F mutation; these interactions are important in forming the S2/S2′ PI binding subsites. When these mechanisms of resistance are combined with distal mutations that influence the interactions of residues 30–32 with inhibitor, PR20 employs a diverse array of mechanisms to evade inhibition.
Mutant HIV protease variants that have been laboratory selected for resistance to clinical inhibitors such as DRV provide a useful method for studying mechanisms of high resistance to a single drug independent of previous treatment [46]. One of these variants with 14 substitutions, designated P51, is resistant to DRV inhibition of precursor autoprocessing as described for PR20 [42]. The crystal structure of ligand-free P51 with inactive D25N substitution exhibits slightly wider flap separation and loss of intersubunit contacts between the flap tip and Pro81 when compared with ligand-free WT PR [47].
A crystal structure of P51 D25N co-crystallized with DRV (PDB: 4NPT) reveals a unique mode of binding for DRV [47]. DRV binds to the open flaps in an orientation almost perpendicular to the standard mode of binding in the active site. The observed unusual binding of DRV is mainly attributed to V32I/V82I/I84V mutations forming hydrophobic interactions with Val47 and shifted intersubunit contacts from the L24I substitution. The P51-DRV structure yields new insights into possible approaches to target the wide open flaps of resistant mutants.
Taken together, these examples show that significant changes in structure via up to 20 coordinated mutations are required for high level resistance to second generation inhibitors like DRV. Crystallographic analysis of several highly resistant mutants share perturbations in the active site, flap regions, 80’s loop, or inter-subunit interactions, which are likely to be mechanisms inducing loss of susceptibility to PIs. In addition, the highly resistant variants are proposed to have increased occupancy of open conformational states relative to wild type enzyme, as assessed by Electron Spin Resonance [48] and Nuclear Magnetic Resonance [49]. These structural and functional insights into HIV PR drug resistant mutants are invaluable in developing novel therapeutics for future AIDS treatment.
Inhibitors designed to bind in the active site
The clinical inhibitors of HIV protease provide a prime example of the success of structure-guided drug design, as discussed in recent reviews [4,50]. The PIs bind in the active site of the protease dimer and compete with substrate binding to the same site, as shown in Figure 4. All the current PIs contain the central hydroxyl group, which mimics the transition state of the reaction. They form interactions with protease similar to those of a peptide substrate, including hydrogen bonds with polar groups like the peptidic amide and carbonyl groups, and hydrophobic groups fitting like amino acid side chains into the protease subsites S2-S2′. The design difficulties can be illustrated by the example of brecanavir, a tyrosyl peptidomimetic with excellent antiviral activity against wild type and resistant virus, which subsequently failed clinical trials due to its poor metabolic profile. Incorporation of deuterium in an attempt to slow drug metabolism did not improve the pharmacokinetic properties of the compound [51]. The two most recently approved PIs, tipranavir and DRV, were designed to target resistant proteases. Here, the structure-guided design of DRV and the newer antiviral inhibitors will be described to illustrate a viable strategy for targeting resistant protease mutants.
Figure 4. Darunavir in the binding site of HIV protease.
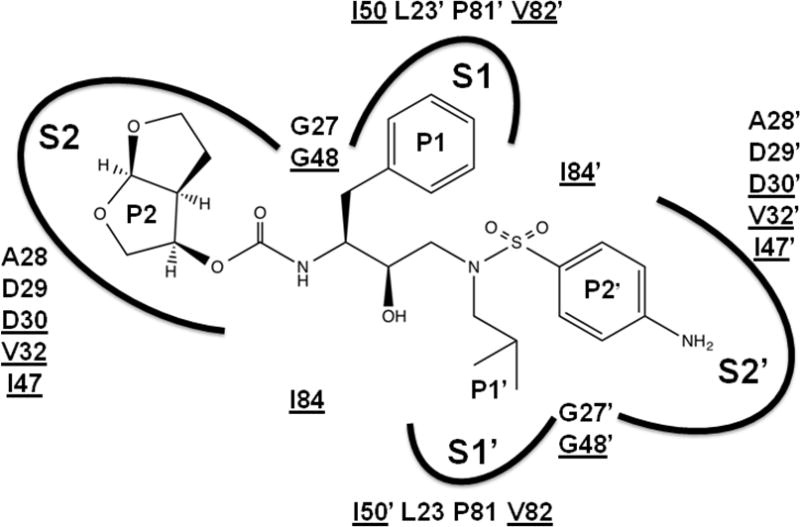
Darunavir is shown with groups labeled P2-P2′. The protease binding site is indicated by arcs for subsites S2, S1, S1′ and S2′ and the amino acid residues contributing to the binding site. Underlined amino acids are mutated in drug resistance.
Darunavir (DRV, TMC114, UIC-94017)
The drug DRV was designed with the bis-tetrahydrofuranylurethane (THF) moiety at P2 (Figure 5) in order to introduce hydrogen bonds with the conserved main chain atoms of the protease, which cannot be easily altered by mutations, unlike the side chains [52]. The interactions of the bis-THF with the protease are illustrated in Figure 6. DRV is effective and well-tolerated in the clinic [53], and shows a high genetic barrier for development of resistance [54]. The effectiveness of DRV is primarily due to its excellent 5–10 pM binding affinity for wild type protease as measured by ITC [55]. In addition, DRV was shown to inhibit protease dimerization [56], and the autoprocessing step, as described in a later section.
Figure 5. Darunavir and selected investigational inhibitors.
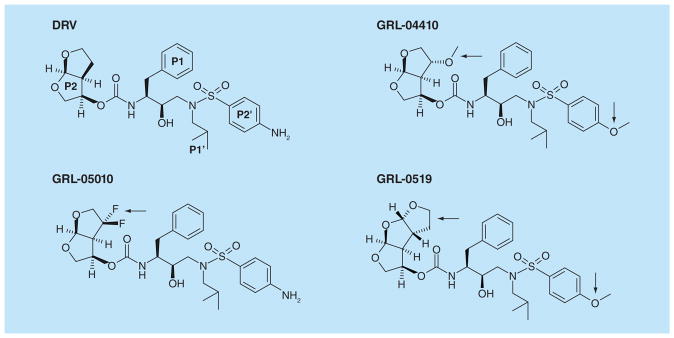
Structures are illustrated in similar conformations to facilitate comparison. Darunavir is shown with the P2, P1, P1’ and P2’ groups labeled. The modifications in the investigational inhibitors are indicated by black arrows. Antiviral inhibitors GRL-04410 and GRL-0519 have a methoxy substitution at P2’, while GRL-0519, GRL-04410 and GRL-05010 have tris-THF, methoxy and gem-difluoro additions at P2, respectively.
Figure 6. Interactions of HIV protease with P2 group of darunavir and selected inhibitors.
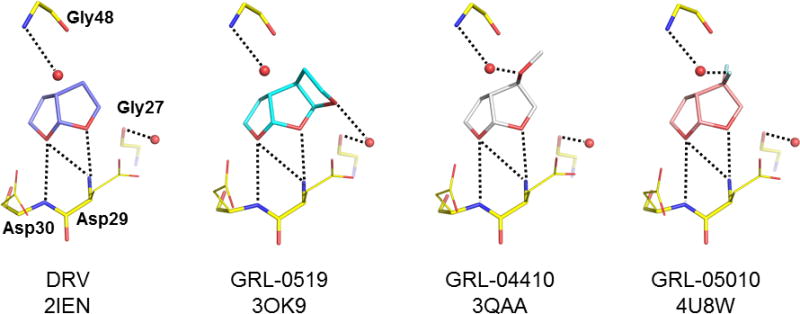
The interactions of the P2 bis-THF group of darunavir with protease residues Asp29, Asp30, Gly27 and Gly48, and the same view of the P2 interactions of antiviral inhibitors GRL-0519, GRL-04410 and GRL-05010. The PDB ID is shown for each structure. Water molecules are shown as spheres. Hydrogen bond interactions are indicated by dotted lines.
The hydrogen bond interactions of DRV are maintained with many resistant mutants, as demonstrated by crystallographic analyses [30]. HIV does evolve resistance to DRV, although generally several mutations are required, as described earlier. Characterized mutants with high level resistance show several orders of magnitude lower affinity for DRV [40–42]. Loss of hydrogen bond interactions with DRV was observed for protease bearing L76V [33], and several resistant mutants isolated from patients with up to 21 substitutions [42,43], while another clinical mutant with 19 mutations showed loss of hydrophobic interactions [45]. These results demonstrate the need for new antiviral inhibitors designed for highly resistant proteases.
Investigational antiviral inhibitors
Collaborative efforts of synthetic chemists, structural biologists, enzymologist and virologists, are needed to design the future-generation protease inhibitors to combat emerging resistant strains. A series of antiviral protease inhibitors with novel chemistry were designed on the DRV scaffold by the strategy of introducing polar interactions with conserved regions of the protease [52]. Structural analysis of protease with resistant mutations has led to an improved strategy for inhibitor design by introducing stronger interactions with the conserved regions around the active site and the flaps. Several compounds introduce new groups at the P2 position of DRV, while modifications of P2′ and P1 have also been tested. Selected examples are shown in Figure 5. In the following paragraphs, this strategy will be described for several antiviral compounds with unusual chemistry and potency against tested strains of resistant virus.
Compound GRL-0519 incorporates tris-THF instead of the bis-THF at the P2 group of DRV, and a methyoxy group instead of the amide on the aniline ring at the P2′ position [57]. GRL-0519 has almost tenfold better in vitro activity than DRV on a wide spectrum of multidrug resistant variants, making this potent inhibitor a prospective candidate for further studies [53,58]. Crystallographic analysis showed that the tris-THF group of GRL-0519 provided additional polar interactions over the bis-THF group of DRV. The interactions of P2 with the protease are shown in Figure 6. The third THF ring formed new water-mediated hydrogen bond interactions with the highly conserved protease residues Thr26, Gly27, Asp29 and Arg87 of one subunit and Arg8’ of the second subunit. Coordinating these residues at the subunit–subunit interface will tend to stabilize the dimer [57]. These interactions of the inhibitor were maintained in the crystal structures of proteases bearing single substitutions of R8Q, D30N, I50V, I54M and V82A [59]. The loss of hydrophobic interactions with protease containing I50V mutation was consistent with 60-fold worse inhibition of catalytic activity, suggesting this mutation will cause resistance to the GRL-0519 inhibitor. The inhibitor was tested with the highly resistant mutant PR20 [44]. Although the binding affinity of 40 nM as measured by isothermal titration calorimetry is similar to that of DRV, the large tris-THF group fits snugly in the enlarged S2 subsite of the mutant, suggesting this strategy is promising for further development.
Another compound developed on the DRV scaffold is GRL-04410, which contains a methoxy substitution at the C4 position of the bis-THF moiety within P2 [60]. At the other end, the P2’ benzyl has the methoxyl group used in TMC-126, instead of the amino group in DRV. The performance of GRL-04410 surpassed that of DRV with a Ki of 2.9 pM and an IC50 of 2.4 nM. A high resolution crystal structure was solved for GRL-04410 in complex with wild type protease. While the DRV-associated backbone hydrogen bonds were preserved, the C4 bis-THF methoxy substitution provided a novel C-H…O interaction and a water-mediated hydrogen bond with the amide of Gly 48 in the flap of the protease (Figure 6). By stabilizing the flexible flaps, inhibitors may retain potency against resistant variants showing highly dynamic flaps.
The unique feature of the new antiviral compound GRL-05010 is the gem-difluoro addition on the C4 position of bis-THF at P2 of DRV. The strategy of introducing fluorine to compounds is employed to increase lipid solubility and improve penetration of the blood–brain barrier [61]. This strategy proved successful in tests on GRL-05010 bearing two fluorines. GRL-05010 has improved lipophilicity and has been shown to penetrate the blood–brain barrier effectively in vitro, an infiltration that is not observed with most of the current PIs [62,63]. This antiviral inhibitor demonstrated excellent inhibition of the protease with a Ki of 5.8 pM [63], and antiviral activity with an EC50 of 3 nM. Furthermore, the antiviral potency remained in the nanomolar range for DRV-resistant strains [62]. Selection using GRL-05010 resulted in resistance due to M46I and I50V mutations, and to a lesser extent, N38K and M36I [62]. The structure of wild type protease in complex with GRL-05010 was solved at a high resolution, and exhibited similar protease-inhibitor interactions as seen for the DRV complex [63]. Halogen interactions between the two fluorine atoms and the carbonyl oxygen of Gly48 might be responsible for the significant improvement in performance (Figure 6). Altogether, these results suggest that GRL-05010 may overcome resistance by stabilizing the flexible flaps and has potential for eradicating virus reservoirs within the central nervous system.
Larger alterations within the DRV scaffold have also generated novel inhibitors. Qiu et al. [64] synthesized DRV-based inhibitors that replaced the bis-THF at P2 with an isosorbide trans-4-hydroxy-L-prolinamide. The isosorbide inhibitors demonstrated a two orders of magnitude improvement in antiviral activity against wild type HIV in comparison to IDV, however, no comparison was reported with DRV and the experimental structure is unknown.
The compound GRL-008 is a derivative of DRV and contains the benzene carboxamide substitution instead of aniline at P2’ [65]. The crystal structure in complex with protease exhibits a new water-mediated connection between the NH of the carboxamide and the backbone carbonyl oxygen of Gly48 in the flexible flap [65]. The structure also forms weaker hydrogen bonds to the backbone of conserved residues Asp29, Asp30/30’. A later study revealed that GRL-008 maintained wild-type polar interactions in the active site of a highly mutated protease from a clinical isolate [66]. It is noteworthy that DRV also conserves polar bonding with the same mutant and outperformed GRL-008 in antiviral activity against wild type and resistant strains.
Compound GS-8374 was designed to contain a paraphosphonic acid moiety in the aromatic ring at the P1 position of the TMC-126 parent structure [67]. The structure in complex with protease revealed that the phosphonate group was exposed to the solvent beyond the protein surface. This phosphonate ‘solvent anchor’ may be responsible for the retention of antiviral activity against resistant variants as measured by ITC. Recently, the structure of GS-8374 was reported in complex with an unusual variant bearing 12 substitutions and a Glu-Glu insertion after Glu35 that is resistant to many drugs [68]. Their analysis suggests that retention of antiviral activity against this mutant is due to the ability of the P1 aromatic phosphonate to rearrange within the expanded cavity walls while remaining anchored to the solvent.
Alternative designs for inhibitors that are not based on DRV/TMC-126 produce distinct binding interactions in the active site of the protease. Another strategy using a pyrrolidine linker was described [69]. C2-symmetric 3,4 disubstituted pyrrolidines with polar benzene carboxamide groups at P2/P2’ inhibited wild type protease at low micromolar (0.26–0.48 μM) values. The most potent inhibitor (3S,4S)-3,4-bis-[4-carbamoyl-benzene-sulfonyl)-(3-methyl-but-2-enyl)-amino]-pyrrolidinium trifluoroacetate engaged the flaps via direct hydrogen bonds between the NH amide of Ile50/50’ and the oxygen of the sulfonamides in each symmetrical component, while the pyrrolidine NH interacted with the carboxylate oxygens of the catalytic Asp25/25’. Furthermore, the crystal structures also displayed P2/P2’ carboxamides forming direct hydrogen bonds to the main chain atoms of Asp30/30’. Overall, these novel inhibitors offer alternative designs for binding within the active site of the closed conformation of the protease dimer.
Many of these compounds have not yet been evaluated for antiviral efficacy against resistant HIV. The most promising of the tested inhibitors are GRL-0519 with tenfold improved antiviral inhibition in vitro against resistant strains of HIV [53,58], and GRL-05010, which retained nanomolar antiviral activity against DRV-resistant virus and showed improved penetration of the blood–brain barrier in vitro [62,63]. Further studies are underway on these inhibitors.
Inhibitors targeting alternative binding sites in the protease
Several strategies have been proposed for inhibitors targeting alternative sites, instead of the competitive inhibitors that bind in the active site cavity of the protease. These include inhibitors that bind to the open conformation of the dimer, to the hinge of the flap, or the intersubunit interface. Inhibition of the open conformation of the protease has been proposed from molecular dynamics simulations, such as the report by [70] based on a crystal structure [71]. Interestingly, a unique binding mode was observed for DRV, where the inhibitor bound to the wide open conformation flap of a highly resistant protease mutant (P51) [47].
A different inhibitor was observed bound to the flaps in an open conformation of the protease dimer. Taking advantage of the nontoxic properties of dibenzodiazepines, a derivative was synthesized and crystallized in complex with protease [72]. Two molecules of the inhibitor occupied the protease active site of a semiopen flap conformation. Each individual molecule made one direct backbone hydrogen bond with the carbonyl oxygen of Gly27, while solvent-mediated connections were established with Asp25, Asp29, Asp30 and Ile50. These solvent bridges would normally be observed as direct hydrogen bonds in protease-DRV complexes, thus explaining the relatively weak micromolar potency of the dibenzodiazepine derivative. Two derivatives of the novel inhibitor were connected via an oxalyl link, which improved the inhibition of protease activity to a more practical nanomolar range. Although crystallography attempts were unsuccessful, a docking study suggests that the improved inhibition might be due to the oxalyl oxygens forming direct hydrogen bonds to the catalytic aspartates instead of the indirect interactions mediated by the conserved waters, as well as favorable van der Waals contacts from the bulkier inhibitor.
Two alternative binding sites were identified in a fragment based crystallographic screen of HIV protease: the loop formed by Gly16-Gly17-Gln18, and a shallow groove on the outside of the flap formed by Trp42, Pro44, Met46, Lys55, Val56, Arg57 [73]. Remarkably, DRV was observed bound to an almost identical location of the flap in crystal structures of the highly resistant PR20 mutant [45]. The amino acid residues contributing to this binding groove are not observed as sites for the common resistance mutations (Table 1), which suggests this groove may be a viable target for inhibitor designs.
Blocking dimerization is a potential, alternative strategy to hinder viral replication. DRV was shown to inhibit both dimerization and proteolytic cleavage by binding to both the monomer and dimer [56]. Hence, DRV has several valuable properties in addition to its binding in the active site cavity.
Inhibitors targeting the Gag or Gag-Pol precursors
Viral maturation, which is the protease-catalyzed processing of viral Gag and Gag-Pol precursors to release individual proteins, is a potential target for inhibitors. This essential process in the viral life cycle must occur in a precise order to produce infectious virus [74,75]. An inhibitor of maturation, bevirimat, was developed to target the final processing step for the Gag precursor (Figure 1). Bevirimat binds at the CA-sp1 site, which is the natural substrate for the protease [76,77]. The antiviral potency of bevirimat led to clinical trials, however, high baseline drug resistance due to the existing polymorphisms halted further development. Mutations in the protease influence the resistance to bevirimat [76]. Resistance mutations were identified around the CA-sp1 cleavage site, at amino acid 201 and the major homology region of CA [78]. New derivatives of bevirimat have been evaluated with limited success to date [79,80].
A critical step in viral maturation is the autoproteolytic cleavage of the protease from its Gag-Pol precursor. Inhibition of this autoprocessing step has been proposed as an alternative target for development, as reviewed by [81]. Two of the current PIs, DRV and SQV, demonstrated significant inhibition of the autoprocessing step that releases mature dimeric protease. DRV and SQV were the most effective of the nine clinical inhibitors in inhibiting precursor autoprocessing at the cleavage site between transframe and the N-terminus of PR [42]. DRV inhibition of this cleavage was reported in mammalian cells [82]. The same two PIs were also shown to be the best inhibitors of proteolysis of the sp1-NC site in Gag-Pol [83], strongly suggesting the unique antiviral properties of these two drugs. In another approach, a monoclonal antibody was found to inhibit autoprocessing at the C-terminus of the protease [84]. The crystal structures of a miniprecursor of HIV-1 protease with four extra N-terminal residues SFNF from the transframe region revealed multiple conformations and unraveling of the N-terminus representing a transient step in autoprocessing [85,86]. These mini-precursor structures provide new targets for design of drugs that may be less susceptible to development of resistance.
Conclusion & future perspective
The rapid mutation of HIV under drug selection to evolve resistant viral strains poses a severe challenge to effective long term AIDS therapy. The virally encoded protease is prone to develop multiple mutations, and highly resistant mutants with around 20 substitutions can show several orders of magnitude worse inhibition by PIs. Strategies to overcome the problem of resistance include the design of competitive inhibitors with very high affinity for wild type enzyme, as exemplified by DRV, inhibitors designed specifically for highly resistant mutants, inhibitors targeting alternative sites on the protease, and compounds binding the precursor substrates of the protease.
Structural and enzymatic studies of highly resistant protease mutants reveal the molecular changes associated with loss of susceptibility to PIs. Mutants with specific combinations of 14–22 amino acids substitutions can show two to four orders of magnitude worse affinity for clinical PIs. Structural analysis reveals the loss of noncovalent interactions of protease with PIs due in part to an expanded binding site. Moreover, the flaps exhibit an increased population of open conformational states. These structural adaptations suggest improved strategies for inhibitor design. First, inhibitors can be designed with larger side groups to fit better in the expanded binding site of the mutants. Second, introducing chemical groups in the inhibitors that form interactions with the flaps will tend to stabilize the protease dimer conformation. This strategy shows promise for new antiviral inhibitors derived on the DRV scaffold. One of these compounds, GRL-05010 includes two fluorides and exhibits improved lipid permeability, which is critical for attacking viral reservoirs in the central nervous system.
Compounds have been identified to bind at alternative sites in the protease structure, such as the interior of the flap in the open conformation or the groove on the outer surface of the closed conformation flap. Little development has been reported for compounds targeting these sites. One problem is that the known compounds are likely to have low affinity for the protease, and may not show effective inhibition of the catalytic activity.
Other potential targets for inhibitors are the rate limiting steps in protease cleavage of its natural substrates, the Gag and Gag-Pol polyprotein precursors. Clinical trials for the maturation inhibitor bevirimat failed due to rapid development of resistant mutations. The cleavage site at the N-terminus of the protease is a promising and little studied target. New structural data for the protease precursor should help in the design of novel inhibitors with affinity for this site. The disadvantage of this target is that the structure of the linker and N-terminal protease residues may have multiple conformations in the precursor.
Overall, the optimal target for inhibitor development is still the active site cavity of the protease dimer. Competitive inhibitors with high affinity for the HIV protease are achievable, and antiviral inhibitors with novel chemistry would be valuable future compounds of HIV/AIDS therapy. The development of new inhibitors specifically designed for the expanded binding site of the highly resistant mutants is an attractive possibility to pursue in future.
Executive summary.
Molecular mechanisms leading to highly resistant protease mutants
Greater knowledge of the mutations and molecular mechanisms resulting in highly resistant proteases will open new possibilities for inhibitors designed to target resistant HIV. Several studied examples of highly resistant proteases show expanded active site cavities and greater variation in flap conformation.
Competitive inhibitors designed for resistant protease variants
Inhibitors designed to fit better in the expanded binding site of highly resistant mutants and/or stabilize the dynamic flaps are being evaluated.
Inhibitors targeting precursor proteins
Autocatalytic cleavage of the HIV protease from the viral Gag-Pol precursor is a critical step in viral replication. Knowledge of the precursor structures involved in protease autoprocessing provides new insights into a key step in viral maturation. Novel inhibitors targeting this step have potential as therapeutic agents that are less susceptible to development of resistance.
Key terms
- Highly active antiretroviral therapy
Uses a combination of drugs that target different stages in the viral life cycle. It is also referred to as combined antiretroviral therapy (cART)
- Protease inhibitor
Clinically approved antiviral inhibitor of HIV-1 protease
- Highly resistant protease mutant
Mutant of HIV protease resulting from drug selection resulting in loss of susceptibility to protease inhibitors by several orders of magnitude as assessed by the binding affinity. A combination of about 20 substitutions is generally observed in highly resistant proteases
Footnotes
For reprint orders, please contact reprints@future-science.com
Financial & competing interests disclosure
The authors’ research is supported in part by the grant U01 GM062920 awarded by the National Institutes of Health. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest;
•• of considerable interest
- 1.Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382(9903):1525–1533. doi: 10.1016/S0140-6736(13)61809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Passaes CP. Sáez-Cirión A. HIV cure research: advances and prospects. Virology. 2014;(454–455):340–352. doi: 10.1016/j.virol.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 3.Clifford DB, Ances BM. HIV-associated neurocognitive disorder. Lancet Infect Dis. 2013;13(11):976–986. doi: 10.1016/S1473-3099(13)70269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber IT, Wang Y-F. HIV protease: role in viral replication, protein-ligand X-ray crystal structures and inhibitor design. In: Ghosh AK, editor. Aspartic Proteases as Therapeutic Targets. Methods and Principles in Medicinal Chemistry. Vol. 45. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2010. pp. 109–137. [Google Scholar]
- 5.Jacobsen H, Yasargil K, Winslow DL, et al. Characterization of Human Immunodeficiency Virus type 1 mutants with decreased sensitivity to proteinase inhibitor Ro 31–8959. Virology. 1995;206(1):527–534. doi: 10.1016/s0042-6822(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 6.Lloyd SB, Kent SJ, Winnall WR. The high cost of fidelity. AIDS Res Hum Retroviruses. 2014;30(1):8–16. doi: 10.1089/aid.2013.0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paterson DL, Swindells S, Mohr J, et al. Adherence to protease inhibitor therapy and outcomes in patients with HIV infection. Ann Intern Med. 2000;133(1):21–30. doi: 10.7326/0003-4819-133-1-200007040-00004. [DOI] [PubMed] [Google Scholar]
- 8.Nolan D, Reiss P, Mallal S. Adverse effects of antiretroviral therapy for HIV infection: a review of selected topics. Expert Opin Drug Saf. 2005;4(2):201–218. doi: 10.1517/14740338.4.2.201. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda JM, Miller C, Currier JS, et al. The correlation between plasma concentrations of protease inhibitors, medication adherence and virological outcome in HIV-infected patients. Antivir Ther. 2004;9(5):753–761. [PubMed] [Google Scholar]
- 10.Harrigan PR, Hogg RS, Dong WWY, et al. Predictors of HIV drug-resistance mutations in a large antiretroviral-naive cohort initiating triple antiretroviral therapy. J Infect Dis. 2005;191(3):339–347. doi: 10.1086/427192. [DOI] [PubMed] [Google Scholar]
- 11.Pham QD, Wilson DP, Law MG, et al. Global burden of transmitted HIV drug resistance and HIV-exposure categories. AIDS. 2014;28(18):2751–2762. doi: 10.1097/QAD.0000000000000494. [DOI] [PubMed] [Google Scholar]
- 12.Bonura F, Tramuto F, Vitale F, et al. Transmission of drug-resistant HIV type 1 strains in HAART-naive patients: a 5 year retrospective study in Sicily, Italy. AIDS Res Hum Retroviruses. 2010;26(9):961–965. doi: 10.1089/aid.2009.0250. [DOI] [PubMed] [Google Scholar]
- 13.Tramuto F, Bonura F, Mancuso S, et al. Detection of a new 3-base pair insertion mutation in the protease gene of Human Immunodeficiency Virus type 1 during highly active antiretroviral therapy (HAART) AIDS Res Hum Retroviruses. 2005;21(5):420–423. doi: 10.1089/aid.2005.21.420. [DOI] [PubMed] [Google Scholar]
- 14.Kozísek M, Sasková KG, Rezácová P, et al. Ninety-nine is not enough: molecular characterization of inhibitor-resistant Human Immunodeficiency Virus type 1 protease mutants with insertions in the flap region. J Virol. 2008;82(12):5869–5878. doi: 10.1128/JVI.02325-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pereira-Vaz J, Duque V, Trindade L, et al. Detection of the protease codon 35 amino acid insertion in sequences from treatment-naïve HIV-1 subtype C infected individuals in the Central Region of Portugal. J Clin Virol. 2009;46(2):169–172. doi: 10.1016/j.jcv.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 16.Shafer RW, Schapiro JM. HIV-1 drug resistance mutations: an updated framework for the second decade of HAART. AIDS Rev. 2008;10(2):67–84. [PMC free article] [PubMed] [Google Scholar]
- 17.Neogi U, Rao SD, Bontell I, et al. Novel tetra-peptide insertion in Gag-p6 ALIX-binding motif in HIV-1 subtype C associated with protease inhibitor failure in Indian patients. AIDS. 2014;28(15):2319–2322. doi: 10.1097/QAD.0000000000000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clavel F, Mammano F. Role of Gag in HIV resistance to protease inhibitors. Viruses. 2010;2(7):1411–1426. doi: 10.3390/v2071411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozísek M, Henke S, Sasková KG, et al. Mutations in HIV-1 Gag and Pol compensate for the loss of viral fitness caused by a highly mutated protease. Antimicrob Agents Chemother. 2012;56(8):4320–4330. doi: 10.1128/AAC.00465-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nijhuis M, van Maarseveen NM, Lastere S, et al. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007;4(1):e36. doi: 10.1371/journal.pmed.0040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21••.Wensing AM, Calvez V, Günthard HF, et al. 2014 Update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22(3):642–650. Current list of mutations associated with drug resistance in HIV. [PMC free article] [PubMed] [Google Scholar]
- 22.Hirsch MS, Günthard HF, Schapiro JM, et al. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society-USA panel. Clin Infect Dis. 2008;47(2):266–285. doi: 10.1086/589297. [DOI] [PubMed] [Google Scholar]
- 23.Friend J, Parkin N, Liegler T, et al. Isolated lopinavir resistance after virological rebound of a ritonavir/lopinavir-based regimen. AIDS. 2004;18(14):1965–1966. doi: 10.1097/00002030-200409240-00016. [DOI] [PubMed] [Google Scholar]
- 24.Mo H, King MS, King K, et al. Selection of resistance in protease inhibitor-experienced, Human Immunodeficiency Virus type 1-infected subjects failing lopinavir- and ritonavir-based therapy: mutation patterns and baseline correlates. J Virol. 2005;79(6):3329–3338. doi: 10.1128/JVI.79.6.3329-3338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kagan RM, Shenderovich MD, Heseltine PNR, Ramnarayan K. Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir. Protein Sci. 2005;14(7):1870–1878. doi: 10.1110/ps.051347405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young TP, Parkin NT, Stawiski E, et al. Prevalence, mutation patterns, and effects on protease inhibitor susceptibility of the L76V mutation in HIV-1 protease. Antimicrob Agents Chemother. 2010;54(11):4903–4906. doi: 10.1128/AAC.00906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babrzadeh F, Varghese V, Pacold M, et al. Collinearity of protease mutations in HIV-1 samples with high-level protease inhibitor class resistance. J Antimicrob Chemother. 2013;68(2):414–418. doi: 10.1093/jac/dks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doherty KM, Nakka P, King BM, et al. A multifaceted analysis of HIV-1 protease multidrug resistance phenotypes. BMC Bioinformatics. 2011;12(1):477. doi: 10.1186/1471-2105-12-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varghese V, Mitsuya Y, Fessel WJ, et al. Prototypical recombinant multi-protease inhibitor resistant infectious molecular clones of Human Immunodeficiency Virus type-1. Antimicrob Agents Chemother. 2013;57(9):4290–4299. doi: 10.1128/AAC.00614-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.Weber IT, Agniswamy J. HIV-1 protease: structural perspectives on drug resistance. Viruses. 2009;1(3):1110–1136. doi: 10.3390/v1031110. Review of the molecular mechanisms suggested by structural and inhibition studies of HIV proteases with single amino acid substitutions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldfarb NE, Ohanessian M, Biswas S, et al. Defective hydrophobic sliding mechanism and active Site expansion in HIV-1 protease drug resistant variant Gly48Thr/Leu89Met: mechanisms for the loss of saquinavir binding potency. Biochemistry. 2014;54(2):422–433. doi: 10.1021/bi501088e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayashi H, Takamune N, Nirasawa T, et al. Dimerization of HIV-1 protease occurs through two steps relating to the mechanism of protease dimerization inhibition by DRV. Proc Natl Acad Sci USA. 2014;111(33):12234–12239. doi: 10.1073/pnas.1400027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Louis JM, Zhang Y, Sayer JM, et al. The L76V drug resistance mutation decreases the dimer stability and rate of autoprocessing of HIV-1 protease by reducing internal hydrophobic contacts. Biochemistry. 2011;50(21):4786–4795. doi: 10.1021/bi200033z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ragland DA, Nalivaika EA, Nalam MNL, et al. Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J Am Chem Soc. 2014;136(34):11956–11963. doi: 10.1021/ja504096m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang YC, Xu Y, Zhang Y, et al. Potent antiviral HIV-1 protease inhibitor GRL-02031 adapts to the structures of drug resistant mutants with its P1’-pyrrolidinone ring. J Med Chem. 2012;55(7):3387–3397. doi: 10.1021/jm300072d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohtaka H, Schön A, Freire E. Multidrug resistance to HIV-1 protease inhibition requires cooperative coupling between distal mutations. Biochemistry. 2003;42(46):13659–13666. doi: 10.1021/bi0350405. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Liu Z, Brunzelle JS, et al. The higher barrier of darunavir and tipranavir resistance for HIV-1 protease. Biochem Biophys Res Commun. 2011;412(4):737–742. doi: 10.1016/j.bbrc.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin P, Vickrey JF, Proteasa G, et al. “Wide-open” 1.3 A structure of a multidrug-resistant HIV-1 protease as a drug target. Structure. 2005;13(12):1887–1895. doi: 10.1016/j.str.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 39.Yedidi RS, Proteasa G, Martinez JL, et al. Contribution of the 80s loop of HIV-1 protease to the multidrug-resistance mechanism: crystallographic study of MDR769 HIV-1 protease variants. Acta Crystallogr D Biol Crystallogr. 2011;67(6):524–532. doi: 10.1107/S0907444911011541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasková KG, Kozísek M, Rezácová P, et al. Molecular characterization of clinical isolates of Human Immunodeficiency Virus resistant to the protease inhibitor DRV. J Virol. 2009;83(17):8810–8818. doi: 10.1128/JVI.00451-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kožíšek M, Lepšík M, Grantz Šašková K, et al. Thermodynamic and structural analysis of HIV to DRV – analysis of heavily mutated patient-derived HIV-1 proteases. FEBS J. 2014;281(7):1834–1847. doi: 10.1111/febs.12743. [DOI] [PubMed] [Google Scholar]
- 42••.Louis JM, Aniana A, Weber IT, Sayer JM. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc Natl Acad Sci USA. 2011;108(22):9072–9077. doi: 10.1073/pnas.1102278108. Darunavir and saquinavir inhibit the autocatalytic cleavage of the wild type protease from its precursor, but are ineffective against autoprocessing of highly resistant mutants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louis JM, Tözsér J, Roche J, et al. Enhanced stability of monomer fold correlates with extreme drug resistance of HIV-1 protease. Biochemistry. 2013;52(43):7678–7688. doi: 10.1021/bi400962r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Agniswamy J, Shen C-H, Wang Y-F, et al. Extreme multidrug resistant HIV-1 protease with 20 mutations is resistant to novel protease inhibitors with P1’-pyrrolidinone or P2-tris-tetrahydrofuran. J Med Chem. 2013;56(10):4017–4027. doi: 10.1021/jm400231v. Highly drug resistant mutant PR20 is assessed with new antiviral inhibitors designed for resistant HIV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agniswamy J, Shen C-H, Aniana A, et al. HIV-1 protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochemistry. 2012;51(13):2819–2828. doi: 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh Y, Amano M, Towata T, et al. In vitro selection of highly DRV-resistant and replication-competent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors. J Virol. 2010;84(22):11961–11969. doi: 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Chang Y-CE, Louis JM, et al. Structures of DRV-resistant HIV-1 protease mutant reveal atypical binding of DRV to wide open flaps. ACS Chem Biol. 2014;9(6):1351–1358. doi: 10.1021/cb4008875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Vera IM, Smith AN, Dancel MC, et al. Elucidating a relationship between conformational sampling and drug resistance in HIV-1 protease. Biochemistry. 2013;52(19):3278–3288. doi: 10.1021/bi400109d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roche J, Louis JM, Bax A. Conformation of inhibitor-free HIV-1 protease derived from NMR spectroscopy in a weakly oriented solution. Chembiochem. 2014;16(2):214–218. doi: 10.1002/cbic.201402585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weber IT, Zhang Y, Tozser J. HIV-1 Protease and AIDS therapy. In: Lendeckel U, Hooper N, editors. Proteases In Biology And Disease, Volume 8. Viral Proteases And Antiviral Protease Inhibitor Therapy. Springer; NY, USA: 2009. pp. 25–46. [Google Scholar]
- 51.Velthuisen EJ, Baughman TM, Johns BA, et al. Synthesis and pharmacokinetic profile of highly deuterated brecanavir analogs. Eur J Med Chem. 2013;63:202–212. doi: 10.1016/j.ejmech.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 52••.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Enhancing protein backbone binding – a fruitful concept for combating drug-resistant HIV. Angew Chem Int Ed Engl. 2012;51(8):1778–1802. doi: 10.1002/anie.201102762. Describes the drug design strategy with examples of a number of new antiviral inhibitors developed with the backbone binding strategy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orkin C, Dejesus E, Khanlou H, et al. Final 192-week efficacy and safety of once-daily DRV/ritonavir compared with lopinavir/ritonavir in HIV-1-infected treatment-naïve patients in the ARTEMIS trial. HIV Med. 2013;14(1):49–59. doi: 10.1111/j.1468-1293.2012.01060.x. [DOI] [PubMed] [Google Scholar]
- 54.De Meyer S, Azijn H, Surleraux D, et al. TMC114, a novel Human Immunodeficiency Virus type 1 protease inhibitor active against protease inhibitor resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother. 2005;49(6):2314–2321. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brower ET, Bacha UM, Kawasaki Y, Freire E. Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use. Chem Biol Drug Des. 2008;71(4):298–305. doi: 10.1111/j.1747-0285.2008.00647.x. [DOI] [PubMed] [Google Scholar]
- 56.Koh Y, Matsumi S, Das D, et al. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J Biol Chem. 2007;282(39):28709–28720. doi: 10.1074/jbc.M703938200. [DOI] [PubMed] [Google Scholar]
- 57.Ghosh AK, Xu CX, Rao KV, et al. Probing multidrug resistance/protein-ligand interaction with new oxatricyclic designed ligands in HIV-1 protease inhibitors. ChemMedChem. 2010;5(11):1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amano M, Tojo Y, Salcedo-Gómez PM, et al. GRL-0519, a novel oxatricyclic ligand-containing nonpeptidic HIV-1 protease inhibitor (PI), potently suppresses replication of a wide spectrum of multi-PI-resistant HIV-1 variants in vitro. Antimicrob Agents Chemother. 2013;57(5):2036–2046. doi: 10.1128/AAC.02189-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang H, Wang YF, Shen CH, et al. Novel P2 tristetrahydrofuran group in antiviral compound 1 (GRL-0519) fills the S2 binding pocket of selected mutants of HIV-1 protease. J Med Chem. 2013;56(3):1074–1083. doi: 10.1021/jm301519z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghosh AK, Martyr CD, Steffey M, et al. Design, synthesis, and X-ray structure of substituted bis-tetrahydrofuran (bis-THF)-derived potent HIV-1 protease inhibitors. ACS Med Chem Lett. 2011;2(4):298–302. doi: 10.1021/ml100289m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filler R, Saha R. Fluorine in medicinal chemistry: a century of progress and a 60 year retrospective of selected highlights. Future Med Chem. 2009;1(5):777–791. doi: 10.4155/fmc.09.65. [DOI] [PubMed] [Google Scholar]
- 62.Salcedo Gómez PM, Amano M, Yashchuk S, et al. GRL-04810 and GRL-05010, difluoride-containing nonpeptidic HIV-1 protease inhibitors (PIs) that inhibit the replication of multi-PI-resistant HIV-1 in vitro and possess favorable lipophilicity that may allow blood–brain barrier penetration. Antimicrob Agents Chemother. 2013;57(12):6110–6121. doi: 10.1128/AAC.01420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ghosh AK, Yashchuk S, Mizuno A, et al. Design of gem-difluoro-bis-tetrahydrofuran as P2 ligand for HIV-1 protease inhibitors to improve brain penetration: synthesis, X-ray studies, and biological evaluation. ChemMedChem. 2015;10(1):107–115. doi: 10.1002/cmdc.201402358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qiu X, Zhao G-D, Tang L-Q, Liu Z-P. Design and synthesis of highly potent HIV-1 protease inhibitors with novel isosorbide-derived P2 ligands. Bioorg Med Chem Lett. 2014;24(11):2465–2468. doi: 10.1016/j.bmcl.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 65.Yedidi RS, Maeda K, Fyvie WS, et al. P2’ benzene carboxylic acid moiety is associated with decrease in cellular uptake: evaluation of novel nonpeptidic HIV-1 protease inhibitors containing P2 bis-tetrahydrofuran moiety. Antimicrob Agents Chemother. 2013;57(10):4920–4927. doi: 10.1128/AAC.00868-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yedidi RS, Garimella H, Aoki M, et al. A conserved hydrogen-bonding network of P2 bis-tetrahydrofuran-containing HIV-1 protease inhibitors (PIs) with a protease active-site amino acid backbone aids in their activity against PI-resistant HIV. Antimicrob Agents Chemother. 2014;58(7):3679–3688. doi: 10.1128/AAC.00107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cihlar T, He G-X, Liu X, et al. Suppression of HIV-1 protease inhibitor resistance by phosphonate-mediated solvent anchoring. J Mol Biol. 2006;363(3):635–647. doi: 10.1016/j.jmb.2006.07.073. [DOI] [PubMed] [Google Scholar]
- 68.Grantz Šašková K, Kozíšek M, Stray K, et al. GS-8374, a prototype phosphonate-containing inhibitor of HIV-1 protease, effectively inhibits protease mutants with amino acid insertions. J Virol. 2014;88(6):3586–3590. doi: 10.1128/JVI.02688-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blum A, Böttcher J, Dörr S, et al. Two solutions for the same problem: multiple binding modes of pyrrolidine-based HIV-1 protease inhibitors. J Mol Biol. 2011;410(4):745–755. doi: 10.1016/j.jmb.2011.04.052. [DOI] [PubMed] [Google Scholar]
- 70.Lexa KW, Carlson HA. Binding to the open conformation of HIV-1 protease. Proteins. 2011;79(7):2282–2290. doi: 10.1002/prot.23054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Böttcher J, Blum A, Dörr S, et al. Targeting the open-flap conformation of HIV-1 protease with pyrrolidine-based inhibitors. Chem Med Chem. 2008;3(9):1337–1344. doi: 10.1002/cmdc.200800113. [DOI] [PubMed] [Google Scholar]
- 72.Schimer J, Cígler P, Veselý J, et al. Structure-Aided Design of Novel Inhibitors of HIV Protease Based on a Benzodiazepine Scaffold. J Med Chem. 2012;55(22):10130–10135. doi: 10.1021/jm301249q. [DOI] [PubMed] [Google Scholar]
- 73.Perryman AL, Zhang Q, Soutter HH, et al. Fragment-based screen against HIV protease. Chem Biol Drug Des. 2010;75(3):257–268. doi: 10.1111/j.1747-0285.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mattei S, Anders M, Konvalinka J, et al. Induced maturation of Human Immunodeficiency Virus. J Virol. 2014;88(23):13722–13731. doi: 10.1128/JVI.02271-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee SK, Potempa M, Kolli M, et al. Context surrounding processing sites is crucial in determining cleavage rate of a subset of processing sites in HIV-1 Gag and Gag-Pro-Pol polyprotein precursors by viral protease. J Biol Chem. 2012;287(16):13279–13290. doi: 10.1074/jbc.M112.339374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fun A, van Maarseveen NM, Pokorná J, et al. HIV-1 protease inhibitor mutations affect the development of HIV-1 resistance to the maturation inhibitor bevirimat. Retrovirology. 2011;8:70. doi: 10.1186/1742-4690-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nguyen AT, Feasley CL, Jackson KW, et al. The prototype HIV-1 maturation inhibitor, bevirimat, binds to the CA-SP1 cleavage site in immature Gag particles. Retrovirology. 2011;8:101. doi: 10.1186/1742-4690-8-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waki K, Durell SR, Soheilian F, et al. Structural and functional insights into the HIV-1 maturation inhibitor binding pocket. PLoS Pathog. 2012;8(11):e1002997. doi: 10.1371/journal.ppat.1002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dang Z, Ho P, Zhu L, et al. New betulinic acid derivatives for bevirimat-resistant Human Immunodeficiency Virus type-1. J Med Chem. 2013;56(5):2029–2037. doi: 10.1021/jm3016969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tang J, Jones SA, Jeffery JL, et al. Synthesis and biological evaluation of macrocyclized betulin derivatives as a novel class of anti-HIV-1 maturation inhibitors. Open Med Chem J. 2014;8:23–27. doi: 10.2174/1874104501408010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81•.Huang L, Chen C. Understanding HIV-1 protease autoprocessing for novel therapeutic development. Future Med Chem. 2013;5(11):1215–1229. doi: 10.4155/fmc.13.89. Review of autocatalytic cleavage of the HIV-1 protease from its precursor and implications for development of novel antiviral agents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang L, Li Y, Chen C. Flexible catalytic site conformations implicated in modulation of HIV-1 protease autoprocessing reactions. Retrovirology. 2011;8:79. doi: 10.1186/1742-4690-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis DA, Soule EE, Davidoff KS, et al. Activity of Human Immunodeficiency Virus type 1 protease inhibitors against the initial autocleavage in Gag-Pol polyprotein processing. Antimicrob Agents Chemother. 2012;56(7):3620–3628. doi: 10.1128/AAC.00055-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sayer JM, Aniana A, Louis JM. Mechanism of dissociative inhibition of HIV protease and its autoprocessing from a precursor. J Mol Biol. 2012;422(2):230–244. doi: 10.1016/j.jmb.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang C, Louis JM, Aniana A, et al. Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease. Nature. 2008;455(7213):693–696. doi: 10.1038/nature07342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agniswamy J, Sayer JM, Weber IT, Louis JM. Terminal interface conformations modulate dimer stability prior to amino terminal autoprocessing of HIV-1 protease. Biochemistry. 2012;51(5):1041–1050. doi: 10.1021/bi201809s. [DOI] [PMC free article] [PubMed] [Google Scholar]