

Abstract
Expression of RAD51, a crucial player in homologous recombination (HR) and DNA double-strand break (DSB) repair, is dysregulated in human tumors, and can contribute to genomic instability and tumor progression. To further understand RAD51 regulation we functionally characterized a long non-coding (lnc) RNA, dubbed TODRA (Transcribed in the Opposite Direction of RA D51), transcribed 69bp upstream to RAD51, in the opposite direction. We demonstrate that TODRA is an expressed transcript and that the RAD51 promoter region is bidirectional, supporting TODRA expression (7-fold higher than RAD51 in this assay, p = 0.003). TODRA overexpression in HeLa cells induced expression of TPIP, a member of the TPTE family which includes PTEN. Similar to PTEN, we found that TPIP co-activates E2F1 induction of RAD51. Analysis of E2F1's effect on the bidirectional promoter showed that E2F1 binding to the same site that promotes RAD51 expression, results in downregulation of TODRA. Moreover, TODRA overexpression induces HR in a RAD51-dependent DSB repair assay, and increases formation of DNA damage-induced RAD51-positive foci. Importantly, gene expression in breast tumors supports our finding that E2F1 oppositely regulates RAD51 and TODRA: increased RAD51 expression, which is associated with an aggressive tumor phenotype (e.g. negative correlation with positive ER (r = -0.22, p = 0.02) and positive PR status (r = -0.27, p<0.001); positive correlation with ki67 status (r = 0.36, p = 0.005) and HER2 amplification (r = 0.41, p = 0.001)), correlates as expected with lower TODRA and higher E2F1 expression. However, although E2F1 induction resulted in TPIP downregulation in cell lines, we find that TPIP expression in tumors is not reduced despite higher E2F1 expression, perhaps contributing to increased RAD51 expression. Our results identify TPIP as a novel E2F1 co-activator, suggest a similar role for other TPTEs, and indicate that the TODRA lncRNA affects RAD51 dysregulation and RAD51-dependent DSB repair in malignancy. Importantly, gene expression in breast tumors supports our finding that E2F1 oppositely regulates RAD51 and TODRA: increased RAD51 expression, which is associated with an aggressive tumor phenotype (e.g. negative correlation with positive ER (r = -0.22, p = 0.02) and positive PR status (r = -0.27, p<0.001); positive correlation with ki67 status (r = 0.36, p = 0.005) and HER2 amplification (r = 0.41, p = 0.001)), correlates as expected with lower TODRA and higher E2F1 expression. However, although E2F1 induction resulted in TPIP downregulation in cell lines, we find that TPIP expression in tumors is not reduced despite higher E2F1 expression, perhaps contributing to increased RAD51 expression. Our results identify TPIP as a novel E2F1 co-activator, suggest a similar role for other TPTEs, and indicate that the TODRA lncRNA affects RAD51 dysregulation and RAD51-dependent DSB repair in malignancy.
Introduction
RAD51 is the central recombinase involved in homologous recombination (HR), a mechanism for high fidelity repair of double-strand breaks (DSBs) that requires an intact, homologous DNA template[1]. DSBs are potentially lethal DNA lesions[2, 3], and disruption of their repair can lead to genomic instability, which plays an important role in both tumor initiation and progression[4]. Indeed, RAD51 expression is often dysregulated in human tumors[5–7], promoting genomic instability[8, 9].
RAD51 is regulated by members of the E2F transcription factor family, which plays a critical role in cell cycle control. E2Fs regulate expression of genes required for cell cycle-progression, DNA replication, mitosis, DNA damage response, checkpoint activation, differentiation, development, apoptosis and autophagy[10–14]. The E2F family is broadly subdivided into 'activator' E2Fs (E2F1-3a) and 'repressor' E2Fs (E2F4-8), based on their predominant effect on target gene expression. Both activator and repressor E2Fs bind the same recognition site in the RAD51 promoter[15–19], and were shown to regulate RAD51 expression during growth stimulation[17], hypoxia[18] and inhibition of poly(ADP-ribose) polymerase, a DNA-repair enzyme[19].
The RAD51 locus contains an annotated long non-coding RNA (lncRNA) transcribed only 69bp upstream of RAD51 in the opposite direction. We investigated whether this lncRNA, dubbed TODRA, regulates RAD51 expression and activity. Our results suggest that TODRA participates in regulation of RAD51 expression through E2F1 and TPIP, a member of the PTEN phosphatase family. To assess the functional effects of this regulatory mechanism we analyzed RAD51-dependent DSB repair as well as formation of RAD51-positive foci following DNA damage. We found that TODRA overexpression induces DSB repair by HR and also increases the fraction of RAD51 foci formed following DNA damage. In breast tumors, expression analysis of RAD51/TODRA, E2F1 and TPIP shows perturbed regulation of RAD51 expression, and the associated increase in RAD51 expression correlates with an aggressive tumor phenotype.
Results
AK125393 (TODRA)–a lncRNA immediately upstream of RAD51 is transcribed in the opposite direction
AK125393 is a putative long non-coding RNA in the RAD51 locus, identified through the FLJ (“full-length long Japan”) effort[20] to sequence full-length human cDNAs. AK125393 lacks significant open reading frames (all <100aa), and was therefore annotated as a non-coding RNA (ncRNA). In the UCSC Genome Browser (assembly Feb. 2009 [GRCh37/Hg19]) AK125393 is shown as a 3-exon non-coding expressed gene transcribed in the opposite direction to RAD51 (Fig 1A), and the transcription start sites (TSSs) of RAD51 and AK125393 are only 69bp apart. We named the AK125393 ncRNA TODRA, for Transcribed in the Opposite Direction of RA D51. To confirm TODRA expression and its TSS, we performed strand specific RT-PCR and 5' and 3'RACE on cDNA. We demonstrated expression of TODRA exons 1–3, (as characterized in the UCSC and NCBI databases) (Fig 1A), and observed splicing of intron 1, indicating this RNA is indeed transcribed and processed (Fig 1B). Other transcription products were also observed, including previously described introns (specifically intron 2) and downstream 3' sequences (Fig 1A). Northern blot analysis using a number of probes did not identify a major transcription product.
Fig 1. Transcriptional analysis of the RAD51/TODRA region.

A. TODRA transcript: Top: Schematic representation of the predicted TODRA (AK125393) gene, as described in the UCSC genome browser. Light grey shaded rectangles depict TODRA exons, the dark grey rectangle depicts RAD51 exon 1, transcribed in the opposite direction. Bottom: Results of TODRA transcript analysis. 5’RACE using capped HeLa mRNA, identified one transcription start site (full arrowhead, +1 corresponds to chr. 15: 40987374, hg19), and 3’RACE identified several possible transcription termini (arrows). The most 5’ end of RAD51 identified using 5’RACE is also shown (empty arrowhead, +1 corresponds to chr. 15: 40987303, hg19). Black bars beneath the diagram indicate confirmed regions of unidirectional transcription determined using strand specific primers for reverse transcription from both HeLa and MCF7 cells. B. Splicing of TODRA exons 1 and 2 is demonstrated in the representative gel. Lane M: pUC Mix Marker, (Fermentas), Lanes 1&2: TODRA strand specific RT-PCR products (F primer located in exon 1, R primer in exon 2). Expected size of product in genomic DNA: 696bp, Expected size of spliced transcript: 480bp, as observed in lanes 1 (cDNA prepared from HeLa cells) and 2 (cDNA prepared from MCF7 cells). C. The RAD51/TODRA region supports transcription in both directions. Top: Schematic representation of the RAD51 and TODRA promoter regions and the fragments cloned into luciferase promoter constructs. Bottom: TODRA putative promoter activity. MCF7 cells were co-transfected with the promoter-less pGL3-basic, pRAD51-UTR or pTODRA and pRL-TK (to normalize for transfection efficiency). Results are shown as fold increase in RLA (relative luciferase activity), compared to pGL3-basic. Values are means ± SE of 4–5 independent transfections performed in duplicates. * p< 0.002, ** p< 0.0001.
To determine if the RAD51 promoter region also supports transcription in the TODRA direction, this intergenic promoter region was cloned upstream of the firefly luciferase gene in the promoter-less pGL3-basic vector, in both forward (RAD51) and reverse (TODRA) orientations. These plasmids, respectively designated pRAD51-UTR and pTODRA (Fig 1C), were transfected into MCF7 cells and assayed for relative promoter activity. Both constructs drove transcription. In this assay, even though the RAD51 construct is larger, and includes all known transcription promoting regions (pRAD51-UTR contains a larger insert in the RAD51 direction, including RAD51 promoter elements located both upstream to TODRA's TSS and in the RAD51 5'UTR), pTODRA activity was 7-fold greater (p<0.0001) than that of pRAD51-UTR (Fig 1C), demonstrating that this region supports bidirectional transcription.
The intergenic E2F site oppositely regulates RAD51 and TODRA transcription
Both E2F1 and E2F4 have previously been shown to bind to the RAD51 proximal promoter and regulate RAD51 expression[17–19]. While E2F1 activates expression[17], E2F4/p130 complexes repress RAD51 [18, 19], through the E2F1/E2F4 binding site located within the shared RAD51/TODRA promoter. To examine its role in TODRA expression, this E2F binding site was mutated based on mutations previously show to abolish the binding site[17] (TTTGGCGGGAAT→TTCGGACGGAAT) and assayed for the effect on promoter activity. In both MCF7 and U2OS cells, abolishing the E2F binding site repressed pTODRA activity by approximately 1.5-fold (p<0.01) (Fig 2A). In contrast, activity of pRAD51, the minimal RAD51 promoter construct, was increased by 30-fold (p<0.003) in MCF7 cells and ~9-fold (p = 0.02) in U2OS cells (Fig 2B). This is consistent with previous studies of RAD51 in MCF7 cells, which showed that under baseline conditions the RAD51-E2F site is occupied mainly by the repressive E2F4 factor[18]. Mutagenesis of the E2F site therefore prevents E2F4 binding and de-represses RAD51. Overexpression of E2F4 did not affect either RAD51 or TODRA promoter activities (S1 Fig). However, E2F1 overexpression resulted in a 10-11-fold increase (p≤0.005) of RAD51 promoter activity (pRAD51-UTR) (Fig 2B), and a 40–80% reduction (p≤0.007) in TODRA promoter activity (Fig 2A). To summarize, our results show that TODRA is indeed regulated by the shared E2F binding site, and that E2F1 has an opposite effect on RAD51 vs. TODRA expression.
Fig 2. The E2F site in the common promoter region has opposite effects on RAD51 and TODRA.

A. Top: Diagram of the core TODRA promoter region cloned into the luciferase reporter vector. Bottom: Effect of mutagenesis of the E2F binding site and E2F1 induction on the TODRA reporter. Wild type TODRA luciferase (reporter) construct, or an E2F binding site mutant (E2F site mut) construct were transfected into MCF7 and U2OS cells. A TODRA luciferase (reporter) construct was also co-transfected with either an E2F1 expression vector or an empty vector control into serum-starved MCF7 and U2OS cells. All experiments included co-transfection with pRL-TK (to normalize for transfection efficiency). Results are depicted as the fold change in RLA compared to the WT construct transfection. Values in all experiments are means ± SE of 3–4 independent transfections performed in duplicate. ** p≤ 0.007, * p≤ 0.02, ^ p = 0.00001. B. Top: Diagram of the core RAD51 promoter region cloned into the luciferase reporter vector. Bottom: Effect of mutagenesis of the E2F binding site and E2F1 induction on the RAD51 reporter. Wild type RAD51 luciferase (reporter) construct, or an E2F binding site mutant (E2F site mut) construct were transfected into MCF7 and U2OS cells. A RAD51 luciferase (reporter) construct was also co-transfected with either an E2F1 expression vector or an empty vector control into serum-starved MCF7 and U2OS cells. All experiments included co-transfection with pRL-TK (to normalize for transfection efficiency). Results are depicted as the fold change in RLA compared to the WT construct transfection. Values in all experiments are means ± SE of 3–4 independent transfections performed in duplicate. ** p≤ 0.007, * p≤ 0.02. C. and D. Top: Diagram of the RAD51/TODRA bidirectional promoter region cloned between the firefly and Renilla luciferase reporter genes (pBDP). C. Mutagenesis of the E2F binding site. E2F site mutant (pBDP E2F site mut) or wild type bidirectional promoter constructs (pBDP) were transfected into MCF7 and U2OS cells. Results are depicted as the fold change in the mutant compared to the WT in the ratio of Firefly/Renilla luciferase activities, which represents the ratio of RAD51/TODRA promoter activities. Values are means ± SE of 3–6 independent transfections performed in duplicate. ** p< 0.0001. D. E2F1 overexpression. pBDP activity was examined in MCF7 and U2OS cells co-transfected with the pBDP construct and either an E2F1 WT, an E2F1 trans-activation domain deletion mutant (ΔTA), or an empty expression vector. Results are depicted as the fold change between each E2F1 expression vector and the empty vector control, in the ratio of Firefly/Renilla luciferase activities, which represents the ratio of RAD51/TODRA promoter activities. Values are means ± SE of 3–6 independent transfections performed in duplicate. ** p< 0.0001. Additional comparisons are indicated above the bars. * p≤ 0.003.
The effect of the E2F site was further analyzed using a bidirectional promoter (BDP) construct mimicking the endogenous bidirectional promoter region. A dual reporter vector was constructed by inserting the overlapping promoter region (analogous to the pTODRA construct) between firefly and Renilla luciferase genes oriented in opposite directions (Fig 2C and 2D). In this pBDP construct, changes in the ratio of Firefly to Renilla luciferase activities represent changes in the ratio of RAD51/TODRA promoter activities. Mutagenesis of the E2F site increased the Firefly/Renilla ratio by 25-fold (p<0.0001) in MCF7 cells and 2-fold (p = 0.001) in U2OS cells, corresponding to an increased ratio of RAD51/TODRA promoter activities (Fig 2C). This is consistent with results from the unidirectional E2F site mutagenized promoter constructs (Fig 2A and 2B). E2F1 overexpression resulted in 12-fold and 5-fold (p≤0.00002) increase in the Firefly/Renilla activity ratios, in MCF7 and U2OS cells respectively, also corresponding to an increased RAD51/TODRA expression ratio (Fig 2D). Overexpression of an E2F1 transactivation domain mutant (E2F1ΔTA) resulted in a milder, 8-fold and 2.6-fold (p<0.0001) induction in MCF7 and U2OS cells respectively (Fig 2D). This suggests that even just the binding of E2F1 to its site is sufficient to modulate the RAD51/TODRA expression ratio. Taken together, these results indicate that in the bidirectional promoter, transcription factor binding to the E2F site results in differential expression of the RAD51 and TODRA genes, and that E2F1, a known RAD51 activator, acts simultaneously as a transcriptional repressor of TODRA. This is only partly explained by E2F1-induced transactivation.
E2F1 regulates endogenous RAD51 and TODRA transcript levels
To determine the effect of E2F1 on activity of the endogenous RAD51/TODRA promoter we utilized a U2OS cell line stably transfected with a conditionally active E2F1 construct (ER-E2F1), in which 4-OHT (4-hydroxytamoxifen) treatment induces E2F1[21]. Elevated E2F1 occupancy of the RAD51 promoter after OHT treatment was confirmed using chromatin immunoprecipitation (ChIP) (Fig 3A). E2F1 induction resulted in a 3-fold increase (p = 0.004) in endogenous RAD51 mRNA levels and a 40% decrease (p = 0.0005) in endogenous TODRA transcript levels (Fig 3B). These results demonstrate that E2F1 oppositely regulates the endogenous RAD51/TODRA bidirectional promoter, providing in vivo evidence for its role in regulating both RAD51 and TODRA expression.
Fig 3. E2F1 induction oppositely affects endogenous RAD51 and TODRA expression.

A. E2F1 induction results in E2F1 binding of the RAD51/TODRA promoter. E2F1 expression was induced in serum starved ER-E2F1 U2OS cells (stably transfected with a constitutively expressed ER-E2F1 fusion protein which upon ligand-dependent activation translocates from the cytoplasm to the nucleus) by treatment with OHT for 8 hours. RAD51/TODRA promoter occupancy was measured with a ChIP assay using E2F1 antibodies (Ab) in lysates of either OHT treated or untreated cells. Real-time PCR was performed to quantitate the RAD51/TODRA template captured by the E2F1 Ab. Promoter occupancy is expressed as fold change relative to that observed in untreated cells. Values are means ± SE of 3 ChIP independent experiments. Real-time reactions were performed in triplicates. ^ p = 0.01. A representative gel of the promoter region PCR amplification products is shown on the left (S2 Fig). B. E2F1 induction and endogenous RAD51 and TODRA transcription. RAD51 and TODRA transcript levels were determined after E2F1 induction (see above), using quantitative real-time RT-PCR normalized to GAPDH. Results are depicted as the fold change in either RAD51 or TODRA transcript levels compared to non-treated cells (time 0). Values are means ± SE of 3–4 independent experiments. Real-time reactions were performed in duplicates. + p≤ 0.04, * p≤ 0.004.
TODRA promotes both RAD51-dependent homologous recombination (HR) repair of an ISceI-induced DSB, and formation of DNA damage-induced RAD51 foci.
To determine the functional relevance of TODRA to RAD51's role in DSB repair, we used a RAD51-dependent DSB repair assay and examined formation of RAD51 positive foci following DNA damage. To assess its effect on DSB repair, we expressed TODRA in HR-inducible (HRind) U2OS cells stably transfected with both a DR-GFP HR reporter cassette and an mCherry-ISceI-GR (Glucocorticoid Receptor) plasmid[22]. Addition of Dexamethasone to the cell media, translocates the ISceI endonuclease to the nucleus, where it generates a unique DSB within the DR-GFP cassette. Functional GFP is produced only upon HR repair of the DSB (Fig 4A). TODRA overexpression significantly increased the number of GFP-positive cells, by 1.6 fold (p<0.04) (Fig 4B). This change is of comparable magnitude to that seen with RAD51 depletion (which decreases HR by ~2.5-fold)[23].
Fig 4. TODRA promotes homologous recombination repair of DSBs.

A. Schematic representation of the HRind cell system . The mCherry-ISceI-GR (Glucoroticoid Receptor) endonuclease is cytoplasmic. Upon addition of Dexamethasone, it rapidly translocates into the nucleus generating a DSB at the ISceI site in the DR-GFP cassette. The DSB can be repaired either by NHEJ (non-homologous end-joining) or HR, but only HR repair reconstitutes functional GFP (green nucleus) from DR-GFP. B. Overexpression of TODRA induces RAD51-dependent HR. HRind cells were transfected with an empty vector (EV) or TODRA minigene and induced with Dexamethasone for 48 hours. GFP expression was measured by FACS. Results are depicted as the fold change in observed HR (as indicated by the number of GFP-positive cells) compared to the empty vector. Values are means ± SE of 3 independent experiments performed in triplicate. * p< 0.04. C. Overepxression of TODRA elevates DNA damage-induced RAD51 foci formation. U2OS cells were transfected with an empty vector (EV) or the TODRA minigene. 48 hrs. post transfection half of each culture was treated for 1 hr. with the DNA damaging agent phleomycin (10μg/ml). Medium was then replaced in all cultures, releasing treated cells from phleomycin exposure. γH2AX and RAD51 foci were imaged either immediately (0 hours) or 6 hours after removal of phleomycin and medium exchange. Top: A representative image of γH2AX (green) and RAD51 (red) foci in empty vector (EV) and TODRA transfected cells 6 hours after removal of phleomycin. DAPI (blue signal in merged images) was used for counterstaining. Scale bars = 10 μm. Bottom: The number of RAD51-positive foci was normalized as the fraction of γH2AX-positive foci per cell and averaged across all samples in each condition. Cells were treated with phleomycin, as indicated, and fixed at the indicated time points post-treatment. Results are depicted as the fold change in the fraction of RAD51 foci in cells overexpressing TODRA compared to the empty vector. Values are means ± SE of 3 independent experiments. * p≤ 0.03.
In addition, we analyzed RAD51 foci formation in U2OS cells treated with the DNA-damaging agent phleomycin. Overexpression of TODRA increased the proportion of RAD51-positive foci by 1.25–1.6 fold in transfected cells (Fig 4C) (p<0.03 vs. empty vector). Thus, TODRA overexpression resulted in similar increases of RAD 51-dependent repair activity using two different assays.
TPIP, a potential TODRA target gene, induces RAD51 expression synergistically with E2F1
To identify genes that may be regulated by TODRA and thus influence RAD51-dependent HR, we attempted to knock-down endogenous TODRA expression. However, we were unable to attain effective TODRA knockdown (we achieved less than 50% reduction in transcript levels). We therefore used the Affymetrix GeneChip Human Gene 1.0 ST expression array to screen for genes affected by overexpression of a TODRA minigene in HeLa cells. We observed no significant change in RAD51 mRNA levels; however, the TPIP transcript was significantly upregulated by TODRA overexpression.
TPIP belongs to the human TPTE (Transmembrane Phosphatase with TEnsin homology) gene family which encodes PTEN-related tyrosine phosphatases[24, 25]. PTEN was shown to transactivate the RAD51 promoter synergistically with E2F1, a member of the E2F transcription family[26]. We therefore investigated whether TPIP affects RAD51 promoter activity. TPIPα TPIPβ (predominant TPIP isoforms) or PTEN expression vectors[25] were co-transfected into MCF7 cells with a RAD51 promoter luciferase reporter (pRAD51-UTR) together with an E2F1 expression vector. Like PTEN, TPIP alone did not activate the RAD51 promoter. However, when co-expressed with E2F1, TPIPα (but not TPIPβ) augmented E2F1 induction of RAD51 promoter activity (by 1.4-fold (p = 0.002)), as compared to E2F1 alone (Fig 5A). These results demonstrate that like PTEN, TPIP acts as a co-factor of E2F1, inducing RAD51 expression.
Fig 5. TPIP regulates RAD51 expression and activity.

A. TPIP co-activates E2F1 induction of RAD51. pRAD51-UTR with an E2F1 expression vector or an empty vector were co-transfected into serum starved MCF7 cells together with either PTEN, TPIPα, TPIPβ or an empty pEGFP-C2 based expression vector and pRL-TK (to normalize for transfection efficiency). Results are depicted as fold change in RLA compared to pRAD51-UTR alone (left bar). Values are means ± SE of 3 independent transfections performed in duplicates. ** p< 0.0001. Additional comparisons are indicated above the bars. * p = 0.002. B. E2F1 induction and endogenous TPIP expression. Endogenous TPIP mRNA levels were determined using quantitative real-time RT-PCR normalized to GAPDH, with and without E2F1 induction in serum starved ER-E2F1 U2OS cells. E2F1 was induced by treatment with OHT for 4 hours. Values are means ± SE of 4 independent experiments. Real-time reactions were performed in duplicates. ** p< 0.00001. C. Overexpression of TPIP reduces HR. HRind cells were transfected with an mOrange2 control vector (CV) or TPIP expression vector (tagged with mOrange2) and induced with Dexamethasone for 48 hours. GFP expression was measured by FACS. Results are depicted as the fold change in observed HR (as indicated by the number of GFP-positive cells among the transfected population [mOrange2 positive cells]) compared to the control vector. Values are means ± SE of 3 independent experiments performed in triplicate. ** p<0.002.
To further investigate the interaction between TPIP and E2F1, we examined whether E2F1 can also regulate TPIP expression. Interestingly, much like for TODRA (Fig 3B), E2F1 overexpression resulted in a 4-fold decrease (p<0.00001) in TPIP mRNA levels (Fig 5B). This suggests that TPIP transcription is regulated in an E2F1-dependent manner, in parallel to TODRA.
Given that TPIP expression is modified by TODRA, and that the TPIP protein co-regulates RAD51 expression, we next asked whether TODRA's effect on RAD51-dependent HR is achieved via TPIP. We overexpressed TPIP in HR-inducible (HRind) U2OS cells described above and analyzed GFP expression in the transfected cells. TPIP overexpression reduced the number of GFP-positive cells, by 1.6 fold (p = 0.001) (Fig 5C). This suggests that TODRA increases RAD51-dependent HR independently of TPIP.
The RAD51/TODRA pathway in breast cancer
We also examined the RAD51-TODRA pathway in vivo, in breast tumors. Because TODRA has been described only recently, it is not represented in expression arrays used in many studies (e.g. the often cited van’t Veer et al. study[27]). We therefore analyzed data obtained by Muggerud et al.[28] who studied global gene expression of 109 breast tumors and 6 normal breast tissues using Agilent Whole Human Genome Oligo Microarrays 44k, which contain probes for RAD51, E2F1, TODRA and TPIP. Correlation of these genes' expression (Table 1) shows a negative correlation between RAD51 and TODRA (p = 0.01), reflecting their opposite regulation by E2F1, which is positively correlated with RAD51 (p< 0.001) and negatively with TODRA (p = 0.002). However, in contrast to our findings in U2OS cells (Fig 5B), E2F1 expression was positively correlated with TPIP expression, and there was no correlation between TODRA and TPIP expression. High expression of RAD51 and E2F1 also correlated with clinico-pathological features, and was associated with young age at onset (p = 0.002), a trend toward lymph node involvement at diagnosis (p = 0.08) and markers of aggressive disease, i.e. high tumor grade, lack of estrogen and progesterone receptor expression, Her2 amplification and ki67 staining (Table 1). TODRA expression was not significantly correlated with clinico-pathological features, but in general its effect was opposite to that of RAD51.
Table 1. The RAD51-TODRA regulatory pathway in breast cancer tumors.
| Correlation of gene-gene expression | ||||
| Transcript 1 | Transcript 2 | Correlation direction | correlation coefficient (r) | p-value |
| RAD51 | TODRA | - | 0.24 | 0.01 |
| E2F1 | RAD51 | + | 0.67 | <0.001 |
| E2F1 | TODRA | - | 0.30 | 0.002 |
| E2F1 * | TPIP * | + | 0.22 | 0.02 |
| TPIP | RAD51 | + | 0.16 | 0.09 (NS) |
| TODRA * | TPIP * | None | - | - |
| Correlation of gene expression and clinico-pathological features | ||||
| Transcript | Clinico-pathological feature | Correlation direction | correlation coefficient (r) | p-value |
| Age at diagnosis | - | 0.30 | 0.002 | |
| Lymph node involvement at diagnosis | + | 0.19 | 0.08 (NS) | |
| RAD51 | Tumor grade | + | 0.43 | <0.001 |
| Positive ER status | - | 0.22 | 0.02 | |
| Positive PR status | - | 0.27 | <0.001 | |
| HER2 amplification | + | 0.41 | 0.001 | |
| ki67 status | + | 0.36 | 0.005 | |
| Age at diagnosis | - | 0.25 | 0.008 | |
| Lymph node involvement at diagnosis | + | 0.21 | 0.04 | |
| E2F1 | Tumor grade | + | 0.37 | <0.001 |
| Positive ER status | - | 0.26 | 0.03 | |
| Positive PR status | - | 0.22 | 0.001 | |
| HER2 amplification | + | 0.22 | 0.03 | |
| ki67 status | + | 0.34 | 0.002 | |
| Tumor grade | + | 0.20 | 0.04 | |
| TPIP | Positive ER status | - | 0.30 | 0.01 |
| Positive PR status | - | 0.34 | 0.004 | |
Relationship between transcript expression levels, along the RAD51-TODRA regulatory pathway, in breast cancer tumors (based on data from Muggerud et al., 2010[28]). + positive correlation,—negative correlation. NS: not significant. All p-values are for 2-tailed analysis. Pearson correlation was used for comparison of continuous variables and Spearman correlation and t-test for non-parametric comparisons.
* Asterisks indicate gene-gene correlations that reflect perturbation of the normal pathway.
Discussion
RAD51 is an essential recombinase, often dysregulated in tumors[5–7], and tight regulation of its expression is pivotal for sustaining genome stability. We hypothesized that a novel lncRNA (AK125393) at the RAD51 locus, transcribed in the opposite direction of RAD51 (namely, TODRA) may play a role in RAD51 regulation. This hypothesis was based on the emerging roles of ncRNAs in general and lncRNAs in particular[29–31]. LncRNAs share features with protein coding genes: they are frequently long (generally >2kb, and some >100kb), spliced and contain canonical polyadenylation signals. In addition, lncRNA promoters are bound and regulated by transcription factors, e.g. Oct3/4, CREB, and p53, and epigenetically marked with specific histone modifications[32]. We found that TODRA's transcription indeed initiates 69bp upstream of the RAD51 TSS, and confirmed that it is transcribed and spliced using strand-specific RT-PCR (Fig 1A and 1B). Northern blotting did not identify a major transcription product, perhaps due to instability or heterogeneity of the transcript, consistent with evidence that lncRNAs are often degraded or processed into small RNAs[33–36]. We determined that the RAD51 promoter region supports bidirectional transcription, acting as a strong promoter in the direction of TODRA as well as in the direction of RAD51 (Fig 1C). Thus, TODRA is a bona fide lncRNA which is driven by the bidirectional RAD51 locus promoter.
Bidirectional promoters are a common organizational motif in the human genome, and notably, a number of DNA-repair genes are arranged in bidirectional pairs separated by less than 300bp[37, 38]. Most bidirectional promoters involving DNA-repair genes have not been thoroughly investigated, but two studied examples include the BRCA1/NRB2 locus[39–41] and the ATM/NPAT promoter[42]. Similar to RAD51 and TODRA, BRCA1 and NBR2 (neighbour of BRCA1 gene 2) are aligned in a head-to-head orientation separated by 218bp, and NRB2, which is annotated as a ncRNA, is transcribed in the opposite direction to BRCA1.
LncRNAs constitute a large portion of the mammalian transcriptome, and play a regulatory role in a range of biological pathways[29–31]. Impaired lncRNA expression and function contribute to disease pathogenesis, including cancer[32, 43–45]. For example, MEG3 (maternally expressed gene 3), is an imprinted, maternally expressed gene, encoding a lncRNA that functions as a tumor suppressor. MEG3 regulates TP53 protein levels and plays a role in tumor cell proliferation, apoptosis and angiogenesis. Its expression is lost in various human tumors (e.g. human pituitary tumors derived from gonadotroph cells and bladder cancer) as well as in tumor cell lines of various origins[46, 47]. Moreover, a more global pattern of altered lncRNA expression has been reported in both breast and ovarian cancers[44, 48].
LncRNA promoters, like protein coding genes, are regulated by transcription factors [32]. The ATM/NPAT bidirectional promoter noted above is regulated by E2F1, which induces expression of both genes[42]. Indeed, E2F1/E2F4 recognition sequences are significantly over-represented in bidirectional compared to unidirectional promoters[49]. The RAD51/TODRA promoter region also contains an E2F binding site that was previously studied in the context of RAD51 regulation[17–19]. In quiescent and hypoxic cells this E2F site is primarily occupied by E2F4 (a component of the repressor complex E2F4/p130) whereas in response to growth stimulation, E2F4 is displaced and RAD51 expression is activated by binding of E2F1 to the same E2F site[17, 18]. We found that the E2F site also regulates TODRA expression, but its effects are opposite to those on RAD51. Abolishing the E2F site, both in uni- and bi-directional constructs, increased RAD51 promoter activity but decreased TODRA promoter activity (Fig 2). The E2F site can bind various members of the E2F family, but since E2F1 and RAD51 are often dysregulated in cancer, we focused on the effect of E2F1 on the bidirectional promoter. We found that while E2F1 overexpression induced RAD51 promoter activity, as previously reported[17], it reduced TODRA promoter activity (Fig 2). In vivo, E2F1 induction increased endogenous RAD51 mRNA levels and decreased endogenous TODRA transcript levels (Fig 3B). Deletion of the transactivating domain of E2F1 attenuated, but did not abolish, these effects (Fig 2D). This suggests that the E2F1 effect at the RAD51/TODRA promoter is mediated via two mechanisms: 1) E2F1 binding displaces E2F4, which is normally bound to this site, thereby de-repressing RAD51 expression and 2) E2F1 actively induces the RAD51 promoter and represses TODRA expression. This dual effect can explain why both mutagenesis of the E2F site and E2F1 overexpression result in RAD51 activation and TODRA repression. It can also explain the partial effect of the E2F1 transactivating mutant, which can displace E2F4, but cannot act as a transcriptional activator. This model is also supported by previous studies which showed alternate occupancy of the RAD51 promoter by E2F1 and E2F4[17, 18].
RAD51 regulation by E2Fs conforms to the common functions of E2F1 as a transcriptional activator and E2F4 as a repressor[10]. However, as our results on TODRA and TPIP expression reveal, this dichotomy is not universal. E2F1 decreases expression of many genes, e.g., BCL3, TGFB2 and INHBA[50, 51], and E2F4 can activate expression of numerous genes, including genes involved in cell cycle, DNA replication and DNA-repair (e.g. RPA1 and SMC3)[52]. Taken together, our results demonstrate that an E2F site in the RAD51/TODRA bidirectional promoter differentially regulates RAD51 and TODRA expression. To the best of our knowledge this is the first example showing opposing effects of E2F1 binding at a single site on transcription of two genes from a bidirectional promoter.
The functional relevance of TODRA with respect to RAD51 function was examined by analysis of RAD51 foci formation following DNA damage as well as a RAD51-dependent DSB repair assay (Fig 4). In these assays, TODRA significantly increased both the fraction of RAD51-positive DNA damage-induced foci and HR repair efficiency (Fig 4B and 4C). To further explore TODRA's role in HR we explored potential targets that might mediate its activity. We found that TODRA overexpression upregulates TPIP (TPTE and PTEN homologous Inositol lipid Phosphatase, MIM #606791), providing a new example of a lncRNA regulating transcription of a protein-coding gene in another locus. TPIP encodes a PTEN-related tyrosine phosphatase, but unlike PTEN, little is known about TPIP's function. TPIP is highly expressed in the testis and at lower levels in the brain and the stomach. Extensive alternative splicing of this gene leads to multiple isoforms. Two predominant forms, TPIPα and TPIPβ, differ at both the N- and C-termini, and only isoform α displays phosphoinositide 3-phosphatase activity[25]. PTEN has been shown to augment E2F1 induction of RAD51[26]. We found that TPIPα, but not TPIPβ, has a similar effect on RAD51 expression (Fig 5A). This raises the possibility that other TPTEs also co-regulate RAD51 expression.
Given our findings that TODRA enhances both TPIP expression and RAD51-dependent DNA repair, and that TPIP co-activates RAD51, the simplest hypothesis was that TODRA increases RAD51-dependent HR via upregulation of TPIP. However, this was not the case; Overexpression of TPIP decreased RAD51-dependent HR (Fig 5C). Even so, we found that both genes now newly implicated in regulating RAD51 expression, affect RAD51-dependent HR. The opposite effects of TODRA and TPIP expression on this process probably reflect its complex regulation. RAD51-dependent double strand break repair is regulated on many non-transcriptional levels, including post-transcriptional modifications such as phosphorylation of both RAD51[53–56] and its partner, BRCA2[57]. Interestingly, a recent study[58] demonstrates that nuclear phosphoinositide signaling may play a role in nuclear export of RAD51 mRNA. Thus, while TPIP can increase RAD51 expression in certain settings (e.g. when E2F1 is expressed), it might also reduce its translation and functional activity by hampering the export of newly transcribed RAD51 transcripts to the cytoplasm.
Finally, we examined the expression of genes associated with the RAD51-TODRA regulatory pathway in breast cancer tumors. RAD51 expression in breast tumors was positively correlated with E2F1 expression and negatively correlated with TODRA (Table 1), indicating that E2F1 indeed regulates the bidirectional promoter in vivo in the malignant state, in the same manner we observed in cell lines. Although in cell lines we found that E2F1 induction reduces TPIP expression, in breast tumors there was a positive correlation between E2F1 and TPIP expression levels. This observation could indicate loss of normal fine-tuning of RAD51 expression in the malignant vs. the normal state. While RAD51 and TODRA expression are directly linked, through a shared E2F-binding site, the effect on TPIP may be less direct, and thus more likely to be affected by additional factors. E2F1 pathways are commonly dysregulated in cancer[10], and increased TPIP expression in the presence of increased E2F1 expression in tumors could reflect dysregulation of TPIP transcription.
Importantly, the positive correlation between E2F1/TPIP and RAD51 expression we show in tumors is consistent with our finding that TPIP is a novel co-activator of E2F1 in RAD51 induction. Increased RAD51 levels may contribute to tumorigenesis, and accordingly we found that elevated RAD51 levels are associated with young age at breast cancer onset, higher tumor grade and characteristics of aggressive tumors (e.g. lack of hormone receptor expression and HER2 amplification).
To summarize, our findings reveal novel and complex regulatory mechanisms of RAD51 expression and activity. We found that a lncRNA, TODRA, regulates both the expression and the activity of a protein coding gene (RAD51), driven, and oppositely regulated, by the same promoter. In a feedback loop (Fig 6), TODRA overexpression increases levels of TPIP, which we subsequently identified as a new RAD51 co-activator. Additionally, both TODRA and TPIP are able to fine-tune homologous recombination, the primary activity of RAD51. Further investigation is needed to determine the mechanism that underlies TODRA regulation of TPIP as well as RAD51 activity, and to determine the role of this pathway in dysregulation of RAD51 expression in malignancy.
Fig 6. TODRA lncRNA plays a role in a new feedback loop regulating RAD51 expression and activity.
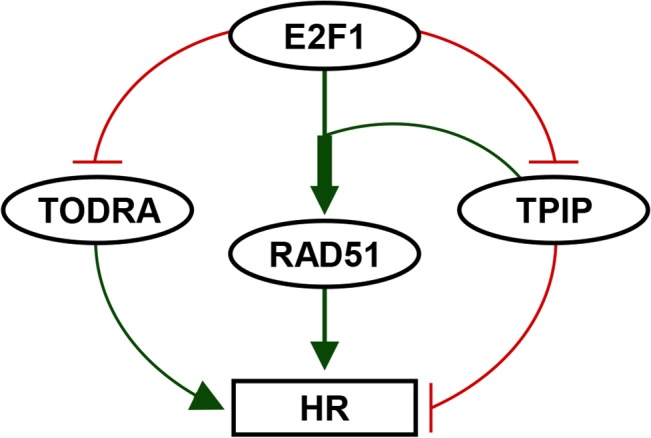
E2F1 induction enhances RAD51 expression (thin green arrow) while simultaneously reducing lncRNA TODRA expression. While E2F1 induction of RAD51 is synergistically enhanced by TPIP (thick green arrow), E2F1 induction also reduces TPIP expression, possibly by affecting TODRA expression, as TODRA expression can increase TPIP expression. This feedback regulation of RAD51 expression can fine-tune RAD51 expression and HR-DSB repair. Green: Enhancement of expression/activity. Red: Suppression of expression/activity.
Materials and Methods
5′ and 3' Rapid Amplification of cDNA Ends (RACE) and reverse transcription.
5′ and 3' ends of RAD51 and TODRA (AK125393) transcripts were determined using First Choice RLM-RACE kit (Ambion, Austin, TX, USA) on de-capped mRNA from HeLa cells, using the gene specific primers: (5'-AGCTGCCTCAGCCAGAATTTTA-3') and (5'-CCACACTGCTCTAACCGTGA-3'); (5′-ATGCGAGTAGGAGGCTCAGA-3′) and (5′-TACTGCCGAAACAAACCACA-3′) and (5′-AATAGTCCAGCTGCGATGGT-3′).
3'RACE used cDNA prepared from total RNA, using the primers: (5′-AGATAAACCTGGCCAACGTG-3′) and (5′-TGAACTCAGGAGGAGGTTGC-3′) or (5′-ATCGCCTTTCCAGTTTCTCA-3′) and (5′-CCCTACATTCCAATAACTCTACCTTC-3′) or (5′-TCTGCTCCTAATTCACCTCCTC-3′) and (5′-AAAACTAAGCCCAGCCGAAC-3′).
Strand-specific Reverse Transcription was performed using specific primers and SuperScript II (Invitrogen, Carlsbad, CA, USA) in the presence of RNase inhibitor (rRNasin, Promega, Madison, WI, USA).
Cell culture
HeLa (ATCC, obtained from Ilana Keshet, the Hebrew University in Jerusalem), MCF7 (ATCC, obtained from Mary-Claire King, University of Washington, Seattle), and U2OS (ATCC) cells were cultured in DMEM, supplemented with 10% Fetal Bovine Serum (FBS), 2mM L-glutamine, 10U penicillin, 10μg streptomycin/ml (Biological Industries, Beit Ha'emek, Israel). Media for U2OS ER-E2F1 stable lines[42] also contained 0.5mg/ml G418 (Alexis Biochemicals, San Diego, CA, USA). Cell lines were maintained at 37°C with 5% CO2 and subcultured 2–3 times weekly.
ER-E2F1 induction of stably transfected U2OS cells, employed 300nM 4-hydroxytamoxifen (OHT) (Sigma-Aldrich, St Louis, MO, USA) following 48 hr. serum starvation (0.1% FBS containing medium).
Plasmid construction
pRAD51-UTR, containing the RAD51 promoter/5'UTR (-663 to +252, relative to the RAD51 TSS) was cloned into pGL3-promoter (Promega). pRAD51, contains the RAD51 core promoter (-114 to +15, relative to the RAD51 TSS). pTODRA, contains the TODRA putative promoter (-320 to +40 relative to the TODRA TSS), in pGL3-basic (Promega). The bidirectional promoter (pBDP) was constructed by inserting the firefly luciferase gene into the Renilla luciferase reporter pGL4.70[hRluc] (Promega) in the opposite orientation. The RAD51/TODRA overlapping promoters were then amplified with the primers: (5'-CGTTCCATGGTACTCGGTCCGCAGCGCTC-3') and (5'-ATTCCATGGTCCCGTCTTGGGTTAGC-3'), and cloned between the two luciferase genes. The TODRA minigene was generated by PCR of gDNA using the primers (5'-CGCGTCGACGTAACGTATC-3') and (5'-ATTGCGGCCGCGAGACAAAGTTTCACTCTTTCG-3') and cloned downstream of an SV40 promoter in a pBluescript SK+ based vector.
Restriction and DNA modification enzymes were acquired from Fermentas MBI (Lithuania) and New England Biolabs (Beverly, MA, USA). QuickChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) was used to introduce a 3bp mutation in the E2F site, confirmed by sequencing.
Antibodies
The following primary antibodies were used in this study: polyclonal rabbit anti-E2F1 (C-20; Santa Cruz Biotechnology Cat# sc-193, RRID:AB_631394); monoclonal mouse anti-phospho-Histone H2A.X (Ser139), clone JBW301 (Millipore Cat# 05–636, RRID:AB_309864); and polyclonal rabbit anti-Rad51 (Ab-1; Merck Cat# PC130-100UL, RRID:AB_10684676). The following secondary antibodies were used in this study: DyLight 488 AffiniPure polyclonal Donkey anti Mouse IgG (H+L); and DyLight 594 monoclonal Donkey anti-rabbit IgG (minimal x-reactivity) (BioLegend Cat# 406405, RRID:AB_1575132).
Transient transfections and luciferase activity measurement
MCF7 and HeLa cells were transfected in 50% confluent 24-well plates using jetPEI cationic polymer transfection reagent (Polyplus Transfection, Illkirch, France). Co-transfections with an E2F1 expression vector were preceded by 48 hr. serum starvation (with 0.1% FBS containing medium). Where indicated PTEN, TPIPα or TPIPβ pEGFP-C2 expression constructs were added (generous gift of Nicholas Leslie, University of Dundee).
Firefly and Renilla luciferase activities were quantitated sequentially using the Dual Luciferase Assay system (Promega) in a MiniLumat LB 9506 luminometer (EG&G Berthold, Germany). Where indicated, the pRL-TK, a Renilla luciferase plasmid driven by the HSV-thymidine kinase (TK) promoter (Promega), was used to normalize for transfection efficiencies by calculating the relative firefly/Renilla luciferase activities (RLA). Results of duplicates were averaged, and corrected for plasmid size differences to reflect equimolar measurements. Statistical analysis was performed using student's t-test.
Quantitative Real Time RT-PCR
Total U2OS RNA was isolated using Tri-Reagent (Molecular Research Center Inc, Cincinnati, OH, USA) and reverse transcribed with ImpromII Reverse Transcriptase (Promega). The TPIP assay included a pre-amplification step using the TaqMan PreAmp Master Mix Kit (Applied Biosystems [ABI], Foster City, CA, USA). Real-time qPCR was performed using universal TaqMan or Power SYBR Green PCR master mix (ABI) in duplicates on the ABI PRISM 7900 Sequence Detector (ABI). Threshold cycle (Ct) values of the amplified genes were normalized to GAPDH levels, and relative expression levels were quantitated using the comparative method (User Bulletin #2, ABI PRISM 7700 Sequence Detection System, 1997) and calculated as 2−ΔΔCt.
All assays targeted RNA specific amplicons using either the TaqMan Gene Expression Assays Hs01685755_m1 for TPIP, or SYBR Green assays using the primers: RAD51: (5′-GCCCACAACCCATTTCAC-3′) and (5′-GGCAACAGCCTCCACAGTAT-3′), TODRA: (5′-TGATCCTGCGCGAGTTTACA-3′) and (5′-GGCCGACGCATTACCTCTT-3′) and GAPDH: (5′-CAGCCTCAAGATCATCAGCA-3′) and (5′-ACAGTCTTCTGGGTGGCAGT-3′).
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using the EZ-ChIP kit (Millipore, Billerica, MA, USA). U2OS cells were cross-linked with 1% formaldehyde and sonicated. Immunoprecipitations used 1.5μg of polyclonal anti-E2F1 antibody (validated for ChIP in [59]) overnight at 4°C in parallel with no-antibody controls.
Quantitative analysis of ChIP assays was performed using real-time PCR in triplicates with Power SYBR Green, using the primers: (5′- GGAGGCGGGGATACGTTAC-3′) and (5′-CTCTCCTTAGGGCTCGGTC-3′). Calculations of Relative Promoter Occupancy were based on a previously described method[60], and calculated as: (1+Eff)ΔCt. Fold change in promoter occupancy was calculated between pairs of E2F1 induction/control: [(1+Eff) ΔCt OHT]/[(1+Eff) ΔCt control].
mRNA expression Microarray
Total RNA extracted from two TODRA overexpressing and two control transfected Hela cell cultures (RNeasy Mini Kit, Qiagen, GmbH, Hilden, Germany) was assessed on a NanoDrop spectrophotometer (Thermo Fisher Scientific) and a 2100 Bioanalyzer (Agilent, Palo Alto, CA USA), amplified, fragmented and biotinylated as cDNA using the Affymetrix GeneChip Whole Transcript (WT) Sense Target Labeling Assay (Affymetrix, Santa Clara, CA) and hybridized to Affymetrix GeneChip Human Gene 1.0 ST expression arrays. The arrays were washed, stained, and scanned using the Affymetrix GeneChip Fluidics Station 400 and GeneChip Scanner 3000 7G.
Microarray data analysis
Raw data was analyzed using the robust multiarray average (RMA) algorithm (Affymetrix Expression Console and Partek Genomics Suite 6.4)[61]. Raw intensity values were background corrected, log2 transformed, quantile normalized and a linear model was fit to the data to obtain an expression summary value for each probe set on each array. The data was analyzed using unsupervised hierarchical cluster analysis (Spotfire DecisionSite for Functional Genomics) and ANOVA (Partek) or student’s t-test. Fold change and p-value were used to identify differentially expressed genes. Complete microarray data are deposited at the Gene Expression Omnibus (GEO) database repository (NCBI) http://www.ncbi.nlm.nih.gov/geo/.
HR Assay
DSB repair by HR was assessed in HR-inducible (HRind) U20S-DR-GFP cells stably transfected with an mCherry-ISceI-GR nuclease, as previously described[22]. In this study, HRind cells were grown in 6 well plates and transiently transfected with either the TODRA minigene, TPIP expression construct (tagged by mOrange2), or an appropriate empty vector. DSBs were induced by adding Dexamethasone to the growth medium 20–24 hours after transfection. Cells that were mCherry-positive (and mOrange2-positive in TPIP transfection experiments) or mCherry+GFP-positive (and mOrange2-positive in TPIP transfections) were counted 48 hours later by flow cytometry using the GACS ARIA III platform (Beckton Dickinson) and the GFP-positive fraction was calculated. Results of triplicates were averaged, and statistical analysis was performed using a student's t-test.
Immunostaining and quantification of RAD51 foci
U2OS cells were grown on coverslips and transfected with either the TODRA minigene (pTODRA) or empty vector (pcDNA3). At 48 hours post-transfection, DNA damage was induced by adding phleomycin (10μg/ml) to the growth medium for 1 hour at 37°C. Cells were then either fixed immediately in 4% formaldehyde (time 0 post treatment, Fig 4C) or allowed to recover for 6 hours in fresh growth medium before fixation. For immunostaining, cells were permeabilized with 0.5% Triton X-100 and blocked in 2% BSA-PBS for 1 hour. After blocking, cells were probed with monoclonal anti-phospho-Histone H2A.X (Ser139)(1:300 dilution; validated for immunofluorescence in [62]) and polyclonal anti-RAD51 (1:300 dilution; validated for immunofluorescence in [63]) antibodies followed by secondary detection with donkey anti-mouse DyLight 488 (1:700 dilution) and donkey anti-rabbit DyLight 594 (1:700 dilution) fluorescent antibodies. For counterstaining, DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) and coverslips were then mounted onto slides with fluorescence mounting medium (Dover Medical). Fluorescent images were captured on an Olympus IX81 inverted microscope and processed off-line for foci counting using FiJi ImageJ software[64]. The number of RAD51-positive foci was normalized as the fraction of gamma-H2AX-positive foci per cell and averaged across all counted cells in each experimental condition. Results of triplicates were averaged, and statistical analysis was performed using student's t-test.
Breast cancer tumors data analysis
We analyzed publicly available gene expression data from Muggerud et al.[28] including tumor samples from 31 cases of pure DCIS, 36 pure invasive cancers, 42 cases of mixed diagnosis (invasive cancer with an in situ component) and 6 normal breast tissue samples. In this study, global gene expression was assayed using the Agilent Whole Human Genome Oligo Microarrays 44k. Available data on samples included clinical information, e.g. age at onset, and pathology data, e.g. tumor grade, and ER, PR, ki67 and HER2 amplification status. For each tumor sample, the relative expression of each gene (RAD51, TODRA, TPIP and E2F1), was normalized to the average expression of the same gene in the normal tissue samples. Correlation between expression levels of the different genes, and between expression levels and pathological and clinical information was performed using Pearson correlation for continuous variables and Spearman correlation and t-test for non-parametric comparisons (PASW Statistics 18).
Supporting Information
pBDP activity was examined in MCF7 cells co-transfected with the pBDP construct and either an E2F4 or an empty expression vector. Results are depicted as the fold change between each E2F4 expression vector and the empty vector control, in the ratio of Firefly/Renilla luciferase activities, which represents the ratio of RAD51/TODRA promoter activities. Values are means ± SE of 3 independent transfections performed in duplicate.
(TIF)
E2F1 expression was induced in serum starved ER-E2F1 U2OS cells (stably transfected with a constitutively expressed ER-E2F1 fusion protein which upon ligand-dependent activation translocates from the cytoplasm to the nucleus) by treatment with OHT for 8 hours. RAD51/TODRA promoter occupancy was measured with a ChIP assay using E2F1 antibodies (Ab) in lysates of either OHT treated or untreated cells. Shown here is an unformatted representative gel of the promoter region PCR amplification products.
(TIF)
Acknowledgments
We thank Ellie and David Werber, who are pleased and honored to have supported the research described in this paper. We thank Nicholas Leslie (University of Dundee) and Hermona Soreq (Hebrew University) for the gift of plasmids.
Data Availability
Complete microarray data are deposited at the Gene Expression Omnibus (GEO) database repository (NCBI) through GEO Series accession number GSE66062 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66062).
Funding Statement
This work was supported by the New York Breast Cancer Research Foundation (IG, ELL), the Israel Science Foundation (Grant Number: 635/06 to DG; 648/13 to MG), and a gift of Ellie and David Werber (IG, ELL). These funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Suwaki N, Klare K, Tarsounas M. RAD51 paralogs: roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin Cell Dev Biol. 2011;22(8):898–905. Epub 2011/08/09. 10.1016/j.semcdb.2011.07.019 [DOI] [PubMed] [Google Scholar]
- 2. Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113. Epub 2008/01/02. 10.1038/cr.2008.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2(3):196–206. Epub 2001/03/21. [DOI] [PubMed] [Google Scholar]
- 4. Kasparek TR, Humphrey TC. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin Cell Dev Biol. 2011;22(8):886–97. Epub 2011/10/27. 10.1016/j.semcdb.2011.10.007 [DOI] [PubMed] [Google Scholar]
- 5. Mitra A, Jameson C, Barbachano Y, Sanchez L, Kote-Jarai Z, Peock S, et al. Overexpression of RAD51 occurs in aggressive prostatic cancer. Histopathology. 2009;55(6):696–704. Epub 2009/12/17. 10.1111/j.1365-2559.2009.03448.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maacke H, Jost K, Opitz S, Miska S, Yuan Y, Hasselbach L, et al. DNA repair and recombination factor Rad51 is over-expressed in human pancreatic adenocarcinoma. Oncogene. 2000;19(23):2791–5. Epub 2000/06/13. [DOI] [PubMed] [Google Scholar]
- 7. Yoshikawa K, Ogawa T, Baer R, Hemmi H, Honda K, Yamauchi A, et al. Abnormal expression of BRCA1 and BRCA1-interactive DNA-repair proteins in breast carcinomas. Int J Cancer. 2000;88(1):28–36. Epub 2000/08/30. [PubMed] [Google Scholar]
- 8. Richardson C, Stark JM, Ommundsen M, Jasin M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene. 2004;23(2):546–53. Epub 2004/01/16. [DOI] [PubMed] [Google Scholar]
- 9. Flygare J, Falt S, Ottervald J, Castro J, Dackland AL, Hellgren D, et al. Effects of HsRad51 overexpression on cell proliferation, cell cycle progression, and apoptosis. Exp Cell Res. 2001;268(1):61–9. Epub 2001/07/20. [DOI] [PubMed] [Google Scholar]
- 10. Polager S, Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008;18(11):528–35. Epub 2008/09/23. 10.1016/j.tcb.2008.08.003 [DOI] [PubMed] [Google Scholar]
- 11. Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene. 2008;27(35):4860–4. Epub 2008/04/14. 10.1038/onc.2008.117 [DOI] [PubMed] [Google Scholar]
- 12. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245–62. Epub 1998/08/08. [DOI] [PubMed] [Google Scholar]
- 13. Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, et al. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16(2):245–56. Epub 2002/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cam H, Dynlacht BD. Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell. 2003;3(4):311–6. Epub 2003/05/03. [DOI] [PubMed] [Google Scholar]
- 15. Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, et al. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21(14):4684–99. Epub 2001/06/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 2002;16(2):235–44. Epub 2002/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iwanaga R, Komori H, Ohtani K. Differential regulation of expression of the mammalian DNA repair genes by growth stimulation. Oncogene. 2004;23(53):8581–90. Epub 2004/10/07. [DOI] [PubMed] [Google Scholar]
- 18. Bindra RS, Glazer PM. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene. 2007;26(14):2048–57. Epub 2006/09/27. [DOI] [PubMed] [Google Scholar]
- 19. Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci U S A. 2010;107(5):2201–6. Epub 2010/01/19. 10.1073/pnas.0904783107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36(1):40–5. Epub 2004/01/01. [DOI] [PubMed] [Google Scholar]
- 21. Chaussepied M, Ginsberg D. Transcriptional regulation of AKT activation by E2F. Mol Cell. 2004;16(5):831–7. Epub 2004/12/03. [DOI] [PubMed] [Google Scholar]
- 22. Shahar OD, Raghu Ram EV, Shimshoni E, Hareli S, Meshorer E, Goldberg M. Live imaging of induced and controlled DNA double-strand break formation reveals extremely low repair by homologous recombination in human cells. Oncogene. 2012;31(30):3495–504. Epub 2011/11/23. 10.1038/onc.2011.516 [DOI] [PubMed] [Google Scholar]
- 23. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature. 2007;450(7169):509–14. Epub 2007/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tapparel C, Reymond A, Girardet C, Guillou L, Lyle R, Lamon C, et al. The TPTE gene family: cellular expression, subcellular localization and alternative splicing. Gene. 2003;323:189–99. Epub 2003/12/09. [DOI] [PubMed] [Google Scholar]
- 25. Walker SM, Downes CP, Leslie NR. TPIP: a novel phosphoinositide 3-phosphatase. Biochem J. 2001;360(Pt 2):277–83. Epub 2001/11/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–70. Epub 2007/01/16. [DOI] [PubMed] [Google Scholar]
- 27. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6. Epub 2002/02/02. [DOI] [PubMed] [Google Scholar]
- 28. Muggerud AA, Hallett M, Johnsen H, Kleivi K, Zhou W, Tahmasebpoor S, et al. Molecular diversity in ductal carcinoma in situ (DCIS) and early invasive breast cancer. Mol Oncol. 2010;4(4):357–68. Epub 2010/07/29. 10.1016/j.molonc.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10(3):155–9. Epub 2009/02/04. 10.1038/nrg2521 [DOI] [PubMed] [Google Scholar]
- 30. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–41. Epub 2009/02/26. 10.1016/j.cell.2009.02.006 [DOI] [PubMed] [Google Scholar]
- 31. Lipovich L, Johnson R, Lin CY. MacroRNA underdogs in a microRNA world: evolutionary, regulatory, and biomedical significance of mammalian long non-protein-coding RNA. Biochim Biophys Acta. 2010;1799(9):597–615. Epub 2010/10/19. 10.1016/j.bbagrm.2010.10.001 [DOI] [PubMed] [Google Scholar]
- 32. Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol. 2010;220(2):126–39. Epub 2009/11/03. 10.1002/path.2638 [DOI] [PubMed] [Google Scholar]
- 33. Carninci P. Molecular biology: The long and short of RNAs. Nature. 2009;457(7232):974–5. Epub 2009/02/20. [DOI] [PubMed] [Google Scholar]
- 34. Project ACSHLET. Post-transcriptional processing generates a diversity of 5'-modified long and short RNAs. Nature. 2009;457(7232):1028–32. Epub 2009/01/27. 10.1038/nature07759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316(5830):1484–8. Epub 2007/05/19. [DOI] [PubMed] [Google Scholar]
- 36. Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, et al. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322(5909):1851–4. Epub 2008/12/06. 10.1126/science.1164096 [DOI] [PubMed] [Google Scholar]
- 37. Adachi N, Lieber MR. Bidirectional gene organization: a common architectural feature of the human genome. Cell. 2002;109(7):807–9. Epub 2002/07/12. [DOI] [PubMed] [Google Scholar]
- 38. Trinklein ND, Aldred SF, Hartman SJ, Schroeder DI, Otillar RP, Myers RM. An abundance of bidirectional promoters in the human genome. Genome Res. 2004;14(1):62–6. Epub 2004/01/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suen TC, Goss PE. Transcription of BRCA1 is dependent on the formation of a specific protein-DNA complex on the minimal BRCA1 Bi-directional promoter. J Biol Chem. 1999;274(44):31297–304. Epub 1999/10/26. [DOI] [PubMed] [Google Scholar]
- 40. Suen TC, Tang MS, Goss PE. Model of transcriptional regulation of the BRCA1-NBR2 bi-directional transcriptional unit. Biochim Biophys Acta. 2005;1728(3):126–34. Epub 2005/03/22. [DOI] [PubMed] [Google Scholar]
- 41. Xu CF, Chambers JA, Solomon E. Complex regulation of the BRCA1 gene. J Biol Chem. 1997;272(34):20994–7. Epub 1997/08/22. [DOI] [PubMed] [Google Scholar]
- 42. Berkovich E, Ginsberg D. ATM is a target for positive regulation by E2F-1. Oncogene. 2003;22(2):161–7. [DOI] [PubMed] [Google Scholar]
- 43. Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38 Epub 2011/04/15. 10.1186/1476-4598-10-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perez DS, Hoage TR, Pritchett JR, Ducharme-Smith AL, Halling ML, Ganapathiraju SC, et al. Long, abundantly expressed non-coding transcripts are altered in cancer. Hum Mol Genet. 2008;17(5):642–55. Epub 2007/11/17. [DOI] [PubMed] [Google Scholar]
- 45. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–8. Epub 2010/06/26. 10.1038/nature09144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ying L, Huang Y, Chen H, Wang Y, Xia L, Chen Y, et al. Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol Biosyst. 2013;9(3):407–11. Epub 2013/01/09. 10.1039/c2mb25386k [DOI] [PubMed] [Google Scholar]
- 47. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. 2012;48(3):R45–53. Epub 2012/03/07. 10.1530/JME-12-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guffanti A, Iacono M, Pelucchi P, Kim N, Solda G, Croft LJ, et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163 Epub 2009/04/22. 10.1186/1471-2164-10-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin JM, Collins PJ, Trinklein ND, Fu Y, Xi H, Myers RM, et al. Transcription factor binding and modified histones in human bidirectional promoters. Genome Res. 2007;17(6):818–27. Epub 2007/06/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, et al. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15(3):267–85. Epub 2001/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Polager S, Kalma Y, Berkovich E, Ginsberg D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene. 2002;21(3):437–46. Epub 2002/02/01. [DOI] [PubMed] [Google Scholar]
- 52. Lee BK, Bhinge AA, Iyer VR. Wide-ranging functions of E2F4 in transcriptional activation and repression revealed by genome-wide analysis. Nucleic Acids Res. 2011;39(9):3558–73. Epub 2011/01/21. 10.1093/nar/gkq1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7(2):195–201. Epub 2005/01/25. [DOI] [PubMed] [Google Scholar]
- 54. Chen G, Yuan SS, Liu W, Xu Y, Trujillo K, Song B, et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J Biol Chem. 1999;274(18):12748–52. Epub 1999/04/23. [DOI] [PubMed] [Google Scholar]
- 55. Yuan ZM, Huang Y, Ishiko T, Nakada S, Utsugisawa T, Kharbanda S, et al. Regulation of Rad51 function by c-Abl in response to DNA damage. J Biol Chem. 1998;273(7):3799–802. Epub 1998/03/28. [DOI] [PubMed] [Google Scholar]
- 56. Shimizu H, Popova M, Fleury F, Kobayashi M, Hayashi N, Sakane I, et al. c-ABL tyrosine kinase stabilizes RAD51 chromatin association. Biochem Biophys Res Commun. 2009;382(2):286–91. Epub 2009/03/17. 10.1016/j.bbrc.2009.03.020 [DOI] [PubMed] [Google Scholar]
- 57. Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, et al. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434(7033):598–604. Epub 2005/04/01. [DOI] [PubMed] [Google Scholar]
- 58. Wickramasinghe VO, Savill JM, Chavali S, Jonsdottir AB, Rajendra E, Gruner T, et al. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol Cell. 2013;51(6):737–50. Epub 2013/10/01. 10.1016/j.molcel.2013.08.031 [DOI] [PubMed] [Google Scholar]
- 59. Maser RS, Mirzoeva OK, Wells J, Olivares H, Williams BR, Zinkel RA, et al. Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol Cell Biol. 2001;21(17):6006–16. Epub 2001/08/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, et al. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005;65(24):11597–604. Epub 2005/12/17. [DOI] [PubMed] [Google Scholar]
- 61. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2). 246–249. [DOI] [PubMed] [Google Scholar]
- 62. Chia J, Goh G, Racine V, Ng S, Kumar P, Bard F. RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. Molecular systems biology. 2012;8:629 Epub 2012/12/06. 10.1038/msb.2012.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Viera A, Rufas JS, Martinez I, Barbero JL, Ortega S, Suja JA. CDK2 is required for proper homologous pairing, recombination and sex-body formation during male mouse meiosis. Journal of cell science. 2009;122(Pt 12):2149–59. Epub 2009/06/06. [DOI] [PubMed] [Google Scholar]
- 64. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9(7):676–82. Epub 2012/06/30. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
pBDP activity was examined in MCF7 cells co-transfected with the pBDP construct and either an E2F4 or an empty expression vector. Results are depicted as the fold change between each E2F4 expression vector and the empty vector control, in the ratio of Firefly/Renilla luciferase activities, which represents the ratio of RAD51/TODRA promoter activities. Values are means ± SE of 3 independent transfections performed in duplicate.
(TIF)
E2F1 expression was induced in serum starved ER-E2F1 U2OS cells (stably transfected with a constitutively expressed ER-E2F1 fusion protein which upon ligand-dependent activation translocates from the cytoplasm to the nucleus) by treatment with OHT for 8 hours. RAD51/TODRA promoter occupancy was measured with a ChIP assay using E2F1 antibodies (Ab) in lysates of either OHT treated or untreated cells. Shown here is an unformatted representative gel of the promoter region PCR amplification products.
(TIF)
Data Availability Statement
Complete microarray data are deposited at the Gene Expression Omnibus (GEO) database repository (NCBI) through GEO Series accession number GSE66062 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66062).