

Abstract
Neuromuscular diseases (NMD) account for a significant proportion of infant and childhood mortality and devastating chronic disease. Determining the specific diagnosis of NMD is challenging due to thousands of unique or rare genetic variants that result in overlapping phenotypes. We present four unique childhood myopathy cases characterized by relatively mild muscle weakness, slowly progressing course, mildly elevated creatine phosphokinase (CPK), and contractures. We also present two additional cases characterized by severe prenatal/neonatal myopathy. Prior extensive genetic testing and histology of these cases did not reveal the genetic etiology of disease. Here, we applied whole exome sequencing (WES) and bioinformatics to identify likely causal pathogenic variants in each pedigree. In two cases, we identified novel pathogenic variants in COL6A3. In a third case, we identified novel likely pathogenic variants in COL6A6 and COL6A3. We identified a novel splice variant in EMD in a fourth case. Finally, we classify two cases as calcium channelopathies with identification of novel pathogenic variants in RYR1 and CACNA1S. These are the first cases of myopathies reported to be caused by variants in COL6A6 and CACNA1S. Our results demonstrate the utility and genetic diagnostic value of WES in the broad class of NMD phenotypes.
Keywords: Arthrogryposis, CACNA1S, central core disease, COL6A3, COL6A6, EMD, exome, muscular dystrophy, myopathy, RYR1
Introduction
Myopathies and muscular dystrophies can be classified into a large heterogeneous subgroup of neuromuscular diseases (NMDs) that are primarily associated with dysfunction of muscle fibers. Identifying the genetic cause of myopathies can be challenging as symptoms overlap and numerous genetic defects in many genes may underlie the clinical pathology of disease. While symptoms can direct successful genetic diagnosis through testing of single genes or small panels of genes, they may also lead to costly, time-consuming, and often unsuccessful attempts at genetic diagnosis. Next-generation sequencing (NGS) can greatly improve the ability to identify pathogenic variants with a single, timely, affordable assay and is beginning to revolutionize genetic testing (Ng et al. 2009, 2010). We thus applied WES to congenital and childhood genetic diagnostic odyssey cases of myopathy/muscular dystrophy (MD). We provide a clinical description of six cases (Table1) and describe the candidate pathogenic variants identified in each (Table2). We first present three cases with Collagen 6 (Col6) myopathies which all carry an identical pathogenic variant in COL6A3 (OMIM# 120250). Importantly, we provide evidence that variants in COL6A6 likely result in myopathy. Next, we present a case with a phenotype very similar to two of the Col6 myopathy cases, but instead was found to have Emery–Dreifuss Muscular Dystrophy (EDMD) caused by a novel variant in EMD (OMIM# 300384) at a known pathogenic genomic position. Finally, we present two cases with calcium channelopathies. We present a dominant case of central core disease (CCD) caused by an insertion in RYR1 (OMIM# 180901), followed by evidence for the first CACNA1S (OMIM# 114208)-related congenital myopathy with ophthalmoplegia. Our results shed light on the genetic etiology of several related myopathy cases and provide evidence for the effectiveness of WES in aiding in resolving the genetic diagnosis.
Table 1.
Case phenotype summary
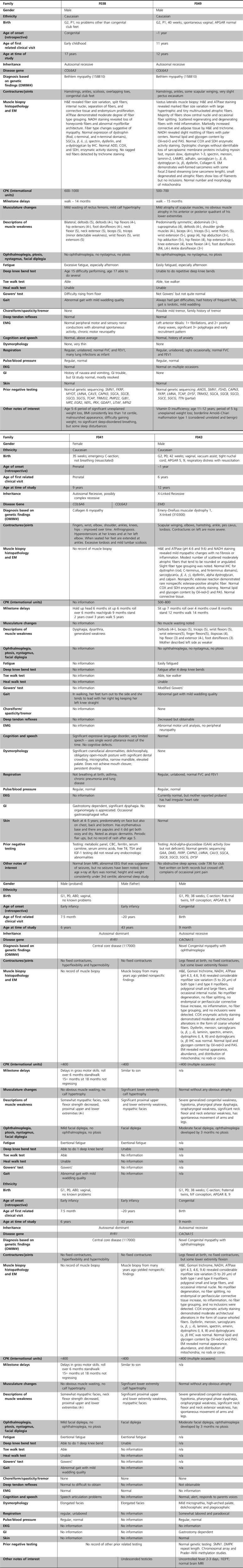 |
Table 2.
Novel myopathy pathogenic variants
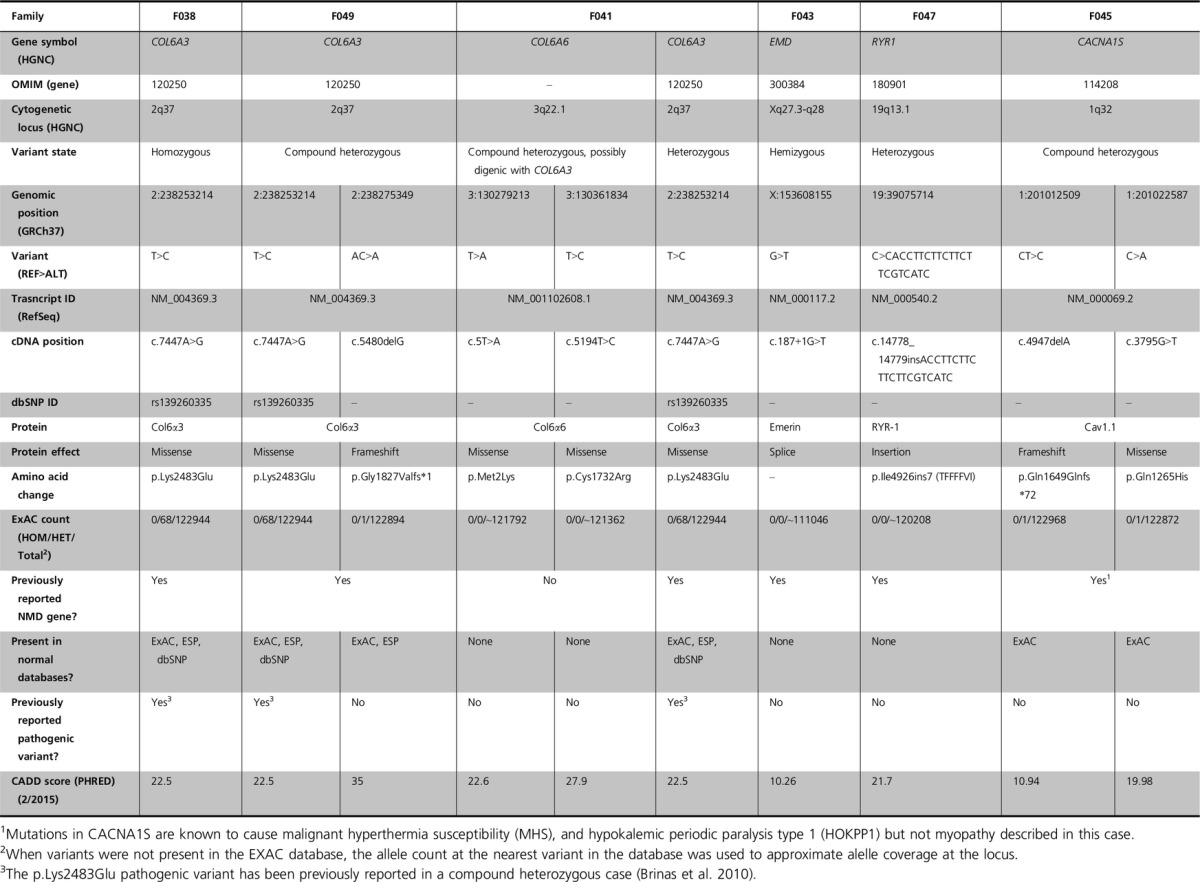
|
Materials and Methods
Patient recruitment and sample collection
Children with clinically diagnosed myopathies and relevant family members were recruited and consented for participation in our research study according to our current Western Institutional Review Board approved protocol (#20120951). Whole blood samples were sent directly to the Dorrance Clinical Laboratory at the Translational Genomics Research Institute (TGen), a CLIA-certified laboratory. DNA was isolated from whole blood and quality and quantity were determined by NanoDrop (ThermoFisher Scientific, Waltham, MA) and Qubit DNA (Life Technologies, Grand Island, NY) assays.
Whole exome library preparation and sequencing
Whole genome (WG) libraries were prepared according to the manufacturer’s protocols using a TruSeq Library Preparation kit (Illumina, San Diego, CA) or a Library Preparation kit (Kapa Biosystems, Wilmington, MA). DNA quantity and quality were determined by analysis on a High Sensitivity DNA Bioanalyzer chip (Agilent Technologies, Santa Clara, CA). Double-stranded DNA concentrations were obtained using a Qubit Hi-sensitivity DNA assay kit (Life Technologies). WG libraries were pooled and exome enrichment was performed using a modified TruSeq Exome Enrichment kit (Illumina) protocol, where all amplification of exome-enriched libraries was performed using the Kapa Biosystems’ library amplification kit. The enriched libraries were quantitated and qualified using Qubit and Bioanalyzer chip assays as described above. The enriched exome libraries were clustered using the Illumina cBot. Sequencing was performed on Illumina HiSeq 2000/2500 systems using Illumina SBS Kit v3 with 83 × 83 or 100 × 100 base pair (bp) paired-end reads with single index reads of 7 bp according to the manufacturer’s protocols.
Bioinformatic analysis
Raw sequence data were converted to FASTQ files using Illumina’s BCL Converter tool which were aligned to build 37 of the human reference genome (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) using the Burrows-Wheeler Alignment tool (BWA) (Li and Durbin 2009) and sorted with SAMtools (Li et al. 2009) to create binary sequence (BAM) files. PCR duplicates were flagged for removal using Picard (http://picard.sourceforge.net), which was also used to evaluate other metrics such as coverage and GC metrics. Figure S1 displays basic sequencing coverage metrics.
Variants were called and BAM files were insertion/deletion realigned and recalibrated using The Genome Analysis Toolkit (GATK) (McKenna et al. 2010). The variant call files were annotated with information from the Genetic variant annotation and effect prediction toolbox (SnpEff) (Cingolani et al. 2012), then further annotated with a custom in-house annotation interface tool with information from numerous databases such as ClinVar (www.ncbi.nlm.nih.gov/clinvar/), Polyphen-2 (Adzhubei et al. 2010), FATHMM (Shihab et al. 2013), SIFT (Kumar et al. 2009), Clinical Genomic Database (Solomon et al. 2013), and The National Heart, Lung, and Blood Institute’s GO Exome Sequencing Project (ESP) (ESP 2013). Allele counts from ExAC (Exome_Aggregation_Consortium 2014) and CADD (Kircher et al. 2014) scores were also obtained. Variants were custom sorted for each family based on inheritance patterns and by annotations. Suspect variants classified as “likely pathogenic” or “pathogenic” based on ACMG guidelines (Richards et al. 2008) were clinically confirmed by Sanger sequencing at GeneDX (Cases 2–6) or to ARUP Laboratories (Case 1).
Results
Collagen 6 myopathies
Case 1. F038 COL6A3
In Family 38, a male child of Canadian descent was born at term with club feet but no additional problems (Table1). He walked somewhat late and since has had a consistently mild abnormal gait. At the age of 5–6 years, he underwent a period of weight loss and lipoatrophy, and at age 17 had a body mass index (BMI) in the first centile, and continued to have a malnourished appearance and difficulty gaining weight. From the age of 8 to age 17, he complained of significant weakness and fatigue. He displayed mild progressive proximal thoracic scoliosis and had significant but nonfixed bilateral lower extremity contractures of hamstrings, ankles, and feet with overlapping toes (Table1). He had no history of skin rash. His respiration and cardiac function were normal. Testing revealed a consistent mild elevation of CPK. Electromyography (EMG) suggested chronic motor neuropathy. Abnormalities revealed by muscle biopsy histology included size variation, split fibers, internal nuclei, connective tissue proliferation, and endomysium proliferation (Fig.1A). ATPase staining revealed moderate Type I and Type II grouping (Fig.1B). NADH staining revealed moth-eaten fibers (Fig.1C). The overall histological diagnosis was abnormal myofibrillar architecture with moderate fiber type grouping of unknown etiology. Genetic sequencing of 15 NMD genes was normal (Table1). Over his 17 years of life, his diagnoses included Charcot–Marie–Tooth disease, myopathy, Pompe disease, MD, limb-girdle muscular dystrophy (LGMD), and spinal muscular atrophy, but without genetic confirmation. There is no other known history of NMD in the family.
Figure 1.
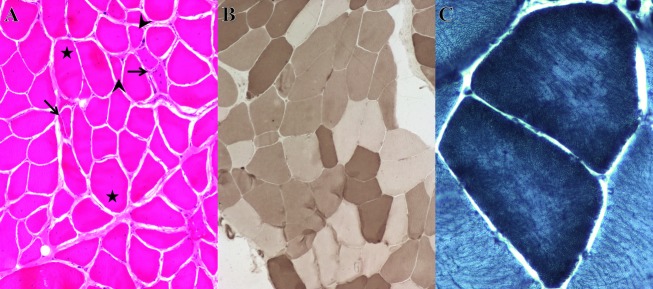
Histopathology images of frozen muscle biopsy cross sections from the affected male child in F038 carrying a homozygous p.Lys2483Glu COL6A3 pathogenic variant. (A) H&E stain reveals atrophic (arrowheads), hypertrophic (stars), and split fibers (arrows). Also present are internal nuclei and connective tissue proliferation. Magnification = 100×. (B) ATPase reaction stain at pH 4.6 demonstrating that both fiber types are affected by hypertrophy and atrophy as well as mild fiber type grouping. Magnification = 100×. (C) NADH oxidative enzyme reaction stain demonstrating myofibrillar architectural disarray (moth-eaten fibers). Magnification = 400×.
WES was performed on samples from the parents, proband, and sibling and revealed a rare homozygous missense variant in COL6A3 (g.chr2:238253214T>C, c.7447A>G, p.Lys2483Glu, NM_004369.3) in the proband (Fig.2 and Table2). The variant was clinically confirmed by Sanger sequencing (data not shown). Each parent and the unaffected female sibling carried this variant in the heterozygous state (Figs.2, 3A). The CADD score for this variant is 22.5. This variant is present in The Single Nucleotide Polymorphism Database (dbSNP) (rs139260335) (Sherry et al. 2001), and ExAC variant databases, but never in the homozygous state. This p.Lys2483Glu variant has been previously identified as pathogenic by Brinas et al. (2010) in a compound heterozygous state, but this is the first homozygous case to our knowledge.
Figure 2.
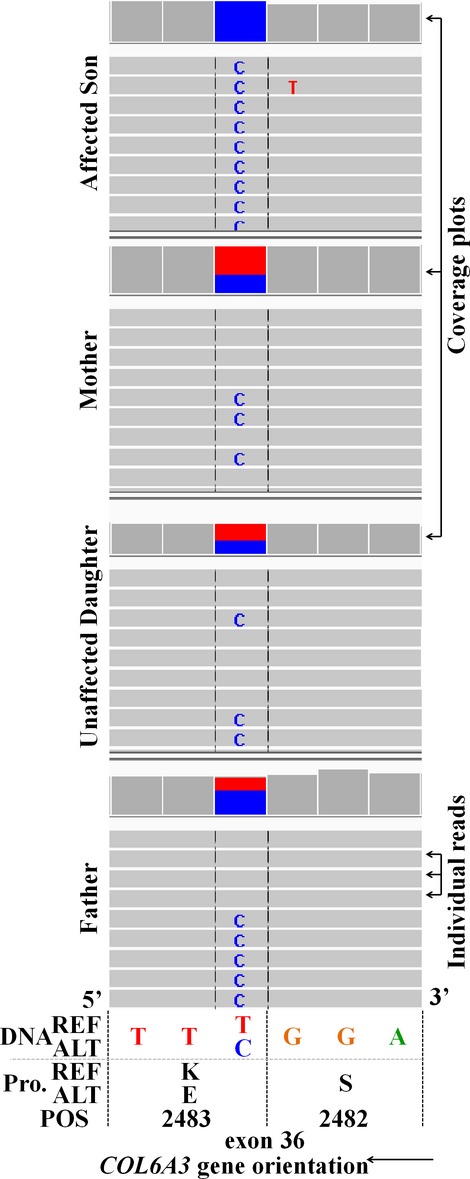
Integrated genomics viewer (IGV) screenshot of F038 WES results of the COL6A3 c.7447A>G missense p.Lys2483Glu pathogenic variant. The affected son is homozygous for the T>C variant and all other family members represented are heterozygous. Note the DNA reference shows the complementary DNA sequence relative to the gene orientation. DNA reference (REF) based on hGRC37. Col6α3 transcript for protein position (Pro. POS) = NM_004369.
Figure 3.
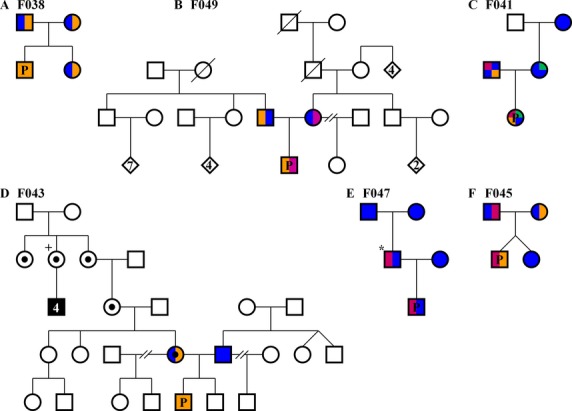
Pedigrees of families. Individuals with color filled symbols underwent exome sequencing. P, proband described in this study.  Represents wild-type alleles for the indicated gene in all pedigrees. (A) Family 38 pedigree.
Represents wild-type alleles for the indicated gene in all pedigrees. (A) Family 38 pedigree.  COL6A3 recessive c.7447A>G variant. (B) Family 49 pedigree.
COL6A3 recessive c.7447A>G variant. (B) Family 49 pedigree.  COL6A3 recessive c.7447A>G variant.
COL6A3 recessive c.7447A>G variant.  COL6A3 recessive c.5480delG variant. (C) Family 41 pedigree.
COL6A3 recessive c.5480delG variant. (C) Family 41 pedigree.  COL6A3 recessive c.7447A>G variant.
COL6A3 recessive c.7447A>G variant.  COL6A6 recessive c.5T>A variant.
COL6A6 recessive c.5T>A variant.  COL6A6 recessive c.5194T>C variant. (D) Family 43 pedigree.
COL6A6 recessive c.5194T>C variant. (D) Family 43 pedigree.  EMD recessive c.187+1G>T splice variant. + Required pacemaker and had four affected sons (black fill). ● Known obligate carrier. (E) Family 47 pedigree.
EMD recessive c.187+1G>T splice variant. + Required pacemaker and had four affected sons (black fill). ● Known obligate carrier. (E) Family 47 pedigree.  RYR1 dominant c.14778_14779insACCTTCTTCTTCTTCGTCATC variant. *De novo event. (F) Family 45 pedigree.
RYR1 dominant c.14778_14779insACCTTCTTCTTCTTCGTCATC variant. *De novo event. (F) Family 45 pedigree.  CACNA1S recessive c.4947delA variant.
CACNA1S recessive c.4947delA variant.  CACNA1S recessive c.3795G>T variant.
CACNA1S recessive c.3795G>T variant.
Case 2 – F049 COL6A3
In Family 49, a male child was born as the product of a 24-year-old, gravida 2 para 1 mother. There were no known complications or health issues at birth. He began walking somewhat late at 14–15 months of age and has always had some gait difficulties (Table1). He was evaluated at the age of 12 years and complains of fatigue and has been unable for several years to keep up with his peers physically. He experienced recent unexplained significant weight loss of 5 kg between the age of 11 and 12. He has no history of rashes or skin abnormalities. He was suspected of having respiratory insufficiency, but had forced expiratory volume 1 (FEV1) and forced volume vital capacity (FVC) of 100 and 105% of predicted. His cognition was normal. In-depth cardiac evaluation was normal. He demonstrated contractures of his hamstrings and ankles and previously wore orthotics to mitigate further ankle contractures. He had no upper extremity contractures but had some protuberance of bilateral scapula. Examination of his strength on multiple occasions by multiple physicians revealed 3–4/5 weakness in all limb and girdle muscles (Table1). He had easily obtainable reflexes, but nerve conduction studies at the age of 9 identified fibrillations and sharp waves in the anterior tibialis. Extensive muscle biopsy histology revealed marked fiber size variation, with large hypertrophic and tiny multinucleated atrophic fibers with a majority showing central nuclei and occasional fiber splitting. Scattered regenerating and degenerating fibers were detected. Both Hematoxylin and Eosin (H&E) and trichrome stains revealed markedly increased connective and adipose tissue (Table1). Of particular interest was the presence of Col6 staining of the endomysial connective tissue and sarcolemmal staining. However, the specific Col6 proteins detected were not reported. NADH reveals slight mottling of fibers with paler centers. Electron microscopy (EM) demonstrated some focal Z-band streaming involving only one sarcomere length. Serial CPK evaluations revealed mildly elevated values in the 500-700 range. His parents and female half sibling are unaffected and no other family history of similar disease was present (Fig.3B).
WES revealed the same COL6A3 pathogenic variant (g.chr2:238253214T>C, c.7447A>G, p.Lys2483Glu) identified in the first case, but only in the heterozygous state in case 2 (Figs.3B, 4A, and Table2). A second COL6A3 frameshift variant (g.chr2:238275349AC>A, c.5480delG, p.Gly1827Valfs*17, NM_004369.3) was also identified in the proband. This variant has been reported in a single allele in ExAC. It is predicted to result in truncation of the protein, and likely does not result in translation of protein due to nonsense-mediated decay (NSMD) (Baker and Parker 2004). Variants were clinically confirmed by Sanger sequencing (Fig.4A and B). The COL6A3 p.Lys2483Glu pathogenic variant was inherited from the father and the p.Gly1827Valfs*17 pathogenic variant inherited from the mother (Fig.3B). Our case is very similar to the case reported by Brinas et al. (2010) which had the p.Lys2483Glu variant with a frameshift pathogenic variant.
Figure 4.
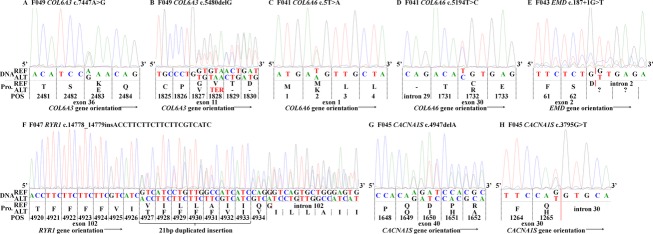
Sanger sequencing traces examples of each pathogenic variant reported from GeneDX clinical confirmation. DNA reference (REF) based on GRCh37. (A) F049 affected male child heterozygous COL6A3 c.7447A>G recessive missense p.Lys2483Glu pathogenic variant. This trace is also similar to the results found in the F041 affected proband (data not shown). (B) F049 affected male child heterozygous COL6A3 c.5480delG recessive frameshift p.Gly1827Valfs*1 pathogenic variant. (C) F041 affected female child heterozygous COL6A6 c.5T>A recessive missense p.Met2Lys likely pathogenic variant. (D) F041 affected female child heterozygous COL6A6 c.5194T>C recessive missense p.Cys1732Arg likely pathogenic variant. (E) F043 affected male child hemizygous EMD c.187+1G>T recessive pathogenic variant. (F) F047 affected male child heterozygous RYR1 c.14778_14779insACCTTCTTCTTCTTCGTCATC dominant duplicated insertion p.Ile4926ins7 (TFFFFVI) pathogenic variant. (G) F045 affected male child heterozygous CACNA1S c.4947delA recessive frameshift p.Gln1649Glnfs*72 pathogenic variant. (H) F045 affected male child heterozygous CACNA1S c.3795G>T recessive missense p.Gln1265His pathogenic variant. Transcripts used for protein position (Pro. POS) for: Col6α3 = NM_004369; Col6α6 = NM_001102608; Emerin = NM_000117; RYR-1 = NM_000540; Cav1.1 = NM_000069. Red solid vertical bars represent splice sites. Dashed black vertical bars represent triplet codon reading frame.
Case 3. F041 COL6A6 and COL6A3
In a third family, an infant female child was discovered during an ultrasound to have arthrogryposis. She was born prematurely at 35 weeks by emergency C-section. At birth, her APGAR score was 1. She was not breathing and was resuscitated. She presented further with micrognathia, dysphagia, dysarthria, and generalized hypotonia (Table1). A feeding tube was implanted at 6 weeks of age. She has had significant milestone delays. She began to hold her head up, sit, smile, laugh, and roll over by ∼6 months. She began to reach and grab for things at ∼9 months. She began to stand at 2 years and began to crawl at 3 years of age. At the age of 5 years, she began walking. She has very limited speech. She has consistent muscle weakness of all muscle groups that does not appear to be improving or progressing. At the time of this study, the child was 9 years of age. She has contractures of the fingers, wrist, elbow, shoulders, ankles, knees, and hips. Contractures present from birth have improved somewhat over the years (Table1).
This family, including the proband, her parents, and her maternal grandmother were enrolled in our study. WES revealed the presence of the same COL6A3 pathogenic variant (g.chr2:238253214T>C, c.7447A>G, p.Lys2483Glu) (Brinas et al. 2010) but only in a heterozygous state in the proband (inherited from her father) with no second pathogenic variant in COL6A3 detected (Fig.3C). Interestingly, we identified compound heterozygous variants in COL6A6 in the proband, including a heterozygous missense g.chr3:130279213T>A, c.5T>A, p.Met2Lys (NM_001102608.1) variant inherited from her father and a heterozygous missense g.chr3:130361834 T>C, c.5194T>C, p.Cys1732Arg (NM_001102608.1) variant from her mother (Fig.3C and Table2). All three COL6 variants in the proband were clinically confirmed by Sanger sequencing (Fig.4C and D). The maternal grandmother did not carry any of these variants (Fig.3C). The COL6A6 variants have not been identified in dbSNP, ESP or ExAC, and received CADD scores of 22.6 (p.Met2Lys) and 27.9 (p.Cys1732Arg). While CADD scores, which take into account many predictors of pathogenicity, suggest these pathogenic variants are damaging, not all predictors suggest pathogenicity. The Residual Variation Intolerance Score (RVIS) of −0.11 (ALL 0.01% category) for the COL6A6 gene as a whole compared to all other genes, is only very slightly intolerant of functional variation (Petrovski et al. 2013). Polyphen2 (Adzhubei et al. 2010) ranks both COL6A6 variants as probably damaging with scores of 0.967 and 1.00 for the p.Met2Lys and p.Cys1732Arg variants, respectively. While we believe that the COL6A6 variants are likely pathogenic, functional studies are warranted and our interpretation must be considered carefully until further COL6A6 pathogenic variants are reported.
Emery–Dreifuss muscular dystrophy
Case 4. EMD F043
We evaluated a fourth case with similar clinical characteristics to the first two COL6A3 cases. The male child was born at full term with mild complications with jaundice and wrapping of the umbilical cord around his neck and respiratory distress (Table1). He began walking at 14 months and at ∼4 years old was a toe walker. He was seen at age 6 for abnormal gait, slowly progressing extremity weakness in his feet and ankles, fatigue on excursion, and equinus foot deformity. Further evaluation at 11 years of age noted bilateral winged scapulae, rather significant progressive contractures at his elbows, moderate hamstring contractures, and moderate flexor contractures of his ankles, but no scoliosis (Table1). He had a somewhat unusual gait with a mild waddling quality. He did not demonstrate any neck flexor/extensor or significant proximal weakness in his shoulder girdles. He did have clear distal weakness in foot dorsiflexors and decreased but obtainable deep tendon reflexes. Recent echocardiogram (EKG) and Holter monitoring revealed no structural heart disease, cardio myopathy, or arrhythmia. Muscle biopsy demonstrated mild myopathic changes without inflammation or significant fibrosis (Table1). CPK values were slightly elevated and ranged from 500 to 800. EMG excluded a peripheral neuropathy and was suggestive of myopathy. His working diagnosis was an undetermined form of LGMD. Initially, no family history of related NMD was noted by the family.
The proband and his parents were enrolled in our study and WES revealed a novel pathogenic splice variant in EMD at g.chrX:153608155G>T, c.187+1G>T (NM_000117.2) for which the mother is heterozygous and the affected son is hemizygous (Fig.3D and Table2). This variant was clinically confirmed by Sanger sequencing (Fig.4E). This c.187+1G>T splice variant has not been identified in dbSNP, or ExAC, but a c.187+1G>A splice variant has been reported to cause EDMD (Yates et al. 1999). After genetic diagnosis of the proband, several male members of the maternal extended family were identified that had previously been diagnosed and genetically confirmed with EDMD or cardiac conduction defects suggesting this variant is X-linked recessive. However, the sister of the maternal great grandmother required a pacemaker, possibly suggestive of a mild dominant effect or haploinsufficiency in heterozygous females (Fig.3D).
Calcium channel myopathies
Case 5. RYR1 F047
In F047, a male child was evaluated beginning at 7.5 months of age with delays in gross motor skills. He was not noted of having any problems at birth and went home from the hospital at a few days of life (Table1). He had some milestone delays and was just beginning to roll over at ∼6 month of age and began to walk at 15 months. He has had some speech articulation problems, but cognitive skills are described as normal. When examined at age 5 years of age he had somewhat elongated myopathic facies with mild facial diplegia. He fatigues somewhat more easily than his peers. He had no fixed contractures or obvious muscle wasting. He was able to do a single deep knee bend with difficulty. He rose from the floor with a significant Gowers’ maneuver. His walking gait was abnormal, manifested by a mild waddling quality and his running gait was also abnormal. His neck flexor strength is decreased. Examination of proximal strength in his upper and lower extremities revealed 4-/5 weakness (Table1). His deep tendon reflexes were difficult to obtain. Approximately a year later at age 6 years, his evaluation was much the same, but it was noted that he was able, with difficulty, to walk on his toes, but was unable to walk on his heels. Also noted was a significant degree of hyperflexibility and hypermobility.
The father, age 43 at the time of this study, had a very similar early childhood history. He underwent an extensive neuromuscular evaluation approximately 20 years ago. The father’s evaluation included EMG, normal plasma CPK, and a muscle biopsy which yielded nonspecific results. The father has a history of having generalized fatigue, and exertional weakness. He had no significant history of medical problems other than undescended testicles. At the time of our current study, he was reexamined and had clear evidence for significant proximal weakness in his upper and lower extremities, and could not do a single deep knee bend unaided (Table1). He also had an elongated face with diplegia and myopathic looking facies. He exhibited significant lower extremity calf hypertrophy. The mother had no known neuromuscular disorder.
The boy, his parents, and paternal grandparents were enrolled in our study and WES revealed a novel small in-frame 21 bp duplicated insertion in RYR1 beginning at g.chr19:39075714, c.14778_14779insACCTTCTTCTTCTTCGTCATC, p.Ile4926ins7 (TFFFFVI) (NM_000540.2) (Fig.5) in the proband and his affected father, but not present in either paternal grandparent indicating that the pathogenic variant is de novo in the father (Figs.3E, 5). This variant was clinically confirmed by Sanger sequencing (Fig.4F). This insertion has not been identified in dbSNP, or ExAC (Table2). Pathogenic variants in RYR1 are the most common cause of CCD (Wu et al. 2006), consistent with the phenotype of the father and affected son.
Figure 5.
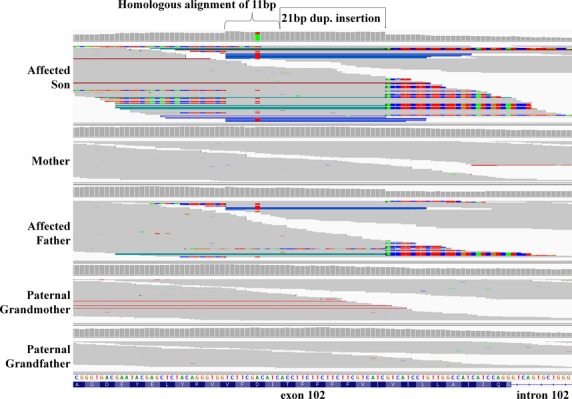
IGV screenshot of the F047 WES results demonstrating a novel 21 bp duplicated RYR1 c.14778_14779insACCTTCTTCTTCTTCGTCATC dominant in frame p.Ile4926ins7 (TFFFFVI) pathogenic variant. Note the homologous alignment of 11 bp causing the insertion to appear to be 32 bp in length. The insertion is present in the affected son and father but not in the paternal grandparents demonstrating a de novo dominant event in the father. RYR-1 transcript = NM_000540.
Case 6. F045 CACNA1S
The last case we describe was the product of a 38 week gestation of male–female twins conceived by IVF. The pair was born via C-section and the boy had APGAR scores of 8 and 9, but was described as being quite hypotonic, severely weak, not very vigorous, and never established the ability to significantly chew, suck, or swallow (Table1). The sister had no noted problems. The boy was hospitalized for 2 months and underwent extensive neurodiagnostic evaluation. During this time, a gastrostomy tube was inserted. His height, weight, and basic vital signs have remained normal throughout his course. At his evaluation at 3 month of age, he was somewhat dolichocephalic and plagiocephalic, for which he wore a helmet. He had a high-arched palate with mild micrognathia. He had clear pharyngeal phase dysphagia, oropharyngeal weakness, significant facial diplegia, but no apparent ptosis. He had no fixed contractures, but did have some flexion in his lower extremities (Table1). He was alert and responded to his parents’ voice and followed them visually, but he moved his head more than his eyes clearly demonstrating ophthalmoplegia. He had spontaneously movement of his hands and feet. Deep tendon reflexes were not obtainable. Muscle biopsy and extensive histology demonstrated moderate myofiber atrophy and hypertrophy of both fiber types with myofiber size variation ranging from 5 to 20 microns (Fig.6A). Fibers were polygonal with no myofiber degeneration or splitting. Occasional internal nuclei were present. No fiber type grouping and no inflammation or inclusions were seen (Fig.6B). Phosphofructokinase (F-6-P) staining revealed course myofibrillar architecture (Fig.6C). Other histological stains did not reveal any further abnormalities (Table1). High-resolution magnetic resonance imaging (MRI) did not reveal any structural abnormalities in the brain, cerebellum, and brainstem.
Figure 6.
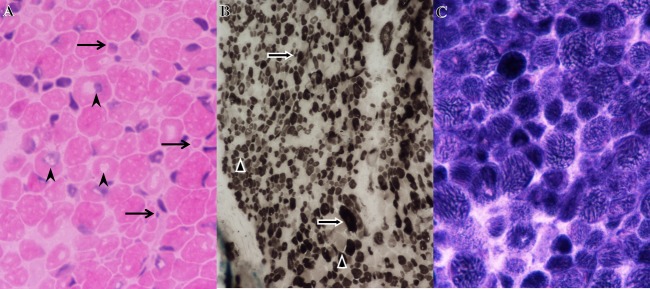
Histopathology images of frozen muscle biopsy cross sections from the affected male child in F045 carrying compound heterozygous p.Gln1649Glnfs*72 and p.Gln1265His CACNA1S pathogenic variants. (A) H&E stain demonstrating marked variability in myofiber size and fiber diameters ranging from 5 to 20 μm. Arrows indicate tiny atrophic fibers. Central nuclei indicated by arrow points. Magnification = 600×. (B) ATPase reaction stain at pH 4.6 demonstrating that both fiber types are affected by hypertrophy and atrophy. Arrows indicate Type I (dark fibers) and arrowheads indicate Type II (light fibers). Magnification = 200×. (C) F-6-P stain demonstrating coarse myofibrillar architecture. Magnification = 600×.
The boy, his parents, and his unaffected fraternal female twin were enrolled in our study. Analysis of variants identified by WES revealed novel pathogenic variants in CACNAS1 in the family. The affected son received a pathogenic single bp deletion at g.chr1:201012509CT>C, c.4947delA, p.Gln1649Glnfs*72 (NM_000069.2) from his father and a missense g.chr1:201022587C>A c.3795G>T, p.Gln1265His (NM_000069.2) from his mother (Fig.3E). These variants were clinically confirmed by Sanger sequencing (Fig.4G and H). The missense p.Gln1265His variant is predicted to be damaging by FATHMM, Polyphen-2, mutationassessor (Reva et al. 2007, 2011), and MutationTaster. No prediction algorithms used assigned this variant as benign. These CACNA1S variants have not been reported in dbSNP, or ESP, but are each present in a single allele in ExAC (Table2). The twin sister of the affected boy was discordant for both CACNAS1 variants (Fig.3E). The clinical features of the disease in Case 6 are consistent with a severe congenital myopathy with ophthalmoplegia. CACNA1S pathogenic variants have been identified as the cause of hypokalemic periodic paralysis type 1 (HOKPP1) (Burge and Hanna 2012; Hanchard et al. 2013), but persistent weakness of the child is somewhat inconsistent with HOKPP1. While it has been suspected that CACNAS1 variants could cause myopathies due to the physical and mechanical coupling between its gene product, the voltage-dependent L-type calcium channel subunit alpha-1S (Cav1.1), and the Ryanodine receptor Ca2+ release channel 1 (RYR-1)(Wu et al. 2006; Rebbeck et al. 2014), no reports prior to this have identified pathogenic variants in CACNAS1 as the cause of congenital myopathy.
Discussion
Congenital and childhood myopathies and muscular dystrophies are a common class of neuromuscular disorders with overlapping phenotypes and heterogeneous genetic etiology. Here, we present the phenotype of 6 myopathy cases that underwent extensive, expensive, and invasive testing including muscle biopsies, EMG, MRI, EKG, and single gene and gene panel sequencing, often over many years, without successful genetic diagnosis prior to enrollment in our research study (Table1). We identified novel pathogenic variants in five genes by WES (Table2).
Collagen 6 myopathies (cases 1-3)
We identified a recessive novel pathogenic variant in COL6A3 and likely pathogenic variants in COL6A6. The family of Col6 proteins are extracellular matrix (ECM) proteins that help maintain tissue integrity of many tissues including muscle, tendon, skin, cartilage, and intervertebral disks (Chu et al. 1988; Knupp and Squire 2001; Bushby et al. 2014). The main forms of Col6 expressed in the ECM are the Collagen 6 alpha 1(Col6α1), 2(Col6α2), and 3(Col6α3) chains coded for by COL6A1, COL6A2, and COL6A3, respectively (Engvall et al. 1986; Bonaldo et al. 1998). Pathogenic variants in COL6A1, COL6A2, and COL6A3 cause two main forms of myopathy; Ullrich congenital muscular dystrophy (UCMD) and Bethlem myopathy (BM) (Bonnemann 2011). Recently, the COL6A6 gene was discovered. It is most similar to COL6A3 as it codes for multiple von Willebrand factor type A (VWFA) domains (Gara et al. 2008; Tagliavini et al. 2014). COL6A6 is expressed in wide range of fetal and adult tissue including brain, heart, and muscle (Fitzgerald et al. 2008; Gara et al. 2011). Col6α3 and Collagen 6 alpha 6(Col6α6) proteins are found in the endomysium and perimysium of skeletal muscle but only Col6α3 is found in the basement membrane (Sabatelli et al. 2011, 2012). Recent evidence demonstrates that Col6α6 is dramatically decreased in skeletal muscle and muscle cell cultures from patients with UCMD and BM independent of clinical phenotype suggesting coregulation of these genes (Tagliavini et al. 2014). Col6α6 was increased in noncollagen myopathies suggesting a significant role in other myopathies as well (Tagliavini et al. 2014).
Characteristic UCMD presents with severe congenital muscle weakness with axial and proximal joint contractures and distal joint hypermobility. BM usually presents with slowly progressive axial and proximal muscle weakness with finger flexion contractures. Skin rashes often accompany BM and UCMD (Bonnemann 2011; Bushby et al. 2014). Many pathogenic variants in Collagen 6 genes (COL6) impair protein expression by disrupting splicing, glycine substitutions required for triple-helix formation, or secretion. However, some pathogenic variants in BM patients have no detectable effect on Col6 assembly and secretion but compromise protein function in the ECM of muscle. Pathogenic variants in COL6 genes have been reported as dominant and recessive (Lampe et al. 2008; Butterfield et al. 2013). The three cases describe here all display recessive phenotype since no carriers have yet displayed a phenotype. Interestingly, all three patients have the pathogenic COL6A3 p.Lys2483Glu variant first reported by Brinas et al. (2010). Case 1 is homozygous for this variant. Case 2 is compound heterozygous for the p.Lys2483Glu variant and a COL6A3 p.Gly1827Valfs*1 frameshift variant that would likely result in truncation and loss of expression. The phenotype of case 1 and case 2 are remarkably similar and indicate a myopathy more akin to the more mild BM form of disease. Their mild phenotype is very similar to that reported by Brinas et al. (2010) with the exception that our case demonstrated contractures. The F041 case had some additional features apart from the other two and is more suggestive of a more severe form of UCMD. The affected child carries novel compound heterozygous variants in COL6A6 (p.Met2Lys and p.Cys1732Arg) in addition to being a heterozygous carrier of the COL6A3 p.Lys2483Glu pathogenic variant. The COL6A6 variants are likely pathogenic and we believe are the primary drivers of the phenotype, but the COL6A3 variant likely contributes to the phenotype due to the overlap in expression and function. While it is clear there is a genetic component to her condition, we cannot rule out that complications at birth did not also contribute to her more severe phenotype.
It is clear that the COL6A3 p.Gly1827Valfs*1 variant in F049 is pathogenic and is predicted to result in NSMD or protein truncation. The p.Lys2483Glu pathogenic variant in COL6A3 causes a change in amino acid structure and opposite charge. It is located in the nonhelical region of the protein, in the VWFA 11 domain (Pan et al. 1998). Missense mutations in VWFA domains have been reported to cause BM (Pan et al. 1998). Changes in COL6A3 expression or function of the p.Lys2483Glu variant at the endomysium and the perimysium is likely related to ECM proliferation identified in histology of F038 and F049 (Sabatelli et al. 2011; Bushby et al. 2014). The histology report for F049 indicates Col6 is present suggesting that the p.Lys2483Glu variant may not result in loss of expression or secretion, however, this must be considered cautiously as the pathology report does not indicate which Col6 proteins are detected. In the Brinas et al. (2010) case, Col6 was detected but altered in fibroblasts and not detected in muscle. In COL6A6, the p.Met2Lys missense pathogenic variant is located in the signal peptide of the protein and may disrupt its secretion. This variant may also disrupt translation of the COL6A6 mRNA as mutations in stem loop structures near the start codon of other collagen genes abolish expression (Manojlovic and Stefanovic 2012). The p.Cys1732Arg pathogenic variant in COL6A6 results in an addition of a charge to the amino acid and a change from hydrophobic to hydrophilic states. This variant is also located at the end of the triple-helical domain and just before the C-terminal VWFA domains (Pan et al. 1998). In general, Cys residues are highly conserved and are critical for formation of collagen oligomer and fibril disulfide bonds (Butterfield et al. 2013).
Of particular interest is the specific period of lipoatrophy and weight loss, and the inability to gain weight in cases 1 and 2, a characteristic of patients not typically described in cases of UCMD or BM (Brinas et al. 2010; Bonnemann 2011; Bushby et al. 2014), but which may be a diagnostic feature of disease caused by these variants. Weight loss and fatigue may be due to nocturnal respiratory insufficiency or apnea common to BM cases with COL6A3 pathogenic variants (Bonnemann 2011). While suggested in case 2, neither case 1 nor 2 demonstrated waking respiratory insufficiency upon testing. Case 2 suffered from intermittent sleeping problems, but a sleep study did not reveal any apnea. Alternatively, several papers demonstrate metabolic changes in UCMD and BM including mitochondrial deficits in mouse models, cell models from UCHM and BM patients, and in patient muscle biopsies (Tagliavini et al. 2013; De Palma et al. 2014; Sorato et al. 2014). While muscle biopsy histology and EM did not reveal defects in morphology or number of mitochondria, respiratory chain deficits may explain the weight loss and fatigue seen in these patients.
Together, these three cases provide substantial evidence of the identification of pathogenic variants in COL6A3 and likely pathogenic variants in COL6A6. While the COL6A3 p.Lys2483Glu pathogenic variant is rare, it has a relatively high heterozygous frequency (Table2) in the population. Therefore, it is critical to report this pathogenic variant and it is very likely that many more genetically undiagnosed cases of BM will be found to have this variant.
Emery–Dreifuss muscular dystrophy (case 4)
In addition to novel Col6 myopathies, we describe a novel c.187+1G>T pathogenic EMD splice variant in a single case of EDMD. EDMD usually manifests in childhood with slowly progressive weakness and limb muscle wasting. Contractures of the elbows, Achilles tendons, and postcervical muscles, are early and characteristic features. Cardiac conduction defects including arrhythmias and risk of sudden death are consistent features as well (Emery 1989; Bonne et al. 1993). The phenotype of the affected child in F043 is consistent with EDMD. The pathogenic variant we identified destroys the required canonical GT nucleotides of the 3′ splice site of exon 2 of the EMD gene. While pathogenic variants have been identified throughout the EMD gene, exon 2 has been reported as a mutational hotspot (Brown et al. 2011). This c.187+1G>T splice variant is also at the exact position of a previously reported c.187+1G>A pathogenic variant in EDMD (Deymeer et al. 1993; Yates et al. 1993, 1999). The G>A and G>T splice variants undoubtedly have very similar if not identical effects on EMD gene splicing and expression. The phenotype of F043 is consistent with phenotypes reported for families with the G>A splice variant (Deymeer et al. 1993; Yates et al. 1993, 1999). The identification of a second variant at the same genomic locus in individuals affected with EDMD confirms both variants as pathogenic.
F043 also demonstrates the importance of obtaining a genetic diagnosis for myopathy patients. In EDMD, cardiac dysfunction is a primary cause of premature mortality if left unmonitored and untreated. As described here, children with similar phenotypes (BM and EDMD) have very different requirements when it comes to cardiac assessment and treatment.
RYR1 and CACNA1S calcium channel myopathies (cases 5-6)
Finally, we describe two calcium channel myopathy cases. The first is a dominant case caused by a novel p.Ile4926ins7 pathogenic variant in the well-known CCD gene RYR1. Second, we describe a novel severe form of congenital myopathy with ophthalmoplegia caused by compound heterozygous pathogenic variants in CACNA1S (p.Gln1649Glnfs*72 and p.Gln1265His). Several calcium channelopathies are associated with pathogenic variants in RYR1 and CACNA1S. Malignant Hyperthermia Susceptibility (MHS) is caused by pathogenic variants in RYR1 or CACNA1S (Monnier et al. 1997; Kim et al. 2013). Pathogenic variants in CACNA1S have been identified as a cause of HOKPP1, but RYR1 have not (Burge and Hanna 2012; Hanchard et al. 2013). Pathogenic variants in RYR1 are the most common causes of CCD, while CACNA1S have not been identified as a cause of CCD (Wu et al. 2006). CCD is most often characterized clinically by a stable or slowly progressive course of congenital hypotonia but can be more severe (Bharucha-Goebel et al. 2013). Most RYR1 pathogenic variants usually result in a dominant phenotype consistent with the dominant heterozygous p.Ile4926ins7 pathogenic variant identified by WES in F047. Of special interest in this case is that the pathogenic variant in the affected father is de novo, which he then passed on to his affected son.
RYR1 and CACNA1S genes code for integral components of excitation-contraction coupling (EC) in skeletal muscle. EC depends on a physical interaction between the skeletal forms of the dihydropyridine receptor L-type Ca2+ channel (DHPR) and RYR-1 (Paolini et al. 2004; Polster et al. 2012). The CACNA1S gene codes for Cav1.1, the main subunit of the DHPR channel (Rebbeck et al. 2014). When a voltage neurostimulus is received, the DHPR channel changes conformation and physically causes RYR-1 to open and release sarcoplasmic reticulum calcium (Rebbeck et al. 2014).
The F047 p.Ile4926ins7 in RYR1 is located in the final hydrophobic transmembrane domain of RYR-1 which has been designated as a pathogenic variant hotspot (Maclennan and Zvaritch 2011). This insertion is in-frame, likely expressed, and is predicted to result in addition of seven amino acids in the final transmembrane helix nearest the Ca2+ pore formed by RYR-1. These extra amino acids undoubtedly alter RYR-1 function and folding. Since this variant shows dominant inheritance, it likely has a dominant negative effect by poisoning RYR-1 tetramer formation and function (Maclennan and Zvaritch 2011).
In F045, the severe congenital myopathy with ophthalmoplegia strongly suggested pathogenic variants in RYR1, but no pathogenic variants in RYR1 were identified. Instead, we identified novel pathogenic frameshift (p.Gln1649Glnfs*72) and missense (p.Gln1265His) variants in CACNA1S. The frameshift variant likely abolishes translation of the mRNA. If the resulting mRNA is translated to protein, it is predicted to result in premature truncation later in the protein. Cav1.1 has four transmembrane domains each with six transmembrane helices that form the Ca2+ channel. The Cav1.1 p.Gln1265His pathogenic variant is located in its fourth repeat domain in the short cytoplasmic loop linking the transmembrane helix S4 and the positively charged transmembrane helix S5. This cytoplasmic loop may be involved in interactions with RYR-1 or other components of the DHPR (Rebbeck et al. 2014). The change in amino acid structure may also interfere with DHPR calcium flux. Due to the phenotypic overlap with RYR1 pathogenic variants in F045, we hypothesize that the p.Gln1265His variant disrupts DHPR and RYR-1 coupling. While pathogenic variants in CACNA1S have been demonstrated as the cause of MHS and HOKPP1, to our knowledge, this is the first reported case of severe congenital myopathy with ophthalmoplegia resulting from pathogenic variants in CACNA1S.
Reporting these cases not only raises awareness to extensive clinical overlap between similar cases with different genetic etiology but most importantly highlights the utility of WES and WGS in providing genetic diagnosis in clinically enigmatic cases.
Acknowledgments
We thank TGen, the Muscular Dystrophy Association (Grant #186435), and the Flinn Foundation for funding. In addition, author C. B. was funded by the Helios Education Foundation and the Freeport-McMoRan Copper & Gold Foundation. We thank Steve Taylor, PA for photomicrography assistance. We are very grateful for the efforts of our project manager, Therese De La Torre, and assistant Kaitlyn Clow.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Exome target coverage statistics. The percent of bases of the targeted exome that had at least 20 high quality reads covering each base.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KE. Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr. Opin. Cell Biol. 2004;16:293–299. doi: 10.1016/j.ceb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Bharucha-Goebel DX, Santi M, Medne L, Zukosky K, Dastgir J, Shieh PB, et al. Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology. 2013;80:1584–1589. doi: 10.1212/WNL.0b013e3182900380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldo P, Braghetta P, Zanetti M, Piccolo S, Volpin D. Bressan GM. Collagen VI deficiency induces early onset myopathy in the mouse: an animal model for Bethlem myopathy. Hum. Mol. Genet. 1998;7:2135–2140. doi: 10.1093/hmg/7.13.2135. [DOI] [PubMed] [Google Scholar]
- Bonne G, Leturcq F. Ben Yaou R. Emery-Dreifuss muscular dystrophy. In: Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors; Pagon RA, Adam MP, editors. GeneReviews. Seattle, WA: University of Washington; 1993. [Google Scholar]
- Bonnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat. Rev. Neurol. 2011;7:379–390. doi: 10.1038/nrneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinas L, Richard P, Quijano-Roy S, Gartioux C, Ledeuil C, Lacene E, et al. Early onset collagen VI myopathies: genetic and clinical correlations. Ann. Neurol. 2010;68:511–520. doi: 10.1002/ana.22087. [DOI] [PubMed] [Google Scholar]
- Brown CA, Scharner J, Felice K, Meriggioli MN, Tarnopolsky M, Bower M, et al. Novel and recurrent EMD mutations in patients with Emery-Dreifuss muscular dystrophy, identify exon 2 as a mutation hot spot. J. Hum. Genet. 2011;56:589–594. doi: 10.1038/jhg.2011.65. [DOI] [PubMed] [Google Scholar]
- Burge JA. Hanna MG. Novel insights into the pathomechanisms of skeletal muscle channelopathies. Curr. Neurol. Neurosci. Rep. 2012;12:62–69. doi: 10.1007/s11910-011-0238-3. [DOI] [PubMed] [Google Scholar]
- Bushby KM, Collins J. Hicks D. Collagen type VI myopathies. Adv. Exp. Med. Biol. 2014;802:185–199. doi: 10.1007/978-94-007-7893-1_12. [DOI] [PubMed] [Google Scholar]
- Butterfield RJ, Foley AR, Dastgir J, Asman S, Dunn DM, Zou Y, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum. Mutat. 2013;34:1558–1567. doi: 10.1002/humu.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu ML, Conway D, Pan TC, Baldwin C, Mann K, Deutzmann R, et al. Amino acid sequence of the triple-helical domain of human collagen type VI. J. Biol. Chem. 1988;263:18601–18606. [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma S, Capitanio D, Vasso M, Braghetta P, Scotton C, Bonaldo P, et al. Muscle proteomics reveals novel insights into the pathophysiological mechanisms of collagen VI myopathies. J. Proteome Res. 2014;13:5022–5030. doi: 10.1021/pr500675e. [DOI] [PubMed] [Google Scholar]
- Deymeer F, Oge AE, Bayindir C, Kaymaz C, Nisanci Y, Adalet K, et al. Emery-Dreifuss muscular dystrophy with unusual features. Muscle Nerve. 1993;16:1359–1365. doi: 10.1002/mus.880161214. [DOI] [PubMed] [Google Scholar]
- Emery AE. Emery-Dreifuss syndrome. J. Med. Genet. 1989;26:637–641. doi: 10.1136/jmg.26.10.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engvall E, Hessle H. Klier G. Molecular assembly, secretion, and matrix deposition of type VI collagen. J. Cell Biol. 1986;102:703–710. doi: 10.1083/jcb.102.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESP. 2013. Seattle, WA NHLBI GO Exome Sequencing Project (ESP). Available at http://evs.gs.washington.edu/EVS/
- Exome_Aggregation_Consortium. Cambridge, MA: 2014. Exome Aggregation Consortium (ExAC) [Google Scholar]
- Fitzgerald J, Rich C, Zhou FH. Hansen U. Three novel collagen VI chains, alpha4(VI), alpha5(VI), and alpha6(VI) J. Biol. Chem. 2008;283:20170–20180. doi: 10.1074/jbc.M710139200. [DOI] [PubMed] [Google Scholar]
- Gara SK, Grumati P, Urciuolo A, Bonaldo P, Kobbe B, Koch M, et al. Three novel collagen VI chains with high homology to the alpha3 chain. J. Biol. Chem. 2008;283:10658–10670. doi: 10.1074/jbc.M709540200. [DOI] [PubMed] [Google Scholar]
- Gara SK, Grumati P, Squarzoni S, Sabatelli P, Urciuolo A, Bonaldo P, et al. Differential and restricted expression of novel collagen VI chains in mouse. Matrix Biol. 2011;30:248–257. doi: 10.1016/j.matbio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Hanchard NA, Murdock DR, Magoulas PL, Bainbridge M, Muzny D, Wu YQ, et al. Exploring the utility of whole-exome sequencing as a diagnostic tool in a child with atypical episodic muscle weakness. Clin. Genet. 2013;83:457–461. doi: 10.1111/j.1399-0004.2012.01951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Jarvik GP, Browning BL, Rajagopalan R, Gordon AS, Rieder MJ, et al. Exome sequencing reveals novel rare variants in the ryanodine receptor and calcium channel genes in malignant hyperthermia families. Anesthesiology. 2013;119:1054–1065. doi: 10.1097/ALN.0b013e3182a8a998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM. Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knupp C. Squire JM. A new twist in the collagen story–the type VI segmented supercoil. EMBO J. 2001;20:372–376. doi: 10.1093/emboj/20.3.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S. Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lampe AK, Zou Y, Sudano D, O’Brien KK, Hicks D, Laval SH, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum. Mutat. 2008;29:809–822. doi: 10.1002/humu.20704. [DOI] [PubMed] [Google Scholar]
- Li H. Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell J, Ruan N, Homer G, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclennan DH. Zvaritch E. Mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum. Biochim. Biophys. Acta. 2011;1813:948–964. doi: 10.1016/j.bbamcr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Manojlovic Z. Stefanovic B. A novel role of RNA helicase A in regulation of translation of type I collagen mRNAs. RNA. 2012;18:321–334. doi: 10.1261/rna.030288.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier N, Procaccio V, Stieglitz P. Lunardi J. Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am. J. Hum. Genet. 1997;60:1316–1325. doi: 10.1086/515454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Nickerson DA, Bamshad MJ. Shendure J. Massively parallel sequencing and rare disease. Hum. Mol. Genet. 2010;19:R119–R124. doi: 10.1093/hmg/ddq390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan TC, Zhang RZ, Pericak-Vance MA, Tandan R, Fries T, Stajich JM, et al. Missense mutation in a von Willebrand factor type A domain of the alpha 3(VI) collagen gene (COL6A3) in a family with Bethlem myopathy. Hum. Mol. Genet. 1998;7:807–812. doi: 10.1093/hmg/7.5.807. [DOI] [PubMed] [Google Scholar]
- Paolini C, Fessenden JD, Pessah IN. Franzini-Armstrong C. Evidence for conformational coupling between two calcium channels. Proc. Natl. Acad. Sci. USA. 2004;101:12748–12752. doi: 10.1073/pnas.0404836101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS. Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster A, Ohrtman JD, Beam KG. Papadopoulos S. Fluorescence resonance energy transfer (FRET) indicates that association with the type I ryanodine receptor (RyR1) causes reorientation of multiple cytoplasmic domains of the dihydropyridine receptor (DHPR) alpha(1S) subunit. J. Biol. Chem. 2012;287:41560–41568. doi: 10.1074/jbc.M112.404194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck RT, Karunasekara Y, Board PG, Beard NA, Casarotto MG. Dulhunty AF. Skeletal muscle excitation-contraction coupling: who are the dancing partners? Int. J. Biochem. Cell Biol. 2014;48:28–38. doi: 10.1016/j.biocel.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Reva B, Antipin Y. Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y. Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- Sabatelli P, Gara SK, Grumati P, Urciuolo A, Gualandi F, Curci R, et al. Expression of the collagen VI alpha5 and alpha6 chains in normal human skin and in skin of patients with collagen VI-related myopathies. J. Invest Dermatol. 2011;131:99–107. doi: 10.1038/jid.2010.284. [DOI] [PubMed] [Google Scholar]
- Sabatelli P, Gualandi F, Gara SK, Grumati P, Zamparelli A, Martoni E, et al. Expression of collagen VI alpha5 and alpha6 chains in human muscle and in Duchenne muscular dystrophy-related muscle fibrosis. Matrix Biol. 2012;31:187–196. doi: 10.1016/j.matbio.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Nguyen AD, Bear KA. Wolfsberg TG. Clinical genomic database. Proc. Natl. Acad. Sci. USA. 2013;110:9851–9855. doi: 10.1073/pnas.1302575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorato E, Menazza S, Zulian A, Sabatelli P, Gualandi F, Merlini L, et al. Monoamine oxidase inhibition prevents mitochondrial dysfunction and apoptosis in myoblasts from patients with collagen VI myopathies. Free Radic. Biol. Med. 2014;75C:40–47. doi: 10.1016/j.freeradbiomed.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini F, Sardone F, Squarzoni S, Maraldi NM, Merlini L, Faldini C, et al. Ultrastructural changes in muscle cells of patients with collagen VI-related myopathies. Muscles Ligaments Tendons J. 2013;3:281–286. [PMC free article] [PubMed] [Google Scholar]
- Tagliavini F, Pellegrini C, Sardone F, Squarzoni S, Paulsson M, Wagener R, et al. Defective collagen VI alpha6 chain expression in the skeletal muscle of patients with collagen VI-related myopathies. Biochim. Biophys. Acta. 2014;1842:1604–1612. doi: 10.1016/j.bbadis.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Ibarra MC, Malicdan MC, Murayama K, Ichihara Y, Kikuchi H, et al. Central core disease is due to RYR1 mutations in more than 90% of patients. Brain. 2006;129:1470–1480. doi: 10.1093/brain/awl077. [DOI] [PubMed] [Google Scholar]
- Yates JR, Warner JP, Smith JA, Deymeer F, Azulay JP, Hausmanowa-Petrusewicz I, et al. Emery-Dreifuss muscular dystrophy: linkage to markers in distal Xq28. J. Med. Genet. 1993;30:108–111. doi: 10.1136/jmg.30.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JR, Bagshaw J, Aksmanovic VM, Coomber E, McMahon R, Whittaker JL, et al. Genotype-phenotype analysis in X-linked Emery-Dreifuss muscular dystrophy and identification of a missense mutation associated with a milder phenotype. Neuromuscul. Disord. 1999;9:159–165. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Exome target coverage statistics. The percent of bases of the targeted exome that had at least 20 high quality reads covering each base.