

Abstract
We describe an alternate approach to protein structure determination that relies on experimental NMR chemical shifts, plus sparse NOEs if available. The newly introduced alignment method, POMONA, directly exploits the powerful bioinformatics algorithms previously developed for sequence-based homology modeling, but does not require significant sequence similarity. Protein templates, generated by POMONA, are subsequently used as input for chemical shift based Rosetta comparative modeling (CS-RosettaCM) to generate reliable full atom models.
High-resolution protein structures, obtained by either X-ray crystallography or NMR spectroscopy, are available for only a small fraction of all known proteins and computational methods are commonly used to model structures for the remainder. Current protein structure prediction methods can be broadly separated into two classes: comparative modeling and de novo methods. Comparative modeling methods rely on detectable similarity between the query sequence and at least one protein of known structure, and can be used to generate models for all proteins in its family using a single representative structure as the starting point1,2. De novo methods, which use no structural template but only the amino acid sequence, rely on an effective conformation searching algorithm and good energy functions, and can be used to build structural models from scratch. However, due to bottlenecks in sampling of a conformational space that exponentially increases with the number of residues, this method remains restricted to small proteins3.
NMR chemical shifts of proteins encode important structural information, and are obtained at the early stage of any NMR structural study, even for quite large proteins4. It has long been recognized that integration of these data or other very limited, “sparse” restraints into structural modeling can be highly beneficial5. These ideas led to development of the powerful and popular de novo protein structure prediction programs, including CHESHIRE6, CS-Rosetta7 and CS23D8, which can generate good quality, all-atom models for proteins of up to ca 125 residues and a variety of folds. Supplementing the input chemical shift data with backbone residual dipolar couplings (RDCs), sparse 1HN-1HN nuclear Overhauser effect (NOE) data9, or distance restraints extracted from remote homology models10, can extend the size limit of the de novo structure generation approach, but the steeply increasing computational cost with protein size poses serious challenges.
Here, we introduce a more direct approach to integrate chemical shift and sparse NOE data into existing, very powerful comparative modeling algorithms. Further refinement of these models is achieved by modification of the previously introduced RosettaCM method11,12, to take advantage of the NMR data when filling in the missing parts and for energetically refining the final structures. Comparative modeling of a protein structure from sequence is used very widely and principally consists of two steps: First, finding related templates from known structures that have some sequence similarity to the query sequence and optimally aligning the query sequence with the sequence of the templates. In a second step, full 3D models are generated guided by information from the aligned templates.
Best alignment between two sequences is usually obtained by optimizing an alignment scoring function, which consists of two components: a matrix of pairwise substitution scores for matching each residue in the database protein to every residue in the query sequence, and a gap penalty function. Given an optimized scoring function, efficient dynamic programming is used to search for the optimal alignment between any pair of sequences. Many excellent comparative modeling methods are available, including the widely used MODELLER program13, I-TASSER 14.
Backbone torsion angles are encoded in NMR chemical shifts, and even though strictly local in character and often not unique, these chemical shifts contain far more information regarding structural homology than sequence alone. Much of the success of the popular chemical-shift-Rosetta (CS-Rosetta) method stems from the fact that chemical shifts facilitate finding of structurally homologous peptide fragments in the protein structure database (PDB) 7,15.
The protocol introduced here relies on a novel chemical-shift-guided protein alignment procedure, POMONA (Protein alignments Obtained by Matching Of NMR Assignments), followed by adaptation of the Rosetta comparative modeling method, RosettaCM12, to take advantage of the available chemical shifts. As a first step in the POMONA-based CS-RosettaCM structure determination protocol (Fig. 1a), experimental 13Cα, 13Cβ, 13C′, 15N, 1Hα, and 1HN chemical shifts are analyzed to generate for each residue a ϕ/ψ probability map. This map, calculated using the neural network based TALOS-N program16, assigns a normalized probability to each 20°×20° voxel of the Ramachandran map. POMONA uses these residue-specific Ramachandran probability maps to search the PDB for structures that are compatible with these ϕ/ψ-probabilities, while allowing for gaps and inserts in the residue sequence. Following an automated clustering and selection procedure, the representative homologues identified by POMONA are used as structural templates to a modified comparative modeling protocol, based on the RosettaCM program12, to generate all atom structures. For details, see Online Methods.
Figure 1.

POMONA/CS-RosettaCM structure generation (a) Flowchart of the POMONA/CS-RosettaCM structure generation protocol. (b–e) Results of POMONA/CS-RosettaCM structure generation for four representative test proteins: nsp1, sensory rhodopsin, maxacal and maltose binding protein (mbp). (b) For each of these, the POMONA alignment scores (H′, Eq. 5) of the top 1000 protein chains in the PDB are plotted versus the Cα-RMSD, calculated over the aligned residues between the query and the database protein. Grey and black dots correspond to sequence identities <20% and ≥20%, respectively, between the query and database protein. After clustering analysis for the alignments with < 20% sequence identity, alignments contained in the ten highest scoring clusters are colored according to the cluster number, i.e., red, green, blue, magenta, dark-green, yellow, cyan, orange, grey and brown for clusters 1–10, respectively. Only the two highest scoring alignments from each of these ten clusters are used as structural templates for CS-RosettaCM modeling. (c) ROSETTA all-atom energy, incl. the experimental chemical shift score, for the CS-RosettaCM models versus their Cα-RMSD relative to the experimental structure. Colors correspond to those of the starting template. For comparison, the horizontal line and the graph at the bottom of each panel represent the lowest Rosetta all-atom energy and the normalized number of structures, respectively, obtained by CS-Rosetta. (d) Same as c but for POMONA/CS-RosettaCM modeling with additional sparse 1H-1H NOE data. (e) Ribbon models of the lowest energy CS-RosettaCM structure (red) (calculated without sparse NOEs) superimposed on the corresponding experimental structure (blue).
To evaluate POMONA’s accuracy and coverage, we rely on the widely used MaxSub score17, which ranges from 1.0 when two aligned structures have a Cα-RMSD of 0 Å for the full length of the query sequence to ~0.0 when sequences lack detectable similarity. Typically, a MaxSub score above ~0.3 is indicative of notable structural similarity (Fig. 2a; Supplementary Fig. 1).
Figure 2.
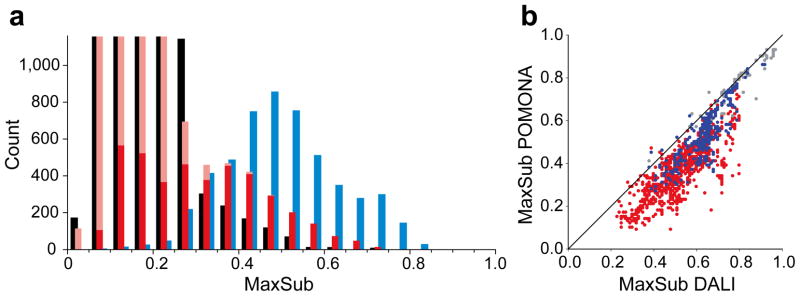
Comparison of protein structure alignments obtained by different methods for the 16 proteins listed in Table 1. (a) Histogram of protein structure alignment quality, represented by a MaxSub score, for the top 1000 alignments identified by POMONA (red bars), the sequence alignment method HHsearch (black), and the structure alignment method DALI (blue). Results are shown only for PDB proteins with < 20% sequence identity to the target protein, and DALI and HHsearch results correspond to default thresholds of Z ≥ 2 and Prob ≥ 10%, respectively, used by these programs to identify homologues. The DALI histogram indicates the limit of how good any search program could possibly function. Positive POMONA alignments are taken from the top ten clusters (solid red bars) within the top 1,000 alignments (solid + transparent red), as identified by the highest H′ score (Eq. 5). (b) Comparison of alignment quality obtained by DALI and POMONA methods. For each of the positive alignments identified by both DALI and POMONA, the MaxSub scores are compared, with color representing sequence identity to the query protein (grey: ≥ 30%, blue: 20–30%, red: < 20%) as observed in the DALI alignments.
When evaluating the performance of POMONA in identifying suitable homologous structures in the PDB, a key question is “how many suitable structures exist?” This can be answered with the program DALI, which is designed to identify structurally similar proteins, regardless of residue sequence18. When comparing to the DALI identified alignments for a set of 16 test proteins, POMONA-identified structural homologues approach the maximum attainable alignment (or MaxSub score) provided by the DALI method (Fig. 2b), performing much better than sequence based alignment by, for example, the powerful HHsearch method19 (Supplementary Fig. 2).
The quality of POMONA alignments roughly correlates with the alignment score (Fig. 1b, Supplementary Fig. 3). However, there also is considerable scatter in this correlation, which means that we cannot simply use the top POMONA alignments as starting templates for CS-RosettaCM. Instead, we find it important to generate a diverse pool of structure templates by subjecting the top scoring alignments to a cluster analysis, and only retaining the two top scoring alignments in each of the first ten clusters (see Online Methods). For most of our 16 test proteins the highest MaxSub score observed for this (up to) 20-member subset is comparable to that obtained for the top 1,000 positive alignments (Table 1). For all but one of the 16 proteins, the best alignment in the selected representative alignments has a MaxSub value in the 0.25–0.69 range, making them useful structural templates for structure generation. Only for protein Mad2 did POMONA fail to find a suitable template. DALI finds three suitable templates in the database, but all contain large gaps (>100 residues), preventing their identification by POMONA.
Table 1.
Performance of POMONA alignment and CS-RosettaCM structure generation for 16 test proteins.
| Name | Size | PDB/BMRB# a | Fold | Homologues & alignments
|
CS-RosettaCM |
csRosetta | ||
|---|---|---|---|---|---|---|---|---|
| DALI b | POMONAc | Rmsdmean d | Rmsdexp d | Rmsdexp d | ||||
| nsp1 | 113 | 2gdtA/7014 | α/β | 2/0/2(0.50) | 0.30/0.30 | 2.18±0.63 | 3.30±0.73 e | 12.1±1.3 |
| HR2876B | 117 | 2ltmAa/18489 | α/β | 2/4/75(0.47) | 0.41/0.41 | 2.89±0.72 | 4.21±0.55 | 6.41±2.76 |
| YR313A | 119 | 2ltlAa/18487 | α/β | 1/2/52(0.45) | 0.26/0.25 | 1.60±0.27 | 3.67±0.45 | 2.80±0.68 f |
| OR36 | 134 | 2lciAa/17613 | α/β | 4/5/799(0.50) | 0.36/0.34 | 2.19±0.73 | 4.32±0.56 | 3.05±0.35 |
| OR135 | 83 | 2ln3Aa/18145 | α/β | 1/1/651(0.70) | 0.52/0.40 | 1.35±0.49 | 1.88±0.42 | 1.21±0.13 e |
| HR2876C | 87 | 2m5oAa/19068 | α/β | 4/4/723(0.57) | 0.35/0.33 | 1.77±0.27 | 2.24±0.42 | 1.17±0.20 e,f |
| MTH1958 | 153 | 1tvgA/6344 | β | 5/14/147(0.76) | 0.53/0.51 | 1.30±0.18 | 2.35±0.17 e | 10.4±4.9 |
| sgr145 | 173 | 3merA/16806 | α/β | 3/43/896(0.72) | 0.66/0.52 | 2.30±0.58 | 3.05±0.74 | 8.2±2.8 |
| fgf2 | 125 | 1basA/4091 | β | 262/23/449(0.83) | 0.74/0.65 | 1.06±0.18 | 1.56±0.19 e | 11.7±1.5 |
| tpx | 167 | 2jszAa/15797 | α/β | 49/376/389(0.70) | 0.69/0.68 | 1.60±0.23 | 2.32±0.22 e | 17.7±2.0 |
| YwIE | 150 | 1zggA/6460 | α/β | 17/66/308(0.65) | 0.69/0.69 | 1.19±0.18 | 1.86±0.23 e | 11.0±3.7 |
| fluA | 184 | 1n0sA/5756 | β/α | 11/38/413(0.63) | 0.51/0.51 | 2.01±0.49 | 3.46±0.34 e | 8.5±1.5 |
| mad2 | 196 | 1go4C | α/β | 42/10/3(0.44) | 0.13/0.11 | 12.74±4.45 | 19.81±1.01 | 15.8±2.6 f |
| s. rhodopsin | 222 | 2ksyAa/16678 | α | 23/153/149(0.64) | 0.62/0.62 | 2.32±0.43 | 3.09±0.51 e | 17.8±3.5 |
| maxacal | 269 | 1svnA | α/β | 273/79/4(0.51) | 0.50/0.50 | 3.29±0.57 | 4.51±0.85 e | 19.4±2.6 |
| mbp | 370 | 1dmbA | α/β | 276/31/182(0.52) | 0.52/0.51 | 2.73±0.50 | 4.24±0.73 e | 26.3±2.1 |
The PDB code for proteins with an NMR-derived structure as the reference.
Number of alignment hits with sequence identity of ≥30%, 30%-20% and <20%, respectively, and a minimum alignment length of at least 2/3 of the total number of target residues; the highest MaxSub value observed for the alignments with a sequence identity of <20% is listed in parentheses.
Highest MaxSub value observed among all top 1,000 POMONA-alignments (sequence identity <20%) and highest MaxSub score among the up to 20 templates used for subsequent CS-RosettaCM modeling.
Cα-RMSD value calculated for all non-flexible residues (as identified by a RCI-S2 ≥0.6 (ref.20)). RMSDmean is the Cα-RMSD between the ten lowest-energy models and their mean coordinates. RMSDexp is the Cα-RMSD between the ten lowest-energy models (derived using database proteins with sequence identity <20%) and the experimental reference structure.
CS-RosettaCM and CS-Rosetta structures that met the acceptance criterion (see Online Methods). To convert a calculated RMSD value to its corresponding RMSD100 value (used in our work to evaluate convergence, see Online Methods), RMSD100 = RMSD/(1+ln(N/100)), where N is the number of residues of the protein.
CS-Rosetta models with a lower Rosetta energy than obtained with the POMONA/CS-RosettaCM approach.
For four representative cases, the database proteins corresponding to the top 1,000 POMONA-derived alignment scores are plotted against their Cα-RMSD relative to the experimental reference structure (Fig. 1b), with database sequences that have <20% sequence identity shown as grey dots, or as colored symbols if they are part of the ten top-scoring clusters. For comparison, POMONA hits for more homologous proteins (≥20% sequence identity) are shown as black symbols, but these are not used in our study as they typically can be identified by standard homology search programs.
When the two highest-scoring members of each cluster are subjected to the CS-RosettaCM protocol, a clear correlation is seen between the lowest total all-atom energy reached for each cluster, and the Cα-RMSD (Fig. 1c). Even though for all four proteins the highest POMONA alignment scores are comparable between the top clusters, the clusters that had the lowest Cα-RMSD relative to the native structure refine to lower total energy during CS-RosettaCM modeling. Correspondingly, the lowest-energy CS-RosettaCM models provide the best match to the query protein. However, because it is by no means guaranteed that a correct solution can be found, especially when there are no proteins with a similar fold in the database (e.g. Mad2, mentioned above), it is useful to compare the total energy with what can be achieved with the standard CS-Rosetta protocol. CS-Rosetta will typically fail for large proteins, and a requirement for accepting a CS-RosettaCM structure therefore is that the total energy, including the chemical shift scoring term, falls well below the lowest values obtained by CS-Rosetta. A second requirement for acceptance is that the ten lowest-energy structures have converged, i.e. cluster within a Cα-RMSD100 of ≤2.5 Å from their average. Both requirements are used to inspect all 16 proteins tested in our study (Table 1).
Immediately following backbone resonance assignment it is usually straightforward to rapidly assign a limited number of unambiguous backbone 1HN-1HN NOEs. These sparse NOEs can be exploited both for guiding POMONA alignment and as restraints during CS-RosettaCM modeling. To evaluate their utility, two sets of such artificial HN-HN NOE distance restraints were generated by randomly selecting N/10 of such distances from the total set that are ≤ 5 Å in the experimental structure and at least five residues apart in sequence, where N is the total number of residues in the protein. In practice, a somewhat larger number of such NOEs is often obtained, in particular when working with perdeuterated proteins4. Inclusion of sparse NOEs enables POMONA to find improved alignments, resulting in better convergence and lower energies during the subsequent CS-RosettaCM modeling stage (Fig. 1d, Supplementary Table 1).
Whereas for proteins larger than ca 100 residues, conventional CS-Rosetta approaches its convergence limits, CS-RosettaCM remains robust in generating converged results, largely because it inherently is a comparative modeling method. Note, however, that the POMONA/CS-RosettaCM protocol is not aimed at reaching maximum convergence, but that the clustering approach used by POMONA emphasizes diversity in the input templates to avoid falsely converging to a wrong solution. As a result, the convergence rate for small structures can actually be higher for standard CS-Rosetta than for our new protocol.
Considering that ~90% of the newly deposited structures already have similar structures present in the PDB, the POMONA/CS-RosettaCM approach is ready to dramatically reduce the workload required for such studies, while extending the size of proteins that easily can be studied by NMR. The approach will fail, however, when no adequate structural template exists in the PDB, or when the only good potential templates have large alignment gaps.
For proteins larger than ca 20 kD, standard protein NMR structure determination typically remains quite labor-intensive, even though chemical shift assignment and collection of amide-amide NOEs is relatively straightforward, and the POMONA/CS-RosettaCM protocol is an enabling technology for such systems. Finding suitable templates is an efficient process which can be completed in a matter of hours, but subsequent CS-RosettaCM modeling is far more computationally intensive. Nevertheless, due to the use of suitable input templates it does not suffer from the combinatorial explosion that restricts conventional Rosetta and CS-Rosetta applications. For large, multi-domain proteins, it is important to note that the NMR chemical shifts do not contain information on relative domain orientation or position, and that this information strictly stems from the PDB template used for modeling. However, the measurement of residual dipolar couplings (RDCs) is often straightforward for larger systems, and readily can be integrated in the modeling procedure to resolve such issues.
The POMONA software and server are at http://spin.niddk.nih.gov/bax/software/POMONA.
ONLINE METHODS
Measurement of local structure similarity
Finding the optimal alignment between two protein sequences typically is based on a residue substitution score for all residue pairs of the two sequences. Such substitution scores, which normally are derived from the amino acid similarity scores, are then used for guiding the alignment procedure to find a set of aligned residues along two sequences that have an optimal overall alignment score. Unlike the sequence based alignment, POMONA aims to align residues of a query protein, which has known NMR chemical shifts, to residues of a database protein with known structure. Structural information encoded in the NMR chemical shifts of the query protein, specifically the ϕ/ψ backbone torsion angles and the secondary structure predicted by TALOS-N16, offers much more definitive information than simply the amino acid type when searching for structural similarity between the query and database proteins. Therefore, these backbone torsion angles and the secondary structure derived from chemical shifts are used as the main terms in deriving substitution scores for the alignment procedure.
In POMONA, a substitution score S(i,j) between residue i in the query protein and residue j in the database protein is defined as:
| [1] |
S(i, j) contains three terms: (1) The ϕ/ψ-fitness score, which has a weighting factor wtorsion, reflects how well the angles of query residue i match to the observed ϕ/ψ angles of database residue j. Here, Di,k (k = 1-324) is the TALOS-N predicted density of voxel k in the 324-voxel ϕ/ψ density map of query residue i, and k(j) is the index number in the 324-voxel Ramachandran map that corresponds to the ϕ/ψ angles of residue j of the database protein. The ϕ/ψ-fitness score, is calculated from Di,k(j) by first subtracting the average of the predicted densities, <Di>, followed by normalization according to the standard deviation, σ(Di), of the predicted densities, which then represents the likelihood that the ϕ/ψ torsion angles of residues i match those of j. (2) The amino acid similarity score between residue i (of amino acid type Ai) and residue j (of type Aj), B(Ai,Aj), which is taken from the BLOSUM62 matrix21. (3) The secondary structure similarity score between the TALOS-N predicted 3-state secondary structure SSi (H, E and L, respectively) for residue i, and the observed secondary structure SSj (as assigned by the program DSSP)22 for residue j:
| [2] |
where conf(i) is the confidence of the TALOS-N predicted secondary structure SSi.
Note that terms (2) and (3) are the principal terms used in conventional, sequence based homology search procedures. In our search, term (2) has a very low weight factor, and term (3) is derived from experimental chemical shifts, which have been shown to considerably increase the accuracy of predicted secondary structure16.
As seen in Eq. 1, our local structure similarity score between residues i and j, includes terms for its two immediate neighbors, i.e., between residues i-1 and j-1, and between residues i+1 and j+1. The weights, wtorsion and wSS, of these terms have been optimized empirically, together with other parameters used by the POMONA alignment method, such that the calculated substitution scores S(i,j) fall in a range of −2.0 to 3.0. The maximum contribution to S(i,j) from residue type similarity (term 2 in Eq. 1) is less than ca 10% when chemical shifts are available. For query residues that lack chemical shifts, only the sequence similarity and secondary structure matching term in Eq. 1, with a comparable weight, are used to calculate a S(i,j) score, which is then scaled to the same range of −2.0 to 3.0.
Protein alignment algorithm
The problem of finding the optimal alignment of two amino acid sequences has been extensively studied and most commonly is solved by means of a dynamic programming algorithm 23,24. POMONA essentially uses the standard Smith-Waterman dynamic programming algorithm25 to find the best alignment between a query protein with ϕ/ψ angle information derived from chemical shifts and a database protein of known structure. Specifically, given a query protein and a database protein of sequence lengths M and N, a substitution scoring matrix S of dimensions M × N is first constructed. Each element of this scoring matrix, S(i,j) (Eq. 1), is derived from the local structural similarity between residue i in the query protein and residue j in the database protein. The aim is to align residues with matching local structure in the two proteins while optimizing the overall alignment score, which is a sum of the substitution scores of all aligned residue pairs (also referred to as equivalent residues) and gap penalties (see Supplementary Note for details) for residues lacking an equivalent residue in either sequence. The recursive dynamic programming equation used here for the local alignment of the two proteins is:
| [3] |
with the initial conditions for the recursion defined by H(M+1, j) = 0 and H(i, N+1) = 0, where M and N again are the sequence lengths of the query and the database protein, G is the VGP gap penalty function (Eq. 1 in the Supplementary Note), and S(i,j) is the residue substitution score for residues i and j in the query and the database proteins, respectively (Eq. 1). The dynamic programming maximum scoring matrix H is calculated for i = M + 1 to 1 and j = N + 1 to 1. For each position [i, j] in H, all previously iterated positions [i′,j′], with i′ = [i+1:M] and j′ = [j+1:N], are evaluated for a maximum value based on the previously calculated H(i′,j′) value for position [i′,j′], while using a gap penalty G(i,j,i′,j′) for opening a gap between positions [i,j] and [i′,j′]. After adding its residue substitution score S(i,j), this maximum value is then assigned to the current position as score H(i,j). After calculating all elements of the H matrix, its largest element, referred to as max(H), corresponds to the optimal alignment score. The residue equivalence assignments are obtained by backtracking in matrix H, starting from the element with the max(H) score and ending with the first element of zero value24. Equivalent residues in this optimal alignment are further evaluated in terms of fitness between their experimental secondary chemical shifts (of the query residues) and those predicted by SPARTA+ 26 (for the database residues), in terms of a χ2 value:
| [4] |
where is the SPARTA+ predicted backbone chemical shift (k = 13Cα, 13Cβ, 13C′, 15N, 1Hα, and 1HN) for a given database residue j, which is aligned to query residue i with experimental chemical shift , and σk,j is the uncertainty of reported by SPARTA+. This , after scaling by a factor c = 1/30, is then added to the optimal alignment score as a penalty to derive a final alignment score for any given alignment of:
| [5] |
Structure alignment with additional NOE data
Some types of NOE data, in particular HN-HN NOEs, often can be obtained relatively easily and unambiguously once the backbone amide signals have been assigned, even for large perdeuterated proteins. Unfortunately, there is no straightforward method for directly integrating such sparse NOE distance information into the Smith-Waterman algorithm. However, the typically very sparse NOE data can be useful to aid the above “chemical-shift-guided” POMONA protein alignment scheme by pre-filtering possible solutions based on these distance constraints, and subsequently evaluating these possible matches by the above described algorithm to generate optimally aligned sequences. The NOE-guided part corresponds to the general problem of finding the optimal alignment of protein structure distance matrices (or protein contact maps)18,27,28, as both the NOEs detected for the query protein and the actual distances measured for the database protein can be converted to contact maps.
Here, we use the method of Wohlers et al.28 to find the optimal overlap between two contact maps derived from the query and the database protein. For the query protein with a NOE list (NOE), a contact map X of size M × M is constructed:
| [6] |
where i and i′ = [1, …, M] and M is the size of query protein. For the database protein, an analogous contact map Y of size N × N is constructed:
| [7] |
where j and j′ = [1, …, N], N is the size of the database protein, and d(j, j′) is the actual HN-HN distance between residues j and j′ in the database protein. Contacting residues i and i′ with X(i, i′) = 1, as well as j and j′ for Y(j, j′) = 1, are stored as lists x and y, respectively. Optimal alignment then corresponds to finding a maximum set of matching [ik, jk] pairs (k = 1 to L, ik ⊂ x and jk ⊂ y, L is the lower of the two numbers of contacting residues, usually the size of list x), between the pairs of contacting residues in the query and database proteins. The largest set of common contacts is based on the objective function:
| [8] |
| [9] |
After finding the optimal match between the query and the database contact maps, the second step of the structure alignment, based on chemical shift data, is restricted to the regions identified by this optimal contact map. For the query and the database proteins with a set of optimally aligned contacting residues [ik, jk], where k = 1 to L, ik ⊂ x and jk ⊂ y, ik < ik+1 and jk < jk+1, the query and the database sequences are divided into L-1 fragment pairs, each of which has a range from ik to ik+1 and from jk to jk+1, respectively. The above described POMONA protein alignment scheme is then applied for each possible pair of query fragment (ik, ik+1) and database fragment (jk, jk+1) [k = 1 to L - 1], now using Eq. 1 as the scoring term. The final overall alignment is then obtained by combining each of the “sub-alignments”, and the final alignment score is taken by summing the POMONA alignment scores from each of the “sub-alignments”, augmented by the penalizing, scaled chemical shift fitness score (Eq. 4).
Training and testing of POMONA
Values for the parameters used by POMONA were obtained iteratively by evaluating the output results for a set of 16 test proteins of varying size and fold complexity (Table 1). POMONA is used to find optimal alignment between the test protein and each of ca 252,000 protein chains in the PDB. POMONA initially retains the 1,000 PDB protein chains that exhibit the highest alignment score. The parameter optimization of POMONA was performed iteratively by monitoring the top 1,000 selected proteins in terms of (1) the ratio of the real structural homologues, as identified by the DALI structure alignment method18 with the actual structure of the target protein, and (2) the accuracy of the POMONA identified alignment to the target protein, expressed in terms of a coordinate RMSD value calculated between the Cα-atoms of the equivalent residues in the target and database protein.
Performance evaluation of POMONA structure alignment
We evaluate the accuracy and coverage achieved by POMONA by using the MaxSub score17. The MaxSub score for two aligned structures (i.e., the query and database proteins) is calculated by first identifying the maximum substructure for which the distances between equivalent residues of two structures after superposition are below a threshold value of 3.5 Å, then computing a normalized score of Σ[1/(1+(di/3.5)2)]/N, where di are the distances between equivalent Cα pairs of two structures in the maximum substructure (after best-fit superposition of the Cα pairs in the maximum substructure), and N is the total length of the query sequence. The spatial information of the aligned structures outside the maximum substructure is not taken into account. The MaxSub score ranges from 1.0, for perfect alignment to near zero when sequences lack structural similarity. Two aligned structures with a 0 Å Cα-RMSD for half of the query sequence length and two aligned structures with a Cα-RMSD of ~3.5 Å for the full length of the query sequence will have the same MaxSub value of 0.5, and a score above ~0.3 is usually indicative of meaningful structural similarity (Fig. 2a; Supplementary Fig. 1).
When comparing to the theoretical limit set by DALI-identified alignments, for our set of 16 test proteins POMONA identifies nearly all of the 2,660 homologues with a sequence identity of ≥20% to the target proteins, missing only ~50 DALI-identified structural homologues. The POMONA-identified homologues show near optimal alignments in terms of MaxSub score compared to DALI (Fig. 2b), performing much better than sequence based alignment (Supplementary Fig. 2), such as the HHsearch method19, one of the best modern sequence-based alignment methods. Structural homologues identified by DALI but missed by POMONA all have long alignment gaps (Supplementary Fig. 4), resulting in depressed POMONA alignment scores that then fall below the detection threshold. Aligning two proteins with long alignment gaps is invariably challenging with a Smith-Waterman based algorithm, as too small a gap penalty would open false gaps whereas too large a gap penalty prevents the opening of gaps. In our work the implemented gap penalty function is tuned to allow the alignment to cross relatively small gaps; the largest lengths for single alignment gaps observed in the POMONA identified alignments are in the 20–30 range.
Not surprisingly, POMONA performs well for finding alignment for homologues with significant sequence identity (> 20–30%), where many other sequence alignment methods also perform well even when solely relying on sequence information. The more important question therefore is how well the program functions in finding structural homologues when there is very little or no significant sequence identity. When parameterized to detect even very weakly homologous structures, HHsearch identifies similarity in a total 10,059 protein chains with a sequence identity of <20% to the target protein for our set of 16 test proteins. Of these, only 8% are consensus with the DALI identified structural homologues that have a sequence identity of <20%, and 85% of the 5,211 DALI-identified structural homologues cannot be identified by HHsearch on the basis of sequence alone. Importantly, POMONA identifies among its positive alignments a large portion (~46%, 2,414/5,211) of DALI-identified homologues target proteins when restricting the search to proteins with <20% sequence identity (Fig. 2a). Structural homologues missed by POMONA nearly all exhibit long alignment gaps (≥ 30) in the DALI identified alignments (Supplementary Fig. 4a). We find that POMONA also missed a number of NMR-determined structures, even in the absence of large gaps. Inspection of these structures indicates that even though the fold of these proteins is close enough to register a DALI alignment, the local backbone deviates too far from ideality to allow their recognition on the basis of chemical shifts.
Clustering and selection of POMONA alignments
Among the top 1,000 alignments identified by POMONA for any given query protein, many will be very similar to one another. Before using these proteins as input for the time-consuming RosettaCM comparative modeling, it therefore is useful to separate this set into a much smaller number (typically ten) of distinct clusters, and only the two best-scoring (cf Eq. 5) models in each cluster are then used as RosettaCM input. Specifically, a hierarchical clustering procedure is used to group the top 1,000 database protein chains, using the normalized Cα-RMSD as a metric. The normalized Cα-RMSD between two database protein chains is calculated only over residues that are commonly aligned to a residue in the query protein, i.e. that do not correspond to inserts or gaps. Subsequently the Cα-RMSD is normalized to the RMS100 value29. A single-linkage algorithm is used for generating the clusters with a Cα-RMS100 ≤4 Å cutoff, and results are sorted by the highest alignment score observed in each cluster. The ten clusters with highest alignment scores are retained and the top two alignments (or one, if there is only a single member in the cluster) are selected as representatives from each of the first ten clusters. Therefore, up to 20 representatives alignments are selected from the first ten clusters and these are used to prepare a pool of structural templates for the subsequent RosettaCM modeling procedure.
When applying this selection criterion to the POMONA alignments with a sequence identity <20%, for most of our 16 test proteins the highest MaxSub score observed for this (up to) 20-member subset is comparable to that obtained for the top 1,000 positive alignments (Table 1). Evaluating the suitability of a given protein alignment as input for RosettaCM comparative modeling is not a straightforward problem, in particular when the protein contains gaps and/or inserts. Alignment accuracy, i.e. the RMSD between the coordinates of equivalent Cα atoms of corresponding residues in the database and query proteins is an important but not the only metric, and coverage can play an equally important role. For example, an aligned database protein chain with a 3-Å RMSD to the target protein and 50% alignment coverage is not necessarily better for structure modeling than one with a 5-Å RMSD but having 90% alignment coverage. An extreme example is seen when comparing the alignments between maltose binding protein, or MBP (PDB and chain id: 1dmbA), and three chains of the engineered protein RG13 (4dxbA, 4dxbB, 4dxcA) (Fig. 1b), which has a high POMONA alignment score but poor alignment accuracy, with a Cα-RMSD value of > 15 Å. RG13 is derived from MBP by substituting its residues 317 and 318 by a 267-residue domain. The MBP domain of RG13 has a sequence identity >99% and a Cα RMSD of only 1.14 Å relative to MBP (Supplementary Fig. 5a). However, due to the large penalty associated with the 267-residue alignment gap, POMONA matches the first 316 residues of these two proteins, or 85% of its total length. The C-terminal 15% fraction of the chain, consisting of three α-helices, are matched by POMONA to the first three helices of the domain inserted in RG13 (Supplementary Fig. 5b), resulting in > 15 Å Cα RMSD. However, despite this large RMSD, the final refined structures are fairly close to the MBP X-ray reference structure, which can be credited to the power of the CS-RosettaCM procedure when provided with the correct secondary structure input.
Structure generation using CS-RosettaCM
The recent RosettaCM protocol12 offers a powerful comparative modeling module within the Rosetta software suite for generating accurate protein models. The inputs to RosettaCM comprise (1) sequence alignments between the query protein and database proteins that serve as structural templates, and (2) standard Rosetta de novo modeling fragments, needed to model the unaligned regions and to explore deviations from the templates in the aligned regions. In our protocol, RosettaCM is used to build 3D protein models, starting from the up to 20 structural templates identified above by POMONA.
Generation of complete, all atom models involves three steps. First, RosettaCM assembles protein backbone topologies by recombining the aligned segments of the query protein and the database template in Cartesian space while building the unaligned regions de novo in torsion angle space. This process uses long fragments (corresponding to secondary structure elements) derived from all template inputs as well as CS-Rosetta de novo fragments (with sizes of 3 and 9 residues), respectively. In the standard RosettaCM implementation, these de novo fragments are selected on the basis of residue sequence, whereas in our work they are picked on the basis of the NMR chemical shifts, using the recently improved chemical shift based Rosetta3 fragment picker15, again excluding all proteins with ≥20% sequence identity from the library. In the second stage, all broken backbone segments are closed by means of a standard loop closure method that combines fragment superposition and structure minimization. The probabilistic distance restraints derived from the alignments, used in standard RosettaCM30, are removed but experimental NOE distance restraints, if available, are included during this stage. Third, the resulting backbone models are optimized using the final all-atom refinement step of standard CS-Rosetta7, but using the most recent parameter set (talaris2013.wts) for scoring the energy.
Selection of all atom models using energies and chemical shifts
Using the above protocol, for each query protein, CS-RosettaCM is parameterized to generate 500 all-atom models from each starting template, for a total of up to 10,000 models. Those models are further evaluated for their fitness with respect to their experimental NMR chemical shifts, using the same method developed for the standard CS-Rosetta protocol7. Specifically, for each all-atom model, a χ2 value is first calculated between the experimental chemical shifts and values predicted by SPARTA+ 26, which is then added to the Rosetta all-atom energy. This chemical shift re-scored Rosetta all-atom energy is used to evaluate and select the final models.
Criteria for convergence and accepting models
The ten models with lowest Rosetta all-atom, chemical shift re-scored energy are retained for inspection of their convergence relative to the lowest energy model, and are accepted as the predicted structure only if (1) these models cluster within less than 2.5 Å, in terms of Cα-RMSD100, from the model with the lowest energy, and (2) the average Rosetta energy of the ten lowest energy models is at least two standard deviations lower than the ten lowest energy models obtained by standard CS-Rosetta (provided with the same inputs and the same all-atom energy scoring scheme).
Performance evaluation of CS-RosettaCM protein structure generation
POMONA-identified alignments offer an accuracy and coverage that approaches the DALI limit. After the clustering and selection procedure, these then are used as input for CS-RosettaCM to generate complete structural models.
For comparison, the standard CS-Rosetta structure generation protocol10 is also performed for all 16 test proteins. However, CS-Rosetta is only able to generate converged (and correct) structures for the smallest of the proteins tested, and for the two proteins with less than 100 residues it actually outperforms the POMONA/CS-RosettaCM method. The latter reaches convergence for 15 out of the 16 proteins tested (after having removed all proteins with ≥ 20% sequence identity from the database), with all of these being close to the target structure (RMSD100 < ~2.5Å; Table 1, Supplementary Fig. 6).
The 16 proteins used to evaluate our method pose different types of challenges. First we focus on four proteins selected from structural genomics projects, incl. HR2876B, YR313A, OR36 and nsp1, that have very few sequence homologues (Fig. 1b, Supplementary Fig. 3) and no good structural homologues in the database (Table 1). Indeed, the best structural homologues identified by DALI for database proteins with <20% sequence identity all have MaxSub scores ≤0.5 (Table 1). Nevertheless, POMONA is able to identify such alignments, and reaches comparable MaxSub scores for the database proteins it selects (Table 1). The resulting CS-RosettaCM models for these proteins all converged quite well, with the ten lowest energy models for each of these clustering within 3Å relative to the model with the lowest Rosetta energy (Table 1, Supplementary Fig. 7). However, within this group of relatively small proteins, only for nsp1 is a considerably lower total Rosetta energy obtained compared to simply using CS-Rosetta (Fig. 1c, Supplementary Fig. 7). Therefore, nsp1 is the only protein in this group for which the CS-RosettaCM model is accepted. For nsp1, the only two structural homologues (3zbdA and 3zbdB; sequence identity ~15%) selected by POMONA have MaxSub scores of 0.30 (Table 1) and a Cα-RMSD of 4.5–5.0 Å (Fig. 1b) for their aligned regions. These input templates suffice for enabling CS-RosettaCM to generate all-atom models with a Cα-RMSD of ~3.3 Å to the experimental structure for its ordered regions. For the other three proteins, the CS-RosettaCM models have a Rosetta energy that is comparable to those of the de novo CS-Rosetta structures. Even though the folds of these CS-RosettaCM models happened to be correct, they could not be accepted as they did not meet the criterion that a substantially lower energy must be reached.
For the other four structural genomics proteins, OR135, HR2876C, sgr145 and MTH1958, DALI identified a substantial number of good structural homologues, with MaxSub scores in the 0.57–0.76 range when considering only database proteins with <20% sequence identity (Table 1, Supplementary Fig. 1). POMONA also identifies many of these alignments, albeit with lower MaxSub scores (Table 1). The low energy RosettaCM structures for these four test proteins all converged to within ~2 Å relative to the structure with the lowest Rosetta energy (Table 1, Supplementary Fig. 7). However, only for proteins sgr145 and MTH1958 does CS-RosettaCM reach energies substantially lower than standard CS-Rosetta (Supplementary Figs. 6 and 7), allowing these models to be accepted. For the small OR135 and HR2876C proteins (< 90 residues), both CS-Rosetta and CS-RosettaCM generate converged and accurate structures, with comparable Rosetta energies.
The remaining eight proteins in our test set are larger (125 to 370 residues), and standard CS-Rosetta fails to converge. For seven of these, many structural homologues with <20% sequence identity and MaxSub scores in the 0.51–0.83 range are identified by DALI (Table 1). POMONA also identifies many of these homologous structures (Fig. 1b, Supplementary Fig. 3), but only for those without large gaps in their DALI-identified alignment. For example, for sensory rhodopsin-II, most of the 149 DALI-identified structures with < 20% identity show gaps of > 100 residues, and POMONA is only able to identify six structural homologues with the shortest gaps (dots with red circles in Fig. 1b). However, this provides an adequate set of starting templates for successful CS-RosettaCM modeling (Table 1). Note that substantial structural rearrangements can occur during the CS-RosettaCM modeling stage. For example, starting from the fifth-ranking cluster with a Cα RMSD of >7.5Å relative to the reference structure, refined models with a backbone RMSD <4Å are obtained by CS-RosettaCM (dark green colors in Fig. 1b,c, right most panels).
For Mad2, only three suitable structural homologues with <20% sequence identity are found by DALI, all with large gaps in their alignments, and these cannot be identified by POMONA. Therefore, without even a single remote structural homologue among the POMONA-obtained templates, subsequent CS-RosettaCM modeling fails to converge (Supplementary Fig. 7). Moreover, none of the models generated even reach an energy as low as the unsuccessful CS-Rosetta approach (Supplementary Figs. 6 and 7), which as expected also fails to converge for this relatively large protein of 196 residues, providing an additional indication that none of the CS-RosettaCM models are of acceptable quality.
The above results demonstrate that the POMONA/CS-RosettaCM protocol performs well, provided that a reasonable structural template can be positively identified by POMONA. In practice, a template with a MaxSub score ≥ 0.3 is needed for successful modeling of all-atom models by CS-RosettaCM. When applying the protocol to a protein of unknown structure, the MaxSub score is not available, and the strict acceptance criteria defined for the CS-RosettaCM approach then are important to ensure correctness of the generated models. Importantly, CS-RosettaCM actually remodels its input template due to the hybrid fragment assembly procedure that is used for both the aligned and unaligned parts of the templates. However, even while this remodeling generally improves the agreement between the template component of the final CS-RosettaCM models and the experimentally determined reference structures, it is insufficient to find or correct the fold of the protein when no adequate structural template is available as input.
The importance of the quality of the input structural templates to CS-RosettaCM is further evaluated by extending the POMONA search to proteins with a sequence identity of up to 30%. Except for nsp1, which has no homologues in the 20–30% range in the database, POMONA identifies virtually all of these more homologous structures it its highest scoring clusters, ensuring that at least some of these will be used as structural templates by CS-RosettaCM. With this improved template quality, reflected in higher MaxSub scores (Supplementary Table 1), CS-RosettaCM then converges for all 16 proteins, yielding improved structural accuracy relative to the experimental structure of the query protein (Supplementary Fig. 8). Comparison of the structural accuracy obtained with the POMONA/CS-RosettaCM protocol when using only the <20% sequence identity templates with those available when using a 30% cutoff confirms that for about half the proteins a structure closer to the experimentally determined structure is obtained, whereas for the other half the final result remains about equal.
Evaluation of modeling performance when including sparse NOEs
Once backbone assignments have been completed it usually is straightforward to measure and assign a limited number of unambiguous backbone 1HN-1HN NOEs. The utility of such sparse NOEs is evaluated by randomly selecting for each N-residue protein several sets of N/10 long range HN-HN NOE distance restraints that are ≤ 5 Å in the experimental structure.
Using again the <20% sequence identity cutoff, the sparse NOEs result in improved POMONA alignments (Supplementary Table 1) for all test proteins, except for Mad2 which has no suitable template available in the database. For example, for nsp1, its two closest structural homologues did not yield the highest alignment scores when no NOE data were used (light green symbols in Fig. 1b, left most panel). With sparse NOEs included, both of these closest structural homologues now fall in the top cluster with the highest score. Subsequent CS-RosettaCM modeling yields somewhat closer agreement to the experimental reference structure (Supplementary Table 1), and the same applies for the other proteins.
Often, some of the residues in the query protein for which an NOE is available will be aligned by POMONA to a gap in the database protein sequence, in which case this sparse NOE will only help in restraining the sampling for such unaligned parts during the CS-RosettaCM modeling procedure, yielding substantial improvement in both convergence and accuracy of the final models (Fig. 1d; Supplementary Figs. 9 and 10, Supplementary Table 1). In cases where the target sequence corresponds to an extreme variant of a known fold, the protocol still permits substantial reorganization of the long template fragments to accommodate these structural differences (such as β-sheet register shifts), guided by the sparse NOEs, thereby offering further improvement of the final models.
Software availability
The POMONA software, including clustering scripts, all required databases and a complete example for ubiquitin, together with the scripts used for the RosettaCM comparative modeling and structure selection procedure, can be freely downloaded from http://spin.niddk.nih.gov/bax/software/POMONA. A public web server (http://spin.niddk.nih.gov/bax/nmrserver/pomona) is also provided, but only for performing the less time-consuming POMONA alignment method for a protein with experimental chemical shift data. Such a search procedure typically requires ca 0.5 h on a 10-CPU desk top work station. By default, this server also generates all inputs and scripts required for running the RosettaCM comparative modeling structure generation. For this purpose, RosettaCM can be downloaded with the Rosetta Software Suite from http://www.rosettacommons.org/software.
Supplementary Material
Acknowledgments
This work was funded by the Intramural Research Program of the NIDDK, US National Institutes of Health (NIH). We thank Y. Song, N. Sgourakis and D. Baker for help and advice on the use of RosettaCM. We also gratefully acknowledge use of the NIH high-performance computational Biowulf Linux cluster.
Footnotes
AUTHOR CONTRIBUTIONS
Y.S. and A.B designed methods and protocols and wrote the manuscript. Y.S. developed the code, optimized the parameterization of the protocol, and analyzed the resulting data.
COMPETING FINANICAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Marti-Renom MA, et al. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 2.Pieper U, et al. Nucleic Acids Res. 2009;37:D347–D354. doi: 10.1093/nar/gkn791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Das R, Baker D. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 4.Tugarinov V, Choy WY, Orekhov VY, Kay LE. Proc Natl Acad Sci USA. 2005;102:622–627. doi: 10.1073/pnas.0407792102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowers PM, Strauss CEM, Baker D. J Biomol NMR. 2000;18:311–318. doi: 10.1023/a:1026744431105. [DOI] [PubMed] [Google Scholar]
- 6.Cavalli A, Salvatella X, Dobson CM, Vendruscolo M. Proc Natl Acad Sci USA. 2007;104:9615–9620. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen Y, et al. Proc Natl Acad Sci U S A. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wishart DS, et al. Nucleic Acids Res. 2008;36:496–502. doi: 10.1093/nar/gkn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raman S, et al. Science. 2010;327:1014–1018. doi: 10.1126/science.1183649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson JM, et al. Proc Natl Acad Sci U S A. 2012;109:9875–9880. doi: 10.1073/pnas.1202485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misura KMS, Chivian D, Rohl CA, Kim DE, Baker D. Proc Natl Acad Sci USA. 2006;103:5361–5366. doi: 10.1073/pnas.0509355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song Y, et al. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Webb B, Sali A. Curr Protoc Bioinform. 2014;47:5.6.1–5.6.32. doi: 10.1002/0471250953.bi0506s47. [DOI] [PubMed] [Google Scholar]
- 14.Xu D, Zhang J, Roy A, Zhang Y. Proteins-Structure Function and Bioinformatics. 2011;79:147–160. [Google Scholar]
- 15.Vernon R, Shen Y, Baker D, Lange OF. J Biomol NMR. 2013;57:117–127. doi: 10.1007/s10858-013-9772-4. [DOI] [PubMed] [Google Scholar]
- 16.Shen Y, Bax A. J Biomol NMR. 2013;56:227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siew N, Elofsson A, Rychiewski L, Fischer D. Bioinformatics. 2000;16:776–785. doi: 10.1093/bioinformatics/16.9.776. [DOI] [PubMed] [Google Scholar]
- 18.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 19.Soding J. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 20.Berjanskii M, Wishart DS. Nat Protoc. 2006;1:683–688. doi: 10.1038/nprot.2006.108. [DOI] [PubMed] [Google Scholar]
- 21.Koonin EV, Galperin MY, editors. Sequence - Evolution - Function: Computational Approaches in Comparative Genomics. Kluwer Academic; Boston: 2003. [PubMed] [Google Scholar]
- 22.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 23.Needleman SB, Wunsch CD. J Mol Biol. 1970;48:443. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 24.Smith TF, Waterman MS. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 25.Madhusudhan MS, Marti-Renom MA, Sanchez R, Sali A. Protein Eng Des Sel. 2006;19:129–133. doi: 10.1093/protein/gzj005. [DOI] [PubMed] [Google Scholar]
- 26.Shen Y, Bax A. J Biomol NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caprara A, Carr R, Istrail S, Lancia G, Walenz B. J Comput Biol. 2004;11:27–52. doi: 10.1089/106652704773416876. [DOI] [PubMed] [Google Scholar]
- 28.Wohlers I, Domingues FS, Klau GW. Bioinformatics. 2010;26:2273–2280. doi: 10.1093/bioinformatics/btq420. [DOI] [PubMed] [Google Scholar]
- 29.Carugo O, Pongor S. Protein Sci. 2001;10:1470–1473. doi: 10.1110/ps.690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson J, Baker D. Proteins. 2011;79:2380–2388. doi: 10.1002/prot.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.