

Abstract
Design and synthesis of a new class of inhibitors for the treatment of osteoporosis and its comparative h17β-HSD2 and m17β-HSD2 SAR study are described. 17a is the first compound to show strong inhibition of both h17β-HSD2 and m17β-HSD2, intracellular activity, metabolic stability, selectivity toward h17β-HSD1, m17β-HSD1 and estrogen receptors α and β as well as appropriate physicochemical properties for oral bioavailability. These properties make it eligible for pre-clinical animal studies, prior to human studies.
Introduction
Osteoporosis is a common, age-related disease, characterized by a systemic impairment of bone mass and microarchitecture, increasing bone fragility and risk of fractures [1]. It has been shown that the drop in 17β-estradiol (E2) and testosterone (T) levels, occurring with ageing, is the main factor driving the onset and progression of this disease [2]. 17β-Hydroxysteroid dehydrogenase type 2 (17β-HSD2) catalyzes the conversion of the highly active E2 and T into the weakly potent 17-ketosteroids estrone (E1) and Δ4-androstene-3,17-dione (Δ4-AD), respectively [3]. It is expressed in osteoblastic cells [4], therefore its inhibition can lead to the desired increase of E2 and T levels in the bone tissue and may thus be a novel approach for the treatment of osteoporosis.
Some steroidal [5–7] and non steroidal [8, 9] 17β-HSD2 inhibitors are already described. In our group we also developed and reported about several classes of non-steroidal 17β-HSD2 inhibitors [10–14], with a strong inhibition of human 17β-hydroxysteroid dehydrogenase type 2 (h17β-HSD2) and a good selectivity toward h17β-HSD1. Since h17β-HSD1 is the biological counterpart of h17β-HSD2, catalyzing the opposite conversion, selectivity toward this enzyme is an important feature to take into consideration. Potent and selective h17β-HSD1 inhibitors have also been described for the treatment of estrogen-dependent diseases [15, 16].
Given that the most commonly used animal model for osteoporosis studies are established in rodents [17, 18], we aimed at the development of new inhibitors displaying a good inhibition of mouse 17β-hydroxysteroid dehydrogenase type 2 (m17β-HSD2) and a reasonable selectivity toward m17β-HSD1. In order to have a compound suitable for animal testing and following human studies, the designed inhibitors should also display h17β-HSD2 inhibitory activity and selectivity toward h17β-HSD1. Other characteristics to be implemented are preferably low affinity to the estrogen receptors (ERs) α and β in order to maximize the E2 local effect and to minimize systemic side effects as well as metabolic stability.
Since the 3D-structure for both human and mouse 17β-HSD2 is up to date not available a ligand based approach was chosen for the design of new inhibitors. A set of h17β-HSD2 inhibitors was selected and tested for m17β-HSD2 inhibitory activity in order to get insight in the SAR and tracking the lead for the rational design of new inhibitors.
Results and Discussion
h17β-HSD2, h17β-HSD1, m17β-HSD2 and m17β-HSD1 cell-free assays were performed similarly, by incubating enzyme, tritiated substrate, cofactor and inhibitor, according to described procedures [19–22].
As starting point 25 previously described h17β-HSD2 inhibitors (Fig 1), belonging to the 2,5-thiophene amide, 1,3-phenyl amide and 1,4-phenyl amide class [12–14], were tested for m17β-HSD2 inhibition, in order to elaborate a comparative SAR and to develop an optimization strategy.
Fig 1. Previously described h17β-HSD2 inhibitors, tested for m17β-HSD2 inhibition.
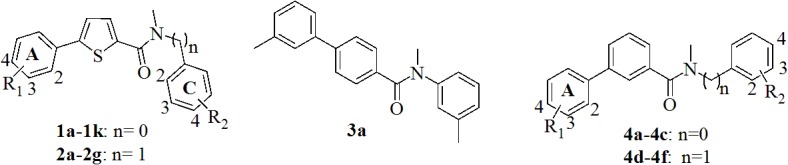
IC50 or percent of inhibition values for both h17β-HSD2 and m17β-HSD2 are given (Table 1, compounds 1a-2g, 3a, 4a-4f) to facilitate comparison.
Table 1. Inhibition of Human and Mouse 17β-HSD2 by 2,5-Thiophene Amide, 1,3-Phenyl Amide and 1,4-Phenyl Amide Derivatives in a Cell-Free Assay.
| IC50 (nM) a or % inh. at 1 μM a , d | ||||
|---|---|---|---|---|
| Cmpd | R1 | R2 | h17β-HSD2 b | m17β-HSD2 c |
| 1a | 3-OMe | 3-OMe | 68 | 29% |
| 1b | 3-Me | 3-Me | 52 | 42% |
| 1c | 3-Me | 3-OMe | 58 | 30% |
| 1d | 2-OMe | 3-OMe | 490 | 19% |
| 1e | 3-OH | 3-OH | 33% | n.i. |
| 1f | 2-OH | 3-OH | 410 | 18% |
| 1g | 3-N(Me)2 | 3-OMe | 170 | 40% |
| 1h | 3-F | 3-OMe | 510 | n.i. |
| 1i | 4-CN | 3-OMe | 48% | n.i. |
| 1j | 2-F, 3-OMe | 3-OMe | 62 | 26% |
| 1k | 2-F, 3-OMe | 3-Me | 62 | 45% |
| 2a | 2-F, 3-OMe | 3-OH | 61 | 65% |
| 2b | 3-OMe | 3-OMe | 370 | 16% |
| 2c | 3-OH | 3-OH | 390 | 26% |
| 2d | 4-OH | 3-OH | 330 | 35% |
| 2e | 3-Me | 3-OH | 160 | 45% |
| 2f | 3-F | 3-OH | 330 | 37% |
| 2g | 4-CN | 3-OH | n.i. | n.i. |
| 3a | 3-Me | 3-Me | 1100 | 50% |
| 4a | 3-OMe | 3-OMe | 520 | 25% |
| 4b | 4-OMe | 3-OMe | 1200 | 11% |
| 4c | 3-OH | 3-OH | 35% | n.i. |
| 4d | 3-OMe | 3-OMe | 11% | n.i. |
| 4e | 3-OH | 3-OH | 640 | 29% |
| 4f | 4-OH | 3-OH | 480 | 22% |
aMean value of at least two determinations, standard deviation less than 20%.
bHuman placental microsomal fraction, substrate E2 [500 nM], cofactor NAD+ [1500 μM].
cMouse liver microsomal fraction, substrate E2 [500 nM], cofactor NAD+ [1500 μM].
dn.i.: no inhibition, i.e., inhibition ≤10%.
In the 2,5-thiophene amide class (compounds 1a-2g), h17β-HSD2 inhibitors [13, 14], a broad range of inhibitory activities was detected, depending on the substitution pattern; the most active compounds show IC50 values around 60 nM (Table 1). Conversely, the inhibitory activity towards m17β-HSD2 was only marginally affected by these changes (inhibitory activity around 30% at 1 μM). Only compound 2a shows a more pronounced m17β-HSD2 inhibition combined with good inhibition of the human enzyme (65% at 1 μM and IC50 = 61 nM). Unfortunately, compound 2a turned out to be metabolically very unstable, with a half-life of only 4 minutes in the human liver S9 fraction [13]. In a further screening, all the tested 2,5-thiophene amide displayed a high metabolic instability [13].
It is striking that neither the nature of substituents on ring A and C or their substitution pattern does appear to exert an effect on m17β-HSD2 inhibition, whereas it is decisive for the h17β-HSD2 one. This result suggests that the inhibitors in this class are likely to have different binding modes in the two enzyme isoforms.
Exchange of the central thiophene by a 1,3-disubstituted phenyl led to compounds 4a-4c for n = 0 and 4d-4f for n = 1, with weak inhibitory activity towards both the human and the mouse enzyme (Table 1).
In contrast, compound 3a (Table 1), bearing a 1,4-disubstituted phenyl moiety as central ring, shows moderate inhibition of both h17β-HSD2 and m17β-HSD2 and also revealed exceptional metabolic stability in the human liver S9 fraction, with a half-life time>120 minutes [13]. It was therefore taken as starting point for the design of a small library of inhibitors where the substitution pattern and the physicochemical nature of substituents on the A and C rings was varied (Fig 2). A larger number of derivatives, bearing substituents with different physicochemical properties on the A ring were prepared, according to their chemical accessibilities. Compounds 25a and 25 were also synthesized to investigate the effect of the methylene linker between the amide function and the C ring.
Fig 2. Chemical Structures of the Designed Compounds.
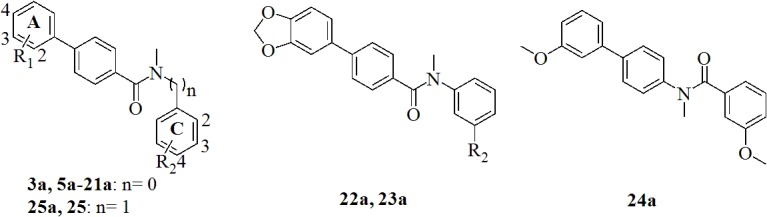
The synthesis of the 1,4-phenyl derivatives 5a-23a, 25a, 7 and 25, depicted in Fig 3, was accomplished following a two- or three-step reaction pathway.
Fig 3. Synthesis of 1,4-Phenyl Derivatives 5a-23a, 25a, 7, 25.

Reagents and conditions: (i) Et3N, CH2Cl2, room temperature, overnight; (ii) DME/EtOH/H2O (1:1:1), Cs2CO3, Pd(PPh3)4, microwave irradiation (150°C, 150W, 20 min); (iii) BF3·SMe2, CH2Cl2, room temperature, overnight.
First, amidation was carried out by reaction of the commercially available 4-bromobenzoyl chloride 5d with substituted anilines 5c or with the 1-(3-methoxyphenyl)-N-methylmethanamine 25c using standard conditions (method A: triethylamine, dichloromethane, from 0C to room temperature, overnight) affording the brominated intermediates 5b, 6b and 11b in almost quantitative yields. Subsequently, Suzuki coupling (Method B) using tetrakis(triphenylphosphine)palladium and cesium carbonate in a mixture of DME/EtOH/H2O (1:1:1,3 mL) as solvent and microwave irradiation (150°C, 150 W for 20 minutes), provided the biphenyl derivatives 5a-21a and 25a, and the 1,3-benzodioxole derivatives 22a and 23a in good yields. Compounds 7a and 25a were submitted to ether cleavage using boron trifluoride-dimethyl sulfide complex, yielding the hydroxylated molecules 7 and 25.
The synthesis of the retroamide 24a, displayed in Fig 4, follows a two-step procedure.
Fig 4. Synthesis of 1,4-Phenyl Retroamide Derivative 24a.
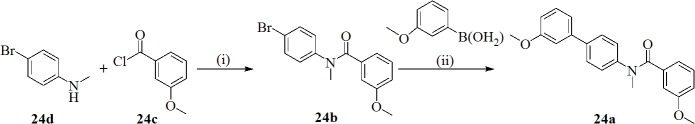
Reagents and conditions: (i) Et3N, CH2Cl2, room temperature, overnight; (ii) DME/EtOH/H2O (1:1:1), Cs2CO3, Pd(PPh3)4, microwave irradiation (150°C, 150W, 20 min).
First the commercially available 3-methoxybenzoyl chloride 24c was reacted with 4-bromo-N-methyl aniline 24d according to method A and afforded the brominated intermediate 24b with 70% yield. Subsequently, Suzuki coupling following method B afforded compound 24a in 68% yield.
Compounds 5a-25a, 7 and 25 were tested for h17β-HSD2, m17β-HSD2 and h17β-HSD1 inhibition (Table 2, results expressed as percentage of inhibition or IC50 value).
Table 2. Inhibition of h17β-HSD2, m17β-HSD2 and h17β-HSD1 by Biphenyl Amide Derivatives with Different Substitution Patterns on the A and C Rings in Cell-Free System.
| IC50 (nM) a or % inh. at 1 μM a , c | IC50 (nM) a or % inh. at 1 μM a , c | |||||
|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | h17β-HSD2 b | h17β-HSD1 c | s. f. d , f | m17β-HSD2 f , g |
| 3a | 3-Me | 3-Me | 1100 | 11500 | 10 | 50% |
| 5a | 3-OMe | 3-Me | 44% | n.i. | n.d. | 56% |
| 6a | 3-Me | 3-OMe | 260 | 6400 | 25 | 260 |
| 7a | 3-OMe | 3-OMe | 330 | 6400 | 20 | 290 |
| 8a | -H | 3-OMe | 51% | 22% | n.d. | 48% |
| 9a | 2-OMe | 3-OMe | 50% | n.i. | n.d. | 33% |
| 10a | 4-OMe | 3-OMe | 710 | 27000 | 38 | 66% |
| 11a | 3-OMe | 4-OMe | 27% | n.i. | n.d. | 58% |
| 12a | 4-OMe | 4-OMe | 19% | n.i. | n.d. | 62% |
| 7 | 3-OH | 3-OH | 36% | 15% | n.d. | 42% |
| 13a | 3-F | 3-OMe | 1000 | 8000 | 8 | 57% |
| 14a | 3-Cl | 3-OMe | 950 | 12600 | 13 | 73% |
| 15a | 3-N(Me)2 | 3-OMe | 650 | 5000 | 8 | 67% |
| 16a | 3-OMe,4-OMe | 3-OMe | 520 | 85500 | 164 | 52% |
| 17a | 3-Me,4-Me | 3-OMe | 300 | 13300 | 44 | 140 |
| 18a | 3-F,4-F | 3-OMe | 31% | n.i. | n.d. | n.d. |
| 19a | 2-F, 3-OMe | 3-OMe | 64% | 35% | n.d. | 67% |
| 20a | 2-F, 3-Me | 3-OMe | 460 | 11300 | 24 | 48% |
| 21a | 2-F, 3-Me | 3-Me | 330 | 3710 | 11 | 56% |
| 22a | - | 3-OMe | 51% | n.i. | n.d. | 56% |
| 23a | - | 3-Me | 560 | 10900 | 20 | 70% |
| 24a | - | - | 11% | n.i. | n.d. | n.d. |
| 25a | 2-F, 3Me | 3-OMe | 310 | 87000 | 283 | 43% |
| 25 | 2-F, 3-Me | 3-OH | 260 | 31000 | 118 | 190 |
aMean value of at least three determinations, standard deviation less than 20% except for 11a (hHSD2): 26%, 7(hHSD1): 25%,.
bHuman placental, microsomal fraction, substrate E2 [500 nM], cofactor NAD+ [1500 μM].
cHuman placental, cytosolic fraction, substrate E1 [500 nM], cofactor NADH [500 μM].
ds.f. (selectivity factor) = IC50(17β-HSD1)/IC50(17β-HSD2).
en.i.: no inhibition (inhibition of <10%).
fn.d.: not determined.
gMouse liver microsomal fraction, substrate E2 [500 nM], cofactor NAD+ [1500 μM].
Compounds 18a and 24a were not tested for inhibition of m17β-HSD2, due to their low inhibitory activity towards the human enzyme. Among the different synthesized 1,4-biphenyl amides without methylene linker (n = 0, Table 2, compounds 5a-21a), the best h17β-HSD2 inhibitory activity and selectivity toward h17β-HSD1 was achieved for compounds 6a, 7a and 17a (Table 2, IC50 values between 260 and 330nM, s.f. between 20 and 44), showing that a 3-OMe-group on ring C in combination with either a 3-OMe- or a 3-Me-group on ring A leads to a maximum in potency and selectivity in this series of compounds.
The presence of a methyl group in 4-position of ring A is tolerated by the h17β-HSD2 (compound 17a, IC50 = 310nM) and increases the selectivity toward h17β-HSD1 (17a: s.f. 44; 6a: s.f. 25). In contrast, compound 16a bearing a 3,4-dimethoxy substituted A ring displays a slight decrease in h17β-HSD2 inhibitory activity if compared to the corresponding compound 7a with only one methoxy group on that ring. The rigidification of the two methoxy substituents by the synthesis of a 1,3-benzodioxole ring (compounds 22a and 23a) could not overcome the drop in potency. Compounds 6a, 7a and 17a displayed the strongest m17β-HSD2 inhibitory activity (IC50 = 260, 290 and 140 nM, respectively).
In general, the introduction of methyl- or methoxy-groups, especially in the 3-positions of rings A and B, had a positive impact on inhibitory activity, which is similar for both 17β-HSD2 isoforms (Table 2). Therefore, inhibitors belonging to the 1,4-phenyl class are likely to bind in a conserved area common to both enzyme, in contrast to the 2,5-thiophene amide class. As 17β-HSD2 belongs to the SDR superfamily, characterized by the conserved Rossmann fold and catalytic triad [23], it is possible that these inhibitors bind in or very close to these regions. Furthermore, since compounds 6a and 17a lack of the two oxygen functions to mimic the E2 interactions with the enzyme, they are likely to bind to the active site in an alternative mode, significantly influenced by the methyl substitution patterns.
Compounds with a methylene linker 25a and 25 (Table 2), showed h17β-HSD2 inhibitory activity in the same order of magnitude as the corresponding derivative 20a lacking the methylene group, but displayed improved selectivities over h17β-HSD1. In contrast to what was observed for human 17β-HSD2, compounds 25a and 25 showed a significant difference in terms of m17β-HSD2 inhibition, indicating that the hydroxy group of compound 25 might function as H-bond donor in the interaction with the enzyme. A similar behavior was observed in the 2,5-thiophene amide class with regard to the h17β-HSD2 inhibitory activity[13]: all the tested 2,5-thiophene amides with a methylene linker achieved the highest h17β-HSD2 inhibition when substituted with an hydroxy group on the C ring. It might be therefore speculated, that the addition of the methylene linker can influence the binding mode of the inhibitors in both classes.
The 1,4-phenyl amides are likely to be competitive inhibitors, as derivatives with a similar structure were found to inhibit the enzyme following this mode of action.
The most potent m17β-HSD2 inhibitors 6a-8a, 10a, 14a, 15a, 17a, 19a, 21a, 23a and 25 were also tested for their m17β-HSD1 inhibitory activity. None of the tested compounds showed any inhibition of m17β-HSD1 at a concentration of 1 μM, indicating a significant selectivity toward this enzyme.
The inhibitory activity of compounds 6a, 17a and 25 was also evaluated in the human mammary cell line MDA-MB-231 containing endogenous 17β-HSD2 (Table 3). The compounds were tested at 250 nM and 1250 nM, representing approximately the IC50 observed in the cell-free assay, and its 5-fold value. As displayed in Table 3, all three compounds showed an inhibition between 60% and 67%at the lower concentration and approximately 90% inhibition at the higher concentration, indicating that the inhibitors can permeate the membrane and are able to inhibit the enzyme in a concentration dependent manner.
Table 3. Human 17β-HSD2 Cellular Inhibition of Compounds 6a, 17a and 25.
aMDA-MB-231 cell line, substrate E2 [200 nM].
bMean value of two determinations, standard deviation less than 15%.
Compounds 6a, 7a, 17a, 21a, 25a and 25 were tested for their affinity toward the ERs α and β according to described methods [24] (assay details are available in the Materials and Methods). Even when applied in a 1000-fold excess relative to E2, no inhibitor was able to displace more than 20% of the steroid from the corresponding receptor, indicating a very low binding affinity to the ERs.
The metabolic stabilities of the most active compounds 6a, 17a and 25 were evaluated using human liver microsomes (S9 fraction). In addition, compounds 5a, 14a, 20a and 25a were also tested in order to investigate whether structure modifications might exert an effect on metabolic stability (Table 4, assay details are available in Materials and Methods). All compounds, except 25, revealed a very high stability, which was not influenced by the nature of the substituents. Compound 25, exhibiting a short half-life, bears a hydroxy group, potentially susceptible to phase ΙΙ metabolism. Interestingly, all the tested inhibitors from the 1,4-phenyl amide class, for n = 0, demonstrated high metabolic stability, which seemingly constitutes a positive feature of the whole class. As species differences for the metabolism of drugs that are not or only partly metabolized are usually small [25], sufficient metabolic stability in species other than human can be anticipated.
Table 4. Half-life in Human Liver Microsomes S9 Fraction of Representative Compounds 3a, 5a, 6a, 14a, 17a, 20a, 25a and 25.
| Cmpd | R1 | R2 | Inhibitor c t1/2(min) a , b |
|---|---|---|---|
| 3a | 3-Me | 3-Me | >120 |
| 5a | 3-OMe | 3-Me | 116 |
| 6a | 3-Me | 3-OMe | 106 |
| 14a | 3-Cl | 3-OMe | 82 |
| 17a | 3-Me,4-Me | 3-OMe | 107 |
| 20a | 2-F, 3-Me | 3-OMe | 104 |
| 25a | 2-F, 3-Me | 3-OMe | 103 |
| 25 | 2-F, 3-Me | 3-OH | 6 |
aMean of at least two determinations, standard deviation less than 25%.
b1 mg/ml pooled mammalian liver S9 fraction (BD Gentest), 2 mM NADPH regenerating system, 1 mM UDPGA and 0.1 mM PAPS at 37°C for 0, 5, 15 and 60 minutes.
cInhibitor tested at a final concentration of 1 μM.
Conclusion
The aim of this work was the design of a compound, which should be suitable for application in both an animal model of osteoporosis and in humans. We report here the discovery of compound 17a, which is the first to show an appropriate profile for this purpose, with strong inhibition of both human and mouse 17β-HSD2 and selectivity toward the respective type 1 enzymes and the ERs. It also displayed good cellular inhibitory activity, high metabolic stability and good physicochemical parameters (MW = 345 and cLogP = 4.75) predictor for good oral bio-availability [26]. A comparative SAR study for h17β-HSD2 and m17β-HSD2 is also described for the 1,4-phenyl and the 2,5-thiophene classes of inhibitors, revealing that only compounds belonging to the first series similarly inhibit the two enzymes, probably through a similar binding mode. The species specific characterization of the thiophene and the phenyl derivatives pointed out the superiority of the latter class of inhibitors, which is able to equally inhibit the two isoenzymes and additionally displays a high metabolic stability. In vivo assays in a mouse osteoporosis model will be carried out soon and the results reported in due course in a specialized journal dealing with bone diseases.
Materials and Methods
Chemical Methods
Chemical names follow IUPAC nomenclature. Starting materials were purchased from Aldrich, Acros, Combi-Blocks or Fluorochem and were used without purification. Column chromatography was performed on silica gel (70–200 μm) and reaction progress was monitored by TLC on Alugram SIL G/UV254 (Macherey-Nagel). Visualization was accomplished with UV light.1H NMR and 13C NMR spectra were measured on a Bruker AM500 spectrometer (at 500 MHz and 125 MHz, respectively) at 300 K and on Bruker Fourier 300 (at 300 MHz and 75 MHz, respectively) at 300K. Chemical shifts are reported in δ (parts per million: ppm), by reference to the hydrogenated residues of deuteriated solvent as internal standard: 2.05 ppm (1H NMR) and 29.8 and 206.3 ppm (13C NMR) for CD3COCD3, 7.26 ppm (1H NMR) and 77.0 ppm (13C NMR) for CDCl3. Signals are described as br (broad), s (singlet), d (doublet), t (triplet), dd (doublet of doublets), ddd (doublet of doublet of doublets), dt (doublet of triplets) and m (multiplet). All coupling constants (J) are given in Hertz (Hz).
Melting points (mp) were measured in open capillaries on a Stuart Scientific SMP3 apparatus and are uncorrected.
Mass spectrometry was performed on a TSQ Quantum (ThermoFisher, Dreieich, Germany). The triple quadrupole mass spectrometer was equipped with an electrospray interface (ESI). The Surveyor-LC-system consisted of a pump, an auto sampler, and a PDA detector. The system was operated by the standard software Xcalibur. A RP C18 NUCLEODUR 100–5 (3 mm) column (Macherey-Nagel GmbH, Dühren, Germany) was used as stationary phase. All solvents were HPLC grade. In a gradient run (acetonitrile/water) the percentage of acetonitrile (containing 0.1% trifluoroacetic acid) was increased from an initial concentration of 0% at 0 min to 100% at 15 min and kept at 100% for 5 min. The injection volume was 15 μL and flow rate was set to 800 μL/min. MS analysis was carried out at a needle voltage of 3000 V and a capillary temperature of 350°C. Mass spectra were acquired in positive mode from 100 to 1000 m/z and UV spectra were recorded at the wave length of 254 nm and in some cases at 360 nm.
All microwave irradiation experiments were carried out in a 507 CEM-Discover microwave apparatus.
All tested compounds exhibited ≥ 95% chemical purity as measured by LC/MS.
The following compounds were prepared according to previously described procedures: 4-bromo-N-methyl-N-(m-tolyl)benzamide 7a[13].
Method A, general procedure for amide formation
To a solution of bromobenzoylchloride (2 mmol) was added the corresponding N-methylaniline (2 mmol) followed by Et3N (2 mmol) in CH2Cl2 (10 mL) at 0°C. After a few minutes, the ice bath was removed and the reaction mixture was warmed up to room temperature and stirred at room temperature overnight. The reaction mixture was extracted twice with CH2Cl2 (2 × 15 mL). The organic layer was dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using hexanes and EtOAc as eluent or by trituration in a mixture of diethyl ether / petroleum ether to afford the desired compound.
Method B, general procedure for Suzuki-Miyaura coupling
In a sealed tube the previously prepared bromo-N-heteroarylcarboxamide derivative (1 eq.) was introduced, followed by the corresponding boronic acid (1.5 eq.), cesium carbonate (3 eq.), tetrakis(triphenylphosphine)palladium (0.02 eq.) and a mixture of DME/EtOH/H2O (1:1:1, v:v:v, 3 mL) as solvent. The reactor was flushed with N2 and submitted to microwave irradiation (150°C, 150 W) for 20 minutes. After cooling to room temperature, a mixture of EtOAc/H2O (1:1, v:v, 2 mL) was added to stop the reaction. The aqueous layer was extracted with EtOAc (3 × 10 mL). The organic layer was washed once with brine and once with water, dried over MgSO4, filtered and the solution was concentrated under reduced pressure. The residue was purified by column chromatography using hexanes and EtOAc as eluent to afford the desired compound.
Detailed synthesis procedures of the compounds
4-Bromo-N-(4-methoxyphenyl)-N-methylbenzamide (11b)
The title compound was prepared by reaction of 4-bromobenzoyl chloride (5c) (1272 mg, 5.8mmol) and 4-methoxy-N-methylaniline (11d) (400 mg, 2.9mmol) according to method A. The residue was purified by silica gel column chromatography (hexanes/EtOAc80:20) to afford the desired product as yellow solid (932 mg, 99%). C15H14BrNO2; MW 320; mp: 61–62°C; MS (ESI) 320, 322 [M]+; IR (cm-1) 1635, 2840, 2850, 2917;1H NMR (C2D6CO, 500 MHz) δ (ppm) 3.36 (s, 3H), 3.74 (s, 3H), 6.82 (d, J = 9.0 Hz, 2H), 7.09 (d, J = 9.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 38.5, 55.7, 115.2, 123.7, 129.3, 131.4, 131.6, 137.1, 138.6, 159.1, 169.5.
4-Bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b)
The title compound was prepared by reaction of 4-bromobenzoyl chloride(5c) (1272 mg, 5.8mmol) and 3-methoxy-N-methylaniline (6d) (400 mg, 2.9mmol) according to method A. The residue was purified by silica gel column chromatography (hexanes/EtOAc80:20) to afford the desired product as yellow solid (935 mg, quantitative). C15H14BrNO2; MW 320; mp: 98–100°C; MS (ESI) 320, 322 [M]+; IR (cm-1) 1638, 2840, 2945, 3012;1H NMR (C2D6CO, 500 MHz) δ (ppm) 3.40 (s, 3H), 3.71 (s, 3H), 6.70–6.72 (m, 1H), 6.74–6.78 (m, 2), 7.17 (t,J = 8.0 Hz, 1H), 7.26–7.28 (m, 2H), 7.39–7.41 (m, 2H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 38.2, 55.7, 113.2, 113.8, 120.2, 123.9, 130.7, 131.3, 131.6, 137.0, 147.0, 161.2, 169.4.
4-Bromo-N-methyl-N-(m-tolyl)benzamide (5b)
The title compound was prepared by reaction of 4-bromobenzoyl chloride(5c) (724 mg, 3.3mmol) and N-methyl-m-toluidine (5d) (200 mg, 1.6mmol) according to method A. The residue was purified by silica gel column chromatography (hexanes/EtOAc80:20) to afford the desired product as white solid (500 mg, 99%). C15H14BrNO; MW 304; mp: 90–92C; MS (ESI) 304, 306 [M]+; IR (cm-1) 1586, 1638, 2853, 2923, 3058;1H NMR (C2D6CO, 300 MHz) δ (ppm) 2.24 (s, 3H), 3.39 (s, 3H), 6.90–6.93 (d, J = 8.0Hz, 1H), 6.99–7.05 (m, 2H), 7.14 (t, J = 8.0 Hz, 1H), 7.23–7.26 (m, 2H), 7.37–7.40 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ (ppm) 21.2, 38.4, 123.9, 125.2, 128.1, 128.5, 129.8, 131.4, 131.6, 137.0, 140.0, 145.8, 169.4.
4-Bromo-N-(3-methoxybenzyl)-N-methylbenzamide (25b)
The title compound was prepared by reaction of 4-bromobenzoyl chloride (5c) (579 mg, 2.6mmol) and 3-methoxy-N-methylbenzylamine (25d) (200 mg, 1.3mmol) according to method A. The residue was purified by silica gel column chromatography (hexanes/EtOAc80:20) to afford the desired product as yellow oil(430 mg, 97%).C16H16BrNO2; MW 334; MS (ESI) 334, 336 [M]+; IR (cm-1) 1630, 2834, 2923;1H NMR (C2D6CO, 500 MHz) δ(ppm) 2.91 (s, 3H), 3.79 (s, 3H), 4.52–4.70 (m, 2H), 6.76–6.80 (m, 1H), 6.86 (dd, J = 8.8 Hz, 5Hz, 1H), 6.95 (s, 1H), 7.28 (t, J = 8.0 Hz, 1H), 7.44 (d, J = 7.6 Hz, 2H), 7.62 (s, 2H); 13C NMR (C2D6CO, 125 MHz) δ(ppm) 37.2, 51.0, 55.5, 113.3, 113.6, 114.4, 119.7, 121.0, 123.9, 129.9, 130.5, 132.3, 137.0, 161.1.
N-(4-bromophenyl)-3-methoxy-N-methylbenzamide (24b)
The title compound was prepared by reaction of 3-methoxybenzoyl chloride (24c) (336 mg, 2.0mmol) and 4-bromo-N-methylaniline(24d) (367 mg, 2.0mmol) according to method A. The residue was purified by silica gel column chromatography (hexanes/EtOAc80:20) to afford the desired product as white solid (440 mg, 70%). C15H14BrNO2; MW 320; mp: 97–100°C; MS (ESI) 320, 322 [M]+; IR (cm-1) 1634, 2834, 2929, 3009;1H NMR (C2D6CO, 300 MHz) δ(ppm) 3.41 (s, 3H), 3.69 (s, 3H), 6.84–6.88 (m, 3H), 7.12–7.17 (m, 3H), 7.44–7.47 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 38.3, 55.6, 114.8, 116.3, 120.1, 121.7, 129.8, 130.0, 133.1, 138.8, 145.7, 160.1, 170.3.
N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (8a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b)(80 mg, 0.25mmol), phenylboronic acid (40 mg, 0.33mmol), cesium carbonate (244 mg, 0.75mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) in DME/EtOH/H2O (1/1/1/, 3 mL) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow solid (77 mg, 97%). C21H19NO2; MW 317; mp: 102–104°C; MS (ESI) 318 [M+H]+; IR (cm-1) 1588, 1636, 2831, 2917, 2960, 3040;1H NMR (C2D6CO, 300 MHz) δ (ppm) 3.44 (s, 3H), 3.70 (s, 3H), 6.72–6.77 (m, 2H), 6.80 (t, J = 2 Hz, 1H), 7.17 (t, J = 8 Hz, 1H), 7.33–3.37 (s, 1H), 7.41–7.46 (m, 4H), 7.51–7.54 (m, 2H), 7.60–7.63 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ (ppm) 38.4, 55.7, 113.0, 113.8, 120.1, 126.9, 127.7, 128.6, 129.8, 130.1, 130.6, 136.7, 140.8, 142.6, 147.4, 161.2, 170.2.
2'-Methoxy-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (9a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (112 mg, 0.35mmol), 2-methoxyphenylboronic acid (70 mg, 0.46mmol), cesium carbonate (342 mg, 1.05mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 8 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as colorless oil (113 mg, 93%). C22H21NO3; MW 347; MS (ESI) 348 [M+H]+; IR (cm-1) 1597, 1640, 2837, 2939;1H NMR (C2D6CO, 500 MHz) δ (ppm) 3.44 (s, 3H), 3.69 (s, 3H), 3.77 (s, 3H), 6.73–6.78 (m, 3H), 6.99 (dt, J = 1.0, 7.3 Hz, 1H), 7.07 (dd, J = 1.0, 8.3 Hz, 1H), 7.18 (dt, J = 1.0, 7.5 Hz, 1H), 7.24 (dd, J = 2.0, 7.5 Hz, 1H), 7.30–7.34 (m, 1H), 7.35–7.39 (m, 4H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 38.4, 55.6, 55.9, 112.5, 113.0, 113.7, 120.0, 121.7, 129.1, 129.5, 130.0, 130.4, 130.6, 131.3, 136.0, 140.7, 147.4, 157.5, 161.1, 170.4.
3'-Methoxy-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (7a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (81 mg, 0.25mmol), 3-methoxyphenylboronic acid (46 mg, 0.3mmol), cesium carbonate (247 mg, 0.75mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as white solid (74 mg, 85%). C22H21NO3; MW 347; mp: 131–134°C; MS (ESI) 348 [M+H]+; IR (cm-1) 1582, 1635, 2837, 2941, 2963;1H NMR (C2D6CO, 500 MHz) δ(ppm) 3.43 (s, 3H), 3.70 (s, 3H), 3.84 (s, 3H), 6.73–6.76 (m, 2H), 6.80–6.81 (m, 1H), 6.92(ddd, J = 1.0, 2.0, 8.0 Hz, 1H), 7.14–7.19 (m, 3H), 7.32–7.35 (m, 1H), 7.41–7.42 (m, 2H), 7.51–7.53 (m, 2H); 13C NMR (C2D6CO, 125 MHz) δ(ppm) 38.4, 55.6, 55.7, 113.0, 113.1, 113.8, 114.3, 120.0, 120.1, 126.9, 130.0, 130.6, 130.8, 136.8, 142.2, 142.5, 147.37, 147.38, 161.2, 170.2.
3'-Hydroxy-N-(3-hydroxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (7)
To a solution of 3'-methoxy-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (7a) (100 mg, 0.29mmol) in CH2Cl2 (10 mL) was added trifluorobromine dimethyl sulphide complex BF3.SMe2 (0.36 mL, 3.46 mM) at room temperature. The reaction mixture was stirred overnight. The reaction was quenched with MeOH (10 mL). The solvent was removed under reduced pressure at 25°C. Water was added to dissolve inorganic salts and the precipitate was filtered off and triturated with diethyl ether to afford the desired product as white solid (80 mg, 87%). C20H17NO3; MW 319; mp: 168–171°C; MS (ESI) 320[M+H]+; IR (cm-1) 1589, 3191;1H NMR (C2D6CO, 300 MHz) δ(ppm) 3.42 (s, 3H), 6.64–6.68 (m, 3H), 6.82–6.86 (m, 1H), 7.07–7.12 (m, 3H), 7.26 (t, J = 8.0 Hz, 1H), 7.39–7.44 (m, 2H), 7.47–7.49 (m, 2H), 8.42 (s, 1H), 8.48 (s, 1H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 38.4, 114.4, 114.6, 115.0, 115.6, 119.0, 119.1, 126.8, 130.1, 130.7, 130.8, 136.6, 142.3, 142.7, 147.4, 158.8, 158.9, 170.1.
4'-Methoxy-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (10a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (123 mg, 0.38mmol), 4-methoxyphenylboronic acid (75 mg, 0.49mmol), cesium carbonate (371 mg, 1.14mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 9 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc80:20) to afford the desired product as white solid (70 mg, 53%). C22H21NO3; MW 347; mp: 128–130°C; MS (ESI) 348 [M+H]+; IR (cm-1) 1601, 1625, 2840, 2935, 2963;1H NMR (C2D6CO, 500 MHz) δ (ppm) 3.43 (s, 3H), 3.70 (s, 3H), 3.82(s, 3H), 6.72–6.75 (m, 2H), 6.78–6.79 (m, 1H), 6.98 (d, J = 2.3 Hz, 1H), 7.00 (d, J = 2.3 Hz, 1H), 7.15–7.18 (m, 1H), 7.37–7.40 (m, 2H), 7.46–7.48 (m, 2H), 7.55–7.58 (m,2H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 38.4, 55.6, 55.7, 112.9, 113.8, 115.2, 120.1, 126.3, 128.8, 130.1, 130.6, 130.1, 135.9, 142.4, 147.5, 160.7, 161.2, 170.3.
3'-Fluoro-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (13a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (100 mg,0.31mmol), 3-fluorophenylboronic acid (57 mg, 0.41mmol), cesium carbonate (303 mg, 0.93mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 7 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow solid (91 mg, 89%). C21H18FNO2; MW 335; mp: 97–100°C; MS (ESI) 336 [M+H]+; IR (cm-1) 1587, 1637, 2834, 2923, 2960;1H NMR (C2D6CO, 300 MHz) δ (ppm) 3.44 (s, 3H), 3.71 (s, 3H), 6.73–6.77 (m, 2H), 6.80–6.81 (m, 1H), 7.09–7.21 (m, 2H), 7.36–7.50 (m, 5H), 7.55–7.57 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ (ppm) 38.4, 55.8, 113.1, 113.9, 114.3, 114.6, 115.2, 115.4, 120.2, 123.7, 127.1, 130.2, 130.8, 131.6, 131.8, 137.4, 141.2, 143.3, 143.4, 147.4, 161.3, 162.6, 165.8, 170.1.
3'-Chloro-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (14a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (80 mg, 0.25mmol), 3-chlorophenylboronic acid (52 mg, 0.33mmol), cesium carbonate (244 mg, 0.75mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as colorless oil (47 mg, 53%). C21H18ClNO2; MW 352; MS (ESI) 352, 354 [M]+; IR (cm-1) 1594, 1639, 2837, 2938;1H NMR (C2D6CO, 500 MHz) δ (ppm) 3.43 (s, 3H), 3.70 (s, 3H), 6.73–6.76 (m, 2H), 6.80–6.81(m, 1H), 7.17 (t,J = 8 Hz, 1H), 7.38–7.40 (m, 1H), 7.43–7.47 (m, 3H), 7.54–7.59 (m, 3H), 7.63–7.64 (m, 1H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 38.3, 55.7, 113.1, 113.8, 120.2, 126.3, 127.0, 127.6, 128.5, 130.1, 130.7, 131.4, 135.3, 137.4, 141.0, 142.9, 147.3, 161.2, 170.0.
N-(3-methoxyphenyl)-N,3'-dimethyl-[1,1'-biphenyl]-4-carboxamide (6a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (77 mg, 0.24mmol), 3-methylphenylboronic acid (42 mg, 0.31mmol), cesium carbonate (235 mg, 0.72mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as colorless oil (56 mg, 71%). C22H21NO2; MW 331; MS (ESI) 332 [M+H]+; IR (cm-1) 1586, 1640, 2840, 2929;1H NMR (C2D6CO, 300 MHz) δ(ppm) 3.38 (s, 3H), 3.44 (s, 3H), 3.70 (s, 3H), 6.74–6.76 (m, 2H), 6.79–6.80 (m, 1H), 7.15–7.20 (m, 2H), 7.31 (t,J = 8 Hz, 1H), 7.39–7.43 (m, 4H), 7.49–7.52 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 21.5, 38.4, 55.7, 113.0, 113.9, 120.2, 124.9, 126.9, 128.5, 129.4, 129.8, 130.1, 130.7, 136.6, 137.4, 140.9, 142.9, 147.5, 161.3, 170.3.
3'-(Dimethylamino)-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (15a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (80 mg, 0.25mmol), 3-(dimethylamino)phenylboronic acid (54 mg, 0.33mmol), cesium carbonate (244 mg, 0.75mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow oil (49 mg, 54%). C23H24N2O2; MW 360; MS (ESI) 361 [M+H]+; IR (cm-1) 1597, 1639, 2797, 2834, 2939;1H NMR (C2D6CO, 300 MHz) δ (ppm) 2.97 (s, 6H), 3.43 (s, 3H), 3.70 (s, 3H), 6.72–6.76 (m, 3H), 6.79 –-6.80 (m, 1H), 6.86–6.93 (m, 2H), 7.17 (t,J = 8.0 Hz, 1H), 7.23 (t, J = 8.0 Hz, 1H), 7.38–7.41 (m, 2H), 7.49–7.52 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ (ppm) 38.5, 40.8, 55.7, 111.8, 113.0, 113.9, 116.0, 120.2, 127.0, 130.0, 130.4, 130.7, 136.5, 141.6, 143.9, 147.5, 152.2, 161.3, 170.4.
N-(3-methoxyphenyl)-N,3',4'-trimethyl-[1,1'-biphenyl]-4-carboxamide (17a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (100 mg, 0.31mmol), 3,4-dimethylphenylboronic acid (62 mg, 0.41mmol), cesium carbonate (303 mg, 0.93mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 7 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow oil (106 mg, 99%). C23H23NO2; MW 345; MS (ESI) 346[M+H]+; IR (cm-1) 1599, 1640, 2840, 2914, 2942;1H NMR (C2D6CO, 500 MHz) δ(ppm) 2.26 (s, 3H), 2.29 (s, 3H), 3.43 (s, 3H), 3.70 (s, 3H), 6.73–6.75 (m, 2H), 6.79 –-6.80 (m, 1H), 7.15–7.19 (m, 2H), 7.32 (dd, J = 2.0Hz, 8.0Hz, 1H), 7.38–7.40 (m, 3H), 7.47–7.49 (m, 2H); 13C NMR (C2D6CO, 125 MHz) δ(ppm) 19.4, 19.8, 38.4, 55.6, 112.9, 113.8, 120.1, 125.0, 126.5, 128.8, 130.0, 130.6, 131.0, 136.2, 137.0, 137.8, 138.3, 142.8, 147.4, 161.2, 170.3.
3',4'-Difluoro-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (18a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (100 mg, 0.31mmol), 3,4-difluorophenylboronic acid (65 mg, 0.41mmol), cesium carbonate (303 mg, 0.93mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 7 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow oil (97 mg, 88%). C21H17F2NO2; MW 353; MS (ESI) 354[M+H]+; IR (cm-1) 1599, 1639, 2837, 2948;1H NMR (C2D6CO, 500 MHz) δ (ppm) 3.43 (s, 3H), 3.70 (s, 3H), 6.73–6.76 (m, 2H), 6.80 –-6.81 (m, 1H), 7.17 (t, J = 8.0 Hz, 1H), 7.35–7.41 (m, 1H), 7.42–7.44 (m, 2H), 7.45–7.48 (m, 1H), 7.52–7.54 (m, 2H), 7.56 (dd, J = 2.0 Hz, 8.0 Hz, 1H), 7.60 (dd, J = 2.0 Hz, 8.0 Hz, 1H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 38.3, 55.7, 113.0, 113.8, 116.6, 116.7, 118.5, 118.7, 120.2, 124.3, 124.4, 126.9, 130.1, 130.7, 137.3, 140.3, 147.3, 161.2, 170.0.
3',4'-Dimethoxy-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (16a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (100 mg, 0.31mmol), 3,4-dimethoxyphenylboronic acid (75 mg, 0.41mmol), cesium carbonate (303 mg, 0.93mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 7 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as white solid (83 mg, 71%). C23H23NO4; MW 377; mp: 153–156°C; MS (ESI) 378[M+H]+; IR (cm-1) 1604, 1635, 2843, 2938, 2969, 3003, 3068;1H NMR (C2D6CO, 300 MHz) δ(ppm) 3.51 (s, 3H), 3.69 (s, 3H), 3.90 (s, 3H), 3.92 (s, 3H), 6.62–6.72 (m, 3H), 6.89–6.91 (d, J = 8.0 Hz, 1H), 7.04–7.17 (m, 3H), 7.38 (s, 4H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 38.6, 55.6, 56.1, 110.4, 111.6, 112.2, 113.0, 119.4, 119.6, 126.1, 129.4, 130.0, 133.2, 134.4, 142.2, 146.3, 149.2, 149.3, 160.2, 170.5.
2'-Fluoro-3'-methoxy-N-(3-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (19a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (80 mg, 0.25mmol), 2-fluoro-3-methoxyphenylboronic acid (56 mg, 0.33mmol), cesium carbonate (244 mg, 0.75mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc 90:10) to afford the desired product as brown solid (43 mg, 47%). C22H20FNO3; MW 365; mp: 125–127°C; MS (ESI) 366[M+H]+; IR (cm-1) 1592, 1644, 2837, 2948, 2975, 3009;1H NMR (C2D6CO, 300 MHz) δ(ppm) 3.44 (s, 3H), 3.70 (s, 3H), 3.90 (s, 3H), 6.72–6.81 (m, 3H), 6.95–7.00 (m, 1H), 7.10–7.20 (m, 3H), 7.39 –-7.45 (m, 4H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 38.3, 55.7, 56.6, 113.1, 113.8, 113.9, 120.1, 122.4, 125.2, 125.3, 129.0, 129.1, 129.6, 130.6, 137.1, 137.5, 147.3, 161.2, 170.1.
2'-Fluoro-N-(3-methoxyphenyl)-N,3'-dimethyl-[1,1'-biphenyl]-4-carboxamide (20a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (72 mg, 0.22mmol), 2-fluoro-3-methylphenylboronic acid (45 mg, 0.29mmol), cesium carbonate (215 mg, 0.66mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 5 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow solid (70 mg, 91%). C22H20FNO2; MW 349; mp: 91–94°C; MS (ESI) 350 [M+H]+; IR (cm-1) 1599, 1635, 2840, 2920, 2963, 3055;1H NMR (C2D6CO, 500 MHz) δ (ppm) 2.29 (d, J = 2.5 Hz, 3H), 3.44 (s, 3H), 3.70 (s, 3H), 6.73–6.77 (m, 2H), 6.79–6.80 (m, 1H), 7.13 (t, J = 8.0 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.39–7.44 (m, 4H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 14.6, 14.7, 38.3, 55.7, 113.1, 113.8, 120.1, 125.0, 125.1, 126.2, 126.3, 128.6, 128.7, 129.0, 129.07, 129.09, 129.6, 130.6, 131.9, 132.0, 137.0, 137.9, 147.3, 157.9, 159.9, 170.1.
4-(Benzo[d][1,3]dioxol-5-yl)-N-(3-methoxyphenyl)-N-methylbenzamide (22a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxyphenyl)-N-methylbenzamide (6b) (80 mg, 0.25mmol), benzo[d][1,3]dioxol-5-ylboronic acid (55 mg, 0.33mmol), cesium carbonate (244 mg, 0.75mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow solid (68 mg, 76%). C22H19NO4; MW 361; mp: 139–141°C; MS (ESI) 362 [M+H]+; IR (cm-1) 1592, 1644, 2837, 2948, 2975, 3009;1H NMR (C2D6CO, 500 MHz) δ(ppm) 3.43 (s, 3H), 3.70 (s, 3H), 6.02 (s, 2H), 6.72–6.73 (m, 1H), 6.74–6.75 (m, 1H), 6.79 –-6.80 (m, 1H), 6.89–6.90 (m, 1H), 7.10–7.12 (m, 2H), 7.17 (t, J = 8.0 Hz, 1H), 7.37–7.39 (m, 2H), 7.44–7.46 (m, 2H); 13C NMR (C2D6CO, 125 MHz) δ(ppm) 38.4, 55.7, 102.3, 108.0, 109.4, 112.9, 113.8, 120.1, 121.4, 126.5, 130.0, 130.6, 135.0, 136.2, 142.4, 147.4, 148.5, 149.3 161.2, 170.2.
3'-Methoxy-N-(4-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (11a)
The title compound was prepared by reaction of 4-bromo-N-(4-methoxyphenyl)-N-methylbenzamide (11b) (100 mg, 0.31mmol), 3-methoxyphenylboronic acid (61 mg, 0.40mmol), cesium carbonate (303 mg, 0.93mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 7 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as colorlessoil (106 mg, 98%). C22H21NO3; MW 347; MS (ESI) 348 [M+H]+; IR (cm-1) 1635, 2840, 2935;1H NMR (C2D6CO, 300 MHz) δ (ppm) 3.40 (s, 3H), 3.75 (s, 3H), 3.85 (s, 3H), 6.82–6.86 (m, 2H), 6.92 (ddd, J = 1.0 Hz, 2.6 Hz, 8.3 Hz, 1H), 7.12–7.19 (m, 4H), 7.32–7.40 (m, 3H), 7.50–7.53 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ (ppm) 38.7, 55.6, 55.7, 113.1, 114.3, 115.2, 120.0, 126.9, 129.2, 130.1, 130.8, 136.9, 139.0, 142.2, 142.3, 159.0, 161.2, 170.2.
4'-Methoxy-N-(4-methoxyphenyl)-N-methyl-[1,1'-biphenyl]-4-carboxamide (12a)
The title compound was prepared by reaction of 4-bromo-N-(4-methoxyphenyl)-N-methylbenzamide (11b) (78 mg, 0.24mmol), 4-methoxyphenylboronic acid (48 mg, 0.32mmol), cesium carbonate (235 mg, 0.72mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc 90:10) to afford the desired product as yellow solid (73 mg, 88%). C22H21NO3; MW 347; mp: 129–132°C; MS (ESI) 348 [M+H]+; IR (cm-1) 1635, 2840, 2932, 2957;1H NMR (C2D6CO, 300 MHz) δ(ppm) 3.39 (s, 3H), 3.74 (s, 3H), 3.83 (s, 3H), 6.82–6.82 (m, 2H), 6.98–7.01 (m, 2H), 7.11–7.14 (m, 2H), 7.34–7.37 (m, 2H), 7.44–7.47 (m, 2H), 7.54–7.57 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 38.7, 55.6, 55.7, 115.2, 126.2, 128.8, 129.2, 130.2, 133.0, 135.9, 139.1, 142.1, 159.0, 160.6, 170.3.
3'-Methoxy-N-methyl-N-(m-tolyl)-[1,1'-biphenyl]-4-carboxamide (5a)
The title compound was prepared by reaction of 4-bromo-N-(3-methylphenyl)-N-methylbenzamide (5b) (123 mg, 0.40mmol), 3-methoxyphenylboronic acid (79 mg, 0.52mmol), cesium carbonate (393 mg, 1.20mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 9 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow oil (101 mg, 77%). C22H21NO2; MW 331; MS (ESI) 332 [M+H]+; IR (cm-1) 1582, 1601, 1639, 2834, 2935, 3034;1H NMR (C2D6CO, 300 MHz) δ(ppm) 2.24 (s, 3H), 3.42 (s, 3H), 3.84 (s, 3H), 6.90–7.00 (m, 3H), 7.08 (s, 1H), 7.11–7.18 (m, 3H), 7.33 (t, J = 8.0 Hz, 1H), 7.38–7.41 (m, 2H), 7.49–7.52 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 21.2, 38.5, 55.6, 113.1, 114.3, 120.0, 125.1, 126.9, 127.9, 128.4, 129.8, 130.1, 130.8, 136.7, 139.9, 142.3, 142.5, 146.2, 161.2, 170.2.
2'-Fluoro-N,3'-dimethyl-N-(m-tolyl)-[1,1'-biphenyl]-4-carboxamide (21a)
The title compound was prepared by reaction of 4-bromo-N-(3-methylphenyl)-N-methylbenzamide (5b) (93 mg, 0.31mmol), 2-fluoro-3-methylphenylboronic acid (62 mg, 0.40mmol), cesium carbonate (303 mg, 0.93mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 7 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as white solid (103 mg, quantitative). C22H20FNO; MW 333; mp: 73–76°C; MS (ESI) 334 [M+H]+; IR (cm-1) 1604, 1634, 2853, 2920;1H NMR (C2D6CO, 500 MHz) δ (ppm) 2.24 (s, 3H), 2.28 (d, J = 2.4 Hz, 3H), 3.43 (s, 3H), 6.94–6.97 (m, 1H), 6.98–7.01 (m, 1H), 7.07 (s, 1H), 7.11–7.16 (m, 2H), 7.23 –-7.26 (m, 2H), 7.38–7.41 (m, 4H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 14.6, 14.7, 21.2, 38.5, 125.05, 125.09, 125.12, 126.2, 126.3, 128.5, 128.6, 128.7, 128.99, 129.01, 129.07, 129.09, 129.6, 129.8, 131.9, 132.0, 136.9, 137.8, 139.9, 146.1, 157.9, 159.9, 170.1.
4-(Benzo[d][1,3]dioxol-5-yl)-N-methyl-N-(m-tolyl)benzamide (22a)
The title compound was prepared by reaction of 4-bromo-N-(3-methylphenyl)-N-methylbenzamide (5b) (79 mg, 0.26mmol), benzo[d][1,3]dioxol-5-ylboronic acid (56 mg, 0.34mmol), cesium carbonate (254 mg, 0.78mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow solid (44 mg, 49%). C22H19NO3; MW 345; mp: 111–113°C; MS (ESI) 346[M+H]+; IR (cm-1) 1602, 1634, 2788, 2911, 2966;1H NMR (C2D6CO, 500 MHz) δ (ppm) 2.24 (s, 3H), 3.42 (s, 3H), 6.02 (s, 2H), 6.89 (d, J = 8.0 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 7.07–7.11 (m, 3H), 7.14 (t, J = 8.0 Hz, 1H), 7.35–7.37 (m, 2H), 7.42–7.44 (m, 2H); 13C NMR (C2D6CO, 125 MHz) δ (ppm) 21.2, 38.5, 102.3, 107.9, 109.4, 121.4, 125.1, 126.5, 127.9, 128.4, 129.8, 130.2, 135.1, 136.2, 139.9, 142.3, 146.3, 148.5, 149.3, 170.2.
2'-Fluoro-N-(3-methoxybenzyl)-N,3'-dimethyl-[1,1'-biphenyl]-4-carboxamide (25a)
The title compound was prepared by reaction of 4-bromo-N-(3-methoxybenzyl)-N-methylbenzamide (25b) (200 mg, 0.60mmol), 2-fluoro-3-methylphenylboronic acid (120 mg, 0.78mmol), cesium carbonate (587 mg, 1.80mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 14 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow oil (200 mg, 87%). C23H22FNO2; MW 363; MS (ESI) 364 [M+H]+; IR (cm-1) 1630, 2834, 2923, 3034;1H NMR (C2D6CO, 500 MHz) δ(ppm) 2.32 (s, 3H), 2.97 (s, 3H), 3.81 (s, 3H), 4.59–4.73 (m, 2H), 6.80–7.00 (m, 3H), 7.17 (t,J = 8 Hz, 1H), 7.27–7.32 (m, 2H), 7.33–7.36 (m, 1H), 7.57–7.59 (m, 2H), 7.62 (s, 2H); 13C NMR (C2D6CO, 125 MHz) δ(ppm) 14.6, 14.7, 37.3, 51.0, 55.5, 113.6, 114.4, 121.0, 125.1, 125.2, 126.2, 126.3, 128.1, 128.7, 128.8, 129.1, 129.2, 129.7, 129.8, 130.5, 132.0, 132.1, 137.1, 138.0, 158.0, 159.9, 161.1.
2'-Fluoro-N-(3-hydroxybenzyl)-N,3'-dimethyl-[1,1'-biphenyl]-4-carboxamide (25)
To a solution of 2'-fluoro-N-(3-methoxybenzyl)-N,3'-dimethyl-[1,1'-biphenyl]-4-carboxamide (25a) (100 mg, 0.28mmol) in CH2Cl2 (10 mL) was added trifluorobromine dimethyl sulphide complex BF3.SMe2 (0.18 mL, 1.68mM) at room temperature. The reaction mixture was stirred overnight. The reaction was quenched with MeOH (10 mL). The solvent was removed under reduced pressure at 25°C. Water was added to dissolve inorganic salts and the precipitated was filtered off and triturated with diethyl ether to afford the desired product as yellow solid (66 mg, 67%). C22H20FNO2; MW 349; mp: 123–126°C; MS (ESI) 350 [M+H]+; IR (cm-1) 1600, 2917, 3181;1H NMR (C2D6CO, 500 MHz) δ (ppm) 2.32 (s, 3H), 2.96 (s, 3H), 4.55–4.69 (m, 2H), 6.72–6.90 (m, 3H), 7.16–7.22 (m, 2H), 7.29 (t, J = 8.0 Hz, 1H), 7.33 –-7.36 (m, 1H), 7.57–7.58 (m, 2H), 7.62 (s, 2H); 13C NMR (C2D6CO, 75 MHz) δ (ppm) 14.6, 14.7, 115.2, 125.1, 125.2, 126.2, 126.4, 128.0, 128.7, 128.9, 129.17, 129.21, 129.7, 129.8, 130.6, 132.0, 132.1, 137.0, 138.0, 157.3, 158.6, 160.6
3-Methoxy-N-(3'-methoxy-[1,1'-biphenyl]-4-yl)-N-methylbenzamide (24a)
The title compound was prepared by reaction of N-(4-bromophenyl)-3-methoxy-N-methylbenzamide (24b) (80 mg, 0.25mmol), 3-methoxyphenylboronic acid (50 mg, 0.33mmol), cesium carbonate (244 mg, 0.75mmol) and tetrakistriphenylphosphine palladium (0.02 eq., 6 mg) according to method B. The residue was purified by silica gel column chromatography (hexanes/EtOAc70:30) to afford the desired product as yellow oil (59 mg, 68%). C22H21NO3; MW 347; MS (ESI) 348 [M+H]+; IR (cm-1) 1640, 2840, 2936;1H NMR (C2D6CO, 300 MHz) δ(ppm) 3.45 (s, 3H), 3.66 (s, 3H), 3.85 (s, 3H), 6.80 –-6.84 (m, 1H), 6.90–6.94 (m, 3H), 7.09–7.19 (m, 3H), 7.24–7.27 (m, 2H), 7.34 (t, J = 8.0 Hz, 1H), 7.57–7.60 (m, 2H); 13C NMR (C2D6CO, 75 MHz) δ(ppm) 38.4, 55.6, 55.7, 113.2, 114.0, 114.9, 116.4, 120.0, 121.9, 128.3, 128.4, 129.7, 130.9, 139.0, 139.6, 142.3, 145.7, 160.1, 161.3, 170.3.
logP Determination
The logP values were calculated from ACD/Labs Percepta 2012 Release program. The logarithm of partition constant P (log P) was calculated using the “GALAS” method (Global Adjusted Locally According to Similarity). The program predicts clogP by comparing the molecule with structurally similar molecules where experimental data are known.
Biological methods
[2,4,6,7-3H]-E2 and [2,4,6,7-3H]-E1 were purchased from Perkin-Elmer, Boston. Quickszint Flow 302 scintillator fluid was bought from Zinsser Analytic, Frankfurt. Other chemicals were purchased from Sigma, Roth or Merck.
Ethics statement
Anonymized placental samples were obtained from Saarbrücken-Dudweiler Hospital’s Department of Gynecology. No author involved in this study has received information about the patients. The microsomal fraction of the mouse enzyme (m17β-HSD2) was obtained from mouse livers, which were bought from Pharmacelsus GmbH (Saarbrücken, Germany).
h17β-HSD1 and h17β-HSD2 enzyme preparation
Cytosolic (h17β-HSD1) and microsomal (h17β-HSD2) fractions were obtained from human placenta according to previously described procedures [20, 27]. Fresh tissue was homogenized and the enzymes were separated from the mitochondria, cell membrane. Nucleus and other rests by fractional centrifugation at 1000 g, 10.000 g and 150.000 g. The pellet fraction containing the microsomal h17β-HSD2 was used for the determination of h17β-HSD2 inhibition, while h17β-HSD1 was obtained after precipitation with ammonium sulfate from the cytosolic fraction for use of testing of h17β-HSD1 inhibition. Aliquots containing h17β-HSD1 or h17β-HSD2 were stored frozen.
m17β-HSD2 enzyme preparation and inhibition
The microsomal fraction (m17β-HSD2) was obtained from mouse liver as described for h17β-HSD2. Inhibitory activities were evaluated by a method identical to the one described for the human enzyme.
m17β-HSD1 enzyme preparation
Recombinant m17β-HSD1 enzyme was produced by transfection of HEK 293 cells with a m17β-HSD1 expression plasmid (coding sequence of NM_010475 in pCMV6Entry vector, OriGene Technologies, Inc.) according to a described procedure[28]. 48 hours after transfection cells were homogenized by sonication (3 x 10 s) in a buffer containing saccharose (40 mMTris, 250 mM saccharose, 5 mM EDTA, 7 mM DTT, 1 mM PMSF, pH 7,5). Cell lysate was centrifuged (1000 g, 15 min, 4°C) and 20% glycerol was added to the supernatant before aliquots were frozen and stored at -70°C.
Inhibition of h17β-HSD2 and m17β-HSD2 in cell-free assay
Inhibitory activities were evaluated following an established method with minor modifications[29–31]. Briefly, the enzyme preparation was incubated with NAD+ [1500 μM] in the presence of potential inhibitors at 37C in a phosphate buffer (50 mM) supplemented with 20% of glycerol and EDTA 1mM. Inhibitor stock solutions were prepared in DMSO. Final concentration of DMSO was adjusted to 1% in all samples. The enzymatic reaction was started by addition of a mixture of unlabelled- and [3H]-E2 (final concentration: 500 nM, 0.11 μCi). After 20 min, the incubation was stopped with HgCl2 and the mixture was extracted with ether. After evaporation, the steroids were dissolved in acetonitrile/water (45:55). E1 and E2 were separated using acetonitrile/water (45:55) as mobile phase in a C18 RP chromatography column (Nucleodur C18, 3μm, Macherey-Nagel, Düren) connected to a HPLC-system (Agilent 1100 Series, Agilent Technologies, Waldbronn). Detection and quantification of the steroids were performed using a radioflow detector (Berthold Technologies, Bad Wildbad). The conversion rate was calculated according to the following equation: %conversion = (%E1/(%E1+%E2))×100. Each value was calculated from at least two independent experiments.
Inhibition of h17β-HSD1 and m17β-HSD1 in cell-free assay
The 17β-HSD1 inhibition assay was performed similarly to the h17β-HSD2 test. The human cytosolic enzyme was incubated with NADH [500 μM] while the mouse recombinant enzyme was reacted with NADPH [500 μM].Test compound and a mixture of unlabelled- and [3H]-E1 (final concentration: 500 nM, 0.15 μCi) were added and mixed for 10 min at 37°C. Further treatment of the samples and HPLC separation was carried out as mentioned above for h17β-HSD2.
Inhibition of h17β-HSD2 in a cellular assay
Cellular h17β-HSD2 inhibitory activity is measured using the breast cancer cell-line MDA-MB-231[32] (17β-HSD1 activity negligible). [3H]-E2 (200 nM) is taken as substrate and is incubated with the inhibitor for 6 h at 37°C. After ether extraction, substrate and product are separated by HPLC and detected with a radioflow detector. Potency is evaluated as percentage of inhibition (inhibitor concentrations used: 1250 nM and 250 nM).
Estrogen receptor affinity in a cell-free assay
The binding affinity of selected compounds to ERα and ERβ was determined according to the recommendations of the US Environmental Protection Agency (EPA) by their Endocrine Disruptor Screening Program (EDSP)[24] using recombinant human proteins. Briefly, 1 nM of ERα and 4 nM of ERβ, respectively, were incubated with [3H]-E2 (3 nM for ERα and 10 nM for ERβ) and test compound for 16–20 h at 4°C.
The potential inhibitors were dissolved in DMSO (5% final concentration). Evaluation of non-specific-binding was performed with unlabeled E2 at concentrations 100-fold of [3H]-E2 (300 nM for ERα and 1000 nM for ERβ). After incubation, ligand-receptor complexes were selectively bound to hydroxyapatite (83.5 g/LinTE-buffer). The bound complex was washed three times and resuspended in ethanol. For radiodetection, scintillator cocktail (Quickszint 212, Zinsser Analytic, Frankfurt) was added and samples were measured in a liquid scintillation counter (1450 LSC & Luminescence Counter, Perkin Elmer).
From these results the percentage of [3H]-E2 displacement by the compounds was calculated. The plot of % displacement versus compound concentration resulted in sigmoidal binding curves. The compound concentrations necessary to displace 50% of the receptor bound [3H]-E2 were determined. Unlabeled E2 IC50 values were determined in each experiment and used as reference. The E2 IC50 determined were 3±20% nM for ERα and 10±20% nM for ERβ.
Relative Binding Affinity was determined by applying the following equation: RBA[%] = (IC50(E2)/IC50(compound)) ∙ 100[24]. This results in a RBA value of 100% for E2.
After the assay was established and validated, a modification was made to increase throughput. Compounds were tested at concentrations of 1000 times the IC50(E2). Compounds with less than 50% displacement of [3H]-E2 at a concentration of 1000 times IC50(E2) were classified as RBA <0.1%.
Metabolic Stability in a cell-free assay
Compounds 7a, 8a, 16a, 19a, 22a, 27a and 27 were tested according to established method[33–35]For evaluation of phase I and II metabolic stability 1 μM compound was incubated with 1 mg/ml pooled mammalian liver S9 fraction (BD Gentest), 2 mM NADPH regenerating system, 1 mM UDPGA and 0.1 mM PAPS at 37°C for 0, 5, 15 and 60 minutes at a final volume of 100 μL. The incubation was stopped by precipitation of S9 enzymes with 2 volumes of cold acetonitrile containing internal standard. Concentration of the remaining test compound at the different time points was analyzed by LC-MS/MS and used to determine half-life (t1/2).
Acknowledgments
We thank Josef Zapp for the NMR measurements, Larissa Müller, Isabella Mang and Martina Jankowski for the biological tests.
Data Availability
All relevant data are within the paper.
Funding Statement
The authors acknowledge the Deutsche Forschungsgemeinschaft (DFG) for financial support (Grants HA1315/12-1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Elexopharm GmbH had no financial interest in this collaboration.
References
- 1. NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. J Am Med Assoc. 2001; 285: 785–795. [DOI] [PubMed] [Google Scholar]
- 2. Vanderschueren D, Gaytant J, Boonen S, Venken K. Androgens and bone. Curr Opin Endocrinol. 2008;15: 250–254. [DOI] [PubMed] [Google Scholar]
- 3. Wu L, Einstein M, Geissler WM, Chan HK, Elliston KO, Andersson S. Expression cloning and characterization of human 17 beta-hydroxysteroid dehydrogenase type 2, a microsomal enzyme possessing 20 alpha-hydroxysteroid dehydrogenase activity. J Biol Chem. 1993;268: 12964–12969. [PubMed] [Google Scholar]
- 4. Dong Y, Qiu QQ, Debear J, Lathrop WF, Bertolini DR, Tamburini PP. 17Beta-hydroxysteroid dehydrogenases in human bone cells. J Bone Miner Res. 1998;13: 1539–1546. [DOI] [PubMed] [Google Scholar]
- 5. Bydal P, Auger S, Poirier D. Inhibition of type 2 17beta-hydroxysteroid dehydrogenase by estradiol derivatives bearing a lactone on the D-ring: structure-activity relationships. 2004; 69: 325–342. [DOI] [PubMed] [Google Scholar]
- 6. Poirier D, Bydal P, Tremblay MR, Sam KM, Luu-The V. Inhibitors of type II 17beta-hydroxysteroid dehydrogenase. Mol Cell Endocrinol. 2001;171: 119–128. [DOI] [PubMed] [Google Scholar]
- 7. Sam KM, Auger S, Luu-The V, Poirier D. Steroidal spiro-gamma-lactones that inhibit 17 beta-hydroxysteroid dehydrogenase activity in human placental microsomes. J Med Chem. 1995;38: 4518–4528. [DOI] [PubMed] [Google Scholar]
- 8. Gunn D, Akuche C, Baryza J, Blue ML, Brennan C, Campbell AM et al. 4,5-Disubstituted cis-pyrrolidinones as inhibitors of type II 17beta-hydroxysteroid dehydrogenase. Part 2. SAR. Bioorg Med Chem Lett. 2005;15: 3053–3057. [DOI] [PubMed] [Google Scholar]
- 9. Wood J, Bagi CM, Akuche C, Bacchiocchi A, Baryza J, Blue ML et al. 4,5-Disubstituted cis-pyrrolidinones as inhibitors of type II 17beta-hydroxysteroid dehydrogenase. Part 3. Identification of lead candidate. Bioorg Med Chem Lett. 2006;16: 4965–4968. [DOI] [PubMed] [Google Scholar]
- 10. Wetzel M, Marchais-Oberwinkler S, Perspicace E, Möller G, Adamski J, Hartmann RW. Introduction of an electron withdrawing group on the hydroxyphenylnaphthol scaffold improves the potency of 17beta-hydroxysteroid dehydrogenase type 2 (17beta-HSD2) inhibitors. J Med Chem. 2011;54: 7547–7557. 10.1021/jm2008453 [DOI] [PubMed] [Google Scholar]
- 11. Wetzel M, Gargano EM, Hinsberger S, Marchais-Oberwinkler S, Hartmann RW. Discovery of a new class of bicyclic substituted hydroxyphenylmethanones as 17beta-hydroxysteroid dehydrogenase type 2 (17beta-HSD2) inhibitors for the treatment of osteoporosis. Eur J Med Chem. 2012;47: 1–17. 10.1016/j.ejmech.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 12. Marchais-Oberwinkler S, Xu K, Wetzel M, Perspicace E, Negri M, Meyer A et al. Structural optimization of 2,5-thiophene amides as highly potent and selective 17beta-hydroxysteroid dehydrogenase type 2 inhibitors for the treatment of osteoporosis. J Med Chem. 2013;56: 167–181. 10.1021/jm3014053 [DOI] [PubMed] [Google Scholar]
- 13. Gargano EM, Perspicace E, Hanke N, Carotti A, Marchais-Oberwinkler S, Hartmann RW. Metabolic stability optimization and metabolite identification of 2,5-thiophene amide 17beta-hydroxysteroid dehydrogenase type 2 inhibitors.Eur J Med Chem. 2014; 87: 203–219. 10.1016/j.ejmech.2014.09.061 [DOI] [PubMed] [Google Scholar]
- 14. Xu K, Al-Soud YA, Wetzel M, Hartmann RW, Marchais-Oberwinkler S. Triazole ring-opening leads to the discovery of potent nonsteroidal 17beta-hydroxysteroid dehydrogenase type 2 inhibitors. Eur J Med Chem. 2011;46: 5978–5990. 10.1016/j.ejmech.2011.10.010 [DOI] [PubMed] [Google Scholar]
- 15. Marchais-Oberwinkler S, Kruchten P, Frotscher M, Ziegler E, Neugebauer A, Bhoga U et al. Substituted 6-phenyl-2-naphthols. Potent and selective nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD1): design, synthesis, biological evaluation, and pharmacokinetics. J Med Chem. 2008; 51: 4685–4698. 10.1021/jm800367k [DOI] [PubMed] [Google Scholar]
- 16. Bey E, Marchais-Oberwinkler S, Kruchten P, Frotscher M, Werth R, Oster A et al. Design, synthesis and biological evaluation of bis(hydroxyphenyl) azoles as potent and selective non-steroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD1) for the treatment of estrogen-dependent diseases. Bioorg Med Chem. 2008;16: 6423–6435. 10.1016/j.bmc.2008.04.073 [DOI] [PubMed] [Google Scholar]
- 17. Turner AS. Animal models of osteoporosis-necessity and limitations. Eur Cell Mater. 2001;1: 66–81. [DOI] [PubMed] [Google Scholar]
- 18. Reinwald S, Burr D. Review of nonprimate, large animal models for osteoporosis research. J Bone Miner Res. 2008;9: 1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kruchten P, Werth R, Marchais-Oberwinkler S, Frotscher M, Hartmann RW. Development of a biological screening system for the evaluation of highly active and selective 17beta-HSD1-inhibitors as potential therapeutic agents. Mol Cell Endocrinol. 2009;301: 154–157. 10.1016/j.mce.2008.09.035 [DOI] [PubMed] [Google Scholar]
- 20. Marchais-Oberwinkler S, Wetzel M, Ziegler E, Kruchten P, Werth R, Henn C et al. New drug-like hydroxyphenylnaphthol steroidomimetics as potent and selective 17beta-hydroxysteroid dehydrogenase type 1 inhibitors for the treatment of estrogen-dependent diseases. J Med Chem. 2011; 54: 534–547. 10.1021/jm1009082 [DOI] [PubMed] [Google Scholar]
- 21. Tremblay MR, Lin SX, Poirier D. Chemical synthesis of 16beta-propylaminoacyl derivatives of estradiol and their inhibitory potency on type 1 17beta-hydroxysteroid dehydrogenase and binding affinity on steroid receptors. Steroids. 2001; 66: 821–831. [DOI] [PubMed] [Google Scholar]
- 22. Perspicace E, Giorgio A, Carotti A, Marchais-Oberwinkler S, Hartmann RW. Novel N-methylsulfonamide and retro-N-methylsulfonamide derivatives as 17beta-hydroxysteroid dehydrogenase type 2 (17beta-HSD2) inhibitors with good ADME-related physicochemical parameters. Eur J Med Chem. 2013; 69: 201–215. 10.1016/j.ejmech.2013.08.026 [DOI] [PubMed] [Google Scholar]
- 23. Kavanagh KL, Jornvall H, Persson B, Oppermann U. Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell Mol Life Sci. 2008;65: 3895–3906. 10.1007/s00018-008-8588-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.U.S. Environmental Protection Agency. Protocol for the In Vitro Estrogen Receptor Saturation Binding and Competitive Binding Assays Using Rat Uterine Cytosol 2015; Available: http://www.epa.gov/endo/pubs/assayvalidation/appendix1_er_ruc.pdf
- 25. Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2: 875–894. [DOI] [PubMed] [Google Scholar]
- 26. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev. 2001;46: 3–26. [DOI] [PubMed] [Google Scholar]
- 27. Zhu DW, Lee X, Breton R, Ghosh D, Pangborn W, Duax WL et al. Crystallization and preliminary X-ray diffraction analysis of the complex of human placental 17 beta-hydroxysteroid dehydrogenase with NADP+. J Mol Biol. 1993;234: 242–244. [DOI] [PubMed] [Google Scholar]
- 28. Sam KM, Boivin RP, Tremblay MR, Auger S, Poirier D. C16 and C17 derivatives of estradiol as inhibitors of 17 beta-hydroxysteroid dehydrogenase type 1: chemical synthesis and structure-activity relationships. Drug Des Discov. Drug 1998;15: 157–180. [PubMed] [Google Scholar]
- 29. Xu K, Wetzel M, Hartmann RW, Marchais-Oberwinkler S. Synthesis and biological evaluation of spiro-δ-lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 2 (17 β-HSD2). Lett Drug Des Discov. 2011;8: 406–421. [Google Scholar]
- 30. Lin SX, Yang F, Jin JZ, Breton R, Zhu DW, Luu-The V et al. Subunit identity of the dimeric 17 beta-hydroxysteroid dehydrogenase from human placenta. J Biol Chem. 1992;267: 16182–16187. [PubMed] [Google Scholar]
- 31. Puranen TJ, Poutanen MH, Peltoketo HE, Vihko PT, Vihko RK. Site-directed mutagenesis of the putative active site of human 17 beta-hydroxysteroid dehydrogenase type 1. Biochem J. 1994;304: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Day JM, Tutill HJ, Newman SP, Purohit A, Lawrence HR, Nicker N et al. 17Beta-hydroxysteroid dehydrogenase Type 1 and Type 2: association between mRNA expression and activity in cell lines. Mol Cell Endocrinol. 2006;248: 246–249. [DOI] [PubMed] [Google Scholar]
- 33. Di L, Kerns EH, Hong Y, Kleintop TA, McConnell OJ, Huryn DM. Optimization of a higher throughput microsomal stability screening assay for profiling drug discovery candidates. J Biomol Screen 2003;4: 453–462. [DOI] [PubMed] [Google Scholar]
- 34. Moreno-Farre JW, P.; Raynud F. Analysis of Potential Drug-Drug Interactions for Anticancer Agents in Human Liver Microsomes by High Throughput Liquid Chromatography/Mass Spectrometry Assay. Aust-Asian J Cancer 2007;6: 55–69. [Google Scholar]
- 35. Hui JP, Stuart Grossert J, Cutler MJ, Melanson JE. Strategic identification of in vitro metabolites of 13-desmethyl spirolide C using liquid chromatography/high-resolution mass spectrometry. Rapid Commun Mass Spectrom. 2012;26: 345–354. 10.1002/rcm.5336 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.