

Abstract
As a sustainable alternative for conventional batch-based synthetic techniques, the concept of continuous-flow processing has emerged in the synthesis of fine chemicals. Systematic tuning of the residence time, a key parameter of continuous-reaction technology, can govern the outcome of a chemical reaction by determining the reaction rate and the conversion and by influencing the product selectivity. This review furnishes a brief insight into flow reactions in which high chemo- and/or stereoselectivity can be attained by strategic residence-time control and illustrates the importance of the residence time as a crucial parameter in sustainable method development. Such a fine reaction control cannot be performed in conventional batch reaction set-ups.
Keywords: chemoselectivity, continuous flow, diastereoselectivity, reactors, residence time
1 Introduction
Continuous-flow (CF) transformations are of considerable current interest, as they offer a substantial number of advantages over conventional batch procedures, such as inherently safer and greener chemistry, outstanding mass and heat transfer, increased parameter space with an unprecedented level of control over the most important reaction conditions, and higher reaction rates.1 Many of these advantages stem from the size of CF reactors, where the diameters of the pores and capillaries are mainly in the micro- or nanometer range. With such short distances, the mass transfer is faster and results in more effective mixing of the substrates.1k-2 The small internal dimensions are accompanied by high surface to volume ratios, which results in effective heat transfer, thereby allowing exact temperature control. The major consequence of these advantages is that the reaction times can be decreased relative to those in conventional batch procedures.
There are numerous fundamental differences between classical batch experiments and CF reaction technology. In a segmentally operated reaction vessel, the definition of reaction time is obvious,3 whereas the concept of reaction time in a continuous process is not so clear, and the term residence time is therefore preferred, that is, the interval spent by a given molecule in the active reactor zone. The reaction rate, conversion, and product selectivity are influenced directly by this specific parameter; the residence time therefore being a crucial determining factor in flow chemistry.
One benefit of CF procedures is the time-resolved scale of the syntheses. In conventional batch reactors, the output depends on the batch size, and the synthesis can become appreciably complicated when unstable reactants are to be handled on a relatively larger scale. In contrast, the scalability of CF reactions is a straightforward function of time: the longer the reactor operates, the larger the scale of the production. Accordingly, CF transformations are time-resolved, and facile industrial scale-up is therefore warranted. The residence time is clearly determined by the flow rate and the volume of the CF reactor and can be fine-tuned precisely via the instrumentation.4 The residence-time distribution of some packed-bed mini-flow reactors has been demonstrated to be influenced crucially by the properties of the filling material.4e Such an extraordinary control is not possible in traditional batch reactions, and flow chemistry therefore allows unprecedented selectivites and reaction rates, and even the development of completely new reaction routes.
Excellent recent reviews have focused on various aspects and applications of CF devices and syntheses in organic chemistry,1a,1k,5 and there are numerous important papers in which conversions and reaction rates have been optimized via residence-time control in CF reactors.6 There are many parameters concerning CF reactors, with which the reaction outcome can be fine-tuned. However, this review focuses on the role of the residence time as a unique tool in the optimization of reaction selectivity. We therefore survey the recent advances in the CF chemistry where high chemo- and/or stereoselectivity is ensured through precise control of the residence time.
2 Residence-Time-Controlled Selectivity
2.1 Selective flash transformations
Yoshida and co-workers have recently introduced the concept of ‘flash chemistry′, for flow reactions involving 1) fast consecutive transformations and reactive intermediates, 2) competing reaction pathways, 3) highly exothermic transformations, 4) undesired byproduct formation, and 5) transformations leading to labile products which readily decompose.7 These reactions are difficult (or sometimes impossible) to accomplish in traditional segmented unit operations, and selectivity issues often reduce their practical applicability. Flash chemical synthesis relies on high-resolution control of the residence time within a flow microreactor. For precise control over the reaction outcome, mixing should take place in a much shorter time than the residence time in the active reactor zone, which necessitates the use of micromixers that allow extremely fast mixing (Figure 1). In the last few years, there have been a huge number of outstanding contributions to the flash chemistry concept in various fields of organic synthesis (such as organolithium chemistry, cross-coupling reactions, and organo fluorine chemistry), and they have recently been reviewed extensively.8 Thus, only several selected examples will be detailed in the present manuscript.
Figure 1.
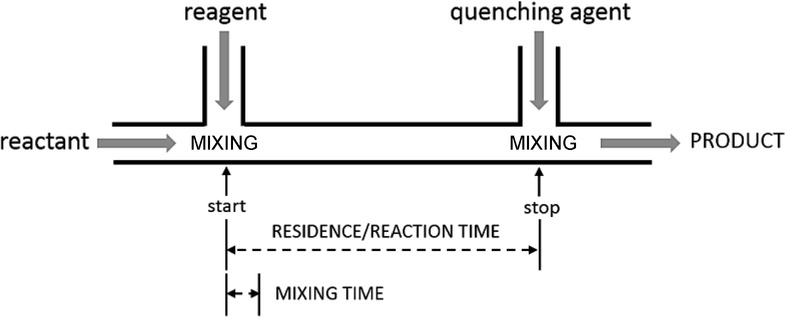
High-resolution residence-time control.7
The synthesis of multisubstituted pyridines has received particular attention in the field of drug discovery.. The reaction of an electrophile with a pyridyllithium species generated from a bromopyridine via bromine–lithium exchange is a popular method for the introduction of substituents into the pyridine ring. However, such transformations need cryogenic conditions (e.g. −78 °C) in order to moderate side reactions (e.g. deprotonation or addition to the pyridine ring), which can lead to undesired byproduct formation and insufficient chemoselectivity.9 From aspects of sustainability and industrial-scale production, the avoidance of cryogenic conditions would be highly beneficial. With CF technology and precise residence-time control, the bromine–lithium exchange becomes readily regulated. The reaction of 2,3-dibromopyridine with n-butyllithium (nBuLi), followed by reaction with an electrophile, was carried out in a flow system consisting of two T-shaped micromixers and two microtube reactors (Figure 2).
Figure 2.
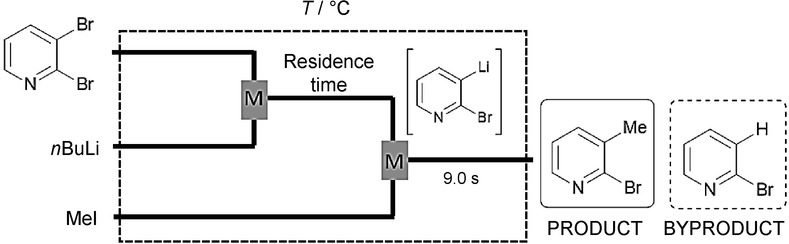
CF set-up for bromine–lithium exchange with 2,3-dibromopyridine, followed by the reaction with MeI (M=mixing unit).9
In an elaborate optimization study, iodomethane (MeI) was applied as electrophile. 2-Bromo-3-methylpyridine was obtained in high yields even at 0 °C if an adequately low residence time (e.g. 0.06 s) was chosen. Increase of the residence time and/or the temperature enhanced the protonation of 2-bromo-3-pyridyllithium, leading to the formation of 2-bromopyridine as byproduct (Figure 3). The methodology was extended to the sequential introduction of two electrophiles by using dibromopyridines as starting materials to obtain disubstituted pyridines without isolation of the monobromopyridine intermediates.
Figure 3.
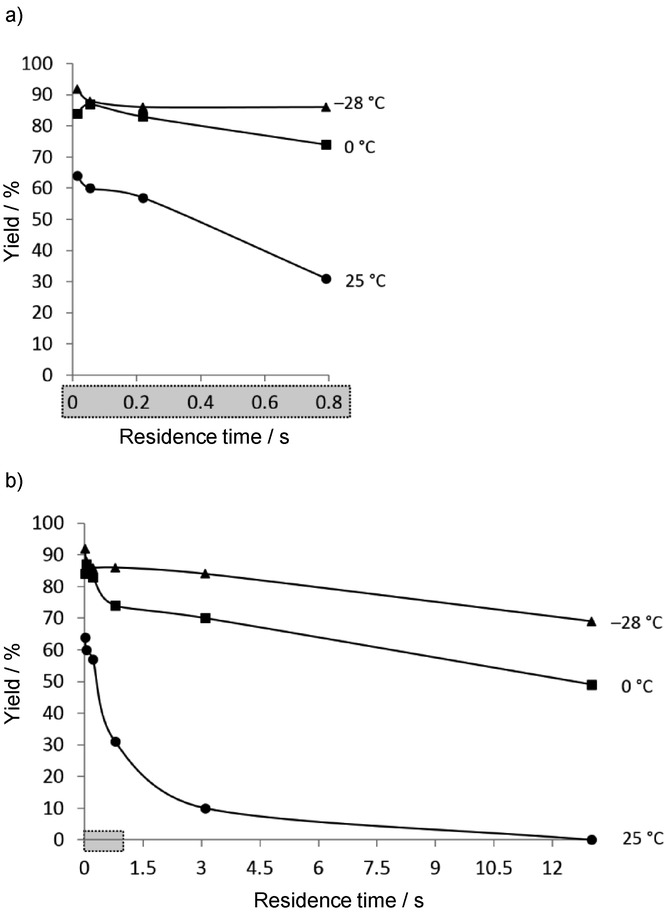
Effect of the residence time (0–0.8 s (a) and 0–13.5 s (b)) on the yield of 2-bromo-3-methylpyridine at different temperatures.9
In the context of ideal synthesis and green sustainable chemistry, protecting-group-free approaches have attracted considerable attention. The next example gives an insight into protecting-group-free CF iodine–lithium exchange reactions. Organolithiums react with ketones rapidly, and ketone carbonyl groups are therefore traditionally protected prior to an organolithium reaction. Nevertheless, free ketone carbonyls can survive for very short intervals through organolithium reactions. It has been demonstrated that, with very accurate control over short residence times, the reaction can be accomplished without protecting group chemistry in a flow microreactor.10 Iodine–lithium exchange reactions of acyliodobenzenes have been investigated (Scheme 1, Figure 4).
Scheme 1.
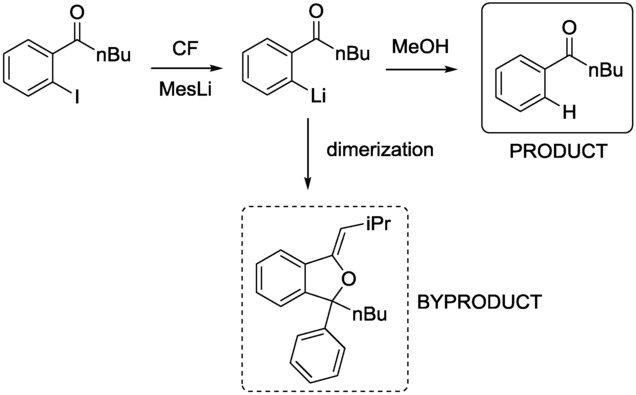
CF reaction of o-pentanoyliodobenzene with MesLi generated in situ. Trapping of the resulting acylphenyllithium with MeOH as electrophile gives the protonated product, or undesired dimerization occurs.10
Figure 4.
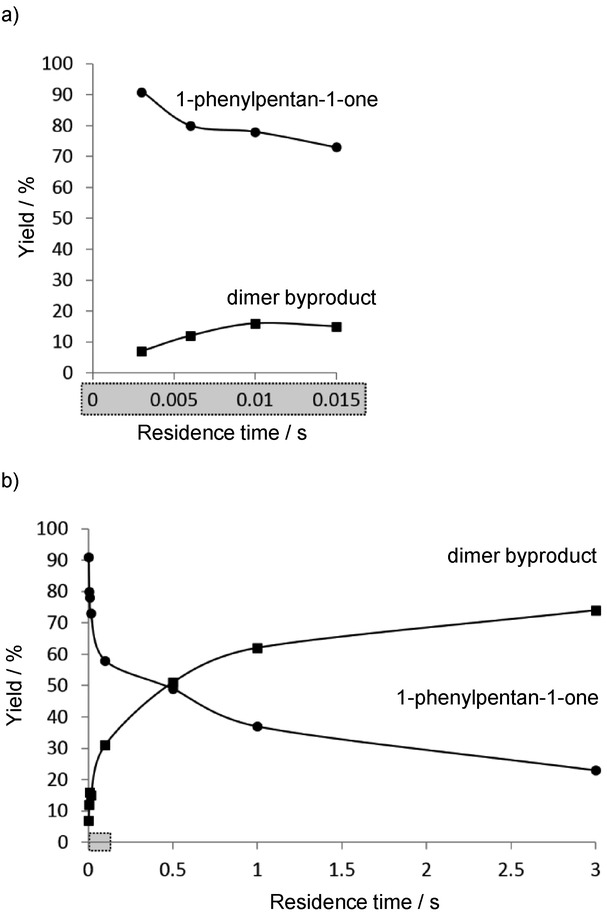
Plots of the yield vs. the residence time (0–0.015 s (a) and 0–3 s (b)) of the iodine–lithium exchange step for the CF reaction outlined in Scheme 1.10
Mesityllithium (MesLi) was generated in-line through the reaction of mesityl bromide and nBuLi at 0 °C. In the same flow, the resulting MesLi was then combined with the acyliodobenzene at −70 °C, and the resulting short-lived acylphenyllithium species was trapped with a quenching electrophile before undesired dimerization could take place (Scheme 1). It was shown that higher reaction times favored byproduct formation, and the residence time of the iodine–lithium exchange step was therefore progressively decreased to as low as 0.003 s so as to obtain the desired compound in high yield without significant formation of the byproduct (Figure 4).
The nitro group reacts vigorously with aryllithiums, and accordingly, the generation of nitro-substituted organolithium compounds in batch macroreactors is difficult; very low temperature is usually needed to gain sufficient control over the reaction selectivity. However, with flash chemistry, this problem can be overcome and nitro-substituted aryllithium reagents can easily be obtained via halogen–lithium exchange reactions in very short residence times.11 Accurate control over the residence time permits the selective use of either the kinetically or the thermodynamically preferred organolithium intermediate in the reaction of 1-bromo-2,5-dimethoxy-3-nitrobenzene with phenyllithium (PhLi) at −48 °C (Scheme 2).
Scheme 2.
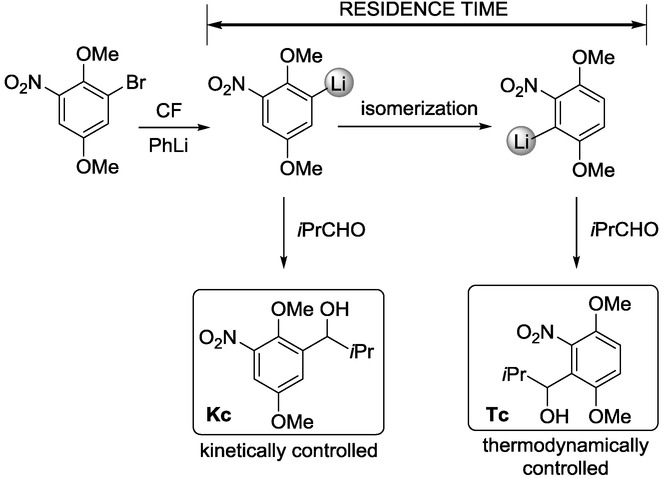
CF reaction of 1-bromo-2,5-dimethoxy-3-nitrobenzene with PhLi, followed by the residence-time-controlled transformation of the organolithium intermediate with isobutyraldehyde.11
Isobutyraldehyde as quenching electrophile is introduced through a mixing unit to give the desired product. The results reveal that, with a residence time of 0.055 s, the Kc product is formed via the kinetically preferred organolithium reagent (Table 1, entry 2). An increase in residence time leads to the formation of the isomeric product derived from isomerization of the aryllithium reagent. With a residence time of 63 s, the thermodynamically controlled product (Tc) was obtained exclusively (Table 1, entry 6). The switch between kinetic and thermodynamic control proved to be an elegant way to fine-tune the reaction outcome.
Table 1.
Switch between kinetic and thermodynamic control by means of the residence time in the CF reaction outlined in Scheme 2.11
| Entry | Residence time [s] | Yield ofKc[a] [%] | Yield ofTc[b] [%] |
|---|---|---|---|
| 1 | 0.014 | 61 | 0 |
| 2 | 0.055 | 84 | 0 |
| 3 | 0.22 | 75 | 2 |
| 4 | 0.76 | 63 | 6 |
| 5 | 6.3 | 35 | 29 |
| 6 | 63 | 0 | 68 |
[a] Kc=kinetically controlled product; [b] Tc=thermodynamically controlled product.
Kappe and co-workers recently reported the highly controlled thermolysis of various substrates in a high-pressure/high-temperature microreactor environment, employing near- or supercritical fluids as reaction media.12 The method relies on exact control of the temperature and the flow rate, and hence, the residence time for the fine-tuning of the chemoselectivity. In the case of 5-benzoyl Meldrums acid, for example, differential scanning calorimetry studies revealed that the thermolysis largely depends on the reaction conditions. At ∼150 °C, a 1,3-dioxin-4-one intermediate is formed, which decomposes to an α-oxoketene dimer at higher temperatures (Scheme 3).
Scheme 3.
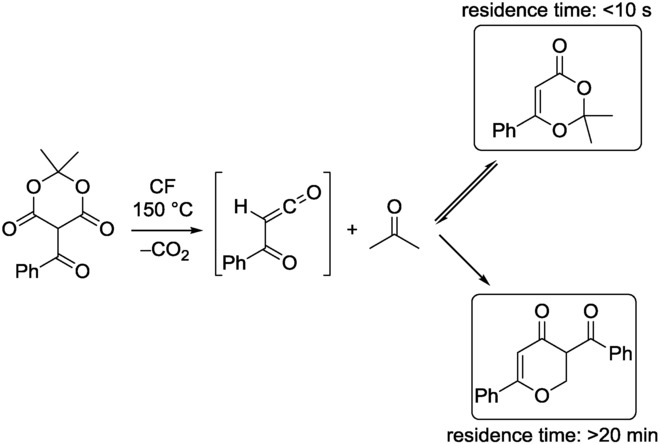
CF thermolysis of 5-benzoyl Meldrums acid leading to different products, depending on the residence time.12
With precise residence-time control, the thermal decomposition of 5-benzoyl Meldrums acid could be nicely tuned to provide both products selectively at 150 °C. With longer residence times (>20 min), the dimer form was obtained with high selectivity, while as the residence time was reduced, the 1,3-dioxin-4-one intermediate became the favored product. As an example, complete conversion of the starting material was observed after 7 s, providing a selectivity of almost 90 % for the 1,3-dioxin-4-one product.
2.2 Photocatalytic peptide fragment coupling
CF operation is highly beneficial for photochemical processes, as the narrow reaction channels and increased surface-to-volume ratio greatly improve light absorption as compared with conventional reaction vessels, and the irradiation-related side reactions and decomposition pathways are minimized as the irradiation period can easily be controlled via the residence time. Conventional methods for the generation of amide bonds suffer from a number of drawbacks (e.g. the need for stoichiometric amounts of activating agents, possible epimerization, high costs, and waste generation), which suggests a need for alternative approaches. For this reason, Jamison et al. developed a versatile CF approach for the construction of amide bonds by means of the photochemical rearrangement of nitrones.13 After verification of the capabilities of the CF method with transformations of simple alkyl-aryl nitrones, its potential concerning dipeptide formation was evaluated. Control of the residence time proved crucial in the fine-tuning of the proportions of the dipeptide product (C), the oxaziridine intermediate (B), and the starting nitrone (A) in the resulting mixture (Table 2).
Table 2.
Peptide fragment coupling in CF with photochemical nitrone–amide rearrangement.13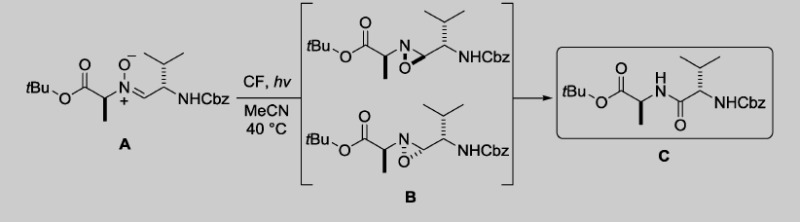
| Entry | Residence time [min] | Concentration [m] | Ratio [A:B:C] |
|---|---|---|---|
| 1 | 1 | 0.05 | 1:9:1 |
| 2 | 2.5 | 0.05 | nd:4:3[a] |
| 3 | 5 | 0.05 | nd:1:5[a] |
| 4 | 10 | 0.05 | nd:nd:1[a] |
| 5 | 10 | 0.1 | nd:1:6[a] |
| 6 | 12.5 | 0.1 | nd:nd:1[a] |
[a] nd: not detected
A quick scan revealed that a 10 min residence time was necessary for quantitative conversion of both the starting nitrone and the oxaziridine intermediates with an initial substrate concentration of 0.05 m (entries 1—4 in Table 2). At the expense of a slight residence time increase (12.5 min), the nitrone concentration could be increased to 0.1 m, thereby achieving higher productivity (entry 6). The scope and applicability of the CF method proved to be wide, but the irradiation time had to be extended by means of a residence-time increase in order to obtain dipeptides in good yields from sterically hindered nitrones.
2.3 Stereoselective synthesis of γ-nitroaldehydes
The synthesis of γ-nitroaldehydes is of considerable current interest as they are useful synthons in organic chemistry and drug discovery.14 The stereoselective CF synthesis of γ-nitroaldehydes has been performed with immobilized peptides as catalysts, as outlined in Scheme 4.15
Scheme 4.
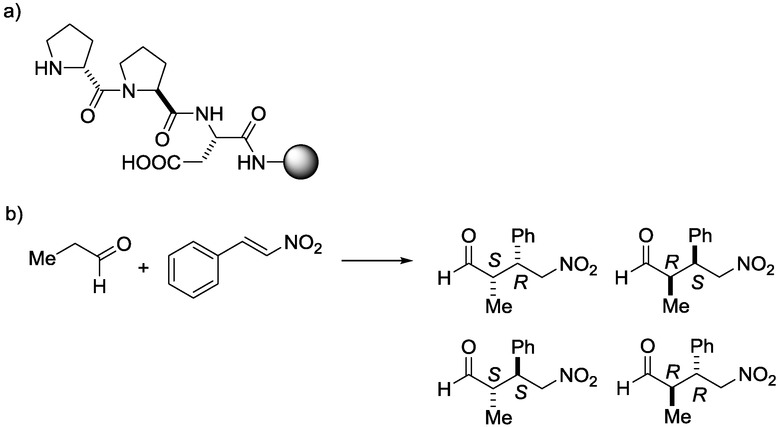
Catalyst structure (a) and reaction scheme (b) relating to the synthesis of γ-nitroaldehydes.15
In this reaction, two stereocenters are generated, and consequently, the formation of four enantiomers is possible. Accordingly, the syn/anti selectivity and the enantioselectivity within an enantiomeric pair can be investigated. One of the major advantages of this methodology in addition to the good selectivities and yields is the simultaneous synthesis and immobilization of the catalyst. The reaction parameters, including the structure of the catalyst, the type of the solid support, the proportions of the reactants, the pressure, the temperature, and the residence time have been optimized. The effects of the residence time on the yield and the diastereoselectivity are depicted in Figure 5.
Figure 5.
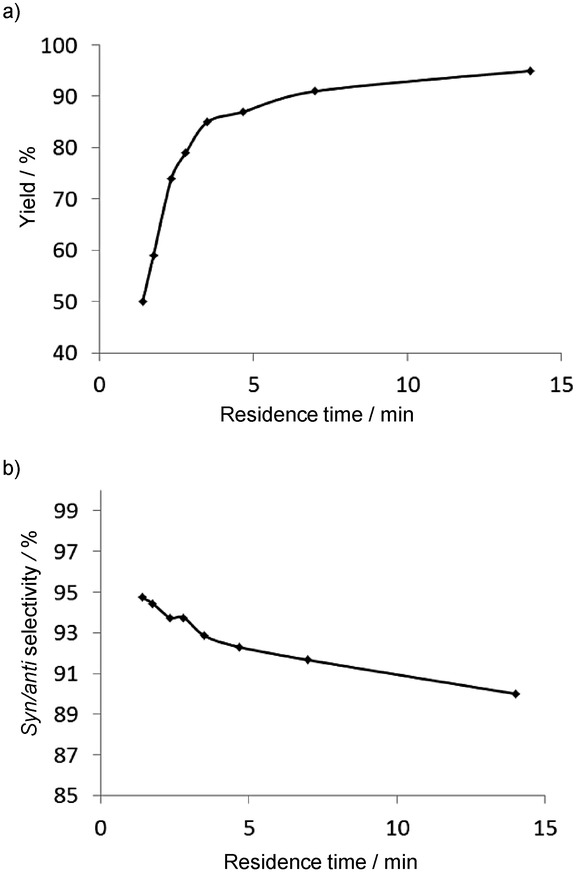
Effects of the residence time on the yield (a) and the syn/anti ratio (b) for the CF stereoselective synthesis of γ-nitroaldehydes.15
Increase of the residence time led to higher yields, but lower diastereoselectivities. The latter effect is explained by the epimerization of the secondary amine functional group of the N-terminal proline of the immobilized catalyst. This was highlighted by recirculation of the γ-nitroaldehyde product on the catalyst, which resulted in a lower syn/anti ratio. CF aldol reactions have also been carried out in the same flow system, but no residence-time dependence of the stereoselectivity was then observed.16 Such a high resolution control between syn/anti selectivity and conversion cannot be achieved in a batch reaction.
2.4 Selective nitration
The nitration of benzene derivatives in conventional batch reactors usually affords a mixture of several products and raises safety issues. Yu et al. employed precise residence-time control and smart CF reactor design to improve the selectivity and safety in the nitration of p-difluorobenzene (Scheme 5).17 The nitrating acid mixture (2.0 equivalents of concentrated sulfuric acid and 1.1 equivalents of fuming nitric acid) and p-difluorobenzene were pumped through a thermostated tubular reactor by separate pumps. The reaction was quenched by ice in the collection vessel.
Scheme 5.
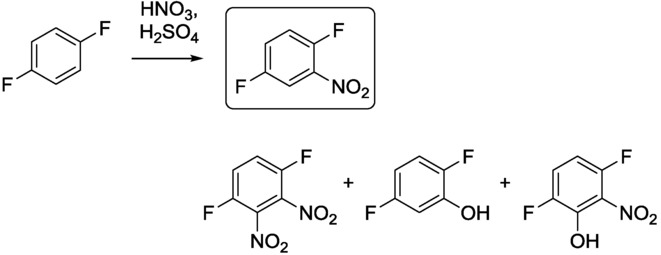
Nitration of p-difluorobenzene.17
Different residence times were explored by adjusting the length of the reaction tube while maintaining a fixed flow rate. It can be seen from Figure 6 that higher residence times led to higher product yields, but also enhanced the formation of 1,4-difluoro-2,3-dinitrobenzene as a byproduct. It is notable that only negligibly small amounts of difluorophenol side products were formed; this is possibly due to the superior heat transfer of the CF reactor. To improve the product selectivity further, two thermostated reactor modules and a cooling module were connected together. The temperatures of the reactor modules were 30∼35 °C and 65∼70 °C, and in each reactor zone the residence time was set precisely to 1 min (0.3 min in the cooling zone). 1,4-Difluoro-2-nitrobenzene was obtained in a yield of 98 % with 99 % gas-chromatography purity, which means that byproducts were hardly generated.
Figure 6.
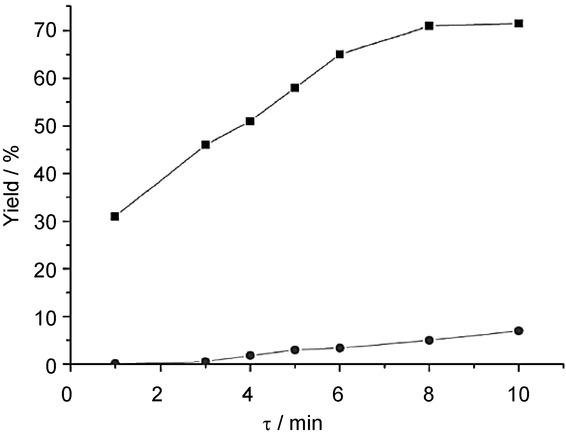
Effect of the residence time on the nitration of p-difluorobenzene. (▪: yield of 1,4-difluoro-2-nitrobenzene, •: yield of 1,4-difluoro-2,3-dinitrobenzene). Reproduced with permission from ref. 17. Copyright 2015, American Chemical Society.
2.5 Residence-time-controlled synthesis of Rufinamide
One of the best-selling five-membered heterocyclic pharmaceuticals is Rufinamide, an antiepileptic drug that contains a 1,2,3-triazole moiety. Hessel and co-workers established a novel and sustainable CF technique for the synthesis of Rufinamide, which relies greatly on precise residence-time control to govern the sensitive balance between the decomposition of the starting materials and the acceleration of the reaction at elevated temperatures.18 The key step in the synthesis of Rufinamide is the 1,3-dipolar cycloaddition of 2,6-difluorobenzyl azide with an appropriate dipolarophile. (E)-Methyl 3-methoxyacrylate was utilized as a nontoxic and inexpensive dipolarophile which results in the selective formation of the desired 1,4-disubsituted 1,2,3-triazole regioisomer without the need for an added catalyst (Figure 7). To attain a higher reaction rate, solvent-free conditions were applied.
Figure 7.
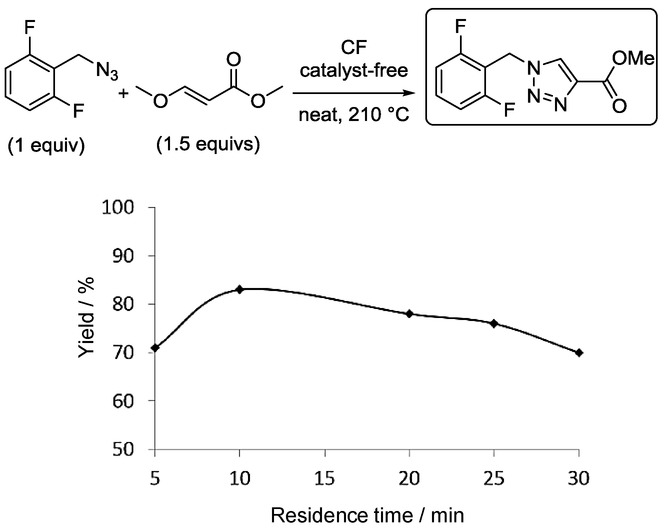
CF synthesis of Rufinamide with precise residence-time control.18
It was found that the use of an elevated temperature is necessary for complete reaction homogeneity and for a sufficiently high reaction rate, but after a point this also leads to the decomposition of the azide, which worsens the yield. 210 °C was chosen as an ideal reaction temperature, and the residence time was then fine-tuned to obtain the optimum balance between the reaction rate and the rate of decomposition of the azide (Figure 7). The highest yield (83 %) was achieved after 10 min. Longer residence times led to lower yields, due to the significant decomposition of the starting material. Without the strategic application of precise residence-time control, the decomposition would lower the yield of the desired compound. The CF procedure lacks most of the drawbacks of the batch synthesis, as it is rapid, safe, and straightforward.
2.6 Selective Heck reactions in CF
Jensen et al. developed an integrated CF system for self-optimization of the Heck reaction of 4-chlorobenzotrifluoride and 2,3-dihydrofuran (Scheme 6).19 The system maximized the yield of the monoarylated product by scanning the equivalents of the alkene and the residence time via the adjustment of the flow rate of the reactants, while the temperature was kept constant at 90 °C. The combination of Pd(OAc)2 and tert-butyl-MePhos as ligand in n-butanol proved to be a sufficiently active and stable catalyst system which did not cause clogging in the reactor channels. A solution containing the aryl chloride, the palladium source, and an amine base was introduced together. The alkene was fed separately, and n-butanol was injected via a third syringe. The optimization was directed by the Nelder–Mead Simplex Method, and the yield was evaluated in every step by in-line HPLC analysis.
Scheme 6.
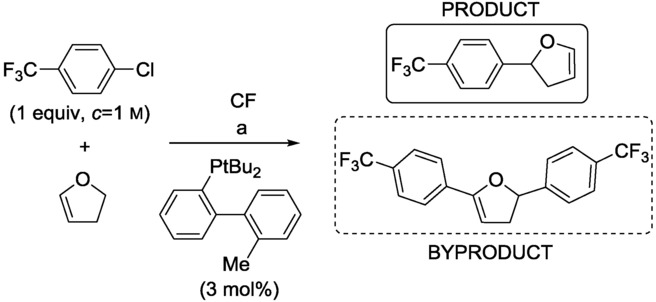
Heck reaction of 4-chlorobenzotrifluoride and 2,3-dihydrofuran in CF. Reagents and conditions: a) Pd(OAc)2 (1 mol%), (cyclohexyl)2NMe (1.2 equivs), n-BuOH, 90 °C.19
The results demonstrated that the yield of the monoarylated product improved significantly with the residence time until a plateau was reached at around 5.5 min. Longer residence times led to decreased yields, as a consequence of diarylated byproduct formation (Figure 8). Increase of the alkene excess resulted in higher yields. Optimum conditions were determined as a residence time of 6 min and an alkene excess of 5.0 equivalents, which corresponded to a yield of 83 %.
Figure 8.
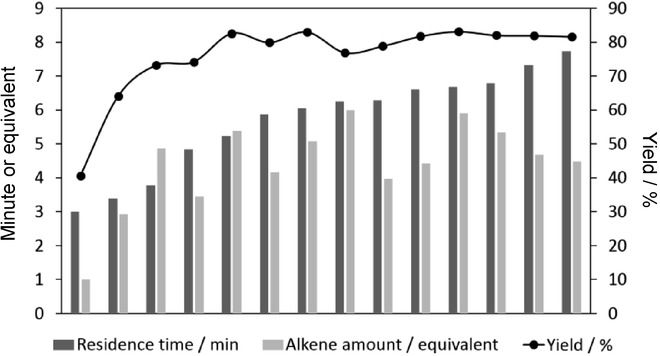
Effects of the residence time and the alkene ratio on the CF Heck reaction of 4-chlorobenzotrifluoride and 2,3-dihydrofuran (Scheme 6).19
To attain sustainable production, the Heck C−C coupling20 followed by hydrogenation for the synthesis of 1,2-diphenylethane was performed in a CF device with a solid-supported Pd as catalyst (Scheme 7).21
Scheme 7.
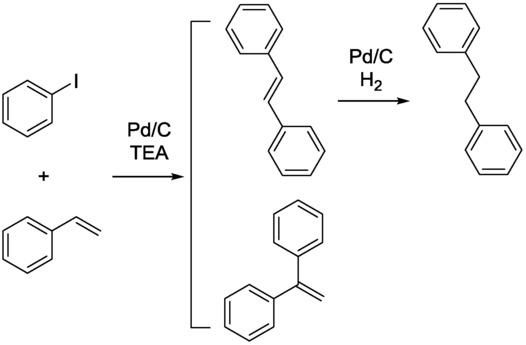
CF Heck reaction followed by hydrogenation for the synthesis of 1,2-diphenylethane (TEA=triethylamine).21
A multichannel compact reactor was constructed for the synthesis of 1,2-diphenylethane. The reaction parameters were optimized, and a methodology orders of magnitudes faster than batch reactions was developed. Under batch conditions, the selectivity for 1,2-diphenylethene was lower than under CF conditions because a substantial amount of 1,1-diphenylethene was detected too. Nevertheless, under CF conditions the formation of cis-1,2-diphenylethene was also observed, and the residence time affected the selectivity and the conversion. On decrease of the residence time, the conversion was lower, as expected, but the selectivity towards 1,2-diphenylethene rather a 1,1-diphenylethene was increased (Figure 9). This observation suggests that the 1,2-disubstituted compound is the kinetic product. Here an inverse relationship was observed between the conversion and the selectivity.
Figure 9.
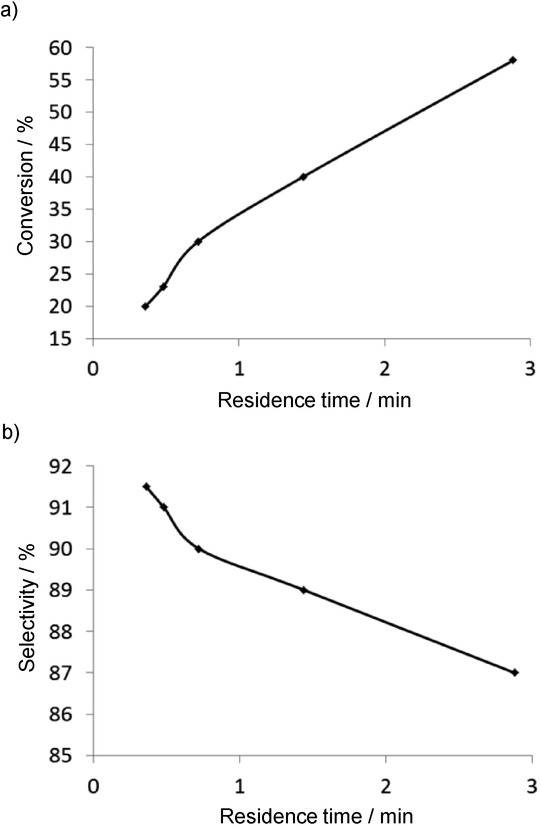
Effects of the residence time on the conversion (a) and the selectivity (b) for the CF Heck reaction.21
2.7 Selective hydrogenations, reductions, and oxidations in CF
Among CF techniques, the most significant progress has been made in the field of CF gas-handling reactions in recent years, mainly driven by CF hydrogenation.22 With CF reactors, the handling of hazardous aggressive gases can be safely performed. Moreover, through the fine-tuning of the reaction parameters, selective transformations can be performed faster and more efficiently as compared with conventional methodologies. The selective reductions of alkynes to alkenes have been achieved in a wall-catalyzed segmented flow reactor by catalytic hydrogenation.23 The commercially available fused-silica capillaries contained a 6 μm thick layer of γ-Al2O3 that was pretreated and impregnated with [Pd(OAc)2] solution, which resulted in nanosized Pd particles evenly dispersed on the coating layer. With the application of this tubing, 3-methyl-1-pentyn-3-ol was selectively reduced to 3-methyl-1-penten-3-ol (Scheme 8).
Scheme 8.
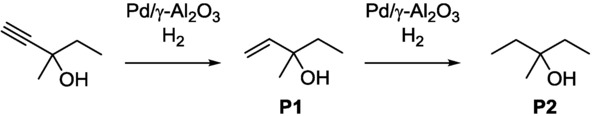
Selective hydrogenation of 3-methyl-1-pentyn-3-ol over a Pd catalyst to the desired 3-methyl-1-penten-3-ol (P1) and overhydrogenation to 3-methyl-3-pentanol (P2).23
Kinetic modelling showed that the maximum possible yield is 78–81 %, which is obtained only at precisely the right residence time. The dependence of the selectivity of the reaction on the residence time has been investigated. There is an optimum value where the formation of P1 displays a maximum value, and the overhydrogenation to P2 is still low. Any further variation of the residence time reduces the yield (Figure 10). At a shorter residence time, the conversion is lower in general, but at longer residence times, the formation of P2 increases, while the concentration of P1 decreases. In a one-day optimization, the theoretical optimum yield of P1 was achieved by using segmented flow.
Figure 10.
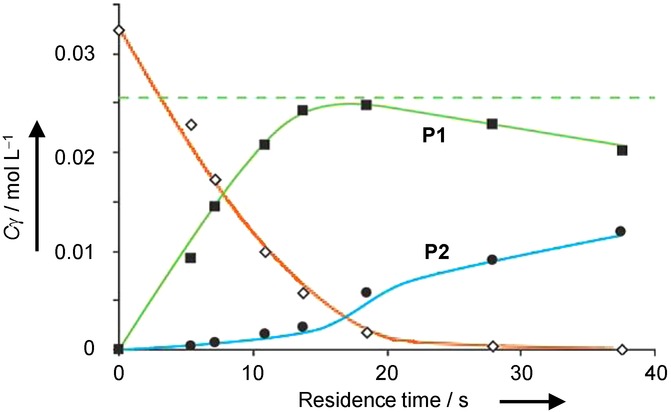
Effluent concentrations as a function of residence time in the hydrogenation of 3-methyl-1-pentyn-3-ol in ethanol over a 0.02 wt % Pd catalyst operated in segmented flow at 24 °C. The dashed line shows the maximum obtainable yield. Reproduced with permission from ref. 23.Copyright 2011, Wiley-VCH Verlag GmbH / Co. KGgA, Weinheim.
The partial reduction of esters with diisobutylaluminium hydride (DIBALH) is not a popular route for aldehyde synthesis, as overreduction to the corresponding alcohol is a significant problem even at low temperatures. Jamison and co-workers exploited the benefits of CF processing together with careful residence-time control and revitalized the DIBALH reduction of esters to yield various aldehydes selectively.24 The residence-time dependence was investigated as a function of the reactor volume at constant flow rate and also as a function of the flow rate at constant reactor volume. The reduction of ethyl hydrocinnamate was initially studied at −78 °C; methanol was introduced separately as a quenching agent to avoid overreduction (Figure 11). Interestingly, the conversion improved when the residence time was shortened at constant reactor volume by increasing the flow rate, and it was observed that at very high flow rates the outcome of the reaction was independent of the residence time (Table 3). These results indicate that the examined reaction proceeds very rapidly, and higher flow rates tend to improve mixing. At such elevated flow rates, the reaction outcome is influenced by the rapid mixing in the reactor channels. It is noteworthy that full conversion and complete selectivity were observed even at very short residence times. The methodology was extended to the selective reduction of a series of further esters.
Figure 11.
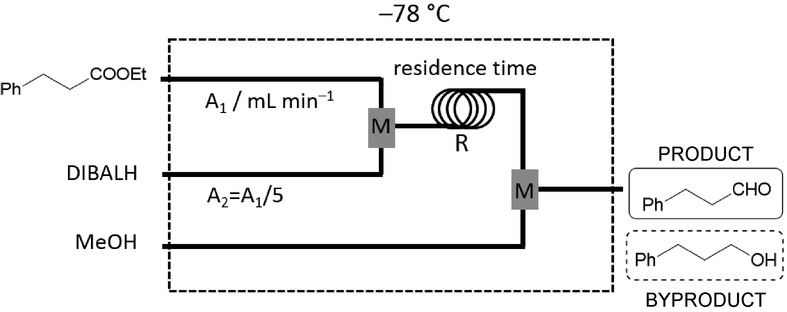
CF set-up for the selective DIBALH reduction of ethyl hydrocinnamate (M=mixing unit, R=tubular reactor).24
Table 3.
Effects of different residence times in the CF DIBALH reduction of ethyl hydrocinnamate (Figure 1).24
| Entry | Volume of R | A1 | Residence time | Selectivity for | ||
|---|---|---|---|---|---|---|
| [μL] | [mL min−1] | [s] | aldehyde | ester | alcohol | |
| 1 | 23 | 5 | 0.23 | 36 | 64 | 0 |
| 2 | 23 | 10 | 0.11 | 50 | 50 | 0 |
| 3 | 23 | 30 | 0.04 | 96 | 4 | 0 |
| 4 | 228 | 5 | 2.28 | 43 | 57 | 0 |
| 5 | 228 | 10 | 1.14 | 61 | 39 | 0 |
| 6 | 228 | 30 | 0.38 | 97 | 3 | 0 |
| 7 | 684 | 5 | 6.84 | 57 | 43 | 0 |
| 8 | 684 | 10 | 3.42 | 85 | 15 | 0 |
| 9 | 684 | 30 | 1.14 | 96 | 3 | 1 |
An early example of CF reduction methodology was the application of diethylzinc and an immobilized chiral catalyst for the enantioselective synthesis of aromatic alcohols (Scheme 9).25
Scheme 9.
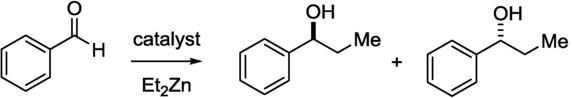
Enantioselective synthesis of phenylpropanol derivatives.25
Ephedrine and camphor immobilized on polystyrene resins were used as catalysts in a bench-top flow apparatus operated with peristaltic pumps. The effects of the flow rate on the yield and enantioselectivity were investigated: lower flow rates resulted in higher yields and higher ee values. Moreover, the flow reactor displayed a better performance than with normal flask operations. The immobilized catalyst in flask operations was recycled six times.
Polymer monoliths containing a chiral 1,2-aminoalcohol derivative were used as catalysts in the reaction outlined in Scheme 9 in a mini-flow reactor.26 The monolithic catalysts proved superior to standard polystyrene catalysts. Furthermore, the utilization of flow conditions resulted in enhanced catalytic activity, providing higher yield and selectivity.
Poliakoff and co-workers investigated the CF selective partial oxidation of methylpyridines, using molecular O2 as oxidant for the selective formation of pyridine carboxylic acids (isonicotinic, nicotinic, and picolinic acids) as precursors for a series of valuable products.27 Manganese(II) bromide was employed as a homogeneous catalyst, and supercritical water (scH2O) served as a cheap and green reaction medium. The oxidation of methylpyridines is known to produce aldehydes and pyridine itself (via decarboxylation) as byproducts (Scheme 10).The effects of the residence time, temperature, pressure, and catalyst concentration were therefore deeply analyzed to achieve selective carboxylic acid formation. It emerged that the residence time has a great impact on the product selectivity. In the case of 3-methylpyridine (at 360 °C), for example, the unwanted decarboxylation to pyridine could be successfully moderated through decrease of the residence time. As aldehyde formation remained negligible, the desired carboxylic acid could be obtained in a good yield of 50 %.
Scheme 10.
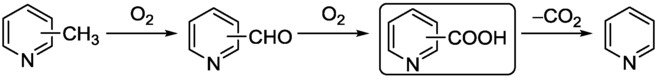
The oxidation of methylpyridines with molecular O2 as oxidant.27
2.8 CF cyclopropanation
Gold catalysis is of considerable current interest because of the massive number of unprecedented transformations,28 for example, highly chemoselective and diastereoselective heterogeneous cyclopropanation reactions. The CF synthesis of cyclopropane derivatives has been achieved through the utilization of Au clusters as catalyst (2.0±0.3 nm) encapsulated in a fourth-generation poly(amidoamine) (G4 PAMAM) dendrimer and loaded onto SBA-15, a mesoporous silica support, with a surface area of 760 m2 g−1.29 The reaction is shown in Scheme 11.
Scheme 11.
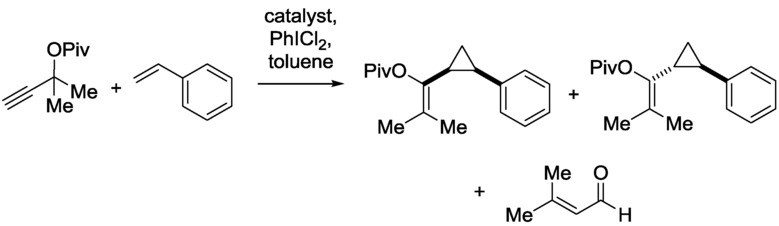
CF synthesis of cyclopropane derivatives.29
Through this CF reaction, the results observed under batch conditions are maintained. Moreover, the catalyst remains active even after 8 h and can be regenerated by reoxidation very effectively, yielding a more active species relative to the starting catalyst. The chemoselectivity of the reaction was investigated as a function of the residence time in a CF reactor. For this purpose, a gold-catalyzed cascade cyclopropanation rearrangement was selected (Scheme 12).
Scheme 12.
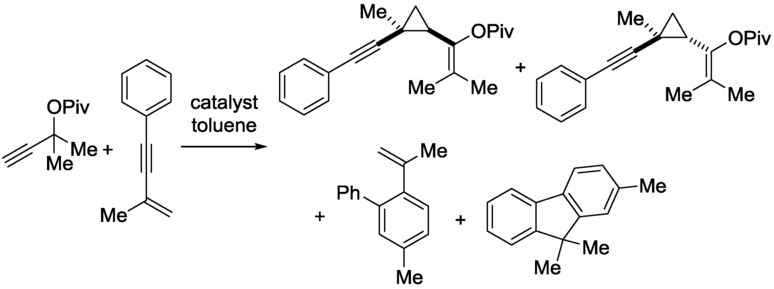
CF gold-catalyzed cascade cyclopropanation rearrangement.29
In this reaction, the cyclopropane adducts can rearrange catalytically into the styrene and fluorene product too. The reaction was also performed in batch mode, when a 40 % yield was achieved, comprising 38 % of the cyclopropane adducts and only 2 % of the rearrangement products, with equal amounts of styrene and fluorene products. However, in CF mode with a flow rate of 5 mL h−1, an 18 % yield of cis-cyclopropane was measured, with 2 % of styrene secondary product formation. In the CF mode, the rearrangement product distribution was changed, because formation of the fluorene product was not observed. Furthermore, through increase of the residence time by switching to a 0.1 mL h−1 flow rate, the conversion was multiplied more than threefold, and only the styrene was formed as secondary product. Continuous change of the residence time of the reactants resulted in a linear increase in the reactivity, coupled with a linear enhancement in the selectivity towards the styrene rearrangement product. In this reaction, the residence time played an important role in the tuning of the reaction selectivity.
Stereoselective cyclopropanation has been carried out by the use of immobilized bisoxazoline as catalyst and styrene and ethyl diazoacetate as starting materials in batch30 and mini-flow reactors (Scheme 13).31
Scheme 13.
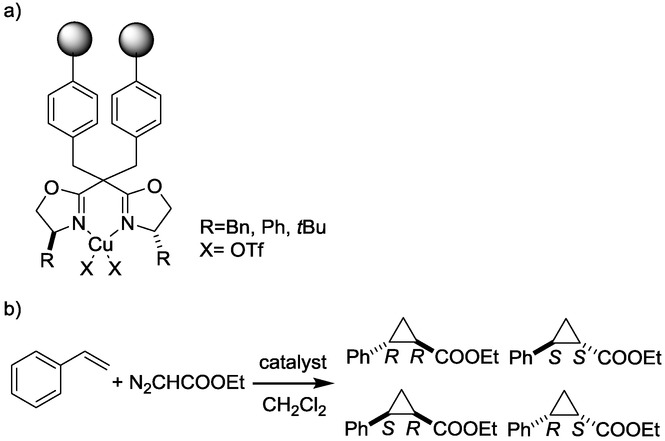
Catalyst (a) used in the cyclopropanation reaction (b).30,31
The conversion of the reaction exhibited a residence-time dependence, that is, the longer the residence time, the higher the conversion. Nonetheless, the chemoselectivity could not be influenced by residence-time control. However, when a different monolithic catalyst was used (Scheme 14), both the conversion and the chemoselectivity displayed a significant dependence on the residence time.32
Scheme 14.
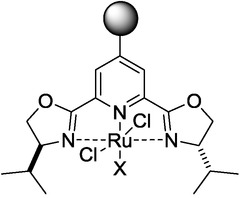
The catalyst used in the cyclopropanation reaction shown in Scheme 3 b.32
The monolithic catalyst was synthetized by the polymerization of the starting 4-vinyl derivative. Through fine-tuning of the residence time, the chemoselectivity of the reaction was changed. At a residence time of 14 min, the yield for trans-cyclopropanes was ∼15 %, while for the cis isomers it was ∼6 %; while at a residence time of 35 min, the corresponding yields were ∼40 % and ∼10 %, respectively. Consequently, upon increase of the residence time by ∼20 min, the yield of the trans-derivatives almost tripled, whereas that of the cis-derivative did not even double. Thus, at longer residence times the selectivity is shifted towards the trans-derivative (Figure 12).
Figure 12.
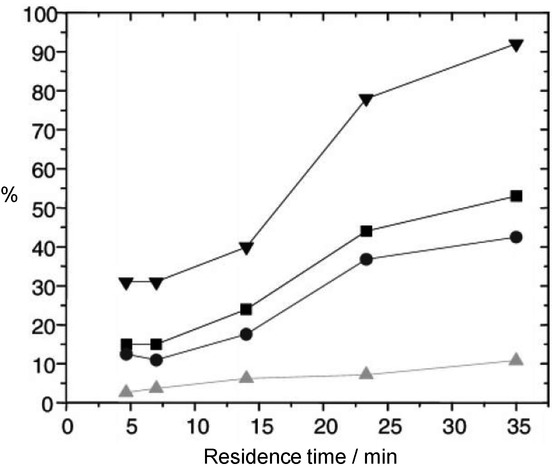
Effects of the residence time on the outcome of the reaction shown on Scheme 3 b with the catalyst shown in Scheme 4. ▼: conversion; ▪: yield of cyclopropanes; •: yield of trans-cyclopropanes; ▲: yield of cis-cyclopropanes. Reproduced with permission from ref. 32. Copyright 2015, American Chemical Society.
2.9 Selective olefin metathesis reactions
Metathesis reactions are of considerable current interest.33 According to green chemical directives, there is a constant need for heterogenized metathesis catalysts. However, with such catalysts, metal leaching always occurs.34 The silica-based mesoporous material MCM-41 was selected for the immobilization of the Grubbs/Hoveyda 2 complex. This catalyst has been used for the ring-opening/ring-closing metathesis (RO-RCM) reaction of cis-cyclooctene, yielding 1,9-cyclohexadecadiene (Scheme 15).35
Scheme 15.
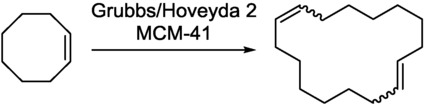
RO-RCM of cis-cyclooctene to 1,9-cyclohexadecadiene.35
This reaction is accompanied by side products such as other oligomers and polymers, compounds formed by double bond isomerization and unsymmetrical metathetical cleavage. To avoid the formation of byproducts, a complete reaction-parameter optimization was carried out, with fine-tuning of the pore diameter of the mesoporous support, the substrate concentration, the temperature, and the residence time. Increase of the residence time led to higher conversions (Figure 13 a). However, when the residence time was decreased, the selectivity towards 1,9-cyclohexadecadiene was improved (Figure 3 b).
Figure 13.
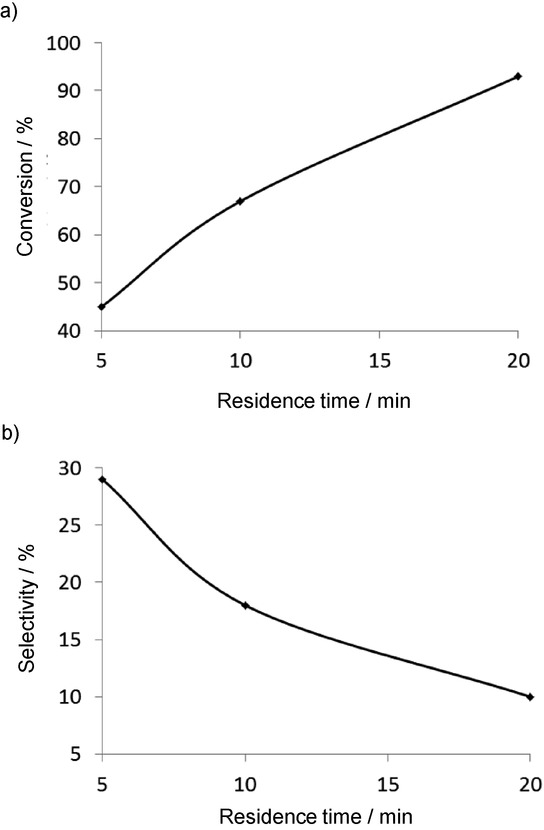
Effects of the residence time on the conversion (a) and the selectivity (b) for the RO-RCM of cis-cyclooctene to 1,9-cyclohexadecadiene.35
This observation implies that the small oligomers are kinetic products and are not formed through backbiting from a larger oligomer or polymer chain. Consequently, a high yield of the dimer product (1,9-cyclohexadecadiene) can be ensured only at low residence times in a CF device and the conversion is inversely proportional to the selectivity.
3 Summary and Outlook
The primary aim of this review has been to show that the strategic application of residence-time control is an efficient tool with which to guide continuous-flow (CF) transformations, and that the residence time can be fine-tuned and readily maintained in CF devices. It can be concluded that the residence time offers an extra advantage with which reactions can be directed. Representative examples have been provided, where the residence time governs the chemoselectivity and the diastereoselectivity. The residence time generally influences the conversion; for example, the longer the residence time, the higher the conversion. Such obvious examples have not been discussed in this review.
Flash chemical transformations, where appropriate residence-time control is crucial, have been discussed. The strategic application of residence-time control allows adequate and easy handling of very labile and reactive reagents. Consequently, merely by means of the fine-tuning of the residence time under general reaction conditions, higher conversions and yields can be achieved than with conventional batch technologies. Novel chemical windows have been opened in the field of organocatalytic and photochemical transformations too by strategic residence-time control. The neat, catalyst-free CF synthesis of Rufinamide, an anticonvulsant drug, has been described. Selected examples have been described where metal catalysts play a vital role, for example, the Heck cross-coupling reaction, selective hydrogenation on Pd surfaces, gold catalysis, and metathesis reactions. Examples have been shown for CF oxidation, reduction, and cyclopropanation reactions, where strategic residence-time control allows the formation of unprecedented reaction products not observed in batch reactions.
Through fine-tuning of the residence time for CF reaction optimization, harsh reaction conditions or aggressive reagents can be avoided. Importantly, the fine-tuning of the residence time can open up significant new fields. CF transformation may have the potential to revolutionize the fine-chemical and pharmaceutical industries. The search for such applications of CF systems started shortly after their discovery, and increasing numbers of studies are currently supporting their utility in drug development. Moreover, efforts in this area are expected to intensify.
Acknowledgments
The authors are grateful to the Hungarian Research Foundation (OTKA) (Nos. NK81371 and PD103994) and the Társadalmi Megújulás Operatív Program (TÁMOP) 4.2.2/B-10/1-2010-0012. I.M.M. acknowledges the award of a János Bolyai scholarship from the Hungarian Academy of Sciences. S.B.Ö. acknowledges a fellowship from the Postdoctoral Research Program of the Hungarian Academy of Sciences.
Biography
 Ferenc Fülöp was born in Szank, Hungary, in 1952. He received his MSc in Chemistry in 1975 and his PhD in 1979, from József Attila University, Szeged, Hungary, under the supervision of Professor Gábor Bernáth. In 1991 he was appointed as full professor at the Institute of Pharmaceutical Chemistry, University of Szeged, and since 1998 has been the head of the Institute. He is a member of Hungarian Academy of Sciences and has a wide range of research interests in synthetic organic chemistry. His recent activities have focused on the use of amino alcohols and β-amino acids in enzymatic transformations, asymmetric syntheses, foldamer construction, and flow chemistry, with a view to the development of pharmacologically active compounds.
Ferenc Fülöp was born in Szank, Hungary, in 1952. He received his MSc in Chemistry in 1975 and his PhD in 1979, from József Attila University, Szeged, Hungary, under the supervision of Professor Gábor Bernáth. In 1991 he was appointed as full professor at the Institute of Pharmaceutical Chemistry, University of Szeged, and since 1998 has been the head of the Institute. He is a member of Hungarian Academy of Sciences and has a wide range of research interests in synthetic organic chemistry. His recent activities have focused on the use of amino alcohols and β-amino acids in enzymatic transformations, asymmetric syntheses, foldamer construction, and flow chemistry, with a view to the development of pharmacologically active compounds.
References
- 1a.Irfan M, Glasnov TN, Kappe CO. ChemSusChem. 2011;4:300–316. doi: 10.1002/cssc.201000354. [DOI] [PubMed] [Google Scholar]
- 1b.Razzaq T, Kappe CO. Chem. Asian J. 2010;5:1274–1289. doi: 10.1002/asia.201000010. [DOI] [PubMed] [Google Scholar]
- 1c.Watts P, Haswell SJ. Drug Discovery Today. 2003;8:586–593. doi: 10.1016/s1359-6446(03)02732-6. [DOI] [PubMed] [Google Scholar]
- 1d.Wiles C, Watts P. Chem. Commun. 2011;47:6512–6535. doi: 10.1039/c1cc00089f. [DOI] [PubMed] [Google Scholar]
- 1e.Wiles C, Watts P. Green Chem. 2012;14:38–54. [Google Scholar]
- 1f.Rasheed M, Wirth T. Angew. Chem. Int. Ed. 50:357–358. doi: 10.1002/anie.201006107. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123 [Google Scholar]
- 1g.Wirth T. ChemSusChem. 2012;5:215–216. doi: 10.1002/cssc.201200061. [DOI] [PubMed] [Google Scholar]
- 1h.Ahmed-Omer B, Brandt JC, Wirth T. Org. Biomol. Chem. 2007;5:733–740. doi: 10.1039/b615072a. [DOI] [PubMed] [Google Scholar]
- 1i.Brandt JC, Wirth T. Beilstein J. Org. Chem. 2009;5:30. doi: 10.3762/bjoc.5.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1j.Hessel V, Kralisch D, Kockmann N, Noel T, Wang Q. ChemSusChem. 2013;6:746–789. doi: 10.1002/cssc.201200766. [DOI] [PubMed] [Google Scholar]
- 1k.Jähnisch K, Hessel V, Lowe H, Baerns M. Angew. Chem. Int. Ed. 43:406–446. doi: 10.1002/anie.200300577. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004;116 [Google Scholar]
- 2a.Schwalbe T, Autze V, Wille G. Chimia. 2002;56:636–646. [Google Scholar]
- 2b.Benaskar F, Patil NG, Rebrov EV, Ben-Abdelmoumen A, Meuldijk J, Hulshof LA, Hessel V, Schouten JC. ChemSusChem. 2013;6:353–366. doi: 10.1002/cssc.201200504. [DOI] [PubMed] [Google Scholar]
- 2c.Osorio-Planes L, Rodríguez-Escrich C, Pericàs MA. Chem. Eur. J. 2014;20:2367–2372. doi: 10.1002/chem.201303860. [DOI] [PubMed] [Google Scholar]
- 2d.Martín-Rapún R, Sayalero S, Pericàs MA. Green Chem. 2013;15:3295–3301. [Google Scholar]
- 2e.Pericàs MA, Herrerías CI, Solà L. Adv. Synth. Catal. 2008;350:927–932. [Google Scholar]
- 2f.Font D, Jimeno C, Pericàs MA. Org. Lett. 2006;8:4653–4655. doi: 10.1021/ol061964j. [DOI] [PubMed] [Google Scholar]
- 2g.Alza E, Pericàs MA. Adv. Synth. Catal. 2009;351:3051–3056. [Google Scholar]
- 3a.Ley SV. Chem. Rec. 2012;12:378–390. doi: 10.1002/tcr.201100041. [DOI] [PubMed] [Google Scholar]
- 3b.Lee CKY, Holmes AB, Ley SV, McConvey IF, Al-Duri B, Leeke GA, Santos RCD, Seville JPK. Chem. Commun. 2005:2175–2177. doi: 10.1039/b418669a. [DOI] [PubMed] [Google Scholar]
- 3c.Qian Z, Baxendale IR, Ley SV. Chem. Eur. J. 2010;16:12342–12348. doi: 10.1002/chem.201002147. [DOI] [PubMed] [Google Scholar]
- 3d.Browne DL, Baumann M, Harji BH, Baxendale IR, Ley SV. Org. Lett. 2011;13:3312–3315. doi: 10.1021/ol2010006. [DOI] [PubMed] [Google Scholar]
- 4a.Ratner DM, Murphy ER, Jhunjhunwala M, Snyder DA, Jensen KF, Seeberger PH. Chem. Commun. 2004:578–580. doi: 10.1039/b414503h. [DOI] [PubMed] [Google Scholar]
- 4b.Moore JS, Jensen KF. Angew. Chem. Int. Ed. 53:470–473. doi: 10.1002/anie.201306468. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014;126 [Google Scholar]
- 4c.McMullen JP, Jensen KF. Annu. Rev. Anal. Chem. 2010;3:19–42. doi: 10.1146/annurev.anchem.111808.073718. [DOI] [PubMed] [Google Scholar]
- 4d.Hartman RL, McMullen JP, Jensen KF. Angew. Chem. Int. Ed. 50:7502–7519. doi: 10.1002/anie.201004637. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011;123 [Google Scholar]
- 4e.Sans V, Karbass N, Burguete MI, García-Verdugo E, Luis SV. RSC Adv. RSC Advances. 2012;2:8721–8728. [Google Scholar]
- 5a.Wiles C, Watts P. Green Chem. 2014;16:55–62. [Google Scholar]
- 5b.Pastre JC, Browne DL, Ley SV. Chem. Soc. Rev. 2013;42:8849–8869. doi: 10.1039/c3cs60246j. [DOI] [PubMed] [Google Scholar]
- 5c.Tsubogo T, Ishiwata T, Kobayashi S. Angew. Chem. Int. Ed. 52:6590–6604. doi: 10.1002/anie.201210066. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- 5d.McQuade DT, Seeberger PH. J. Org. Chem. 2013;78:6384–6389. doi: 10.1021/jo400583m. [DOI] [PubMed] [Google Scholar]
- 5e. Muñoz JdeM, Alcázar J, de La Hoz A, Díaz-Ortiz A. Adv. Synth. Catal. 2012;354:3456–3460. [Google Scholar]
- 5f.Wegner J, Ceylan S, Kirschning A. Chem. Commun. 2011;47:4583–4592. doi: 10.1039/c0cc05060a. [DOI] [PubMed] [Google Scholar]
- 5g.Zhao D, Ding K. ACS Catal. 2013;3:928–944. [Google Scholar]
- 5h.Vaccaro L, Lanari D, Marrocchia A, Strappavecciaa G. Green Chem. 2014;16:3680–3704. [Google Scholar]
- 5i.Newman SG, Jensen KF. Green Chem. 2013;15:1456–1472. [Google Scholar]
- 5j.Hintermair U, Francio G, Leitner W. Chem. Commun. 2011;47:3691–3701. doi: 10.1039/c0cc04958a. [DOI] [PubMed] [Google Scholar]
- 5k.Burguete MI, García-Verdugo E, Luis SV. Beilstein J. Org. Chem. 2011;7:1347–1359. doi: 10.3762/bjoc.7.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5l.Wegner J, Ceylan S, Kirschning A. Adv. Synth. Catal. 2012;354:17–57. [Google Scholar]
- 6a.Cyr P, Deng ST, Hawkins JM, Price KE. Org. Lett. 2013;15:4342–4345. doi: 10.1021/ol4018134. [DOI] [PubMed] [Google Scholar]
- 6b.Kurahashi K, Takemoto Y, Takasu K. ChemSusChem. 2012;5:270–273. doi: 10.1002/cssc.201100373. [DOI] [PubMed] [Google Scholar]
- 6c.Bou-Hamdan FR, Seeberger PH. Chem. Sci. 2012;3:1612–1616. [Google Scholar]
- 6d.Petersen TP, Polyzos A, OBrien M, Ulven T, Baxendale IR, Ley SV. ChemSusChem. 2012;5:274–277. doi: 10.1002/cssc.201100339. [DOI] [PubMed] [Google Scholar]
- 6e.Pastre JC, Browne DL, OBrien M, Ley SV. Org. Process Res. Dev. 2013;17:1183–1191. [Google Scholar]
- 6f.Naber JR, Buchwald SL. Angew. Chem. Int. Ed. 49:9469–9474. doi: 10.1002/anie.201004425. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010;122 [Google Scholar]
- 6g.Noël T, Maimone TJ, Buchwald SL. Angew. Chem. Int. Ed. 50:8900–8903. doi: 10.1002/anie.201104652. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011;123 [Google Scholar]
- 6h.Strompen S, Weiß M, Gröger H, Hilterhaus L, Liese A. Adv. Synth. Catal. 2013;355:2391–2399. [Google Scholar]
- 7.Yoshida J, Nagaki A, Yamada T. Chem. Eur. J. 2008;14:7450–7459. doi: 10.1002/chem.200800582. [DOI] [PubMed] [Google Scholar]
- 8a.Yoshida J, Takahashi Y, Nagaki A. Chem. Commun. 2013;49:9896–9904. doi: 10.1039/c3cc44709j. [DOI] [PubMed] [Google Scholar]
- 8b.Yoshida J, Kim H, Nagaki A. ChemSusChem. 2011;4:331–340. doi: 10.1002/cssc.201000271. [DOI] [PubMed] [Google Scholar]
- 9.Nagaki A, Yamada S, Doi M, Tomida Y, Takabayashi N, Yoshida J. Green Chem. 2011;13:1110–1113. [Google Scholar]
- 10.Kim H, Nagaki A, Yoshida J. Nat. Commun. 2011;2:264. doi: 10.1038/ncomms1264. [DOI] [PubMed] [Google Scholar]
- 11.Nagaki A, Kim H, Yoshida J. Angew. Chem. Int. Ed. 48:8063–8065. doi: 10.1002/anie.200904316. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009;121 [Google Scholar]
- 12.Cantillo D, Sheibani H, Kappe CO. J. Org. Chem. 2012;77:2463–2473. doi: 10.1021/jo3001645. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Blackman ML, Leduc AB, Jamison TF. Angew. Chem. Int. Ed. 52:4251–4255. doi: 10.1002/anie.201300504. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013;125 [Google Scholar]
- 14.Wiesner M, Revell JD, Wennemers H. Angew. Chem. Int. Ed. 47:1871–1874. doi: 10.1002/anie.200704972. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008;120 [Google Scholar]
- 15.Ötvös SB, Mándity IM, Fülöp F. ChemSusChem. 2012;5:266–269. doi: 10.1002/cssc.201100332. [DOI] [PubMed] [Google Scholar]
- 16.Ötvös SB, Mándity IM, Fülöp F. J. Catal. 2012;295:179–185. [Google Scholar]
- 17.Yu Z, Lv Y, Yu C, Su W. Org. Process Res. Dev. 2013;17:438–442. [Google Scholar]
- 18.Borukhova S, Noël T, Metten B, de Vos E, Hessel V. ChemSusChem. 2013;6:2220–2225. doi: 10.1002/cssc.201300684. [DOI] [PubMed] [Google Scholar]
- 19.McMullen JP, Stone MT, Buchwald SL, Jensen KF. Angew. Chem. Int. Ed. 49:7076–7080. doi: 10.1002/anie.201002590. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010;122 [Google Scholar]
- 20a.Beletskaya IP, Cheprakov AV. Chem. Rev. 2000;100:3009–3066. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 20b.Phan NTS, Van Der Sluys M, Jones CW. Adv. Synth. Catal. 2006;348:609–679. [Google Scholar]
- 21.Fan XL, Manchon MG, Wilson K, Tennison S, Kozynchenko A, Lapkin AA, Plucinski PK. J. Catal. 2009;267:114–120. [Google Scholar]
- 22.Mason BP, Price KE, Steinbacher JL, Bogdan AR, McQuade DT. Chem. Rev. 2007;107:2300–2318. doi: 10.1021/cr050944c. [DOI] [PubMed] [Google Scholar]
- 23.Bakker JJW, Zieverink MMP, Reintjens RWEG, Kapteijn F, Moulijn JA, Kreutzer MT. ChemCatChem. 2011;3:1155–1157. doi: 10.1002/cctc.201100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webb D, Jamison TF. Org. Lett. 2012;14:568–571. doi: 10.1021/ol2031872. [DOI] [PubMed] [Google Scholar]
- 25.Hodge P, Sung DWL, Stratford PW. J. Chem. Soc. Perkin Trans. 1. 1999:2335–2342. [Google Scholar]
- 26.Burguete MI, García-Verdugo E, Vicent MJ, Luis SV, Pennemann H, Graf von Keyserling N, Martens J. Org. Lett. 2002;4:3947–3950. doi: 10.1021/ol026805o. [DOI] [PubMed] [Google Scholar]
- 27.Fraga-Dubreuil J, Garcia-Verdugo E, Hamley PA, Vaquero EM, Dudd LM, Pearson I, Housley D, Partenheimer W, Thomas WB, Whiston K, Poliakoff M. Green Chem. 2007;9:1238–1245. [Google Scholar]
- 28.Hashmi ASK, Hutchings GJ. Angew. Chem. Int. Ed. 45:7896–7936. doi: 10.1002/anie.200602454. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006;118 [Google Scholar]
- 29.Gross E, Liu JHC, Toste FD, Somorjai GA. Nat. Chem. 2012;4:947–952. doi: 10.1038/nchem.1465. [DOI] [PubMed] [Google Scholar]
- 30.Burguete MI, Fraile JM, García-Verdugo E, Luis SV, Martínez-Merino V, Mayoral JA. Ind. Eng. Chem. Res. 2005;44:8580–8587. [Google Scholar]
- 31.Burguete MI, Cornejo A, García-Verdugo E, García J, Gil MJ, Luis SV, Martínez-Merino V, Mayorald JA, Sokolova M. Green Chem. 2007;9:1091–1096. doi: 10.1021/jo070119r. [DOI] [PubMed] [Google Scholar]
- 32.Burguete MI, Cornejo A, García-Verdugo E, Gil MJ, Luis SV, Mayoral JA, Martínez-Merino V, Sokolova M. J. Org. Chem. 2007;72:4344–4350. doi: 10.1021/jo070119r. [DOI] [PubMed] [Google Scholar]
- 33.Trnka TM, Grubbs RH. Acc. Chem. Res. 2001;34:18–29. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]
- 34.Leadbeater NE, Marco M. Chem. Rev. 2002;102:3217–3273. doi: 10.1021/cr010361c. [DOI] [PubMed] [Google Scholar]
- 35.Bru M, Dehn R, Teles JH, Deuerlein S, Danz M, Müller IB, Limbach M. Chem. Eur. J. 2013;19:11661–11671. doi: 10.1002/chem.201203893. [DOI] [PubMed] [Google Scholar]