

Abstract
p53 plays an important role in drug responses by regulating cell cycle progression and inducing programmed cell death. The C-terminal of p53 self-regulates the protein negatively; however, whether it affects the sensitivity of cancer cells to anticancer drugs is unclear. In this study, two experimental methods were used to compare the sensitivity to anticancer drugs of human lung 801D cancer cells transfected with adenovirus bearing either full-length p53 or the deleted-C-terminal p53 in vivo. Adenovirus-mediated deliveries of full-length or deleted-C-terminal p53 were performed after development of tumors (the first method) or by infection into cells before xenotransplantation (the second method). The results showed that infection with the deleted-C-terminal p53 increased 801D cell sensitivity to anticancer drugs in the second, but not in the first method, as indicated by greater tumor-inhibition rates. In addition, compared with the first method, the second method resulted in viruses with more uniformly infected cells and the infection rates between groups were similar. This yielded smaller within-group variations and greater uniformity among transplanted tumors. The second method could circumvent the difficulties associated with intratumoral injection.
Keywords: drug sensitivity, experimental method, in vivo, p53
Introduction
Cisplatin and paclitaxel have been used widely for the treatment of patients with lung cancer 1–3; however, their efficiencies are only 25–54%. This low efficiency is partly because of the development of drug resistance of cancer cells. The resistance mechanism is complex and remains unclear. Recent studies have shown that many factors affect the tolerance and sensitivity of cancer cells to chemotherapeutic drugs 4,5. Among these factors, the tumor-suppressor gene p53 plays an important role in regulating the sensitivity of cancer cells to drugs by blocking cell cycle progression and inducing programmed cell death 2,6. As a transcription factor, the central domain of p53 targets specific DNA sequences and regulates the transcription of downstream target genes. Conversely, the central domain is regulated by many factors, including the C-terminus, which plays a negative role. Previous results have shown that lung cancer cells transfected with a deleted-C-terminal p53 grow more slowly than those transfected with full-length p53 in vitro 7. Similar findings have also been reported 8. However, the studies of the effect of p53 deleting C-terminal on the sensitivity of cancer cells to drugs have not been reported.
The xenograft in nude mice refers to human cells or primary tissues that are inoculated into immunodeficient nude mice subcutaneously. The method plays an important role in the research of tumor biology, drugs and gene therapy, etc. and has been used for decades 9. This method has some shortcomings after it has long been used and evaluated. For example, the experimental results are not completely consistent with the clinical outcomes; the research fields have some limitations; and some difficulties exist in the operation process so that they affect the accuracy of experimental results. In view of these existing problems, the method is also improving 10–13. To solve the problem of difficulties in the operation process, the method was improved.
In this study, we compared the sensitivities of cancer cells transfected with either adenovirus-packaged deleted-C-terminal p53 or full-length p53 to cisplatin and paclitaxel in vivo. Two techniques were used to deliver adenoviruses, referred to as the ‘first’ and ‘second’ methods. In the first method, adenovirus-packaged full-length p53 or deleted-C-terminal p53 were injected into xenograft tumors, respectively, and the drugs were then administered. In the second method, tumor cells were infected with three adenoviruses before xenotransplantation and the drugs were then administered when the tumors became palpable. The results of the two methods were not exactly compatible. Infection with p53 deleting the C-terminal increased the sensitivity of 801D cells to anticancer drugs in the second, but not the first method. In addition, the second method showed more advantages.
Materials and methods
801D cell line and p53 gene status
801D cells (kindly provided by the People’s Liberation Army General Hospital) were cultured in 1640 medium supplemented with 10% fetal bovine serum. The p53 gene in these cells shows loss of heterozygosity in the 248th codon and a CGG→CTT transversion 7.
BALB/c nude mice and chemotherapeutic drugs
All animal experiments and maintenance conformed to the guidelines of both the Animal Care and Use Committee and the American Association of Laboratory Animal Care. Female BALB/c nude mice (Vital River, Beijing, China) aged 4–6 weeks (average weight 20 g) were used in this study. These mice were raised in a pathogen-free environment at a temperature of 21±2°C and a relative humidity of 30–70%. Specialized personnel were responsible for their feeding.
Cisplatin was purchased from Qilu Pharmaceutical Factory (Jinan, China) and paclitaxel was purchased from Beijing Sihuan Medical Science and Technology Company (Beijing, China). The two drugs were dissolved in saline to a concentration of 0.3 mg/ml.
Construction of the recombinant p53 adenovirus
Recombinant p53 adenoviral plasmids were constructed using the Ad-Track-Easy transgenic system as described previously 6. Two deficient adenoviruses carrying either full-length p53 [p53(wt)] or the deleted-C-terminal p53 [p53(del)] were prepared. The two recombinants, plus the empty vector, were then infected into 293 packaging cells, together with the defective adenovirus, to generate the control adenovirus (Ad), Ad-p53(del), and Ad-p53(wt) 14. Virus concentrations were determined by multiplicity of infection (MOI) 6.
Preparation of the p53 recombinant adenovirus
Two recombinants were constructed using the Track-Easy plasmid vector: Track-Easy-p53(del) and Track-Easy-p53(wt). Track-Easy alone served as an empty-vector control. Track-Easy-p53(del) contained a truncated form of p53 in which the amino acids 356–393 of its C-terminal and all noncoding sequences after the stop codon were deleted. Track-Easy-p53(wt) contained wild-type p53. The two recombinants plus the empty vector were transfected into 293 cells using lipofectamine to prepare three deficient adenoviruses: Ad-p53(del), Ad-p53(wt), and Ad (control) 14.
Examination of the infected efficiencies of p53 reagents
Cell proliferation assay
Tumor cells (3000/well) were seeded in flat-bottom 96-well plates. Cell proliferation was evaluated using a 3-(4, 5-dimethyl-thiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; Promega, Madison, Wisconsin, USA) assay, which was performed at a fixed time every day for the next 5 days. 20 μl MTS was added to each well, followed by incubation for 3 h. The absorbance was recorded at 490 nm using an EL-800 universal microplate reader (Bio-Tek Instruments, Winooski, Vermont, USA). This experiment was repeated three times.
Cell apoptotic analysis
Cells were inoculated in a 96-well plate and grown to 75% confluence. Cells in the logarithmic growth phase were collected. After washing with RPMI-1640 medium without serum, cells were incubated in 100 μl RPMI-1640 medium without serum [50 mg/ml propidium iodide (Sigma-Aldrich, St Louis, Missouri, USA), 5 μg/ml Hoechest 33342 (Thermo, Hom Bridge City, Massachusetts, USA)]. Cells were incubated for 10 m at 37°C and the percentage of apoptotic cells was determined by Thermo Scientific ArrayScan VTI HCS (Thermo). Cellquest software (Becton Dickinson, Franklin, New Jersey, USA) was used for analysis. This experiment was repeated three times.
Western blot
Protein of 50–100 mg was subjected to 12% polyacrylamide gel electrophoresis. The separated protein was transferred to a polyvinylidene difluoride membrane by electrotransfer. The blots were blocked with 5% milk (Protifar; Nutricia, Zoetermeer, the Netherlands) in Tris-buffered saline–Tween 20 (TBS–T: 10 mmol/l Tris, pH 8.0, 150 mmol/l NaCl, 0.001% Tween 20) at room temperature for 1 h and incubated overnight at 4°C with specific antibodies against p53 (SantaCruz, Huissen, the Netherlands) and GAPDH (Kandcheng, Shanghai, China) diluted in 5% milk/TBS–T.
Drug sensitivity experiments
Determination of virus concentration in infected 801D cells
The three viruses, Ad, Ad-p53(wt) and Ad-p53(del), infected 801D cells at MOIs of 25, 50, and 100. Infection status was confirmed using fluorescence microscopy and detecting the expression of p53. The colony formation and inhibition rates were calculated after 2 weeks.
The second experimental method
Nude mice were divided randomly into four groups. There were six mice in each group. These groups were the Ad control group (drug untreated group) and the Ad, Ad-p53(del), Ad-p53(wt) groups (drug treatment groups). The three viruses, Ad, Ad-p53(del), and Ad-p53(wt), infected 801D cells at a MOI of 50. After 24 h, cells were harvested and washed twice with saline. Cells were resuspended at a density of 1.0×107 cells/ml. A suspension (2.0×106 cells) of 0.2 ml (equal to 1.0×108 virus) was injected subcutaneously into the flank of BALBc (nu/nu) mice. When tumors became palpable, cisplatin or paclitaxel was injected into the abdominal cavity at 3 μg/g body weight. Control groups did not receive drug treatment. After 3 days, cisplatin or paclitaxel was injected again at the same dose, respectively. After a further 5 days, paclitaxel alone was injected at the same dose. All mice were killed after 45 days and the number of tumors, average tumor weight, and inhibition rates were calculated. Animal care and experiments were conducted in accordance with the Animal Research Committee Guidelines of Beijing Chest Hospital, Capital Medical University.
The first experimental method
801D cells were harvested, washed twice with saline, and resuspended at a density of 1.0×107 cells/ml. 0.2 ml suspension (2.0×106 cells) was then injected subcutaneously into the flanks of BALBc (nu/nu) mice. Nude mice were divided randomly into four groups. There were six mice in each group. These groups were the Ad control group (drug untreated group) and the Ad, Ad-p53(wt), Ad-p53(del) groups (drug treatment groups). Ad, Ad-p53(wt) and Ad-p53(del) were injected into the inoculation sites of the experimental groups at a concentration of 2.0×109 per tumor, with an additional Ad group as a nondrug control. The experimental groups were administered either cisplatin or paclitaxel, as described in the second experimental method section.
Statistical analysis
All data were analyzed using the SPSS, 13.0 software (IBM Corp., Armonk, New York, USA). The inhibition rates of colony formation and tumor weights among the different groups were analyzed using one-way analysis of variance. Differences were considered to be significant at P value less than or equal to 0.05.
Results
Optimization of adenovirus-mediated growth inhibition of human lung cancer 801D cells in vitro
From the time-related curve, the infection rate increased gradually from 0 to 24 h and reached a plateau at 24 h (Fig. 1a). Therefore, the infection time was determined in 24 h. The optimal dose of virus required to affect colony formation was determined by infecting 801D cells with different concentrations of the three prepared adenoviruses (Fig. 1b). A virus concentration of 100 MOI resulted in colony-formation inhibition rates of 80, 92, and 97% in Ad-infected, Ad-p53(wt)-infected, and Ad-p53(del)-infected cells, respectively. The inhibition rates were 40, 70, and 92%, respectively, at 50 MOI and 38, 48, and 85%, respectively, at 25 MOI. These results suggest that a viral dose of 100 MOI was toxic because the colony-formation inhibition rates of all three viruses, including the Ad control, were similarly high. In contrast, viral loads of 25 and 50 MOI resulted in lower inhibition rates for the control Ad virus (40 and 38%, respectively), but the colony-formation inhibition rate of Ad-p53(wt) was also low at a 25 MOI. We therefore used viral loads of 50 MOI in subsequent experiments.
Fig. 1.
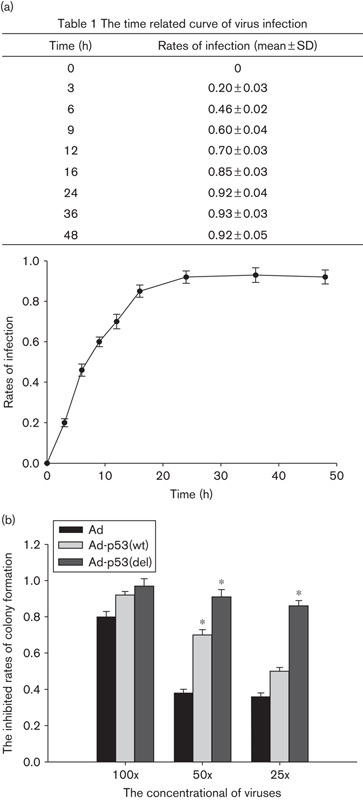
The optimal time and concentration of virus infection (a) the time-related curve of virus infection; (b) colony formation inhibition rates in 801D cells infected with three different recombinant viruses as indicated. *P<0.05 (compared with the Ad group).
p53 expression in the infected lines
For the detection of p53, 50 mg protein of human lung cancer cell lines was subjected to western blot (Fig. 2a). Compared with 801D cells, p53 protein levels of 801D-p53(wt) and 801D-p53(del) cells increased. p53 expression was increased two times and 1.96 times in 801D-p53(wt) and 801D-p53(del), respectively. To determine the function of the truncated p53, we performed a cell proliferation assay. As indicated in Fig. 2b, 801D-p53(wt) and 801D-p53(del) cells showed decreased growth beginning on the second day. From the third day, proliferation of 801D-p53(wt) and 801D-p53(del) cells showed differences. The proliferation of 801D-p53(del) cells was slower than that of 801D-p53(wt) cells. In addition, apoptosis was analyzed by calculating the apoptosis ratios by HCS. Compared with 801D cells, more apoptosis cells appeared in 801D-p53(wt) and 801D-p53(del) cells (Fig. 2c). Compared with 801D cells, the apoptosis ratio of 801D-p53(wt) cells increased 1.99-fold (P<0.05) and that of 801D-p53(del) cells increased 2.65-fold (P<0.05).
Fig. 2.
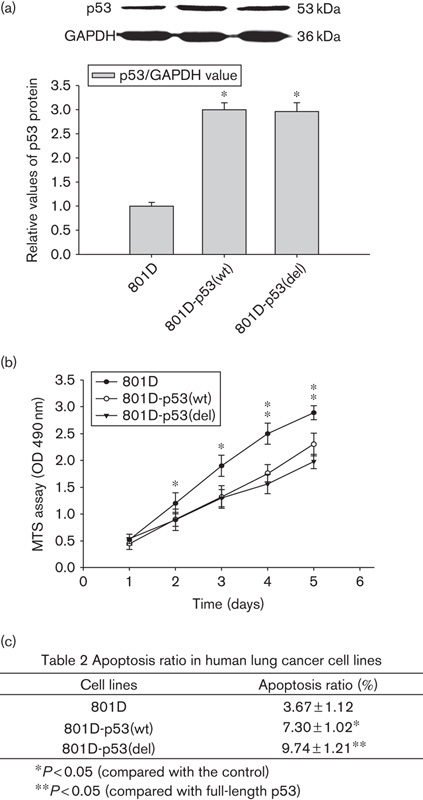
The identification of p53 virus infection (a) the identification of p53 protein; (b) the growth curve of 801D and infected cell strains; (c) apoptosis condition of 801D and infected cell strains. MTS, 3-(4, 5-dimethyl-thiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium.
Drug sensitivity experiments in nude mice
Deleted-C-terminal p53 increases 801D cell sensitivity to anticancer drugs by the second method
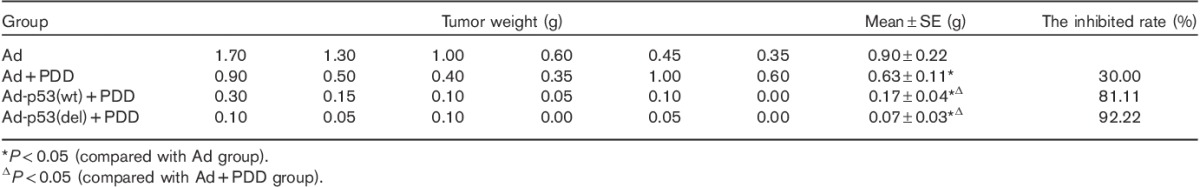
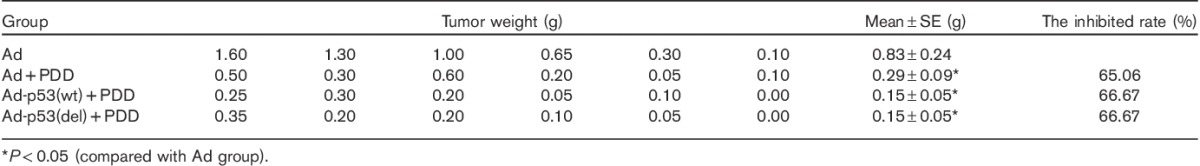
The effect of deleting the C-terminal of p53 on the sensitivity of lung cancer cells to cisplatin was tested using the second method (Table 1). Tumor weights differed among the various treatments. Tumor weights in the untreated Ad group were significantly greater than those of the Ad+PDD, Ad-p53(del)+PDD, and Ad-p53(wt)+PDD groups (P<0.05), suggesting that cisplatin suppressed tumor growth. The average tumor weight in the Ad+PDD group was significantly greater than that in the Ad-p53(del)+PDD group and the Ad-p53(wt)+PDD group (P<0.05). Although there was no significant difference in the tumor weights between the Ad-p53(wt)+PDD and the Ad-p53(del)+PDD groups (P>0.05), the average tumor weight of the Ad-p53(del)+PDD group was smaller than that of the Ad-p53(wt)+PDD group. Compared with the Ad+PDD group, the tumor-inhibition rates were increased by 51.11% in the Ad-p53(wt)+PDD group and by 62.22% in the Ad-p53(del)+PDD group. These results suggest that p53 deleting the C-terminal significantly increased the sensitivity of 801D cells to cisplatin.
Table 1.
PDD sensitivity assay of the second experimental method
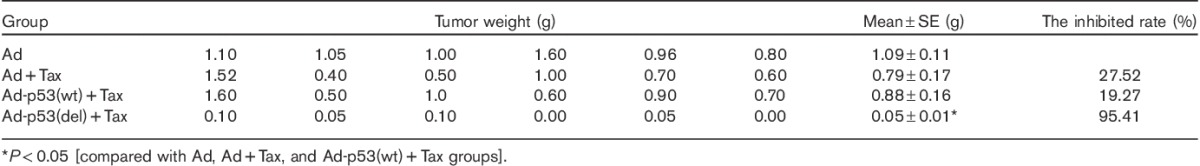
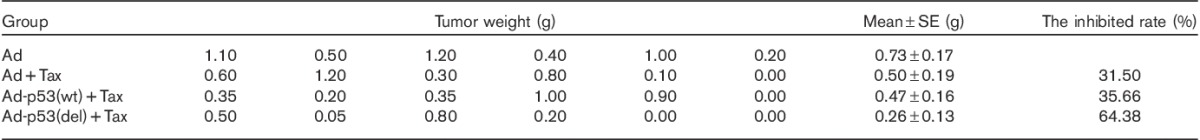
The effect of p53 deleting the C-terminal on the sensitivity of lung cancer cells to paclitaxel (Tax) was also examined using the second method (Table 2). The average tumor weight in the Ad-p53(del)+Tax group was significantly smaller than those in the Ad, Ad+Tax, and Ad-p53(wt)+Tax groups (P<0.05). There were no significant differences among the Ad, Ad+Tax, and Ad-p53(wt)+Tax groups (P>0.05). Compared with the Ad+Tax group, the tumor-inhibition rate was increased by 57.89% in the Ad-p53(del)+Tax group. These results suggest that deleted-C-terminal p53, but not full-length p53, increased the sensitivity of 801D cells to paclitaxel.
Table 2.
Paclitaxel sensitivity assay of the second experimental method
Deleted-C-terminal p53 does not increase 801D cell sensitivity to anticancer drugs by the first method
The sensitivity of lung cancer cells with C-terminal-deleted p53 to cisplatin was also examined using the first method (Table 3). The average tumor weight in the Ad group was significantly greater than those in the Ad+PDD, Ad-p53(del)+PDD, and Ad-p53(wt)+PDD groups (P<0.05). However, there were no significant differences among the Ad+PDD, Ad-p53(del)+PDD, and Ad-p53(wt)+PDD groups (P>0.05). Compared with the Ad+PDD group, the tumor-inhibition rates in the Ad-p53(wt) and Ad-p53(del) groups were increased by 1.61 and 1.61%, respectively, after cisplatin treatment. These results suggest that, under the first method, neither deleted-C-terminal p53 nor full-length p53 increased the sensitivity of 801D cells to cisplatin.
Table 3.
PDD sensitivity assay of the first experimental method
The sensitivity of lung cancer cells to paclitaxel was detected under the conditions of the first method (Table 4). There were no significant differences in the average tumor weights among the Ad, Ad+Tax, Ad-p53(wt)+Tax, and Ad-p53(del)+Tax groups (P>0.05). Similarly, there were no significant differences in tumor-inhibition rates after paclitaxel treatment in either the Ad-p53(wt) or the Ad-p53(del) groups compared with paclitaxel-treated Ad alone. These results suggest that neither deleted-C-terminal p53 nor full-length p53 increased the sensitivity of 801D cells to paclitaxel in the first method.
Table 4.
Paclitaxel sensitivity assay of the first experimental method
Comparison of two experimental methods
Interclass variations becomes small and central tendency is better in the second method
p53 deletion at the C-terminal increased the sensitivity of 801D cells to anticancer drugs with the second, but not the first method, as indicated by significant differences between several groups. We therefore analyzed the data further (Table 5 and Fig. 3). The SEs in the Ad-p53(del) and Ad-p53(wt) groups with cisplatin treatment in the second method were fewer than those in the first method, whereas the SE in the Ad group with cisplatin treatment in the second method was greater than that of the first method. The ratios of SEs and mean in Ad-p53(del) and Ad-p53(wt) groups with cisplatin treatment in the second method were greater than those in the first method, whereas the ratio of SE and mean in the Ad group with cisplatin treatment in the second method was less than that in the first method.
Table 5.
Comparison results of two methods in the chemotherapy drugs susceptibility test
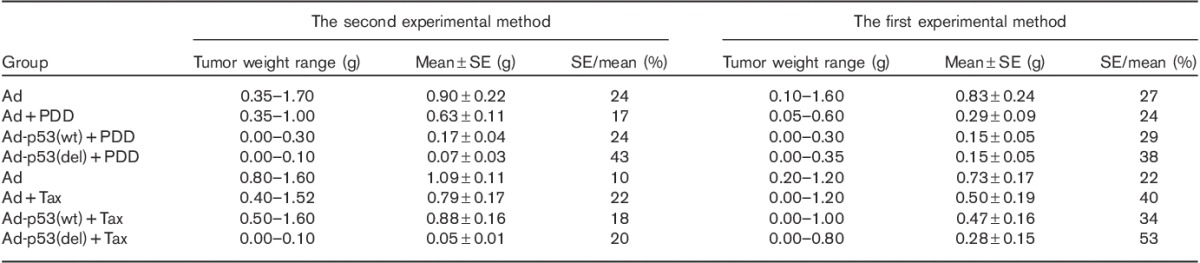
Fig. 3.
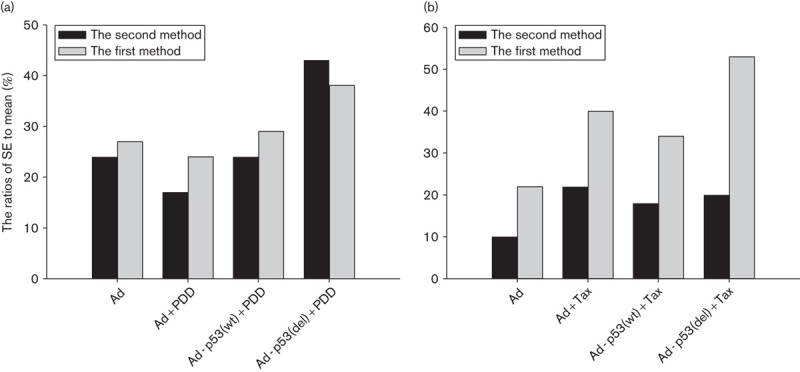
The SE/mean comparisons of two methods in chemotherapy drugs susceptibility test. (a) The SE/mean comparison of two methods in the PDD susceptibility test. (b) The SE/mean comparison of two methods in the Tax susceptibility test.
The results of paclitaxel experiments were analyzed under two experimental methods (Table 5 and Fig. 3). The analytic results showed that the SEs of the Ad, Ad+Tax, Ad-p53(wt)+Tax, and Ad-p53(del)+Tax groups in the second method were less than those in the first method. The ratios of SEs and mean of the Ad, Ad+Tax, Ad-p53(wt)+Tax, and Ad-p53(del)+Tax groups in the second method were less than those in the first method. This analysis indicates that the data showed lower interclass variations and better central tendency in the second method.
Discussion
Recent studies have shown that the drug sensitivity of cancer cells is related to drug response. Cytotoxic drugs kill tumor cells by a variety of ways and mechanisms 4,5, and further work is required to improve the efficacy of drug responses. The genetic background and molecular abnormalities of cancer cells affect their sensitivity to anticancer drugs. The p53 tumor-suppressor gene plays an important regulatory role in multiple complex gene networks 15. Notably, p53 is regulated negatively by its C-terminal. p53 is often targeted by anticancer agents; for example, the anticancer agent cisplatin stimulates the molecular sensors ataxia telangiectasia mutated, ataxia telangiectasia and Rad3-related, and the DNA-dependent protein kinase, which can activate p53 phosphorylation through a negative-feedback loop that promotes dissociation from MDM2, and thereby increases p53 expression and stability 16,17.
p53 gene expression has been shown to increase the sensitivity of cancer cells to anticancer drugs in vitro, in vivo, and in clinical trials 18–22. Because the C-terminal of p53 plays a negative regulatory role, we examined the sensitivity of human lung cancer cells with p53 deleting the C-terminal to chemotherapeutic drugs with two methods. The experimental results showed that tumor-inhibition rates increased in mice treated with cisplatin in the deleted-C-terminal and full-length p53 groups. However, significant tumor inhibition was only achieved in tumors expressing p53 deleting the C-terminal and only in mice treated using the second method after cisplatin treatment. The inhibition rate in cells with p53 deleting the C-terminal, but not in those with full-length p53, was significantly increased compared with the Ad control group with or without paclitaxel treatment in the second, but not in the first method. These results provide direct evidence that p53 deletion in the C-terminal increases the sensitivity of tumor cells to chemotherapeutic drugs. In this study, p53 deletion of the C-terminal removed 37 amino acids (356–393) of the C-terminal and its noncoding region. These sequences regulate p53 function negatively and include sequences that can inhibit p53 transcription or induce its degradation when combined with microRNA. This is the first study to report a relationship between p53 deleting the C-terminal and chemotherapeutic drug responses.
The C-terminal of p53 mainly includes the functional domains. The C-terminal includes the nuclear localization signal (NLS), the nuclear export signal, the tetramerization domain, and the C-terminal regulatory domain. The tetramerization domain is essential for DNA binding, protein–protein interactions, post-translational modifications, and p53 degradation. Nuclear p53 enables its activity as a transcription factor 23–25 and so NLS is essential. In the truncated p53, we reserve the NLS and the tetramerization domain and remove the other part of the C-terminal. We proved that the truncated p53 negatively regulated tumor cell sensitivity to anticancer drugs. However, the mechanisms remain unclear and require further investigation: first, it should be clarified as to what elements on these sequences are responsible for the negative regulation. Is it possible that some miRNA or lncmRNA targets these sequences to inhibit p53 function or the 3′ noncoding mRNA sequence is self-regulated? Second, the key genes involved in the signaling pathways through which those repressor sequences regulated drug sensitivity should be identified. It is very important to understand the role of 3′ sequences of p53 in tumor suppression and to elucidate the mechanisms underlined, which would benefit both basic science research and new anticancer drug development.
Furthermore, the first and second methods were analyzed in terms of their abilities to deliver robust results. The second method produced statistically significant findings, whereas the first method did not. Tumor-inhibition rates increased in the Ad-p53(del)+drug groups, but not in the Ad-p53(wt)+drug groups compared with the Ad+drug groups, which indicates that p53 deletion of the C-terminal increased the response of 801D cells to cisplatin and paclitaxel.
As for the stability and reliability of the two experimental methods, the second method produced smaller SEs than the first method. The SE reflects the size of sampling error, that is, a smaller SE indicates greater stability and reliability of the mean. Thus, the result of the analysis of the SE suggests that the second method is preferable to the first method. Because the size of SE is related to the mean, we also analyzed the ratios of SE and mean. However, the ratio of SE to mean in the Ad-p53(del)+PDD group was greater for the second method than for the first method in the cisplatin-sensitivity experiments. The result is inconsistent with that of the analysis of SE alone. This discrepancy may be because the SE and mean in the Ad-p53(del)+PDD group were smaller for the second method compared with the first method, given that it is difficult to decrease the SE beyond a certain value in small samples, in brief, whether SEs or the ratios of SEs and mean indicated that the second method was preferable to the first method. Similarly, the ratios of SE and mean in the Ad+Tax, Ad-p53(wt)+Tax, and Ad-p53(del)+Tax groups were lower for the second method compared with the first method in the paclitaxel-sensitivity experiments, which also suggests superiority of the second method.
In addition to its reliability, the second method also has several other advantages. First, the ratio and division of infected cells can be observed under a fluorescence microscope. Second, cells are infected more uniformly and the infection rates are similar among groups. Finally, this method overcomes the restrictions associated with injecting virus into the inoculation sites, such as uneven infection of the tumor cells. Overall, the second method therefore represents a more efficient method for conducting experiments to test the effects of adenovirus-mediated factors on tumor drug sensitivity in vivo.
Conclusion
The second experimental method is recommended for performing drug sensitivity tests in vivo. In other words, viruses infect cells before cells are inoculated into nude mice and drugs are used once the tumors become palpable. In addition, the results of this study show that p53 deletion of the C-terminal can increase the sensitivity of 801D cells to chemotherapeutic drugs compared with full-length p53.
Acknowledgements
The work was financed by the Beijing Municipal Science and Technology Committee grant (No. 954830600) and the Beijing Natural Science Foundation (7153162).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Persons DL, Yazlovitskaya EM, Pelling JC. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J Biol Chem 2000; 275:35778–35785. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg B, vanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature 1969; 222:385–386. [DOI] [PubMed] [Google Scholar]
- 3.Braeuning A. The connection of beta-catenin and phenobarbital in murine hepatocarcinogenesis: a critical discussion of Awuah et al., PLoS ONE 7(6):e39771, 2012. Arch Toxicol 2013; 87:401–402. [DOI] [PubMed] [Google Scholar]
- 4.Bagnyukova TV, Serebriiskii IG, Zhou Y, Hopper-Borge EA, Golemis EA, Astsaturov I. Chemotherapy and signaling: How can targeted therapies supercharge cytotoxic agents? Cancer Biol Ther 2010; 10:839–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu A, Krishnamurthy S. Cellular responses to cisplatin-induced DNA damage. J Nucleic Acids 2010; 2010:201367–201382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 1998; 95:2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Li JZ, Lai BT, Yang XH, Zhang CY, Yue WT, Zhan XP. Inhibitory effect of p53 with deletion of C-terminal 356–393 amino acids on malignant phenotype of human lung cancer cell line. Zhonghua Zhong Liu Za Zhi 2003; 25:527–530. [PubMed] [Google Scholar]
- 8.Gohler T, Reimann M, Cherny D, Walter K, Warnecke G, Kim E, Deppert W. Specific interaction of p53 with target binding sites is determined by DNA conformation and is regulated by the C-terminal domain. J Biol Chem 2002; 277:41192–41203. [DOI] [PubMed] [Google Scholar]
- 9.Kelland LR. Of mice and men: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur J Cancer 2004; 40:827–836. [DOI] [PubMed] [Google Scholar]
- 10.Morgan RA. Human tumor xenografts: the good, the bad, and the ugly. Mol Ther 2012; 20:882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.John T, Kohler D, Pintilie M, Yanagawa N, Pham NA, Li M, et al. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin Cancer Res 2011; 17:134–141. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Fu X, Hoffman RM. A new patient-like metastatic model of human lung cancer constructed orthotopically with intact tissue via thoracotomy in immunodeficient mice. Int J Cancer 1992; 51:992–995. [DOI] [PubMed] [Google Scholar]
- 13.Caysa H, Hoffmann S, Luetzkendorf J, Mueller LP, Unverzagt S, Mader K, Mueller T. Monitoring of xenograft tumor growth and response to chemotherapy by non-invasive in vivo multispectral fluorescence imaging. PLoS One 2012; 7:e47927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H, Lai B, Li W, Yang X, Zhang C, Wei P, Li J. Preparation of two types p53 recombinant adenovirus and quantitative exogenous expression of green fluorescence protein by flow cytometry. Zhongguo Fei Ai Za Zhi 2010; 13:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis – the p53 network. J Cell Sci 2003; 116 (Pt 20):4077–4085. [DOI] [PubMed] [Google Scholar]
- 16.Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 2000; 20:3224–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wetzel CC, Berberich SJ. p53 binds to cisplatin-damaged DNA. Biochim Biophys Acta 2001; 1517:392–397. [DOI] [PubMed] [Google Scholar]
- 18.Choi JH, Ahn KS, Kim J, Hong YS. Enhanced induction of Bax gene expression in H460 and H1299 cells with the combined treatment of cisplatin and adenovirus mediated wt-p53 gene transfer. Exp Mol Med 2000; 32:23–28. [DOI] [PubMed] [Google Scholar]
- 19.Guntur VP, Waldrep JC, Guo JJ, Selting K, Dhand R. Increasing p53 protein sensitizes non-small cell lung cancer to paclitaxel and cisplatin in vitro. Anticancer Res 2010; 30:3557–3564. [PubMed] [Google Scholar]
- 20.Huang Y, Jeong JS, Okamura J, Sook-Kim M, Zhu H, Guerrero-Preston R, Ratovitski EA. Global tumor protein p53/p63 interactome: making a case for cisplatin chemoresistance. Cell Cycle 2012; 11:2367–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Zhang Y, Zhao J, Kong F, Chen Y. Overexpression of miR-22 reverses paclitaxel-induced chemoresistance through activation of PTEN signaling in p53-mutated colon cancer cells. Mol Cell Biochem 2011; 357:31–38. [DOI] [PubMed] [Google Scholar]
- 22.Tokalov SV, Abramyuk AM, Abolmaali ND. Protection of p53 wild type cells from taxol by genistein in the combined treatment of lung cancer. Nutr Cancer 2010; 62:795–801. [DOI] [PubMed] [Google Scholar]
- 23.Li Q, Falsey RR, Gaitonde S, Sotello V, Kislin K, Martinez JD. Genetic analysis of p53 nuclear importation. Oncogene 2007; 26:7885–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang SH, Clarke MF. A bipartite nuclear localization signal is required for p53 nuclear import regulated by a carboxyl-terminal domain. J Biol Chem 1999; 274:32699–32703. [DOI] [PubMed] [Google Scholar]
- 25.Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell 2006; 125:1041–1053. [DOI] [PubMed] [Google Scholar]