

Abstract
Histone deacetylase inhibitors (HDACi) are promising therapeutics for cancer. HDACi alter the epigenetic state of tumors and provide a unique approach to treat cancer. Although studies with HDACi have shown promise in some cancers, variable efficacy and off-target effects have limited their use. To overcome some of the challenges of traditional HDACi, we sought to use a tumor-specific dendrimer scaffold to deliver HDACi directly to cancer cells. Here we report the design and evaluation of tumor-specific dendrimer–HDACi conjugates. The HDACi was conjugated to the dendrimer using an ester linkage through its hydroxamic acid group, inactivating the HDACi until it is released from the dendrimer. Using a cancer cell model, we demonstrate the functionality of the tumor-specific dendrimer–HDACi conjugates. Furthermore, we demonstrate that unlike traditional HDACi, dendrimer–HDACi conjugates do not affect tumor-associated macrophages, a recently recognized mechanism through which drug resistance emerges. We anticipate that this new class of cell-specific epigenetic therapeutics will have tremendous potential in the treatment of cancer.
Keywords: cancer, dendrimers, epigenetics, histone deacetylase (HDAC) inhibitors, nanoparticles
Introduction
Cancer is characterized by uncontrolled cell proliferation because of genetic and epigenetic abnormalities.1 Epigenetics refers to the heritable changes in gene expression caused by histone or DNA modifications as opposed to alterations in the DNA primary sequence.2 Unlike genetic changes, epigenetic changes are potentially reversible through modulation of epigenetic machinery which lends itself towards therapeutic manipulation. The appreciation that cancer can be driven above the DNA level has created an opportunity to develop cancer therapeutics that modulate aberrant epigenetic states characteristic of tumor cells.
Histone modifications regulate the local chromatin architecture surrounding DNA sequences and are important epigenetic regulators of cell growth and proliferation.3 Histone acetylation is a common epigenetic modification and is controlled by histone acetyltransferases and deacetylases. Histone deacetylases (HDACs) are responsible for the deacetylation of histones which leads to compaction of the surrounding DNA regions and repression of gene transcription. Aberrant HDAC activity has been associated with numerous cancers, making HDAC inhibition a promising therapeutic target.4 HDAC inhibition induces apoptosis in malignant cells through various mechanisms, including alterations in cell cycle regulation and activation of oxidative stress pathways.
There are four classes of HDACs: class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), and class IV (HDAC11) represent Zn2+-dependent amidohydrolases, whereas class III is composed of the mechanistically distinct NAD+-dependent sirtuins.5 Class I, II, and IV HDAC inhibitors (HDACi) consist of a capping group that is exposed to the environment, an alkyl linker that is surrounded by a hydrophobic tunnel, and a metal-binding moiety that is buried in the protein active site.6 Because the metal-binding moiety, which is critical for HDACi activity, is structurally similar across Class I, II, and IV HDACi, modifications of this structural element can be applied to many HDACi.
Dendrimers are branched, synthetic macromolecules with controllable chemical topology which have been used for a broad range of biomedical applications, including drug delivery, gene transfection, and tissue engineering.7 Our early studies demonstrated that folic acid (FA) conjugated to a generation 5 (G5) polyamidoamine (PAMAM) dendrimer could target the folate receptor (FR) on cancer cells to deliver therapeutics in vitro and in vivo.8 These studies demonstrated that targeting cancer cells greatly enhanced the efficacy and reduced the toxicity of the dendrimer therapeutics compared with therapeutics alone. Building on these studies, we have improved the synthetic approach by using the highly efficient copper-catalyzed azide–alkyne 1, 3-dipolar cycloaddition (CuAAC) as well as exploring different targeting ligands and therapeutics to optimize the efficacy of targeted drug delivery.9 In this report, we describe the synthesis and biological evaluation of tumor-specific dendrimer–HDACi conjugates. We demonstrate the cell specificity and efficacy of dendrimer–HDACi conjugates in a human epithelial cancer cell model and also show that the tumor-specific HDACi have no effects on tumor-associated macrophages.
Results and Discussion
Synthesis and characterization of linker-modified suberoylanilide hydroxamic acid (SAHA) compounds and dendrimer–SAHA conjugates
In this report, we used suberoylanilide hydroxamic acid (SAHA) as a representative HDACi.10 SAHA has antitumor activity and structural elements that are shared by many Zn-dependent HDACi. To conjugate SAHA to the dendrimer scaffold, we modified SAHA with an azido-linker through the hydroxamic acid group. The modified SAHA was attached to the alkyne-modified G5 PAMAM dendrimer scaffold using the CuAAC reaction. Based on our prior work on cleavable linkers, we sought to evaluate the effects of two different linkages, an ester and an amide, on the functional properties of SAHA.7b The linker-modified SAHA compounds were synthesized as shown in Scheme 1. We modified SAHA with an azido-linker through either an ester bond (SAHA-eAzide 1) or an amide bond (SAHA-aAzide 2). The control dendrimer–HDACi conjugate (G5-eSAHA-Alkyne 5) and targeted dendrimer–HDACi conjugate (G5-FA-eSAHA-Alkyne 6) (Scheme 2) were synthesized using the CuAAC reaction of SAHA-eAzide 1 with the corresponding alkyne-modified dendrimers 3 and 4, respectively (Scheme 3).
Scheme 1.
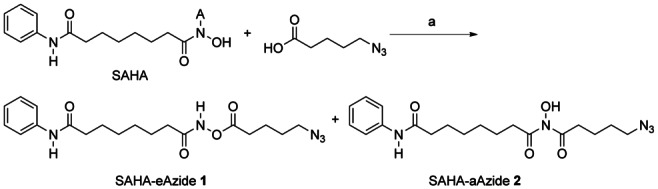
Synthesis of SAHA-eAzide 1 and SAHA-aAzide 2. Reagents and conditions: a) 5-azidopentanoic acid, CMPI, DMAP, DMF, rt, 6 h, 14 % (1), 20 % (2).
Scheme 2.
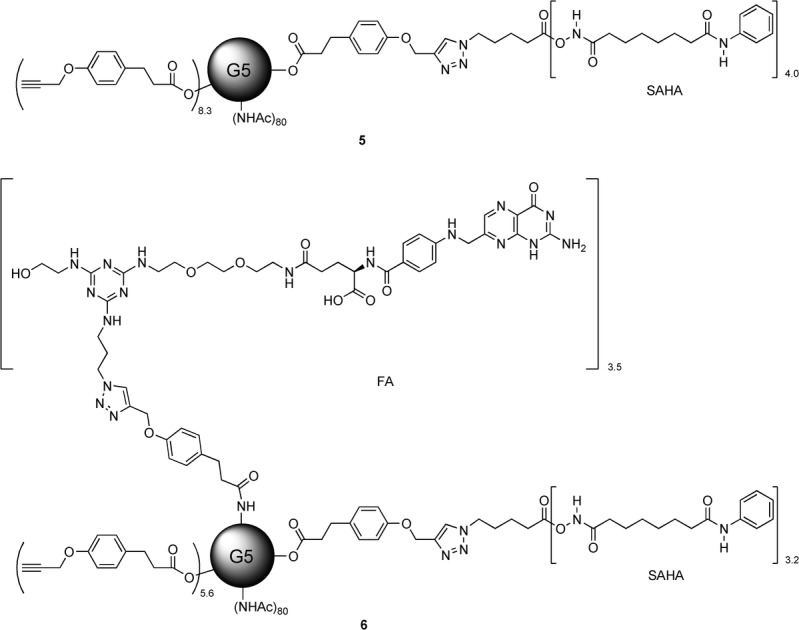
Structures of PAMAM G5 dendrimer conjugates 5 and 6.
Scheme 3.
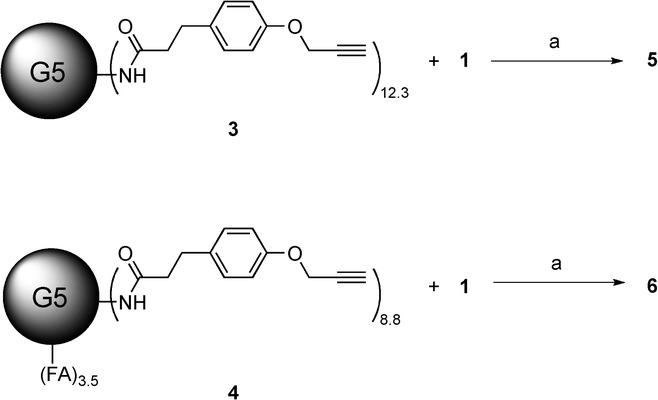
Synthesis of PAMAM G5 dendrimer conjugates 5 and 6. Reagents and conditions: a) 1) CuSO4, sodium ascorbate, SAHA-eAzide 1, under N2, o/n, 2) glycidol, rt, 6 h, 84 % (5), 85 % (6).
The conjugates and intermediates were analyzed by matrix-assisted laser desorption ionization/time-of-flight mass spectrometry (MALDI-TOF MS), high-performance liquid chromatography (HPLC), and NMR as we have previously described.11 The number of ligands (FA and SAHA) attached to the dendrimer scaffold was obtained from the integration of the methyl protons of the terminal acetyl groups to the aromatic protons on the conjugated ligands. The number of acetyl groups per dendrimer was determined by computing the total number of end groups from the number average molecular weight from gel permeation chromatography (GPC) and potentiometric titration data for G5-NH2 (100 %) as we have previously described.12 The total number of end groups was applied to the ratio of primary amines to acetyl groups, obtained from the 1H NMR of the partially acetylated dendrimer, to compute the average number of acetyl groups per dendrimer.
Biological evaluation of linker-modified SAHA compounds
To evaluate the functionality of the linker-modified SAHA compounds, we used the annexin V/7-aminoactinomycin D (An/7AAD) apoptosis assay and the folate-receptor-overexpressing KB human cancer cell line. KB cells were incubated with 100 nm of the linker-modified SAHA compounds 1 or 2 or free SAHA. After 24 h, the cells treated with the ester-linker-modified SAHA (SAHA-eAzide 1) and free SAHA had evidence of apoptosis, while the amide-linker-modified SAHA (SAHA-aAzide 2) did not show any evidence of apoptosis (Figure 1). To verify that the KB cell apoptosis was due to alteration of histones, we probed for histone acetylation using protein immunoblotting. We found that free SAHA and the SAHA-eAzide 1 not only increased apoptosis, but also induced hyperacetylation of histones confirming that SAHA-eAzide 1 was still active (Figure 1). In contrast, the amide-modified SAHA (SAHA-aAzide 2) did not induce apoptosis or histone hyperacetylation, suggesting that this modification inactivated the SAHA molecule.
Figure 1.
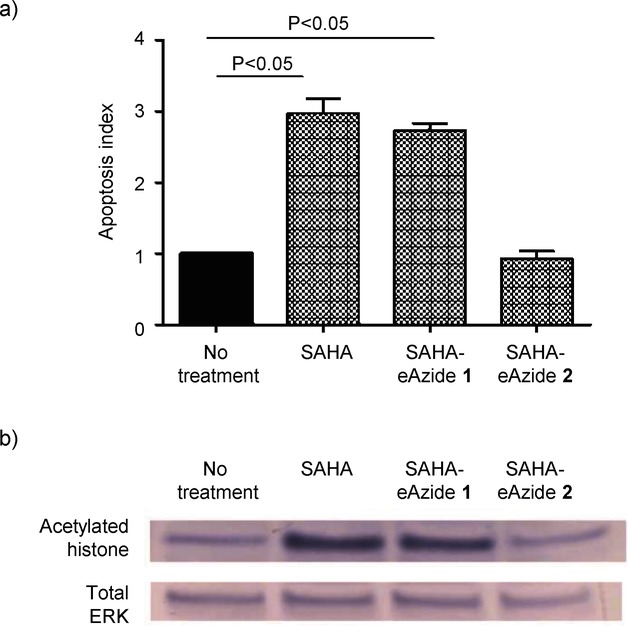
Biological evaluation of linker-modified SAHA. a) Apoptosis index demonstrating apoptosis induction with free SAHA and the ester-modified SAHA. The amide-modified SAHA did not demonstrate any evidence of apoptosis at the time points and concentrations tested. KB cells were incubated with 100 nm of the linker-modified SAHA compounds and free SAHA for 24 h. Values represent the mean ± S.E.M. for n=3 independent experiments. b) Protein immunoblots demonstrate that both free SAHA and eSAHA induce hyperacetylation of histones consistent with their known epigenetic mechanism of action. In agreement with the apoptosis data, the amide-modified SAHA compound did not demonstrate any hyperacetylation of histones. KB cells were incubated with 10 μm of the SAHA compounds 2 h and cell lysates were collected. Total ERK was used as the protein loading control.
Tumor-specific uptake of dendrimer–SAHA conjugates
Because only the SAHA-eAzide 1 demonstrated therapeutic efficacy, further evaluations were conducted using only the ester-modified SAHA compound. Next, we wanted to compare the cell specificity of the tumor-specific dendrimer–SAHA conjugates. We and others have shown that FA-functionalized dendrimers internalize through the FR in vitro and in vivo.14 Using our recently described in situ click reporter strategy, we monitored the uptake of the FA-targeted and nontargeted control dendrimer–SAHA conjugates by flow cytometry.14b This strategy allows us to track dendrimer therapeutics after they have been delivered to cells by using CuAAC to conjugate fluorescent reporters in situ. We incubated G5-FA-eSAHA-Alkyne 6 and the nontargeted control G5-eSAHA-Alkyne 5 with KB cells for one hour at 37 °C. After incubation, the cells were washed and processed, and Alexa Fluor 647-azide (AF647) was conjugated to the internalized dendrimer conjugates using the CuAAC reaction as we have previously described.14b The G5-FA-eSAHA-Alkyne 6 demonstrated a dose-dependent uptake into KB cells while the nontargeted G5-eSAHA-Alkyne 5 did not demonstrate any uptake (Figure 2). Importantly, the uptake of G5-FA-eSAHA-Alkyne 6 could be blocked by pretreatment with 100 μm FA, confirming the FR-specificity of the tumor-targeted dendrimer–HDACi (Figure 2). To further confirm the cell specificity of the dendrimer conjugates, we used our in situ click reporter strategy and confocal microscopy to demonstrate the FR-dependent uptake of the targeted-dendrimer SAHA conjugates. KB cells were treated as described above for one hour. After incubation, the KB cells were processed, and Alexa Fluor 555-azide (AF555) was conjugated to the internalized dendrimer conjugates using the CuAAC reaction. The G5-FA-eSAHA-Alkyne 6 had significantly more uptake at one hour compared with the nontargeted G5-eSAHA-Alkyne 5 (Figure 2).
Figure 2.
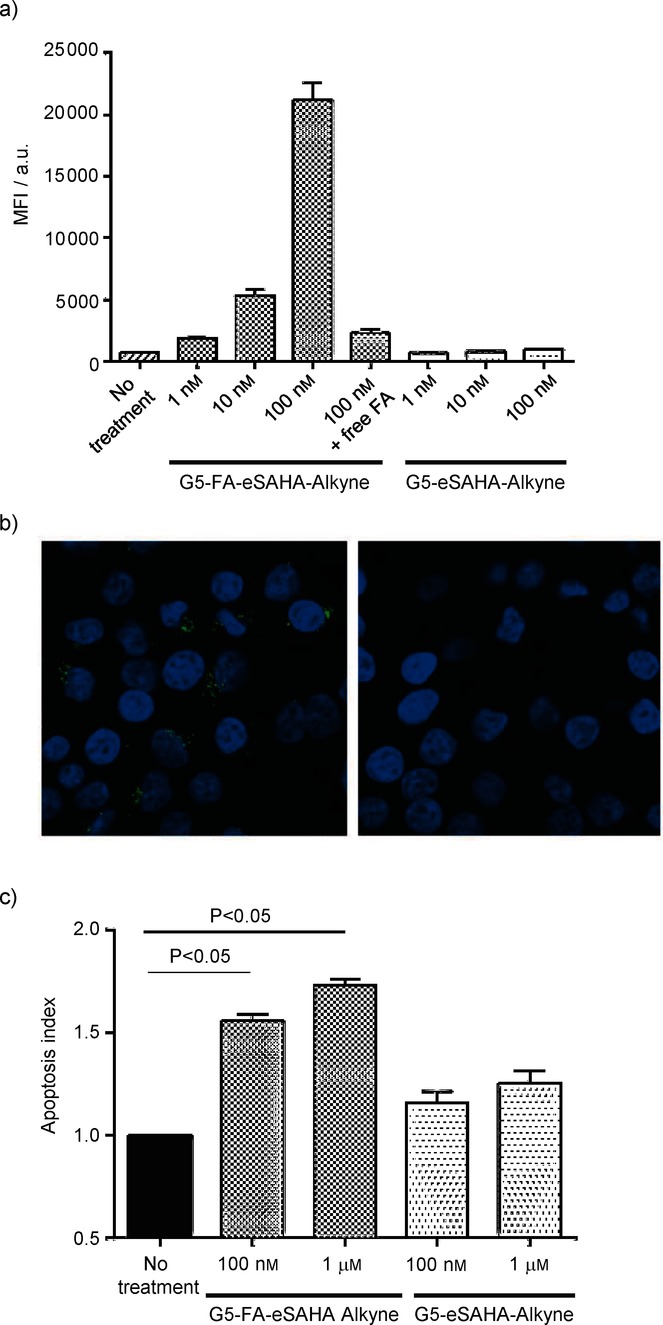
Biological evaluation of the dendrimer SAHA conjugates. a) Mean fluorescence intensity (MFI) demonstrates concentration-dependent uptake of the G5-FA-eSAHA compared to other conjugates. Pretreatment of KB cells with free FA blocked this uptake confirming the FR specificity of the targeted dendrimer conjugates. KB cells were incubated with dendrimer SAHA conjugates for 1 h and analyzed by flow cytometry for cell-specific uptake. Values represent the mean ± S.E.M. for n=3 independent experiments. b) Confocal images using the in situ click detection methodology confirm the cell specificity of the FA-targeted dendrimer conjugates (punctate, green cytoplasmic signal; left panel) compared with the nontargeted controls (no cytoplasmic signal; right panel). KB cells were incubated with 100 nm of the dendrimer SAHA conjugates for 1 h and subsequently analyzed using confocal microscopy. c) KB cells treated with the targeted G5-FA-eSAHA showed significantly greater levels of apoptosis than KB cells treated with the nontargeted G5-eSAHA. KB cells were treated with dendrimer–SAHA conjugates for 5 d and assessed for apoptosis using the annexin/7AAD assay. Values represent the mean ± S.E.M. for n=3 independent experiments.
Biological evaluation of dendrimer–SAHA conjugates
After confirming the activity of the ester-modified SAHA compound and the cell specificity of the G5-FA-eSAHA-Alkyne 6, we then evaluated the therapeutic efficacy of the dendrimer–SAHA conjugates. The activity of HDACi depends upon its hydroxamic acid moiety and its ability to block zinc-dependent histone deacetylases. Based on previous studies, we hypothesized that the G5-FA-eSAHA-Alkyne 6 conjugates would target FR-overexpressing cancer cells, internalize, and hydrolyze in the endosome, allowing free SAHA to inhibit histone deacetylases and to induce apoptosis. We evaluated the cytotoxicity of the dendrimer–SAHA conjugates 5 and 6 at five days. As expected, the G5-FA-eSAHA-Alkyne 6 was more cytotoxic compared with the nontargeted G5-eSAHA-Alkyne 5 (Figure 2).
One of the advantages of tumor-specific therapeutics is that many cancer drugs can also affect the immune system, reducing its ability to combat tumor initiation and progression. Macrophages contribute a large proportion of the cells in the tumor microenvironment and increased numbers are associated with a worse prognosis.15 Several studies have shown that HDACi are immunosuppressive through their effects on macrophages.16 Because of these macrophage effects, avoidance of HDAC inhibition in macrophages is desirable. To evaluate the capacity of the tumor-specific HDACi to avoid clearance and effects on macrophages, we examined the cytotoxic effects of free SAHA and the dendrimer–SAHA conjugates in the macrophage cell model RAW264.7. RAW264.7 cells were treated with free SAHA and the dendrimer–SAHA conjugates for 72 hours and evaluated for evidence of apoptosis using the An/7AAD assay mentioned above. At 72 hours, free SAHA induced apoptosis in RAW264.7 cells, while the dendrimer–SAHA conjugates had no effect, further confirming the cell specificity of the tumor-specific dendrimer–SAHA conjugates (Figure 3).
Figure 3.
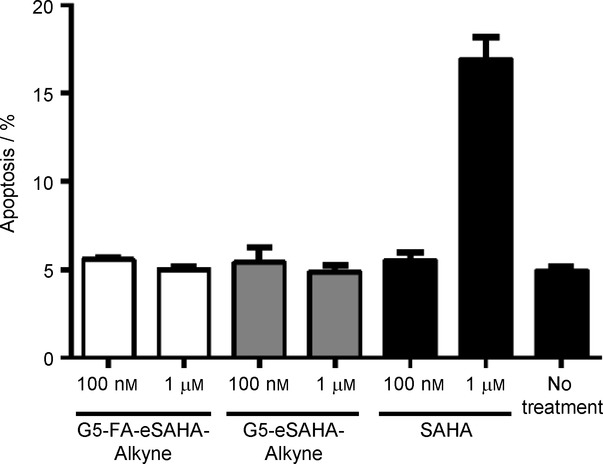
Biological evaluation of the dendrimer SAHA conjugates in tumor-associated macrophages. RAW264.7 cells treated with 1 μm free SAHA showed significantly greater levels of apoptosis than RAW264.7 cells treated with the dendrimer–SAHA conjugates confirming the tumor-specificity of targeted dendrimer–SAHA conjugates. RAW264.7 cells were incubated with dendrimer SAHA conjugates and free SAHA for 3 d and assessed for apoptosis using the annexin/7AAD assay. Values represent the mean ± S.E.M. for n=3 independent experiments.
In this study, we used the CuAAC reaction to engineer tumor-specific dendrimer HDACi and evaluated their functionality in a cancer cell model. To our knowledge, this is the first description of a tumor-specific HDACi—an approach that has the potential to greatly improve the efficacy and reduce the toxicities associated with this class of epigenetic therapeutics. Additionally, we evaluated the functional effects of modifying the metal-binding domain of HDACi with different linkers. These results provide important insights on HDACi that will help inform the rational optimization of this promising class of therapeutics.
Most HDACi are composed of three functional elements: a capping group, a carbon linker, and a metal-binding domain. We have previously demonstrated that a cleavable linker to attach a therapeutic to the dendrimer scaffold increases the efficacy of the therapeutic once it is delivered to the target cell.7b Building on these studies, we chose to modify the hydroxamic acid metal-binding domain for the dual purpose of providing a chemical handle to conjugate the HDACi to the dendrimer scaffold, as well as to inactivate the HDACi while it is attached to the dendrimer scaffold. As expected, the stable amide-linker modified HDACi was nonfunctional; however, the ester-linker modified HDACi was functional and led to hyperacetylation of histones and triggered apoptosis.
Some of the biggest challenges facing traditional HDACi are limitations in efficacy and toxicities, both of which are at least partially due to the lack of site-specific delivery of HDACi to the desired biological targets. We and others have used biological and synthetic ligands with PAMAM dendrimer scaffolds to target drugs to diseased cells and tissues. This approach not only improves the pharmacologic properties of the therapeutic by enhancing its circulating half-life and decreasing the off-target effects, but this approach also increases the local concentration of the therapeutic in diseased cells, further enhancing the therapeutic efficacy. As our molecular understanding of cancer evolves, the toolbox of receptor–ligand combinations that can be used to facilitate tumor-specific interactions will expand, further improving our ability to develop site-specific therapeutics to treat cancer and other diseases.
Conclusions
In summary, we have developed a tumor-specific histone deacetylase inhibitor (HDACi) using a polyamidoamine (PAMAM) dendrimer scaffold and a representative HDACi that shows cell specificity and therapeutic efficacy in a cancer cell model. Specifically, we have shown that 1) click chemistry can be used to efficiently functionalize tumor-specific dendrimer scaffolds with HDACi, 2) the ester-modified HDACi compounds and the ester-linked dendrimer–HDACi conjugates lead to increased histone acetylation and apoptosis, and 3) the tumor-specific HDACi have no effects on tumor-associated macrophages thereby increasing their therapeutic efficacy on cancer cells. Further studies are ongoing to evaluate the efficacy of this tumor-specific epigenetic therapeutic in vivo.
Experimental Section
1H NMR spectra were obtained using a Varian Inova 500 MHz (Palo Alto, USA). MALDI-TOF mass spectra were recorded on a PE Biosystems Voyager System 6050 (Waltham, USA), using 2,5-dihydroxybenzoic acid (DHB) as the matrix. Electrospray ionization mass spectra (ESI-MS) were recorded using a Micromass Quattro II Electronic HPLC/MS/MS mass spectrometer (Waters, Milford, USA).
Materials: All solvents and chemicals were of reagent grade quality, purchased from Sigma–Aldrich (St. Louis, USA), and used without further purification unless otherwise noted. Thin-layer chromatography (TLC) and column chromatography were performed with 25 DC-Plastikfolien Kieselgel 60 F254 (Merck, Kenilworth, USA), and Baxter silica gel 60 Å (230–400 mesh, Baxter International, Deerfield, USA), respectively.
Synthesis of G5-NHAc-Alkyne 3 and G5-NHAc-FA-Alkyne 4. Compounds 3 and 4 were synthesized as we have previously described.9,11,13 G5-NHAc-Alkyne 3 was obtained as a white solid; MALDI-TOF MS m/z: 32774 [M]+; the 1H NMR-integration-determined mean number of acetyl groups per dendrimer is 80.1, and the mean number of alkyne ligands per dendrimer is 12.3. G5-NHAc-FA-Alkyne 4 was obtained as a brown solid; MALDI-TOF MS m/z: 33059[M]+; the 1H NMR-integration-determined mean number of FA per dendrimer is 3.5, and the mean number of free alkyne ligands per dendrimer is 8.8.
Synthesis of N1-((5-azidopentanoyl)oxy)-N8-phenyloctanediamide (SAHA-eAzide, 1) and N1-(5-azidopentanoyl)-N1-hydroxy-N8-phenyloctanediamide (SAHA-aAzide, 2). SAHA (50 mg, 0.19 mmol) in dimethylformamide (DMF, 2 mL) was added to a solution of 5-azidopentanoic acid (41 mg, 0.28 mmol), 2-chloro-1-methyl-pyridinium iodide (CMPI) (97 mg, 0.38 mmol), and 4-(dimethylamino)pyridine (DMAP) (95 mg, 0.78 mmol) in DMF (3 mL). The reaction mixture was stirred at rt for 6 h. The solvent was removed under vacuum. The residue was dissolved in CH2Cl2, washed with water, dried over Na2SO4, and rotary evaporated. The resulting residue was purified by column chromatography on silica gel (CH3OH/CH2Cl2 5:95) to give two isomers SAHA-eAzide 1 and SAHA-aAzide 2 as white solids, 1 (10 mg, 14 %) and 2 (14 mg, 20 %); 1H NMR (500 MHz, CDCl3) 1: δ=0.89 (t, J=7.0 Hz, 2 H), 1.44 (m, 2 H), 1.62–1.83 (m, 8 H), 2.28 (t, J=7.0 Hz, 2 H), 2.39 (t, J=7.5 Hz, 2 H), 2.54 (t, J=7.5 Hz, 2 H), 3.32 (t, J=6.5 Hz, 2 H), 7.12 (t, J=7.5 Hz, 1 H), 7.25 (s, 1 H), 7.33 (t, J=8.0 Hz, 2 H), 7.53 (d, J=8.0 Hz, 2 H), 9.00 ppm (s, 1 H), 2: δ=0.89 (t, J=7.0 Hz, 2 H), 1.13 (m, 2 H), 1.65–1.78 (m, 8 H), 1.94 (t, J=7.0 Hz, 2 H), 2.36 (t, J=7.5 Hz, 2 H), 2.58 (t, J=7.5 Hz, 2 H), 2.97 (s, 1 H), 3.33 (t, J=6.5 Hz, 2 H), 7.10 (t, J=7.5 Hz, 1 H), 7.27 (s, 1 H), 7.32 (t, J=8.0 Hz, 2 H), 7.52 ppm (d, J=8.0 Hz, 2 H); ESI-MS m/z [M+H]+calcd for C19H28N5O4: 390.2, found 390.2.
Synthesis of G5-eSAHA-Alkyne 5. G5-NHAc-Alkyne 3 (10 mg, 0.31 μmol) was dissolved in CuSO4 (10 mol % per SAHA-eAzide, 1.0 mg mL−1 H2O) and sodium ascorbate (60 mol % per SAHA-eAzide, 1.0 mg mL−1 H2O solution) solution. SAHA-eAzide 1 (7.5 azide mole ratio to G5-NHAc-Alkyne 3, 5.0 mg mL−1 DMSO solution) was added. The reaction mixture was stirred at rt under N2 o/n. Glycidol (10 μL) was added to cap the remaining free amino groups on the dendrimer surface, and the reaction mixture was stirred at rt for 6 h. Samples were purified using 10 000 MWCO centrifugal filtration devices. The purification consisted of ten cycles (5×PBS and 5× deionized H2O) at 4800 rpm for 20 min each. The purified dendrimer conjugates were lyophilized to yield 5 as white solids (9.1 mg, 84 %); MALDI-TOF MS m/z: 34722 [M]+; the 1H NMR-integration-determined mean number of SAHA-eAzide molecules is 4.0.
Synthesis of G5-FA-eSAHA-Alkyne 6. G5-NHAc-FA-Alkyne 4 (10 mg, 0.30 μmol) was dissolved in CuSO4 (10 mol % per SAHA-azide, 1.0 mg mL−1 H2O) and sodium ascorbate (60 mol % per SAHA-azide, 1.0 mg mL−1 H2O solution) solution. SAHA-eAzide 1 (7.5 azide mole ratio to G5-NHAc-FA-Alkyne 4, 5.0 mg mL−1 DMSO solution) was added. The reaction mixture was stirred at rt under N2 o/n. Excess glycidol (10 μL) was added to cap the remaining free amino group on the dendrimer surface. The reaction mixture was stirred at rt for an additional 6 h. Samples were purified using 10 000 MWCO centrifugal filtration devices. The purification consisted of ten cycles (5×PBS and 5× deionized H2O) at 4800 rpm for 20 min each. The purified dendrimer samples were lyophilized to yield 6 as brown solids (9.1 mg, 84 %); MALDI-TOF MS m/z: 35921 [M]+; the 1H NMR-integration-determined mean number of SAHA-eAzide molecules is 3.2.
Cell culture: KB cells, a human cancer cell model (American Type Culture Collection, ATCC, Manassas, USA), were maintained as a monolayer cell culture in FA-free Roswell Park Memorial Institute (RPMI) 1640 medium (Life Technologies, Grand Island, USA) supplemented with 10 % fetal bovine serum (FBS), 100 U mL−1 penicillin, and 100 mg mL−1 streptomycin. The cell cultures were incubated at 37 °C with 5 % CO2. RAW264.7 cells were cultured at 37 °C in a humidified atmosphere of 5 % CO2 and 95 % air, in RPMI 1640 medium containing 10 % FBS supplemented with penicillin (100 units mL−1) and streptomycin (100 g mL−1).
Apoptosis assay and flow cytometry: In preparation for flow cytometry, KB cells were plated in 12-well plates and grown to 50 % confluency. Cells were washed with warm divalent cation-free PBS (1 mL) before the addition of new medium. All cells treatments were performed using the culture medium. After the indicated incubation times, the cells were washed with warm divalent cation-free PBS (1 mL) and immediately resuspended in culture medium, centrifuged at 1500 rpm for 5 min, and washed with FBS-free media. The resulting pellet was resuspended in annexin binding buffer (100 uL, BioLegend, San Diego, USA) containing a 1:100 dilution of both stock Alexa Fluoro 647 annexin (BioLegend, San Diego, USA) and 1 mg mL−1 7-aminoactinomycin D (Sigma Aldrich, St. Louis, USA) for 15 min in the dark. Cells were washed twice with cell culture media to remove any nonspecifically bound dye, followed by resuspending the pellet in annexin V binding buffer (200 uL) for flow cytometric? analysis. 50 000 events were collected using an Accuri C6 Flow Cytometer (Accuri Instruments, Inc., Ann Arbor, USA) and analyzed using FlowJo software version 7.6.5 (Tree Star Inc., Ashland, USA).
Protein immunoblotting: Cells were harvested and lysed using radioimmunopreciptation assay (RIPA) buffer containing 25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 % Nonidet P-40, 1 % sodium deoxycholate, 1 mm Na3VO4 and 10 mm NaF (phosphatase inhibitors), and 0.1 % SDS with 10 μL of Halt Protease Inhibitor Cocktail (Pierce, Rockford, USA) added for each 1 mL of buffer. Samples were separated using sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidede fluoride (PVDF) membrane. PVDF membranes were blocked with 0.1 % BSA/0.1 % Tween-20 in tris-buffered saline (TBS, blocking buffer) and washed with blocking buffer. PVDF membranes were then probed with rabbit antibodies against acetylated histone 3 (1:500 for 1 h) or total extracellular-signal-regulated kinase 1 (ERK1, 1:1000 for 1 h, Santa Cruz Biotechnology; Santa Cruz, USA). Alkaline-phosphatase-conjugated goat anti-rabbit secondary antibodies (Thermo Scientific, Waltham, USA) were used at a dilution of 1:20000 and incubated for 1 h.
Statistics: Data were analyzed by Students t-test using GraphPad 6.01 statistical software (GraphPad Software Inc., San Diego, USA).
Acknowledgments
The authors thank J. D. Kratz (University of Michigan) for technical support. S .N. G. was supported by a Samuel and Jean Frankel Cardiovascular Center Inaugural Grant and by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award number K08HL123621.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2a.Esteller M. Nat. Rev. Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 2b.Baylin SB, Jones PA. Nat. Rev. Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z, Bates SE. Mol. Pharm. 2011;8:2021–2031. doi: 10.1021/mp200329f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolden JE, Peart MJ, Johnstone RW. Nat. Rev. Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 5a.Insinga A, Minucci S, Pelicci PG. Cell Cycle. 2005;4:741–743. doi: 10.4161/cc.4.6.1717. [DOI] [PubMed] [Google Scholar]
- 5b.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S, Pelicci PG. Nat. Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 6a.Frey RR, Wada CK, Garland RB, Curtin ML, Michaelides MR, Li J, Pease LJ, Glaser KB, Marcotte PA, Bouska JJ, Murphy SS, Davidsen SK. Bioorg. Med. Chem. Lett. 2002;12:3443–3447. doi: 10.1016/s0960-894x(02)00754-0. [DOI] [PubMed] [Google Scholar]
- 6b.Curtin ML, Garland RB, Heyman HR, Frey RR, Michaelides MR, Li J, Pease LJ, Glaser KB, Marcotte PA, Davidsen SK. Bioorg. Med. Chem. Lett. 2002;12:2919–2923. doi: 10.1016/s0960-894x(02)00622-4. [DOI] [PubMed] [Google Scholar]
- 7a.Goonewardena SN. Curr. Atheroscler. Rep. 2012;14:247–253. doi: 10.1007/s11883-012-0249-9. [DOI] [PubMed] [Google Scholar]
- 7b.Goonewardena SN, Kratz JD, Zong H, Desai AM, Tang S, Emery S, Baker JR, Jr, Huang B. Bioorg. Med. Chem. Lett. 2013;23:2872–2875. doi: 10.1016/j.bmcl.2013.03.088. [DOI] [PubMed] [Google Scholar]
- 8a.Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR., Jr Cancer Res. 2005;65:5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- 8b.Thomas TP, Majoros IJ, Kotlyar A, Kukowska-Latallo JF, Bielinska A, Myc A, Baker JR., Jr J. Med. Chem. 2005;48:3729–3735. doi: 10.1021/jm040187v. [DOI] [PubMed] [Google Scholar]
- 9.Zong H, Thomas TP, Lee KH, Desai AM, Li MH, Kotlyar A, Zhang Y, Leroueil PR, Gam JJ, Banaszak Holl MM, Baker JR., Jr Biomacromolecules. 2012;13:982–991. doi: 10.1021/bm201639c. [DOI] [PubMed] [Google Scholar]
- 10.Choi SE, Weerasinghe SV, Pflum MK. Bioorg. Med. Chem. Lett. 2011;21:6139–6142. doi: 10.1016/j.bmcl.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mullen DG, Fang M, Desai A, Baker JR, Orr BG, Holl MMB. ACS Nano. 2010;4:657–670. doi: 10.1021/nn900999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majoros IJ, Thomas TP, Mehta CB, Baker JR. J. Med. Chem. 2005;48:5892–5899. doi: 10.1021/jm0401863. [DOI] [PubMed] [Google Scholar]
- 13a.Zhang Y, Thomas TP, Lee K-H, Li M, Zong H, Desai AM, Kotlyar A, Huang B, Banaszak Holl MM, Baker JR. Bioorg. Med. Chem. 2011;19:2557–2564. doi: 10.1016/j.bmc.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14a.Thomas TP, Goonewardena SN, Majoros IJ, Kotlyar A, Cao ZY, Leroueil PR, Baker JR. Arthritis Rheum. 2011;63:2671–2680. doi: 10.1002/art.30459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b.Goonewardena SN, Zong H, Leroueil PR, Baker JR. ChemPlusChem. 2013;78:430–437. [Google Scholar]
- 15.Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD, Bast MA, Rosenwald A, Muller-Hermelink HK, Rimsza LM, Campo E, Delabie J, Braziel RM, Cook JR, Tubbs RR, Jaffe ES, Lenz G, Connors JM, Staudt LM, Chan WC, Gascoyne RD. N. Engl. J. Med. 2010;362:875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabiec AM, Krausz S, de Jager W, Burakowski T, Groot D, Sanders ME, Prakken BJ, Maslinski W, Eldering E, Tak PP, Reedquist KA. J. Immunol. 2010;184:2718–2728. doi: 10.4049/jimmunol.0901467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary