

Abstract
This is the first report of 5-styryl-oxathiazol-2-ones as inhibitors of the Mycobacterium tuberculosis (Mtb) proteasome. As part of the study, the structure–activity relationship of oxathiazolones as Mtb proteasome inhibitors has been investigated. Furthermore, the prepared compounds displayed a good selectivity profile for Mtb compared to the human proteasome. The 5-styryl-oxathiazol-2-one inhibitors identified showed little activity against replicating Mtb, but were rapidly bactericidal against nonreplicating bacteria. (E)-5-(4-Chlorostyryl)-1,3,4-oxathiazol-2-one) was most effective, reducing the colony-forming units (CFU)/mL below the detection limit in only seven days at all concentrations tested. The results suggest that this new class of Mtb proteasome inhibitors has the potential to be further developed into novel antitubercular agents for synergistic combination therapies with existing drugs.
Keywords: antitubercular agents, 5-styryl-oxathiazolones, Mtb proteasome inhibitor, Mycobacterium tuberculosis, nonreplicating Mtb, rapid bactericidal activity
Introduction
Tuberculosis (TB) has been declared a global health emergency by the World Health Organization (WHO). It is estimated that about one third of the worlds population is currently infected with the responsible actinobacteria Mycobacterium tuberculosis (Mtb).1 Resistance of Mtb to available antibiotics has increased drastically, and multidrug-resistant (MDR) strains of Mtb as well as extensively drug-resistant (XDR) strains are becoming a major health problem worldwide.1 Furthermore, cases have recently been reported where no available first- and second-line drugs are effective, and this has led to the term totally drug-resistant (TDR) TB.2 Resistant strains of Mtb require longer treatment, which may lead to interruption of therapy and, in turn, lead to further drug resistance. The usual drug regimen for TB at present consists of antibiotics discovered more than 60 years ago, and includes isoniazid, rifampicin, pyrazinamide, and ethambutol.3 These antibiotics act by disruption of cell wall synthesis4 and inhibition of RNA synthesis.4c-5 It was quickly discovered that monotherapy led to resistance in the bacteria, and therefore the recommended TB treatment today utilizes a combination of the four first-line drugs for two months, and thereafter a pharmacotherapy with rifampicin and isoniazid for an additional four months.3 Considering the long treatment time, the adverse effects associated with the drugs, and the problems with resistant strains, it is clear that there is a great need for novel antibiotics that possess new antitubercular modes of action and that can be used in combination with existing drugs.
Proteasomes are responsible for degrading proteins, and so help maintain intracellular protein homeostasis.6 The human proteasome consists of a cylindrical 26S particle composed of a 20S core catalytic component capped at one or both ends with a 19S regulatory subunit, which recognizes and binds the substrate protein. In eukaryotic proteasomes, the 20S core particle is composed of four heptameric rings, of which the two inner rings are made up of seven different β-subunits, with only three of them responsible for the proteolytic activity (β1, β2, β5, with peptidyl-glutamyl-peptide-hydrolyzing, trypsin-like and chymotrypsin-like activities, respectively).7 The Mtb proteasome displays the same overall structure as the proteasomes of eukaryotic systems.8 However, prokaryotic 20S proteasomes, including those from Mtb, have been shown to have open cylindrical ends, in contrast to the tighter ends of eukaryotic proteasomes, and possess only one type of β-subunit, usually resembling the eukaryotic β5 chymotrypsin-like structure.8,9 Proteolytic activity involves the β-hydroxyl group of the N-terminal threonine, which is present in the active site of proteasome subunits.7a–7d
In many diseases, such as cancer, auto-immune diseases, and neurodegenerative diseases, cells accumulate various proteins, and the human proteasome has thus emerged as a promising therapeutic target.6a,7a The first human proteasome inhibitor, Bortezomib (Velcade), was approved for clinical use in the USA in 2003 and in Europe in 2004 for the treatment of multiple myeloma (Figure 1). The X-ray structure of the yeast proteasome in complex with the compound was reported in 2006, giving valuable insights into its binding interactions.10 Serious toxicity associated with peptidyl boronates, such as Bortezomib, is mainly due to the inhibition of the β5 subunit of the human proteasome,11 precluding the use for treatment of, for example, infectious diseases. Therefore, the search for alternative classes of inhibitors with greater selectivity towards prokaryotic proteasomes is ongoing.
Figure 1.
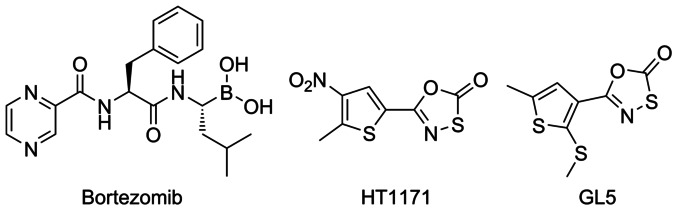
Structures of the proteasome inhibitors Bortezomib and oxathiazol-2-one compounds HT1171 and GL5.
Proteasomes have been shown to be of great importance to Mtb; they help the bacteria survive the nitrosative stress caused by the hosts immune response12 and increase their ability to persist in infected mice.13 Furthermore, the actinobacteria are unusual among prokaryotes in that they express proteasomes, meaning that selective action against bacteria of this phylum should be possible.6b,14 It is particularly interesting that proteasome inhibitors have been shown to be active on nonreplicating Mtb. This is important since nonreplicating Mtb (which to a large extent underlie the need for long treatment times), display reduced protein synthesis, and are left unaffected by most antitranscription or antitranslation agents currently used for treatment.15 Only a few classes of compounds have proven to be active on nonreplicating Mtb, for example, oxathiazol-2-ones, quinolones, and allylic thiocyanates.7c,16 Mtb proteasome inhibitors displaying species selectivity, thus low associated toxicity, are promising candidates for the development of new antitubercular drugs.
Lin et al. have previously reported that the 1,3,4-oxathiazol-2-one compounds HT1171 and GL5 (Figure 1) act as suicide-substrate inhibitors of the Mtb proteasome displaying high selectivity over the human proteasome and other proteases, including trypsin, cathepsin B, and matrix metalloproteases.7c They demonstrated that the oxathiazol-2-one moiety acts as an electrophilic warhead, such that the active-site nucleophile, the N-terminal threonine residue, is effectively cyclocarbonylated by an irreversible covalent attack on the heterocycle.7c,8b
Moreover, oxathiazolones have been reported to have antimycobacterial activity when they were exploited as carboxylic acid bioisosteres in substituted pyridines and pyrazines.17 In an attempt to identify a new generation of species-selective proteasome inhibitors, Gryder et al. investigated the effect of replacing the boronate group on the dipeptide backbone of Bortezomib with an oxathiazol-2-one moiety. Unfortunately, the resulting compound was not active on the Mtb proteasome and only slightly active on the human 20S proteasome.18 More recently, Yang et al. assessed the antitubercular activity of a series of GL5-type oxathiazol-2-ones, together with their dithiazol-3-one analogues, on the virulent Mtb H37Rv strain, achieving a lowest minimum inhibitory concentration (MIC) value of 4 μg mL−1 (15 μm) for HT1171.16g These compounds also possessed mycobacterial cell wall permeation properties, and were active against nonreplicating Mtb. They have therefore been proposed as highly interesting antitubercular agents for the synergistic combination treatment of TB.7c
In the present study, we further investigate the structure–activity relationships of 1,3,4-oxathiazol-2-one derivatives with respect to both their potency as Mtb proteasome inhibitors and their selectivity over the chymotrypsin-like catalytic activity of the human proteasome as an early assessment of potential toxicity. Furthermore, the compounds were optimized with regard to solubility and stability. Finally, activity against replicating and nonreplicating Mtb and cytotoxicity to mammalian cells are reported for a collection of selective 5-styryl-1,3,4-oxathiazol-2-one inhibitors.
Results and Discussion
In a first effort to investigate the chemical space, a wide variety of oxathiazol-2-ones were prepared from the corresponding commercially available amides by treatment with chlorocarbonyl sulfenyl chloride at 100 °C for 15 min with microwave (MW) heating (Method A) or at room temperature overnight (Method B). Isolated products were thereafter evaluated for proteasome inhibition based on the assay procedures described by Lin et al.7c (Table 1, entries 1–23). Compounds were tested for chymotrypsin-like peptidase activity against both the Mtb 20S open-gate proteasome (in which the alpha subunit has an 8-residue deletion at the N-terminus, as described by Lin et al.8a) and the human 20S proteasome, tracked by cleavage of the fluorogenic substrate suc-LLVY-7-amino-4-methylcoumarin.
Table 1.
Activity, solubility, and stability of proteasome inhibitors.
| Entry | Structure | Cmpd | Method, Yield | Mtb proteasome IC50 [nM] | Human proteasome IC50 [nM] | Solubility [μM] | Stability [%][a] |
|---|---|---|---|---|---|---|---|
| 1 | 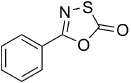 |
1 | A, 43 % | 1350 | 6200 | 63.8 | 97.4 |
| 2 | 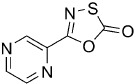 |
2 | B, 23 % | 2300 | 4500 | 35.1 | 3.6 |
| 3 | 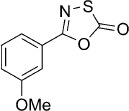 |
3 | B, 23 % | 665 | 15 000 | 30.9 | 15.4 |
| 4 | 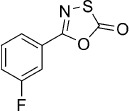 |
4 | B, 83 % | 630 | 11 000 | 86.2 | 33.5 |
| 5 | 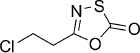 |
5 | A, 60 % | >100 000 | >100 000 | — | — |
| 6 | 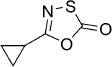 |
6 | A, 27 % | >100 000 | >100 000 | — | — |
| 7 | 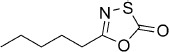 |
7 | A, 56 % | >100 000 | >100 000 | — | — |
| 8 |  |
8 | B, 24 % | 80 500 | 1400 | — | — |
| 9 | 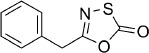 |
9 | A, 43 % | >100 000 | 1900 | — | — |
| 10 | 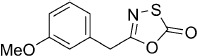 |
10 | A, 27 % | >100 000 | >100 000 | — | — |
| 11 |  |
11 | A, 39 % | >100 000 | >100 000 | — | — |
| 12 | 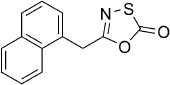 |
12 | A, 51 % | >100 000 | >100 000 | — | — |
| 13 | 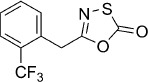 |
13 | B, 56 % | >100 000 | 8700 | — | — |
| 14 | 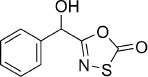 |
14 | B, 40 % | >100 000 | 20 000 | — | — |
| 15 | 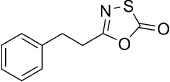 |
15 | B, 71 % | >100 000 | 6700 | — | — |
| 16 | 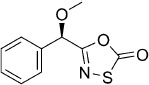 |
16 | B, 58 %[b] | >100 000 | 5800 | — | — |
| 17 | 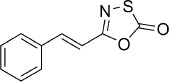 |
17 | B, 40 % | 420 | >100 000 | 19.1 | 38.7 |
| 18 | 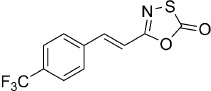 |
18 | B, 42 % | 640 | >100 000 | 0.1 | n.d. |
| 19 | 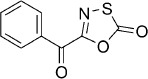 |
19 | 57 %[c] | 2250 | 2950 | 5.7 | 46.8 |
| 20 | 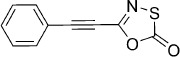 |
20 | B, 24 % | 1035 | 4200 | 24.9 | 89.6 |
| 21 | 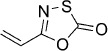 |
21 | B, 8 % | 27 000 | 9100 | — | — |
| 22 | 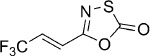 |
22 | B, 81 % | 38 000 | >100 000 | 44.1 | 79.9 |
| 23 |  |
23 | B, 90 % | >100 000 | 14 000 | — | — |
| 24 | 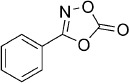 |
24 | 49 %[d] | 31 500 | >100 000 | 7.6 | 8.5 |
| 25 | 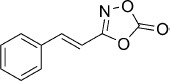 |
25 | 51 %[d,e] | 54 500 | — | 12.3 | — |
| 26 | 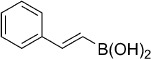 |
26 | —[f] | >100 000 | >100 000 | — | — |
| 27 | 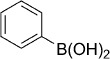 |
27 | —[f] | >100 000 | >100 000 | — | — |
| 28 | Bortezomib | — | — | 135 | (2.56)[g] | — | — |
General method for the synthesis of 5-substituted oxathiazol-2-ones. Method A reagents and conditions: amide (1.0 equiv), chlorocarbonyl sulfenyl chloride (1.5 equiv), 1,4-dioxane (4.0 mL mmol−1), 100 °C MW, 15 min. Method B: amide (1.0 equiv), chlorocarbonyl sulfenyl chloride (2.0 equiv), THF (4.0 mL mmol−1), rt, o/n. [a] Chemical stability in PBS pH 7.4 at 25 °C as % remaining after 24 h. [b] The amide was prepared, and the crude sample was used without further purification. [c] Reagents and conditions: 14 (1.0 mmol), Dess–Martin periodinane (1.1 mmol), CH2Cl2 (5 mL), 0 °C, 2 h. [d] General method for the synthesis of 3-substituted dioxazolones. Reagents and conditions: starting hydroxamic acid (1.0 equiv), 1,1′-carbonyldiimidazole (1.1 equiv), CH3CN (4.0 mL mmol−1), 0 °C, 1 h. [e] The hydroxamic acid was prepared and used as starting material for the dioxazolone synthesis without further purification. [f] Commercially available compound. [g] Literature value.[18] n.d.=not determined.
The 5-aryl-substituted 1,3,4-oxathiazol-2-ones in entries 1–4 (Table 1, 1–4) have previously been reported as inhibitors of the Mtb proteasome.19 To further develop the core structure, we decided to find out if elongated compounds with different spacers could have a positive impact on the activity. For this purpose, various tethers between the aryl and the heterocyclic warhead were introduced in the 5-position, for example, alkene, alkyne, and methylene linkers. We found that an sp2-hybridized carbon directly coupled to the oxathiazol-2-one ring in the 5-postion was crucial for the compounds activity. Additionally, the introduction of the (E)-alkene linker (entries 17–18) gave inhibitors equally or more potent (17 and 18; IC50=420 and 640 nm, respectively) toward the Mtb proteasome compared to when the aryl was substituted directly onto the oxathiazol-2-one (1–4). More importantly, this type of compound was not active on the human proteasome (IC50>100 000 nm). For compounds with an alkyne- or carbonyl-containing spacer (19 and 20), the activities on the Mtb proteasome were slightly lower compared to olefinic inhibitors 17 and 18. Moreover, these linkers provided less selective inhibitors. When an sp3-hybridized carbon was used as a linker atom, no activity was obtained against the Mtb proteasome, and little or no inhibition was seen towards the human proteasome (entries 9–16). Interestingly, when an ethyl bridge was introduced instead of the double bond between the phenyl and oxathiazol-2-one (compare moieties 15 and 17), no inhibition was observed for the Mtb proteasome, but the saturated compound instead gained some activity toward the human proteasome. This suggests that the (E)-double bond is important both for the mycobacterial inhibition and the selectivity over the human proteasome. However, the allowed substitution pattern of the (E)-alkene linker seems to be slightly constrained; when an additional phenyl group was introduced onto the ethylene linker, no activity was detected against the Mtb proteasome (entry 23). Instead, compound 23 became a more potent inhibitor of the human proteasome.
The aryl group of the styryl moiety also seems to be important for the interaction with the Mtb protein. When a vinyl group was substituted onto the oxathiazol-2-one, the activity decreased from 420 nm for 17 to 27 000 nm for 21. This pattern was also observed when aliphatic substituents were introduced (5–8). The effect is probably due to the lack of an aryl group as well as the effect of the hybridization of the carbon directly attached to the oxathiazol-2-one.
To summarize the results depicted in Table 1, it appears that an aryl group conjugated with the oxathiazolone scaffold is highly beneficial for the inhibition of the Mtb proteasome (see products 1–4 and 17–22), and in particular high selectivity over the human proteasome is caused by the presence of the (E)-alkene linker (17 and 18). This may also be coupled to the loss of activity for the unconjugated 15.
To increase the scope of the investigation, we wished to evaluate other heterocyclic warheads such as the dioxazolones (Table 1, entries 24–25), but these compounds suffered from poor stability due to rapid hydrolysis. The oxathiazol-2-one was also replaced with a boronic acid functionality (Table 1, entries 26–27) in an attempt to mimic the reversible covalently-bonded warhead used in Bortezomib. However, the simple boronic acid analogues were completely inactive on both the Mtb and human proteasomes, suggesting that the oxathiazol-2-one moiety is a more useful electrophile for the inactivation of the target.
A series of 5-styryl-oxathiazol-2-ones was synthesized to further investigate the effect of the phenyl substitution on the structure–activity relationship of proteasome inhibitors. For this, we developed a synthetic route where aryl iodides or bromides were used as starting material in a microwave-assisted Mizoroki–Heck cross-coupling reaction with acrylamide to yield the corresponding 3-substituted acrylamides.20, 21 The final 5-substituted-oxathiazolones were obtained by treating the corresponding amides with chlorocarbonyl sulfenyl chloride (Scheme 1).
Scheme 1.
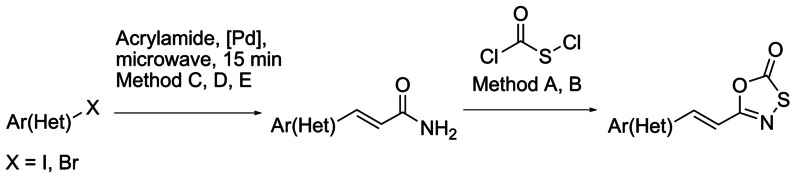
General synthetic route for the preparation of 5-styryl-oxathiazolones.
A diverse set of para-substituted 5-styryl-oxathiazolones were chosen to explore the structural requirements of the active site (Table 2, entries 1–12). The majority of the compounds displayed the same range of inhibiting activity against the Mtb proteasome as 17 and 18 (IC50 values between 415–1700 nm). The results showed no clear trend in activity for electron-rich and electron-poor aryl substituents, although electron-poor phenyl groups gave slightly better inhibition of the Mtb proteasome compared to electron-rich phenyls. The inhibition of the human proteasome was unfortunately also affected, giving a poorer selectivity profile. However, based on the relative activities for nonstyryl containing 21 and 22 (Table 1, entries 21–22), it is evident that the phenyl group is important for inhibition of the Mtb proteasome.
Table 2.
Structure–activity relationship of 5-styryl-oxathiazolone inhibitors with the Mtb and human proteasome.
| Entry | Structure
|
Cmpd | Method, Yield | Mtb proteasome IC50 [nM] | Human proteasome IC50 [nM] | Solubility [μM] | Stability [%][a] |
|---|---|---|---|---|---|---|---|
| 1 | 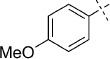 |
28 | C, 75 %; B, 71 % | 1350 | 8500 | 3.9 | 1.5 |
| 2 | 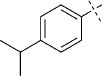 |
29 | C, 65 %; B, 66 % | 1700 | 14 500 | 0.4 | 99.8 |
| 3 | 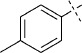 |
30 | C, 59 %; B, 43 % | 1000 | >100 000 | 1.1 | 96.0 |
| 4 | 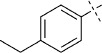 |
31 | C, 50 %; B, 43 % | 1080 | >100 000 | 0.1 | 98.2 |
| 5 | 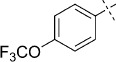 |
32 | D; B, 64 %[b] | 780 | >100 000 | 0.2 | 94.8 |
| 6 | 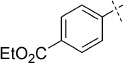 |
33 | D, 67 %; B, 69 % | 475 | >100 000 | 0.3 | 91.1 |
| 7 | 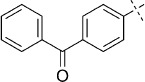 |
34 | D, 63 %; B, 80 % | 1190 | 87 000 | <2.5 | 96.1 |
| 8 | 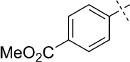 |
35 | D, 60 %; B, 68 % | 415 | 27 500 | 0.8 | 97.3 |
| 9 | 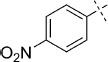 |
36 | D, 49 %; B, 80 % | 1400 | 26 000 | 2.4 | 96.9 |
| 10 | 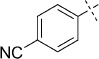 |
37 | D; B, 20 %[b] | 495 | 41 500 | 5.2 | 95.4 |
| 11 | 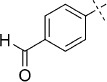 |
38 | D; B, 15 %[b] | 695 | 32 500 | 4.7 | 91.4 |
| 12 | 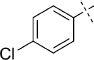 |
39 | D, 68 %; B, 73 % | 565 | >100 000 | 0.3 | 98.2 |
| 13 |  |
40 | D; B, 11 %[b] | 785 | 50 000 | 0.1 | 98.5 |
| 14 | 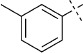 |
41 | D; A, 35 %[b] | 1600 | 39 000 | 0.7 | 98.1 |
| 15 | 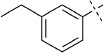 |
42 | E; A, 17 %[b] | 1450 | 32 500 | 5 | 96.4 |
| 16 | 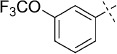 |
43 | D; A, 61 %[b] | 1800 | 39 500 | 0.2 | 96.2 |
| 17 | 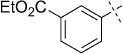 |
44 | D; A, 43 %[b] | 9900 | >100 000 | 7.1 | 96 |
| 18 |  |
45 | D; A, 32 %[b] | 22 000 | >100 000 | 1.2 | 94 |
| 19 | 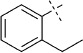 |
46 | E; A, 19 %[b] | 68 500 | >100 000 | 0.8 | 98 |
| 20 | 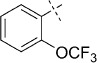 |
47 | D; A, 38 %[b] | >100 000 | >100 000 | 0.3 | 95.7 |
| 21 |  |
48 | E; A, 32 %[b] | >100 000 | >100 000 | 0.4 | 97.7 |
General method for the synthesis of 2-substituted acrylamide. Method C reagents and conditions: starting aryl iodide (1.0 equiv), acrylamide (2.0 equiv), Pd(OAc)2 (0.05 equiv), tri-tert-butylphosphonium tetrafluoroborate (0.10 equiv), Et3N (3.0 equiv), CH3CN (4.0 mL mmol−1), 120 °C MW, 15 min. Method D reagents and conditions:. starting aryl iodide (1.0 equiv), acrylamide (2.0 equiv) and Pd(OAc)2 (0.05 equiv), Et3N (3.0 equiv), CH3CN (4.0 mL mmol−1), 120 °C MW, 15 min. Method E reagents and conditions: starting aryl bromide (1.0 equiv), acrylamide (1.5 equiv) and trans-bis(acetato)bis[o-(di-o-tolylphosphino)benzyl]dipalladium(II) (0.05 equiv), NaOAc (3.0 equiv), DMF (4.0 mL mmol−1), 140 °C MW, 15 min. [a] Chemical stability in PBS pH 7.4 at 25 °C; as % remaining after 24 h. [b] Isolated yield over two steps.
Four of the compounds with the best inhibition and selectivity profiles (Table 2, 30–33) were chosen for further investigation. For these compounds, the corresponding ortho- and meta-analogues were synthesized and evaluated in the enzyme activity assay (Table 2, entries 14–21). The para-substituted analogues (Table 2, entries 3–6) were more active than ortho- and meta-substituted analogues on the Mtb proteasome, with IC50 values in the range of 475–1080 nm. Furthermore, no inhibition of the human proteasome was detected for these compounds. When substituents were introduced in the meta-position (Table 2, entries 14–17), inhibitory activity of the Mtb proteasome decreased (IC50 1450–9900 nm) and poorer selectivity was observed. The ortho-substituted analogues (entries 18–21), despite the same mesomeric contribution as the para-analogues on the styryl moiety, showed almost no inhibition of the Mtb proteasome (IC50 22 000– >100 000 nm). The same trend was observed for chloro-functionalized 39 and disubstituted 40 (Table 2, entries 12–13); the para-substituted compound was more active on the Mtb proteasome and had superior selectivity with regard to the human proteasome.
Overall, the styryl-analogues 28–48 in Table 2 displayed good stability but poor solubility (0.1–7.1 μm). To hopefully improve the solubility of the compounds, a small series of nitrogen-, oxygen- and sulfur-containing heterocyclic derivatives were prepared (Table 3). The introduction of five-membered, six-membered, and bicyclic heterocycles into the scaffold generally improved the solubility with retained chemical stability. Furthermore, the heterocyclic analogues were active on the Mtb proteasome, although slightly higher IC50 values were obtained compared to the compounds in Table 2. However, for the five-membered heterocycle-substituted oxathiazolones, the compounds were not as selective with regard to the human proteasome (Table 3, entries 3–5 and 9).
Table 3.
Optimization of 5-styryl-oxathiazolone inhibitors with regard to solubility.
| Entry | Structure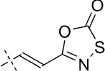
|
Cmpd | Method, Yield | Mtb proteasome IC50 [nM] | Human proteasome IC50 [nM] | Solubility [μM] | Stability [%][a] |
|---|---|---|---|---|---|---|---|
| 1 | 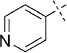 |
49 | D; B, 7 %[b] | 2750 | 77 000 | 84.7 | 91.6 |
| 2 | 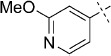 |
50 | D; A, 39 %[b] | 15 000 | 29 500 | 2.1 | 97.7 |
| 3 | 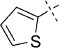 |
51 | C, 46 %; B, 45 % | 2100 | 18 500 | 22.6 | 97.6 |
| 4 | 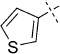 |
52 | C, 75 %; B, 53 % | 1150 | 11 300 | 19.7 | 99.1 |
| 5 | 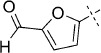 |
53 | C; B, 5 %[b] | 735 | 3300 | 56.1 | 88.9 |
| 6 | 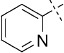 |
54 | E; A, 3 %[b] | 2350 | >10 000[c] | 64.6 | n.d. |
| 7 | 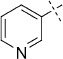 |
55 | E; A, 15 %[b] | 1350 | >10 000[c] | 70.6 | n.d. |
| 8 |  |
56 | E; B, 4 %[b] | 2100 | >10 000[c] | 2.4 | n.d. |
| 9 | 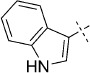 |
57 | –, 31 %;[d] B, 12 % | 1200 | 5000 | 4.1 | n.d. |
General method for the synthesis of 2-substituted acrylamides. Method C reagents and conditions: starting aryl iodide (1.0 equiv), acrylamide (2.0 equiv), Pd(OAc)2 (0.05 equiv), tri-tert-butylphosphonium tetrafluoroborate (0.10 equiv), Et3N (3.0 equiv), CH3CN (4.0 mL mmol−1), 120 °C MW, 15 min. Method D reagents and conditions:. starting aryl iodide (1.0 equiv), acrylamide (2.0 equiv) and Pd(OAc)2 (0.05 equiv), Et3N (3.0 equiv), CH3CN (4.0 mL mmol−1), 120 °C MW, 15 min. Method E reagents and conditions: starting aryl bromide (1.0 equiv), acrylamide (1.5 equiv) and trans-bis(acetato)bis[o-(di-o-tolylphosphino)benzyl]dipalladium(II) (0.05 equiv), NaOAc (3.0 equiv), DMF (4.0 mL mmol−1), 140 °C MW, 15 min. [a] Chemical stability in PBS pH 7.4 at 25 °C; as % remaining after 24 h. [b] Isolated yield over two steps. [c] The compound interferes with the assay at 10 μm, therefore it was not possible to measure human proteasome inhibition. [d] Reagents and conditions: 1) (2E)-3-(1H-Indolyl-3-yl)acrylic acid (2.0 mmol), 1,1′-carbonyldiimidazole (2.0 mmol), DMF (10 mL), rt, 30 min, 2) NH4HCO3 (4.0 mmol), rt, o/n.
Based on these promising results for both inhibition and selectivity of the 5-styryl-oxathiazol-2-ones, a collection of synthesized compounds was selected for evaluation of biological activity. Active Mtb proteasome inhibitors displaying full to moderate selectivity were tested against a virulent strain of Mtb (H37Rv). Four 5-aryl-oxathiazol-2-ones (Table 4, entries 1–4) were also tested for comparison, as well as three known antitubercular agents as a reference (entries 30-32). Cytotoxicity for mammalian cells was investigated using the Vero cell line (African green monkey kidney cells); the results are reported in Table 4.
Table 4.
Activity of compounds against actively-replicating Mtb.
| Entry | Cmpd | OD-MIC [μm] | OD-inhib. at 20 μm [%] | RFU-MIC [μm] | RFU-inhib. at 20 μm [%] | Vero cell cytotoxicity TC50 [μm] |
|---|---|---|---|---|---|---|
| 1 | 1 | >20 | 33 | >20 | 32 | 12 |
| 2 | 2 | inactive | inactive | 18 | ||
| 3 | 3 | >20 | 27 | >20 | 30 | 11 |
| 4 | 4 | >20 | 22 | >20 | 24 | 21 |
| 5 | 17 | >20 | 22 | >20 | 27 | 22 |
| 6 | 18 | >20 | 28 | >20 | n.d. | 41 |
| 7 | 19 | inactive | inactive | 19 | ||
| 8 | 22 | inactive | inactive | 21 | ||
| 9 | 28 | >20 | 33 | >20 | 30 | n.d. |
| 10 | 29 | inactive | inactive | 17 | ||
| 11 | 30 | >20 | 41 | >20 | 43 | 21 |
| 12 | 31 | >20 | 41 | >20 | 46 | 17 |
| 13 | 32 | inactive | inactive | 20 | ||
| 14 | 33 | >20 | 45 | >20 | 45 | n.d. |
| 15 | 34 | >20 | 83 | >20 | 85 | 16 |
| 16 | 35 | >20 | 33 | >20 | 30 | n.d. |
| 17 | 36 | >20 | 48 | >20 | 45 | n.d. |
| 18 | 37 | >20 | 36 | >20 | 35 | n.d. |
| 19 | 38 | >20 | 32 | >20 | 32 | n.d. |
| 20 | 39 | >20 | 36 | >20 | 36 | n.d. |
| 21 | 40 | inactive | inactive | n.d. | ||
| 22 | 41 | >20 | 35 | >20 | 32 | n.d. |
| 23 | 42 | >20 | 26 | >20 | 24 | n.d. |
| 24 | 43 | >20 | 24 | >20 | 27 | n.d. |
| 25 | 44 | >20 | 27 | >20 | 39 | n.d. |
| 26 | 45 | inactive | inactive | n.d. | ||
| 27 | 46 | inactive | inactive | n.d. | ||
| 28 | 47 | inactive | inactive | n.d. | ||
| 29 | 48 | inactive | inactive | n.d. | ||
| 30 | Isoniazid[a] | 0.3 | 0.3 | n.d. | ||
| 31 | Ethambutol[a] | 6.5 | 6.6 | n.d. | ||
| 32 | Ofloxacin[a] | 1.2 | 1.3 | n.d. |
The minimum inhibitory concentration (MIC) was determined against aerobically-grown Mtb H37Rv using a 10-point serial dilution by optical density (OD) and relative fluorescence units (RFU) measurement. The highest concentration of compound tested was 20 μm. The % growth was plotted, and a curve was fitted using the Gompertz fit. The MIC was defined as the minimum concentration at which growth was completely inhibited and was calculated from the inflection point of the fitted curve to the lower asymptote (zero growth). The % inhibition of growth at 20 μm is given. Compounds defined as inactive gave % inhibition <20 at 20 μm. Cytotoxicity was measured against the Vero cell line; the TC50 is the concentration required to inhibit growth by 50 % over 48 h. n.d.=not determined. [a] Known antitubercular agents were used as reference compounds.3
The oxathiazolones had little or no activity against replicating bacteria when compared to the known antitubercular agents used as reference (entries 30-32 in Table 4). A few compounds showed >20 % inhibition of growth at the maximum concentration tested (20 μm), but none had an MIC≤20 μm. There was no clear difference between the 5-styryl-oxathiazol-2-ones (entries 5–29) and the previously reported 5-aryl-analogues (entries 1–4) or with respect to the electronic nature and/or position of the ring substitution. This finding is consistent with the MIC values (15–299 μm) for a series of 5-aryl-oxathiazol-2-ones recently tested on the same Mtb strain.16g Compounds demonstrated some cytotoxicity against the Vero cell line, with TC50 values (concentration required to inhibit growth by 50 % over 48 h) in the range of 11–41 μm (when tested). However, these results indicate that our new inhibitors are generally 1000/4000-fold less toxic than the approved human proteasome inhibitor Bortezomib (TC50 less than 10 nm).7c The observed cytotoxicity might be related to the inhibition of the trypsin-like and caspase-like activity of the mammalian proteasome.22 Alternatively, it might be due to the non-proteasome-related activity of the styryl oxathiazolone compounds towards other cellular enzymes that catalyze bond cleavage via the same mechanism as the Mtb-proteasome (i.e. nucleophilic attack). On the other hand, a previous report from Lin et al. demonstrated generally reduced inhibition potency for the parent aryl-oxathiazolone inhibitors (e.g. compounds 1–4 included in our study) toward β1 and β2 non-chymotrypsin-like active sites of the proteasome.7c Additionally high selectivity towards other human proteases, including trypsin, cathepsin B, and matrix metalloproteases was reported for the same proteasome inhibitors.7c However, recent research by Bassett et al. has revealed cytotoxicity towards Vero cells for a number of proteasome inhibitors reported as antitubercular agents.23
Previous work suggested that the proteasome plays a key role during oxidative and nitrosative stress,12 and that proteasome inhibitors are bactericidal against nonreplicating Mtb subjected to nitrosative stress.7c We therefore determined whether the 5-styryl-oxathiazolone class of compounds had activity against Mtb under nonreplicating conditions. For this we used a starvation model, in which bacteria are nutrientdeprived for 14 days,24 to induce the nonreplicating state, after which they are exposed to compounds over 21 days in this state (Figure 2).
Figure 2.

5-Styryl-oxathiazol-2-ones are bactericidal against nonreplicating Mycobacterium tuberculosis (Mtb). Mtb was grown aerobically, washed, and resuspended in PBS-tyloxapol for 14 d. Compounds were added with the indicated concentrations. Colony forming units (CFU) were determined by plating serial dilutions. The lower limit of detection (LoD) is indicated.
All four 5-styryl-oxathiazol-2-ones tested (17, 33, 37, and 39) were bactericidal against nonreplicating Mtb, displaying a very rapid kill (>4 logs in 7–14 days). All compounds gave complete sterilization of cultures within 14 days at the lowest concentration tested (20 μm). The 5-(4-chloro)styryl-oxathiazol-2-one inhibitor (39) was most effective, reducing the colony-forming units (CFU)/mL below the detection limit in only seven days at all concentrations. These data confirm the selectivity of proteasome inhibitors for nonreplicating over replicating bacteria.
Conclusions
Our results demonstrate that 5-styryl-oxathiazol-2-ones provide a promising scaffold for Mtb proteasome inhibitors. In the current study we have further evaluated the structure–activity relationship of oxathiazolones as proteasome inhibitors. We found that conjugated 5-styryl-oxathiazolones are equally or more potent inhibitors of the Mtb proteasome compared to the previously reported 5-aryl-oxathiazolones. As part of this investigation we prepared a series of novel 5-styryl-oxathiazolones from the corresponding aryl iodides or bromides by a microwave-assisted Mizoroki–Heck coupling with acrylamide. Subsequent cyclization using chlorocarbonyl sulfenyl chloride yielded the oxathiazolones. Our enzyme activity results showed that para-substituted 5-styryl-oxathiazol-2-ones were superior to ortho- and meta-substitution with regard to Mtb proteasome IC50 values, as well as their relative lack of effects against the chymotrypsin-like peptidase activity of human proteasomes. We also produced a small series of oxathiazolones in which solubility-enhancing heterocyclic groups were introduced. However, we gained solubility at the expense of selectivity regarding the human proteasome. Finally, a selection of the synthesized inhibitors showed rapid bactericidal activity against nonreplicating Mtb, although they were not very active against actively replicating cells.
The present results suggest that this new class of Mtb proteasome inhibitors can be used as the basis for development of novel antitubercular drugs that are effective when protein synthesis of the mycobacteria is drastically reduced, such as in the nonreplicative state or during antibiotic treatment. Further development will include the improvement of the safety profile of this class of Mtb proteasome inhibitors.30
Experimental Section
General Chemistry. Analytical thin-layer chromatography (TLC) was performed on silica gel 60 F-254 plates (Merck) and visualized with UV light. Flash column chromatography was performed on columns prepacked with PHARMA-SIL® (5 g/10 g, hydrophilic high surface activity silica, UCT, Inc., Bristol, USA). 1H and 13C NMR spectra were recorded on Varian Mercury Plus instruments (Palo Alto, USA); 1H NMR spectra at 399.9 MHz and 13C NMR spectra at 100.5 MHz. The chemical shifts for 1H NMR and 13C NMR were referenced to tetramethylsilane (TMS) via residual solvent signals (1H, CDCl3 at 7.26 ppm and [D6]-dimethylsulfoxide (DMSO) at 2.50 ppm; 13C, CDCl3 at 77.0 ppm and [D6]DMSO at 39.5 ppm), while chemical shifts for 19F NMR were referenced to CFCl3 used as internal standard (0.0 ppm). The microwave reactions were performed in a Biotage Initiator (Uppsala, Sweden) producing controlled irradiation at 2450 MHz with a power of 0–300 W. The reaction temperature was determined using the built-in on-line infrared (IR) sensor. All reactions were performed in sealed microwave-transparent process vials designed for 0.5–2, 2—5, or 10–20 mL reaction volumes. Gas chromatography/electron ionization mass spectrometry (GC/EI-MS) was performed on a Varian Saturn 3900/2100 system equipped with a CP-Sil 8 CB capillary column (30 m×0.25 mm, 0.25 μm) operating at an EI potential of 70 eV. The oven temperature (GC) was 70–300 °C. Analytical high-performance liquid chromatography/electrospray ionization mass spectrometry (HPLC/ESI-MS) was performed on a Gilson HPLC system (Middleton, USA) with a Finnigan AQA ESI quadropole mass spectrometer with electrospray ionization (Thermo Fisher Scientific, Waltham, USA) and using an Onyx Monolithic C18 column 4.6×50 mm (Phenomenex, Torrance, USA) with CH3CN/H2O in 0.05 % HCOOH as mobile phase at a flow rate of 4 mL min−1 or on a Dionex UltiMate 3000 HPLC system (Sunnyvale, USA) with a Bruker amaZon SL ion trap mass spectrometer (Billerica, USA), using a Phenomenex Kinetex C18 column (50×3.0 mm, 2.6 μm particle size, 100 Å pore size) with CH3CN/H2O in 0.05 % HCOOH as mobile phase at a flow rate of 1.5 mL min−1. Detection was by UV (diode array detector) and MS (ESI+ mode). Preparative HPLC purification was performed by UV-triggered (254 nm) fraction collection with a Dionex UltiMate 3000 HPLC system, using an Agilent PrepHT Zorbax SB-C8 column (21.2×150 mm, 5 μm particle size) (Santa Clara, USA) with CH3CN/H2O in 0.05 % trifluoroacetic acid (TFA) as mobile phase. High-resolution mass spectrometry (HRMS) was performed on a Micromass Q-Tof2 mass spectrometer (Waters, Milford, USA) equipped with an electrospray ion source. HRMS data are provided for all novel compounds, unless low ionization level, intrinsic to the physicochemical properties of the specific molecules, did not permit the measurement of the corresponding molecular mass data. However, in some cases, it was possible to determine the low resolution molecular mass by the Varian Saturn 3900/2100 GC/MS system and/or by the Bruker amaZon SL ion trap mass spectrometer during the LC/MS analysis. All starting materials and reagents are commercially available and were used as received.
General method for the synthesis of 5-substituted oxathiazol-2-one. Method A: A mixture of starting amide (1.0 equiv) and chlorocarbonyl sulfenyl chloride (1.5 equiv) in 1,4-dioxane (4.0 mL mmol−1) was irradiated with microwaves at 100 °C for 15 min. The mixture was allowed to cool down to rt, and the solvent evaporated at reduced pressure. The crude material was purified by silica gel column chromatography and/or preparative HPLC to give the final compound with purity>95 % by HPLC analysis (λ=254 nm). Method B: A mixture of starting amide (1.0 equiv) and chlorocarbonyl sulfenyl chloride (2.0 equiv) in freshly distilled THF (4.0 mL mmol−1) was stirred o/n at rt. The solvent was evaporated at reduced pressure, and the crude material was purified by silica gel column chromatography and/or preparative HPLC to give the final compound with purity>95 % by HPLC analysis (λ=254 nm).
5-Phenyl-1,3,4-oxathiazol-2-one (1).7c According to Method A, benzamide (121 mg, 1.0 mmol) was used to give, after preparative HPLC purification, 77 mg (43 % isolated yield) of the title compound as a white powder. TLC: Rf=0.53 (isohexane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=7.99–7.96 (m, 2 H), 7.60–7.55 (m, 1 H), 7.52–7.47 (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=173.8, 157.4, 132.6, 129.0, 127.4, 125.8; HRMS m/z for C8H5NO2S as [M+H]+ adduct not found due to low ionization level.
5-(Pyrazin-2-yl)-1,3,4-oxathiazol-2-one (2).17 According to Method B, pyrazinamide (123 mg, 1.0 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after preparative HPLC purification, 42 mg (23 % isolated yield) of the title compound as white crystals. TLC: Rf=0.29 (pentane/EtOAc 1:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=9.29 (s, 1 H), 8.79 (s, 1 H), 8.75 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=172.2, 154.6, 147.2, 144.7, 144.3, 140.2 ppm; ESI-MS m/z: 182 [M+H]+; HRMS m/z calcd for C6H3N3O2S [M+H]+ 182.0024, found 182.0031.
5-(3-Methoxyphenyl)-1,3,4-oxathiazol-2-one (3).7c According to Method B, 3-methoxybenzamide (151 mg, 1.0 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 80 mg (38 % isolated yield) of the title compound as a white powder. LC purity (254 nm): 97 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.53–7.49 (m, 2 H), 7.41–7.39 (m, 1 H), 7.27–7.20 (m, 1 H), 3.84 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.7, 159.5, 156.6, 130.6, 126.8, 119.5, 118.9, 111.7, 55.5 ppm; ESI-MS m/z: 210 [M+H]+; HRMS m/z for C9H7NO3S as [M+H]+ adduct not found due to low ionization level.
5-(3-Fluorophenyl)-1,3,4-oxathiazol-2-one (4).7c According to Method B, 3-fluorobenzamide (139 mg, 1.0 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column purification (pentane/EtOAc 100:0 → 100:1), 163 mg (83 % isolated yield) of the title compound as a white solid. TLC: Rf=0.79 (pentane/ EtOAc 100:3); LC purity (254 nm): 97 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.78 (dt, J=7.6, 1.2 Hz, 1 H), 7.70 (ddd, J=9.2, 2.8, 1.6 Hz, 1 H), 7.65 (td, J=8.0, 5.9 Hz, 1 H), 7.53 ppm (tdd, J=8.8, 2.8, 0.8 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.5, 162.1 (d, J=244.0 Hz), 155.6 (d, J=3.1 Hz), 131.7 (d, J=8.4 Hz), 127.6 (d, J=9.2 Hz), 123.4 (d, J=3.0 Hz), 119.7 (d, J=21.4 Hz), 113.8 ppm (d, J=23.7 Hz); 19F NMR (376 MHz, [D6]DMSO): δ=−111.4 ppm (td, J=9.3, 5.3 Hz); EI-MS m/z: 197 [M]+; HRMS m/z for C8H4FNO2S as [M+H]+ adduct not found due to low ionization level.
5-(2-Chloroethyl)-1,3,4-oxathiazol-2-one (5). According to Method A, 3-chloropropionamide (81 mg, 0.75 mmol) was used to give, after silica column purification (pentane/EtOAc 100:2), 75 mg (60 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.31 (isohexane/EtOAc 10:1); LC purity (254 nm):99 %; 1H NMR (400 MHz, CDCl3): δ=3.84 (t, J=6.4 Hz, 2 H), 3.11 ppm (t, J=6.4 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=173.5, 157.9, 38.4, 33.7 ppm; EI-MS m/z: 166/168 [M+1]+; ESI-MS m/z: 166/168 [M+H]+; HRMS m/z for C4H4ClNO2S as [M+H]+ adduct not found due to low ionization level.
5-Cyclopropyl-1,3,4-oxathiazol-2-one (6). According to Method A, cyclopropanecarboxamide (85 mg, 1.0 mmol) was reacted to give, after silica column purification (pentane/EtOAc 100:1), 38 mg (27 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.51 (isohexane/EtOAc 10:1); LC purity (254 nm) 99 %; 1H NMR (400 MHz, CDCl3): δ=1.98–1.90 (m, 1 H), 1.18–1.12 (m, 2 H), 1.11–1.04 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=174.2, 162.7, 11.2, 8.4 ppm; EI-MS m/z: 143 [M]+; HRMS m/z for C5H5NO2S as [M+H]+ adduct not found due to low ionization level.
5-Pentyl-1,3,4-oxathiazol-2-one (7). According to Method A, hexanamide (86 mg, 0.75 mmol) was used to give, after silica column purification (pentane/EtOAc 100:1), 73 mg (56 % isolated yield) of the title compound as colorless oil. TLC: Rf=0.65 (isohexane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=2.61 (t, J=7.0 Hz, 2 H), 1.73 (quin, J=7.0 Hz, 2 H), 1.40–1.30 (m, 4 H), 0.91 ppm (t, J=7.0 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ=174.4, 162.0, 30.9, 30.4, 25.0, 22.1, 13.8 ppm; EI-MS m/z: 174 [M+1]+; ESI-MS m/z: 174 [M+H]+; HRMS m/z for C7H11NO2S as [M+H]+ adduct not found due to low ionization level.
5-(Fluoromethyl)-1,3,4-oxathiazol-2-one (8). According to Method B, fluoroacetamide (154 mg, 2.0 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column purification (pentane/EtOAc 100:0 → 100:2), 64 mg (24 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.55 (pentane/EtOAc 10:1); LC purity (254 nm)=96 %; 1H NMR (400 MHz, [D6]DMSO): δ=5.39 ppm (d, J=45.6 Hz, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.4, 155.7 (d, J=21.5 Hz), 76.9 ppm (d, J=171.0 Hz); 19F NMR (376 MHz, [D6]DMSO): δ=0.96 ppm (t, J=45.5 Hz); EI-MS m/z: 135 [M]+, 196; HRMS m/z for C3H2FNO2S as [M+H]+ adduct not found due to low ionization level.
5-Benzyl-1,3,4-oxathiazol-2-one (9).25 According to Method A, 2-phenylacetamide (135 mg, 1.0 mmol) was reacted to yield, after preparative HPLC purification, the title compound as a white powder (83 mg, 43 %). TLC: Rf=0.39 (isohexane/EtOAc 10:1); LC purity (254 nm): 97 %; 1H NMR (400 MHz, CDCl3): δ=7.40–7.28 (m, 5 H), 3.93 ppm (s, 2 H); 13C NMR (100 MHz, CDCl3): δ=174.0, 160.0, 132.3, 129,1, 129.0, 127.9, 37.0 ppm; HRMS m/z for C9H7NO2S as [M+H]+ adduct not found due to low ionization level.
5-(3-Methoxybenzyl)-1,3,4-oxathiazol-2-one (10). According to Method A, 4-methoxyphenylacetamide (83 mg, 0.50 mmol) was used to yield, after preparative HPLC purification, 30 mg (27 % isolated yield) of the title compound as a white powder. TLC: Rf=0.35 (isohexane/EtOAc 10:1); LC purity (254 nm): 97 %; 1H NMR (400 MHz, CDCl3): δ=7.28 (t, J=7.8 Hz, 1 H), 6.90–6.82 (m, 3 H), 3.90 (s, 2 H), 3.81 ppm (s, 3 H); 13C NMR (100 MHz, CDCl3): δ=174.0, 160.0, 133.7, 130.0, 121.3, 114.9, 113.2, 55.3, 37.0 ppm; EI-MS m/z: 184, 147; ESI-MS m/z: 224 [M+H]+; HRMS m/z calcd for C10H9NO3S [M+H]+ 224.0381, found 224.0380.
4-((2-Oxo-1,3,4-oxathiazol-5-yl)methoxy)benzaldehyde (11). According to Method A, 2-(4-Formylphenoxy)acetamide (90 mg, 0.50 mmol) was used to yield, after preparative HPLC purification, 47 mg (39 % isolated yield) of the title compound as white powder. TLC: Rf=0.10 (isohexane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=9.92 (s, 1 H), 7.89 (d, J=8.8 Hz, 2 H), 7.09 (d, J=8.8 Hz, 2 H), 5.05 ppm (s, 2 H); 13C NMR (100 MHz, CDCl3): δ=190.5, 172.6, 161.9, 155.8, 132.1, 131.3, 115.0, 63.2 ppm; EI-MS m/z: 238 [M+1]+; ESI-MS m/z: 238 [M+H]+; HRMS m/z calcd for C10H7NO4S [M+H]+ 238.0174, found 238.0175.
5-(Naphthalen-1-ylmethyl)-1,3,4-oxathiazol-2-one (12). According to Method A, 1-naphthylacetamide (93 mg, 0.50 mmol) was used to yield, after preparative HPLC purification, 62 mg (51 % isolated yield) of the title compound as a white powder. TLC: Rf=0.48 (isohexane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=8.03 (d, J=7.8 Hz, 1 H), 7.92–7.83 (m, 2 H), 7.61–7.51 (m, 2 H), 7.50–7.45 (m, 2 H), 4.38 ppm (s, 2 H); 13C NMR (100 MHz, CDCl3): δ=173.9, 159.9, 133.9, 131.6, 129.0, 128.5, 128.3, 126.9, 126.1, 125.5, 123.2, 34.6 ppm; EI-MS m/z: 228, 167; ESI-MS m/z: 244 [M+H]+; HRMS m/z calcd for C13H9NO2S [M+H]+ 244.0432, found 244.0427.
5-(2-(Trifluoromethyl)benzyl)-1,3,4-oxathiazol-2-one (13). According to Method B, 2-(trifluoromethyl)phenylacetamide (152 mg, 0.75 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after two silica column purification (1st: pentane/EtOAc 100:0 → 10:1, 2nd: pentane/EtOAc 100:0 → 100:2), 110 mg (56 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.53 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=7.72 (d, J=7.8 Hz, 1 H), 7.58 (t, J=7.8 Hz, 1 H), 7.49–7.41 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=173.7, 158.9, 132.3, 132.0, 130.5 (q, J=1.5 Hz), 129.1 (q, J=30.7 Hz), 128.3, 126.6 (q, J=5.4 Hz), 124.0 (q, J=273.7 Hz), 33.8 ppm (q, J=2.3 Hz); 19F NMR (376 MHz, CDCl3) δ: −59.8 ppm; ESI-MS m/z: 262 [M+H]+; HRMS m/z calcd for C10H6F3NO2S [M+H]+ 262.0150, found 262.0154.
5-(Hydroxy(phenyl)methyl)-1,3,4-oxathiazol-2-one (14). According to Method B, 2-hydroxy-2-phenylacetamide (151 mg, 1.0 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column (pentane/EtOAc 100:0 → 1:1) and preparative HPLC purification, 84 mg (40 % isolated yield) of the title compound as a pale yellow oil. TLC: Rf=0.17 (pentane/EtOAc 5:1); LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.48–7.32 (m, 5 H), 6.73 (d, J=5.5 Hz, 1 H), 5.65 ppm (d, J=5.5 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=174.0, 161.9, 138.3, 128.5, 128.4, 126.7, 69.9 ppm; ESI-MS m/z: 232 [M+Na]+; HRMS m/z for C9H7NO3S as [M+H]+ adduct not found due to low ionization level.
5-Phenethyl-1,3,4-oxathiazol-2-one (15). 3-Phenylpropionic acid (150 mg, 1.0 mmol) and 1,1′-carbonyldiimidazole (357 mg, 2.2 mmol) were dissolved in DMF (5 mL), and the mixture was stirred at rt. After 30 min, NH4HCO3 (316 mg, 4.0 mmol) was added, and the resulting suspension was stirred at rt for 3 d. EtOAc (50 mL) was added, and the organic phase was washed with H2O (2×20 mL) and brine (20 mL). Evaporation of solvent gave 171 mg of the crude corresponding amide, purified by precipitation from EtOAc/pentane to afford 92 mg (62 % isolated yield) of 3-phenylpropionamide as a white solid. TLC: Rf=0.28 (pentane/EtOAc 1:1); GC/MS purity: 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.35–6.98 (m, 6 H), 6.76 (bs, 1 H), 2.81 (t, J=8.0 Hz, 2 H), 2.36 ppm (t, J=8.0 Hz, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.4, 141.5, 128.2, 128.1, 125.8, 36.7, 30.9 ppm; EI-MS m/z: 149 [M]+. According to Method B, 3-phenylpropionamide (150 mg, 1.0 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column purification (pentane/EtOAc 100:0 → 100:1), 147 mg (71 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.79 (pentane/EtOAc 10:1); LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.35–7.20 (m, 5 H), 2.98 ppm (s, 4 H); 13C NMR (100 MHz, [D6]DMSO): δ=174.3, 161.2, 139.6, 128.4, 128.3, 126.4, 31.2, 30.4 ppm; ESI-MS m/z: 208 [M+H]+; HRMS m/z for C10H9NO2S as [M+H]+ adduct not found due to low ionization level.
(R)-5-(Methoxy(phenyl)methyl)-1,3,4-oxathiazol-2-one (16). (R)-(−)-2-Methoxyphenylacetic acid (332 mg, 2.0 mmol) and 1,1′-carbonyldiimidazole (649 mg, 4.0 mmol) were dissolved in DMF (10 mL), and the mixture was stirred at rt. After 30 min, NH4HCO3 (632 mg, 8.0 mmol) was added and the resulting suspension was stirred at rt for 3 d. EtOAc (50 mL) was added, and the organic phase was washed with H2O (2×30 mL) and brine (30 mL). Evaporation of solvent gave 180 mg (55 % yield) of the crude (R)-2-methoxy-2-phenylacetamide as a white powder, used for the next step without further purification. GC/MS purity: 99 %; EI-MS m/z: 166 [M+1]+. According to Method B, (R)-2-methoxy-2-phenylacetamide (180 mg, 1.1 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column purification (pentane/EtOAc 100:0 → 100:1), 140 mg (58 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.64 (pentane/EtOAc 10:1); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.48–7.38 (m, 5 H), 5.44 (s, 1 H), 3.43 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.7, 159.4, 135.4, 129.0, 128.7, 127.2, 78.8, 57.3 ppm; ESI-MS m/z: 164; HRMS m/z calcd for C10H9NO3S [M+H]+ 224.0381, found 224.0379.
(E)-5-Styryl-1,3,4-oxathiazol-2-one (17).26 According to Method B, trans-cinnamamide (147 mg, 1.0 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column purification (pentane/EtOAc 100:0 → 10:1) and trituration in pentane/EtOAc/MeOH 10:1:1 drop, 82 mg (40 % isolated yield) of the title compound as a pale yellow powder. TLC: Rf=0.76 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=7.56–7.49 (m, 2 H), 7.50 (d, J=16.4 Hz, 1 H), 7.44–7.39 (m, 3 H), 6.64 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=173.4, 157.6, 142.0, 134.1, 130.6, 129.1, 127.8, 112.8 ppm; EI-MS m/z: 191, 161, 129; ESI-MS m/z: 206 [M+H]+; HRMS m/z calcd for C10H7NO2S [M+H]+ 262.0276, found 262.0273.
(E)-5-(4-(Trifluoromethyl)styryl)-1,3,4-oxathiazol-2-one (18). According to Method B, 4-(trifluoromethyl)cinnamamide (183 mg, 0.85 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 97 mg (42 % isolated yield) of the title compound as a white powder. TLC: Rf=0.57 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.01 (d, J=8.2 Hz, 2 H), 7.79 (d, J=8.2 Hz, 2 H), 7.63 (d, J=16.4 Hz, 1 H), 7.17 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.3, 157.0, 139.5, 138.2 ppm (q, J=1.5 Hz), 129.9 (q, J=32.2 Hz), 128.8, 125.7 (q, J=3.8 Hz), 124.0 (q, J=272.2 Hz), 116.0; 19F NMR (376 MHz, [D6]DMSO): δ=−61.3 ppm; ESI-MS m/z: 274 [M+H]+; HRMS m/z for C11H6F3NO2S as [M+H]+ adduct not found due to low ionization level.
5-Benzoyl-1,3,4-oxathiazol-2-one (19).25 In a round-bottom flask, Dess–Martin periodinane (467 mg, 1.1 mmol) was added to a precooled solution (T=0 °C) of 5-(hydroxy(phenyl)methyl)-1,3,4-oxathiazol-2-one (14) (209 mg, 1.0 mmol) in anhydrous CH2Cl2 (5 mL). The resulting reaction mixture was stirred 2 h at 0 °C and then filtered to remove the formed solid. The mother liquor was washed with H2O (10 mL) and dried on anhydrous Na2SO4. Evaporation of the solvent under reduced pressure gave 347 mg of the crude material that was triturated with EtOAc for 30 min at rt. Filtration of the formed solid and evaporation of the solvent gave 117 mg of a colorless oil that was purified by silica column purification (pentane/EtOAc 100:0 → 10:1) to give 118 mg (57 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.48 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.18–8.13 (m, 2 H), 7.78–7.72 (m, 1 H), 7.63–7.57 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=177.4, 172.3, 153.3, 134.5, 133.5, 130.6, 128.6 ppm; EI-MS m/z: 208 [M+1]+; ESI-MS m/z: 208 [M+H]+; HRMS m/z calcd for C9H5NO3S [M+H]+ 208.0068, found 208.0070.
5-(Phenylethynyl)-1,3,4-oxathiazol-2-one (20). In a round-bottom flask, SOCl2 (714 mg, 6.0 mmol) was added to a solution of phenylpropiolic acid (730 mg, 5.0 mmol) in CHCl3 (10 mL) at rt After addition of DMF (1 drop), the reaction mixture was stirred 1 h at rt To the resulting solution was then added 25 % aq NH4OH (2.5 mL) at −10 °C. The reaction mixture was allowed to return to rt and stirred 30 min at this temperature. CHCl3 (10 mL) and H2O (10 mL) where added, the aqueous layer was extracted with CHCl3 (2×20 mL) and the combined organic phases were dried over Na2SO4. Evaporation of the solvent under reduced pressure gave 377 mg (52 % isolated yield) of the crude 3-phenylpropiolamide as a white solid, which was used for the next step without further purification. TLC: Rf=0.72 (EtOAc); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.14 (bs, 1 H), 7.66 (bs, 1 H), 7.61–7.52 (m, 2 H), 7.51–7.42 ppm (m, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=153.9, 132.0, 130.2, 128.9, 119.9, 84.2, 82.9 ppm; ESI-MS m/z: 146 [M+H]+, 291 [2 M+H]+. According to Method B, 3-phenylpropiolamide (150 mg, 1.03 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by silica column (pentane/EtOAc 100:1 → 10:1) followed by preparative HPLC purification, 51 mg (24 % isolated yield) of the title compound as a white powder. TLC: Rf=0.80 (pentane/EtOAc 9 :1); LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.75–7.71 (m, 2 H), 7.64–7.58 (m, 1 H), 7.56–7.50 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=172.3, 141.5, 132.6, 131.6, 129.2, 118.1, 92.3, 75.7 ppm; ESI-MS m/z: 204 [M+H]+; HRMS m/z for C10H5NO2S as [M+H]+ adduct not found due to low ionization level.
5-Vinyl-1,3,4-oxathiazol-2-one (21).27 According to Method B, acrylamide (107 mg, 1.5 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after two silica column purification (1 st: pentane/EtOAc 100:0 → 10:1, 2nd: pentane/EtOAc 100:0 → 100:2), 15 mg (8 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.73 (pentane/EtOAc 10:1); LC purity (254 nm): 98 %; 1H NMR (400 MHz, CDCl3): δ=6.32 (dd, J=17.6, 10.9 Hz, 1 H), 6.23 (d, J=17.6 Hz, 1 H), 5.92 ppm (d, J=10.5 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=173.1, 157.0, 127.9, 122.9 ppm; EI-MS m/z: 129 [M]+; HRMS m/z for C4H3NO2S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(3,3,3-Trifluoroprop-1-en-1-yl)-1,3,4-oxathiazol-2-one (22). According to Method B, 4,4,4-trifluorocrotonamide (306 mg, 2.2 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column purification (pentane/EtOAc 100:0 → 100:2), 350 mg (81 % isolated yield) of the title compound as a colorless oil. TLC: Rf=0.69 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.12–7.01 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=172.7, 154.2, 127.1 (q, J=34.3 Hz), 124.0 (q, J=6.9 Hz), 122.5 ppm (q, J=268.4 Hz); 19F NMR (376 MHz, [D6]DMSO): δ=−63.5 ppm (d, J=5.6 Hz); HRMS m/z for C5H2F3NO2S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(1,2-Diphenylvinyl)-1,3,4-oxathiazol-2-one (23). 2-Phenyl-trans-cinnamic acid (224 mg, 1.0 mmol) and 1,1′-carbonyldiimidazole (357 mg, 2.2 mmol) were dissolved in DMF (5 mL) and the mixture was stirred at rt. After 30 min, NH4HCO3 (316 mg, 4.0 mmol) was added and the resulting suspension was stirred at rt for 3 d. EtOAc (50 mL) was added, and the organic phase was washed with H2O (2×20 mL) and brine (20 mL). Evaporation of solvent gave 212 mg of the crude corresponding amide, purified by silica column chromatography (pentane/EtOAc 1:1 → 3:1) to afford 150 mg (67 % isolated yield) of 2-phenyl-trans-cinnamamide as white crystals. TLC: Rf=0.63 (pentane/EtOAc=1:1); GC/MS purity: 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.46–7.35 (m, 4 H), 7.30 (bs, 1 H), 7.24–7.15 (m, 5 H), 7.03–6.97 (m, 2 H), 6.90 ppm (bs, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=170.0, 136.7, 136.6, 135.0, 133.9, 129.7, 129.3, 128.8, 128.2, 128.1, 127.8 ppm; EI-MS m/z: 223 [M]+. According to Method B, 2-phenyl-trans-cinnamamide (256 mg, 1.1 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after silica column purification (pentane/EtOAc 100:0 → 100:1), 290 mg (90 % isolated yield) of the title compound as a white solid. TLC: Rf=0.77 (pentane/EtOAc 10:1); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.65 (s, 1 H), 7.50–7.43 (m, 3 H), 7.35–7.29 (m, 2 H), 7.28–7.20 (m, 3 H), 7.13–7.08 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.6, 158.5, 137.6, 134.1, 134.0, 130.3, 129.9, 129.4, 128.9, 128.6, 128.4, 127.8 ppm; ESI-MS m/z: 282 [M+H]+; HRMS m/z calcd for C16H11NO2S [M+H]+ 282.0589, found 282.0587.
3-Phenyl-1,4,2-dioxazol-5-one (24). Benzhydroxamic acid (137 mg, 1.0 mmol) was reacted with 1,1′-carbonyldiimidazole to give, after silica column purification (pentane/EtOAc 100:0 → 100:5), 86 mg of the crude dioxazolone which was purified by precipitation from EtOAc/pentane to give 80 mg (49 % isolated yield) of the title compound as a white solid. TLC: Rf=0.87 (pentane/EtOAc=9:1); LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.87–7.83 (m, 2 H), 7.76–7.71 (m, 1 H), 7.67–7.61 ppm (m, 2 H); 13C NMR (100 MHz, [D6]DMSO): δ=163.0, 153.9, 133.7, 129.6, 126.4, 120.3 ppm; HRMS m/z for C8H5NO3 as [M+H]+ adduct not found due to low ionization level.
(E)-3-Styryl-1,4,2-dioxazol-5-one (25). trans-Cinnamic acid (296 mg, 2.0 mmol) and 1,1′-carbonyldiimidazole (486 mg, 3.0 mmol) were dissolved in THF (8 mL), and the mixture was stirred at rt. After 1 h, hydroxylamine hydrochloride (278 mg, 4.0 mmol) was added, and the resulting suspension was stirred at rt for 2 d. 5 % aq KHSO4 (30 mL) was added, and the mixture was extracted with EtOAc (2×30 mL). The collected organic phases were washed with brine (30 mL) and dried over Na2SO4. Evaporation of solvent gave 280 mg (86 % isolated yield) of the corresponding N-hydroxycinnamamide as a pale pink solid, used in the next step without further purification. LC purity (254 nm)=95 %; ESI-MS: m/z 164 [M+H]+. The obtained N-hydroxycinnamamide (140 mg, 0.86 mmol) was reacted with 1,1′-carbonyldiimidazole to give, after silica column purification (CH3CN) and evaporation of the solvent by freeze-drying, 123 mg of the crude dioxazolone which was purified by preparative HPLC to give 83 mg (51 % isolated yield) of the title compound as a white solid. TLC: Rf=0.64 (pentane/EtOAc 10:1); LC purity (254 nm): 97 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.84–7.78 (m, 2 H), 7.56 (d, J=16.4 Hz, 1 H), 7.49–7.43 (m, 3 H), 7.15 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=163.2, 153.6, 142.2, 133.8, 130.8, 129.0, 128.4, 106.9 ppm; ESI-MS m/z: 190 [M+H]+; HRMS m/z for C10H7NO3 as [M+H]+ adduct not found due to low ionization level.
General method for the synthesis of 2-substituted acrylamide. Method C (Mizoroki–Heck reaction, electron-rich iodide) A suitable microwave vial was loaded with solid reactants and reagents: starting iodide (1.0 equiv), acrylamide (2.0 equiv), palladium(II) acetate (0.05 equiv), and tri-tert-butylphosphonium tetrafluoroborate (0.10 equiv). CH3CN (4.0 mL mmol−1) was added, followed by liquid reactants: starting iodide (1.0 equiv, in case). Oxygen was removed by bubbling gaseous nitrogen into the resulting mixture for 15 min, then triethylamine (3.0 equiv) was added, the vial was capped, and the reaction mixture was irradiated with microwaves at 120 °C for 15 min. The mixture was allowed to cool down to rt, and the collected crude material was purified by silica gel column chromatography to give the desired 3-substituted acrylamide. Method D (Mizoroki–Heck reaction, electron-poor iodide) A suitable microwave vial was loaded with solid reactants and reagents: starting iodide (1.0 equiv), acrylamide (2.0 equiv), and palladium(II) acetate (0.05 equiv). CH3CN (4.0 mL mmol−1) was added, followed by liquid reactants: starting iodide (1.0 equiv, in case). Oxygen was removed by bubbling gaseous nitrogen into the resulting mixture for 15 min, then triethylamine (3.0 equiv) was added, the vial was capped, and the reaction mixture was irradiated with microwaves at 120 °C for 15 min. The mixture was allowed to cool down to rt, and the collected crude material was purified by silica gel column chromatography to give the desired 3-substituted acrylamide. Method E (Mizoroki–Heck reaction, electron-poor bromide) A suitable microwave vial was loaded with solid reactants and reagents: starting bromide (1.0 equiv), acrylamide (1.5 equiv), trans-bis(acetato)bis[o-(di-o-tolylphosphino)benzyl]dipalladium(II) (0.05 equiv), and NaOAc (3.0 equiv). Dry DMF (4.0 mL mmol−1) was added, followed by liquid reactants: starting bromide (1.0 equiv, in case). The vial was capped, and the reaction mixture was irradiated with microwaves at 140 °C for 15 min. The mixture was allowed to cool down to rt, and the collected crude material was purified by silica gel column chromatography to give the desired 3-substituted acrylamide.
(E)-5-(4-Methoxystyryl)-1,3,4-oxathiazol-2-one (28). According to Method C, 4-iodoanisole (234 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum to give 139 mg of the crude corresponding acrylate, that was purified by silica column chromatography (EtOAc/MeOH 100:5 → 100:10) to afford 133 mg (75 % isolated yield) of (E)-3-(4-methoxyphenyl)acrylamide as a white powder. TLC: Rf=0.34 (EtOAc); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.52 (dm, J=8.6 Hz, 2 H), 7.45 (bs, 1 H), 7.38 (d, J=16.0 Hz, 1 H), 7.03–6.97 (m, 3 H), 6.47 (d, J=15.6 Hz, 1 H), 3.80 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.9, 160.3, 138.8, 129.1, 127.4, 119.8, 114.4, 55.2 ppm; ESI-MS m/z: 178 [M+H]+. According to Method B, (E)-3-(4-methoxyphenyl)acrylamide (256 mg, 1.4 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 240 mg (71 % isolated yield) of the title compound as a pale yellow powder. LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.73 (dm, J=8.6 Hz, 2 H), 7.49 (d, J=16.0 Hz, 1 H), 6.99 (dm, J=8.6 Hz, 2 H), 6.83 (d, J=16.4 Hz, 1 H), 3.81 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.6, 161.1, 157.7, 141.2, 130.0, 126.8, 114.4, 110.6, 55.4 ppm; ESI-MS m/z: 236 [M+H]+; HRMS m/z calcd for C11H9NO3S [M+H]+ 236.0381, found 236.0383.
(E)-5-(4-Isopropylstyryl)-1,3,4-oxathiazol-2-one (29). According to Method C, 1-iodo-4-isopropylbenzene (246 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum to give 150 mg of the crude corresponding acrylate, which was purified by silica column chromatography (EtOAc/MeOH 100:0 → 100:10) to afford 118 mg (62 % isolated yield) of (E)-3-(4-isopropylphenyl)acrylamide as an off-white powder. TLC: Rf=0.42 (EtOAc); LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.49 (bs, 1 H), 7.47 (d, J=8.0 Hz, 2 H), 7.37 (d, J=16.0 Hz, 1 H), 7.28 (d, J=8.0 Hz, 2 H), 7.05 (bs, 1 H), 6.55 (d, J=16.0 Hz, 1 H), 2.90 (sep, J=7.0 Hz, 1 H), 1.20 ppm (d, J=7.0 Hz, 6 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.8, 150.0, 139.1, 132.5, 127.6, 126.9, 121.3, 33.3, 23.7 ppm; ESI-MS m/z: 190 [M+H]+, 231 [M+CH3CN+H]+, 379 [2 M+H] +. According to Method B, (E)-3-(4-isopropylphenyl)acrylamide (118 mg, 0.62 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by silica column chromatography (pentane/EtOAc 100:0 → 100:2) and preparative HPLC purification, 102 mg (66 % isolated yield) of the title compound as a white solid. TLC: Rf=0.60 (pentane/EtOAc 10 :1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.69 (d, J=8.1 Hz, 2 H), 7.50 (d, J=16.3 Hz, 1 H), 7.31 (d, J=8.1 Hz, 2 H), 6.93 (d, J=16.3 Hz, 1 H), 2.92 (sep, J=6.8 Hz, 1 H), 1.21 ppm (d, J=6.8 Hz, 6 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.5, 157.5, 151.2, 141.4, 131.9, 128.3, 126.9, 112.2, 33.4, 23.6 ppm; ESI-MS m/z: 248 [M+H]+, 289 [M+CH3CN+H]+; HRMS m/z for C13H13NO2S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(4-Methylstyryl)-1,3,4-oxathiazol-2-one (30). According to Method C, 1-iodo-4-methylbenzene (218 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum to give 126 mg of the crude corresponding acrylate, which was purified by silica column chromatography (EtOAc/MeOH 100:0 → 100:10) to afford 95 mg (59 % isolated yield) of (E)-3-(p-tolyl)acrylamide as a white powder. TLC: Rf=0.36 (EtOAc); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.48 (bs, 1 H), 7.44 (d, J=8.0 Hz, 2 H), 7.37 (d, J=16.0 Hz, 1 H), 7.22 (d, J=8.0 Hz, 2 H), 7.04 (bs, 1 H), 6.55 (d, J=16.0 Hz, 1 H), 2.32 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.8, 150.8, 139.1, 132.1, 129.5, 127.5, 121.3, 20.9 ppm; ESI-MS m/z: 162 [M+H]+, 203 [M+CH3CN+H] +, 323 [2 M+H]+. According to Method B, (E)-3-(p-tolyl)acrylamide (95 mg, 0.59 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 41 mg (32 % isolated yield) of the title compound as white crystals. LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.66 (d, J=8.1 Hz, 2 H), 7.49 (d, J=16.4 Hz, 1 H), 7.25 (d, J=8.1 Hz, 2 H), 6.92 (d, J=16.4 Hz, 1 H), 2.34 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.5, 157.5, 141.4, 140.5, 131.4, 129.6, 128.2, 112.2, 21.0 ppm; ESI-MS m/z: 220 [M+H]+, 261 [M+CH3CN+H]+; HRMS m/z for C11H9NO2S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(4-Ethylstyryl)-1,3,4-oxathiazol-2-one (31). According to Method C, 1-ethyl-4-iodobenzene (232 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum to give 170 mg of the crude corresponding acrylate, which was purified by silica column chromatography (EtOAc/MeOH 100:0 → 100:10) to afford 88 mg (50 % isolated yield) of (E)-3-(4-ethylphenyl)acrylamide as a white powder. TLC: Rf=0.40 (EtOAc); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.49 (bs, 1 H), 7.47 (d, J=8.0 Hz, 2 H), 7.38 (d, J=16.0 Hz, 1 H), 7.25 (d, J=8.0 Hz, 2 H), 7.04 (bs, 1 H), 6.55 (d, J=16.0 Hz, 1 H), 2.62 (q, J=7.6 Hz, 2 H), 1.18 ppm (t, J=7.6 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.8, 145.4, 139.1, 132.4, 128.3, 127.6, 121.3, 28.1, 15.4 ppm; ESI-MS m/z: 176 [M+H]+, 217 [M+CH3CN+H] +, 351 [2 M+H]+. According to Method B, (E)-3-(4-ethylphenyl)acrylamide (88 mg, 0.50 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 50 mg (43 % isolated yield) of the title compound as yellow crystals. LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.68 (d, J=8.1 Hz, 2 H), 7.50 (d, J=16.4 Hz, 1 H), 7.28 (d, J=8.1 Hz, 2 H), 6.93 (d, J=16.4 Hz, 1 H), 2.63 (q, J=7.7 Hz, 2 H), 1.18 ppm (t, J=7.7 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.5, 157.5, 146.7, 141.4, 131.7, 128.4, 128.3, 112.2, 28.1, 15.3 ppm; ESI-MS m/z: 234 [M+H]+, 275 [M+CH3CN+H] +; HRMS m/z for C12H11NO2S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(4-(Trifluoromethoxy)styryl)-1,3,4-oxathiazol-2-one (32). According to Method D, 1-iodo-4-trifluoromethoxy-benzene (288 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (double purification: 1st EtOAc; 2nd pentane/EtOAc 1:1 → 0:1) to afford 221 mg of a 1:1 mixture of (E)-3-(4-(trifluoromethoxy)phenyl)acrylamide and starting acrylamide as a white powder. TLC: Mizoroki–Heck product Rf=0.48, acrylamide Rf=0.31 (EtOAc); ESI-MS m/z: 232 [M+H]+. According to Method B, the obtained Mizoroki–Heck reaction mixture (221 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.25 mL) to give, after purification by silica column chromatography (pentane/EtOAc 100:0 → 100:2), 185 mg (64 % isolated yield after two steps) of the title compound as a white powder. TLC: Rf=0.55 (pentane/EtOAc 10:1); LC purity (254 nm): 94 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.93 (dm, J=9.0 Hz, 2 H), 7.58 (d, J=16.4 Hz, 1 H), 7.43 (dm, J=8.0 Hz, 2 H), 7.05 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.4, 157.2, 149.4, 139.7, 133.5, 130.2, 121.3, 120.0 (q, J=256.9 Hz), 114.4 ppm; ESI-MS m/z: 290 [M+H]+; HRMS m/z for C11H6F3NO3S as [M+H]+ adduct not found due to low ionization level.
(E)-Ethyl 4-(2-(2-oxo-1,3,4-oxathiazol-5-yl)vinyl)benzoate (33). According to Method D, ethyl 4-iodobenzoate (276 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum. The crude corresponding acrylate was purified by silica column chromatography (EtOAc/MeOH 1:0 → 10:1) to afford 147 mg (67 % isolated yield) of (E)-ethyl 4-(3-amino-3-oxoprop-1-en-1-yl)benzoate as an off-white powder. TLC: Rf=0.53 (EtOAc); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.98 (dm, J=8.3 Hz, 2 H), 7.69 (dm, J=8.3 Hz, 2 H), 7.61 (bs, 1 H), 7.46 (d, J=16.0 Hz, 1 H), 7.20 (bs, 1 H), 6.73 (d, J=16.0 Hz, 1 H), 4.31 (q, J=6.9 Hz, 2 H), 1.33 ppm (t, J=6.9 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.2, 165.3, 139.4, 137.8, 130.2, 129.7, 127.7, 124.9, 60.8, 14.1 ppm; ESI-MS m/z: 220 [M+H]+, 439 [2 M+H]+. According to Method B, (E)-ethyl 4-(3-amino-3-oxoprop-1-en-1-yl)benzoate (147 mg, 0.67 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 129 mg (69 % isolated yield) of the title compound as a white powder. TLC: Rf=0.39 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.98 (bd, J=8.6 Hz, 2 H), 7.92 (bd, J=8.6 Hz, 2 H), 7.59 (d, J=16.4 Hz, 1 H), 7.13 (d, J=16.4 Hz, 1 H), 4.33 (q, J=7.1 Hz, 2 H), 1.33 ppm (t, J=7.1 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.3, 165.2, 157.0, 139.9, 138.6, 130.9, 129.5, 128.4, 115.7, 60.9, 14.1 ppm; ESI-MS m/z: 278 [M+H]+; HRMS m/z calcd for C13H11NO4S [M+H]+ 278.0487, found 278.0482.
(E)-5-(4-Benzoylstyryl)-1,3,4-oxathiazol-2-one (34). According to Method D, 4-iodobenzophenone (308 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum. The crude corresponding acrylate was purified by silica column chromatography (EtOAc/MeOH 1:0 → 10:1) to afford 158 mg (63 % isolated yield) of (E)-3-(4-benzoylphenyl)acrylamide as an off-white powder. TLC: Rf=0.47 (EtOAc); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.80–7.66 (m, 7 H), 7.63 (bs, 1 H), 7.61–7.55 (m, 2 H), 7.50 (d, J=15.8 Hz, 1 H), 7.21 (bs, 1 H), 6.75 ppm (d, J=15.8 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=156.0, 127.1, 99.8, 98.8, 98.1, 97.8, 93.6, 91.1, 90.4, 89.5, 88.5, 85.8 ppm; ESI-MS m/z: 252 [M+H]+, 503 [2 M+H]+. According to Method B, (E)-3-(4-benzoylphenyl)acrylamide (158 mg, 0.63 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 157 mg (80 % isolated yield) of the title compound as a white powder. TLC: Rf=0.26 (pentane/EtOAc 10:1); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.96 (dm, J=8.8 Hz, 2 H), 7.80–7.74 (m, 4 H), 7.73–7.67 (m, 1 H), 7.64 (d, J=16.4 Hz, 1 H), 7.61–7.55 (m, 2 H), 7.16 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=195.1, 173.4, 157.1, 140.0, 138.1, 138.0, 136.8, 132.9, 130.1, 129.6, 128.6, 128.2, 115.6 ppm; ESI-MS m/z: 310 [M+H]+; HRMS m/z calcd for C17H11NO3S [M+H]+ 310.0538, found 310.0535.
(E)-Methyl 4-(2-(2-oxo-1,3,4-oxathiazol-5-yl)vinyl)benzoate (35). According to Method D, methyl 4-iodobenzoate (262 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum. The crude corresponding acrylate was purified by silica column chromatography (EtOAc/MeOH 1:0 → 10:1) to afford 124 mg (60 % isolated yield) of (E)-methyl 4-(3-amino-3-oxoprop-1-en-1-yl)benzoate as an off-white powder. TLC: Rf=0.47 (EtOAc); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.98 (bd, J=8.3 Hz, 2 H), 7.70 (bd, J=8.3 Hz, 2 H), 7.61 (bs, 1 H), 7.46 (d, J=15.9 Hz, 1 H), 7.20 (bs, 1 H), 6.73 (d, J=15.9 Hz, 1 H), 3.86 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.2, 165.8, 139.5, 137.8, 129.9, 129.7, 127.8, 125.0, 52.2 ppm; ESI-MS m/z: 206 [M+H]+, 411 [2 M+H]+. According to Method B, (E)-methyl 4-(3-amino-3-oxoprop-1-en-1-yl)benzoate (124 mg, 0.60 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 108 mg (68 % isolated yield) of the title compound as white powder. TLC: Rf=0.32 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.98 (bd, J=8.6 Hz, 2 H), 7.92 (bd, J=8.6 Hz, 2 H), 7.60 (d, J=16.4 Hz, 1 H), 7.13 (d, J=16.4 Hz, 1 H), 3.87 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.3, 165.7, 157.0, 139.8, 138.7, 130.6, 129.6, 128.4, 115.7, 52.3 ppm; ESI-MS m/z: 264 [M+H]+; HRMS m/z calcd for C12H9NO4S [M+H]+ 264.0331, found 264.0336.
(E)-5-(4-Nitrostyryl)-1,3,4-oxathiazol-2-one (36). According to Method D, 1-iodo-4-nitrobenzene (249 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum. The crude corresponding acrylate was purified by silica column chromatography (EtOAc/MeOH 1:0 → 10:1) to afford 95 mg (49 % isolated yield) of (E)-3-(4-nitrophenyl)acrylamide as a yellow powder. TLC: Rf=0.53 (EtOAc); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.26 (bd, J=9.0 Hz, 2 H), 7.83 (bd, J=9.0 Hz, 2 H), 7.66 (bs, 1 H), 7.52 (d, J=15.9 Hz, 1 H), 7.27 (bs, 1 H), 6.80 ppm (d, J=15.9 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.9, 147.5, 141.5, 136.8, 128.6, 126.7, 124.1 ppm; ESI-MS m/z: 193 [M+H]+. According to Method B, (E)-3-(4-nitrophenyl)acrylamide (95 mg, 0.49 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 99 mg (80 % isolated yield) of the title compound as a pale yellow powder. TLC: Rf=0.24 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.26 (bd, J=8.9 Hz, 2 H), 8.07 (bd, J=8.9 Hz, 2 H), 7.67 (d, J=16.4 Hz, 1 H), 7.24 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.3, 156.8, 147.9, 140.7, 138.7, 129.2, 124.0, 117.3 ppm; ESI-MS m/z: 251 [M+H]+; HRMS m/z for C10H6N2O4S as [M+H]+ adduct not found due to low ionization level.
(E)-4-(2-(2-Oxo-1,3,4-oxathiazol-5-yl)vinyl)benzonitrile (37). According to Method D, 4-iodobenzonitrile (229 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (double purification: 1st EtOAc, 2nd pentane/EtOAc 1:1 → 0:1) to afford 123 mg of a 1:2 mixture of (E)-3-(4-cyanophenyl)acrylamide and starting acrylamide as white a powder. TLC: Mizoroki–Heck product Rf=0.37, acrylamide Rf=0.31 (EtOAc); ESI-MS: m/z 173 [M+H]+. According to Method B, the obtained Mizoroki–Heck reaction mixture (123 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.21 mL) to give, after purification by precipitation from EtOAc/pentane, 47 mg (20 % isolated yield after two steps) of the title compound as an off-white powder. TLC: Rf=0.21 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.98 (bd, J=8.4 Hz, 2 H), 7.90 (bd, J=8.4 Hz, 2 H), 7.61 (d, J=16.4 Hz, 1 H), 7.20 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.3, 156.9, 139.3, 138.8, 132.8, 128.8, 118.6, 116.6, 112.2 ppm; ESI-MS m/z: 231 [M+H]+; HRMS m/z calcd for C11H6N2O2S [M+H]+ 231.0228, found 231.0226.
(E)-4-(2-(2-Oxo-1,3,4-oxathiazol-5-yl)vinyl)benzaldehyde (38). According to Method D, 4-iodobenzaldehyde (232 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (double purification: 1st EtOAc, 2nd pentane/EtOAc 1:1 → 0:1) to afford 100 mg of a 2:3 mixture of (E)-3-(4-formylphenyl)acrylamide and starting acrylamide as a white powder. TLC: Mizoroki–Heck product Rf=0.37, acrylamide Rf=0.31 (EtOAc); ESI-MS m/z: 176 [M+H]+. According to Method B, the obtained Mizoroki–Heck reaction mixture (100 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.10 mL) to give, after purification by precipitation from EtOAc/pentane, 36 mg (15 % isolated yield after two steps) of the title compound as a pale yellow powder. TLC: Rf=0.18 (pentane/EtOAc 10:1); LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=10.03 (s, 1 H), 8.01 (bd, J=8.4 Hz, 2 H), 7.95 (bd, J=8.4 Hz, 2 H), 7.62 (d, J=16.4 Hz, 1 H), 7.19 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=192.6, 173.3, 157.0, 139.9, 139.8, 136.8, 129.9, 128.8, 116.2 ppm; ESI-MS m/z: 234 [M+H]+; HRMS m/z for C11H7NO3S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(4-Chlorostyryl)-1,3,4-oxathiazol-2-one (39). According to Method D, 1-chloro-4-iodobenzene (238 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to 0 °C, and the collected precipitate was washed with cold CH3CN (3 mL) and dried by vacuum. The crude corresponding acrylate was purified by silica column chromatography (EtOAc/MeOH 1:0 → 10:1) to afford 123 mg (68 % isolated yield) of (E)-3-(4-chlorophenyl)acrylamide as a yellow powder. TLC: Rf=0.60 (EtOAc); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.58 (dm, J=8.4 Hz, 2 H), 7.54 (bs, 1 H), 7.47 (dm, J=8.4 Hz, 2 H), 7.40 (d, J=15.9 Hz, 1 H), 7.13 (bs, 1 H), 6.61 ppm (d, J=15.9 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=166.4, 137.8, 133.9, 133.8, 129.2, 128.9, 123.2 ppm; ESI-MS m/z: 182/184 [M+H]+, 363/365 [2 M+H]+. According to Method B, (E)-3-(4-chlorophenyl)acrylamide (123 mg, 0.68 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after purification by precipitation from EtOAc/pentane, 118 mg (73 % isolated yield) of the title compound as a white powder. TLC: Rf=0.68 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.81 (dm, J=8.4 Hz, 2 H), 7.54 (d, J=16.3 Hz, 1 H), 7.50 (dm, J=8.4 Hz, 2 H), 7.03 ppm (d, J=16.3 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.4, 157.2, 140.0, 134.9, 133.2, 129.9, 129.0, 114.0 ppm; ESI-MS m/z: 240/242 [M+H]+; HRMS m/z for C10H6ClNO2S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(2,5-Dichlorostyryl)-1,3,4-oxathiazol-2-one (40). According to Method D, 1,4-dichloro-2-iodobenzene (273 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (double purification: 1st EtOAc; 2nd pentane/EtOAc 1:1 → 0:1) to afford 184 mg of a 1:1 mixture of (E)-3-(2,5-dichlorophenyl)acrylamide and starting acrylamide as a white powder. TLC: Mizoroki–Heck product Rf=0.69, acrylamide Rf=0.31 (EtOAc); ESI-MS m/z: 216/217 [M+H]+. According to Method B, the obtained Mizoroki–Heck reaction mixture (184 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.24 mL) to give, after purification by precipitation from EtOAc/pentane, 31 mg (11 % isolated yield after two steps) of the title compound as an off-white powder. TLC: Rf=0.79 (pentane/EtOAc 10:1); LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.18 (d, J=2.5 Hz, 1 H), 7.63 (d, J=16.2 Hz, 1 H), 7.61 (d, J=8.7 Hz, 1 H), 7.53 (dd, J=8.7, 2.5 Hz, 1 H), 7.25 ppm (d, J=16.2 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.2, 156.6, 134.4, 133.5, 132.7, 132.1, 131.6, 131.4, 127.5, 117.6 ppm; ESI-MS m/z: 274/276 [M+H]+; HRMS m/z for C10H5Cl2NO2S as [M+H]+ adduct not found due to low ionization level.
(E)-5-(3-Methylstyryl)-1,3,4-oxathiazol-2-one (41). According to Method D, 1-iodo-3-methylbenzene (218 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude to (E)-3-(m-tolyl)acrylamide, ESI-MS m/z: 162 [M+H]+. According to Method A, the crude 3-substituted acrylamide was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.34 mL) to give, after silica column chromatography (CH2Cl2/isohexane 1:9) and recrystallization in CH3CN, 76 mg (35 % isolated yield after step two) as a white solid. ESI-MS m/z: 220 [M+H]+; LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=7.47 (d, J=16.2 Hz, 1 H), 7.34–7.20 (m, 4 H), 6.63 (d, J=16.2 Hz, 1 H), 2.39 ppm (m, 3 H); 13C NMR (100 MHz, CDCl3): δ=173.4, 157.7, 142.1, 138.8, 134.0, 131.4, 129.0, 128.5, 125.0, 112.6, 21.3 ppm; HRMS m/z calcd for C11H9NO2S [M+H]+ 220.0432, found 220.0437.
(E)-5-(3-Ethylstyryl)-1,3,4-oxathiazol-2-one (42). According to Method E, 1-bromo-3-ethylbenzene (370 mg, 2.0 mmol) was used as starting bromide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, filtered through a plug of cotton, and concentrated under reduced pressure. The residue was dissolved in EtOAc (25 mL) and washed with H2O (25 mL), and the organic phase was dried over MgSO4 and evaporated to yield the crude (E)-3-(3-ethylphenyl)acrylamide as a pale yellow solid. ESI-MS m/z: 176 [M+H]+. According to Method A, the crude 3-substituted acrylamide was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.67 mL) to give, after silica column chromatography (EtOAc/isohexane 2:100 → 5:100) and recrystallization in CH3CN, 77 mg (17 % isolated yield after two steps) as a white solid. ESI-MS m/z: 234 [M+H]+; LC purity (254 nm): 99 %; 1H NMR (400 MHz, CDCl3): δ=7.48 (d, J=16.2 Hz, 1 H), 7.36–7.23 (m, 4 H), 6.63 (d, J=16.2 Hz, 1 H), 2.68 (q, J=7.6 Hz, 2 H), 1.27 ppm (t, J=7.6 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ=173.4, 157.8, 145.1, 142.2, 134.1, 130.3, 129.1, 127.4, 125.3, 112.6, 28.7, 15.5 ppm; HRMS m/z calcd for C12H11NO2S [M+H]+ 234.0589, found 234.0591.
(E)-5-(3-(Trifluoromethoxy)styryl)-1,3,4-oxathiazol-2-one (43). According to Method D, 1-iodo-3-(trifluoromethoxy)benzene (288 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (EtOAc/isohexane 50:50 → 75:25) to (E)-3-(3-(trifluoromethoxy)phenyl)acrylamide as a pale yellow solid. TLC: Rf=0.47 (EtOAc:iso-hexane, 75:25); ESI-MS m/z: 232 [M+H]+. According to Method A, the crude 3-substituted acrylamide was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.22 mL) to give, after silica column chromatography (etylacetate/isohexane 1:100), 176 mg (61 % isolated yield after two steps) as a white solid. ESI-MS m/z: 299 [M+H]+; LC purity (254 nm): 98 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.81 (m, 1 H), 7.78, (dm, J=8.0 Hz, 1 H), 7.55 (d, J=16.4 Hz, 1 H), 7.54 (m, 1 H), 7.38 (m, 1 H), 7.01 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.8, 157.5, 149.3, 140.0, 137.1, 131.3, 127.7, 122.9, 121.0, 120.5 (q, J=257 Hz), 115.6 ppm; HRMS m/z calcd for C11H6F3NO3S [M+H]+ 299.0099, found 299.0103.
(E)-Ethyl 3-(2-(2-Oxo-1,3,4-oxathiazol-5-yl)vinyl)benzoate (44). According to Method D, ethyl-3-iodobenzoate (276 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (EtOAc/isohexane 50:50 → 75:25) to afford 206 mg of (E)-3-(3- ethoxycarbonyl)acrylamide as pale yellow solid. TLC: Rf=0.58, (EtOAc); ESI-MS m/z: 220 [M+H]+. According to Method A, the crude 3-substituted acrylamide (206 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.17 mL) to give, after recrystallization in ethylacetate/isohexane, 118 mg (43 % isolated yield after two steps) as a white solid, 43 % yield. ESI-MS m/z: 278 [M+H]+; LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.24 (m, 1 H), 8.05, (dm, J=7.7 Hz, 1 H), 7.97 (dm, J=7.8 Hz, 1 H), 7.60 (d, J=16.4 Hz, 1 H), 7.58 (dd, J=7.7, 7.8 Hz, 1 H), 7.05 (d, J=16.4 Hz, 1 H), 4.32 ppm (q, J=7.2 Hz, 2 H), 1.32 (t, J=7.2 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.9, 165.8, 157.6, 140.7, 135.2, 132.6, 131.2, 131.1, 129.9, 129.4, 114.9, 61.5, 14.6 ppm; HRMS m/z calcd for C13H11NO4S [M+H]+ 278.0487, found 278.0486.
(E)-5-(2-Methylstyryl)-1,3,4-oxathiazol-2-one (45). According to Method D, 1-iodo-2-methylbenzene (218 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (EtOAc/isohexane 50:50 → 75:25) to (E)-3-(o-tolyl)acrylamide as a pale yellow solid, 149 mg. TLC: Rf=0.48 (EtOAc); ESI-MS m/z: 162 [M+H]+. According to Method A, the crude 3-substituted acrylamide was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.26 mL) to give, after silica column chromatography (EtOAc/isohexane 1:100), 70 mg (32 % isolated yield after two steps) as a white solid. ESI-MS m/z: 220 [M+H]+; LC purity (254 nm): 95 %; 1H NMR (400 MHz, CDCl3): δ=7.69 (d, J=16.2 Hz, 1 H), 7.48 (dm, J=7.8 Hz, 1 H), 7.25–7.13 (m, 3 H), 6.49 (d, J=16.2 Hz, 1 H), 2.37 ppm (m, 3 H); 13C NMR (100 MHz, CDCl3): δ=173.5, 157.8, 139.5, 137.6, 133.0, 131.0, 130.3, 126.5, 125.9, 113.8, 19.8 ppm; HRMS m/z calcd for C11H9NO2S [M+H]+ 220.0432, found 220.0436.
(E)-5-(2-Ethylstyryl)-1,3,4-oxathiazol-2-one (46). According to Method E, 1-bromo-2-ethylbenzene (218 mg, 1.0 mmol) was used as starting bromide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and filtered through a plug of cotton. The reaction mixture was then extracted between EtOAc (25 mL) and H2O (25 mL), and the organic phase was dried over MgSO4 and evaporated to yield the crude (E)-3-(2-ethylphenyl)acrylamide as a pale yellow solid, 117 mg. ESI-MS m/z: 176 [M+H]+. According to Method A, the crude 3-substituted acrylamide was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.23 mL) to give, after silica column chromatography (EtOAc/isohexane 2:100), 90 mg (19 % isolated yield after two steps) as a white solid. ESI-MS m/z: 234 [M+H]+; LC purity (254 nm): 98 %; 1H NMR (400 MHz, CDCl3): δ=7.80 (d, J=16.2 Hz, 1 H), 7.57 (m, 1 H), 7.35 (m, 1 H), 7.28–7.22 (m, 2 H), 6.58 (d, J=16.2 Hz, 1 H), 2.79 (q, J=7.5 Hz, 2 H), 1.24 ppm (t, J=7.5 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ=173.5, 157.8, 143.8, 139.3, 132.3, 130.6, 129.4, 126.5, 126.1, 113.9, 26.4, 15.9 ppm; HRMS m/z calcd for C12H11NO2S [M+H]+ 234.0589, found 234.0584.
(E)-5-(2-(Trifluoromethoxy)styryl)-1,3,4-oxathiazol-2-one (47). According to Method D, 1-iodo-2-(trifluoromethoxy)benzene (288 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite. Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (EtOAc/isohexane 50:50 → 75:25) to (E)-3-(2-(trifluoromethoxy)phenyl)acrylamide as a pale yellow solid, 229 mg. TLC: Rf=0.49 (EtOAc/iso-hexane 75:25); ESI-MS m/z: 232 [M+H]+. According to Method A, the crude 3-substituted acrylamide was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.20 mL) to give, after precipitation from EtOAc/isohexane, 110 mg (38 % isolated yield after two steps) as a white solid. ESI-MS m/z: 299 [M+H]+; LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.04 (d, J=7.8 Hz, 1 H), 7.54 (m, 1 H), 7.47 (d, J=16.4 Hz, 1 H), 7.45–7.38 (m, 2 H), 7.08 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.7, 157.1, 146.7, 132.9, 132.6, 128.7, 128.6, 127.5, 122.1, 120.6 (q, J=258 Hz), 110.0 ppm; HRMS m/z calcd for C11H6F3NO3S [M+H]+ 299.0099, found 299.0096.
(E)-Ethyl 2-(2-(2-Oxo-1,3,4-oxathiazol-5-yl)vinyl)benzoate (48). According to Method E, ethyl 2-bromobenzoate (506 mg, 2.2 mmol) was used as starting bromide in the Mizoroki–Heck reaction. After microwave irradiation, the product was filtered and washed with EtOAc (20 mL) give (E)-ethyl 2-(3-amino-3-oxoprop-1-en-1-yl)benzoate as a pale white solid, 391 mg. According to Method A, the crude 3-substituted acrylamide was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.60 mL) to give, after filtration and washing with EtOAc (10 mL), 198 mg (32 % isolated yield after two steps) as a white solid. ESI-MS m/z: 278 [M+H]+; LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.18 (d, J=16.2 Hz, 1 H), 7.95 (dm, J=8.0 Hz, 1 H), 7.90 (dd, J=7.8, 1.6 Hz, 1 H), 7.65 (ddd, J=8.0, 7.6, 1.6 Hz, 1 H), 7.54 (ddm, J=7.8, 7.6 Hz, 1 H), 6.92 (d, J=16.2 Hz, 1 H), 4.33 (q, J=7.2 Hz, 2 H), 1.33 ppm (t, J=7.2 Hz, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.8, 166.7, 157.6, 139.7, 125.1, 133.0, 130.9, 130.5, 130.3, 128.1, 116.0, 61.7, 14.5 ppm; HRMS m/z calcd for C13H11NO4S [M+H]+ 278.0487, found 278.0485.
(E)-5-(2-(Pyridin-4-yl)vinyl)-1,3,4-oxathiazol-2-one (49). According to Method D, 4-iodopyridine (205 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through silica gel (CH3CN). Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (pentane/EtOAc 1:1 → 0:1, then EtOAc/MeOH 9:1) to afford 120 mg of a 1:1 mixture of (E)-3-(pyridin-4-yl)acrylamide and starting acrylamide as a pale yellow solid. TLC: Mizoroki–Heck product Rf=0.25, acrylamide Rf=0.72 (EtOAc/MeOH 9:1); ESI-MS m/z: 149 [M+H]+. According to Method B, the obtained Mizoroki–Heck reaction mixture (120 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.34 mL) to give, after purification by preparative HPLC, 15 mg (7 % isolated yield after two steps) of the title compound as an off-white powder. TLC: Rf=0.32 (pentane/EtOAc=1:1); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.66–8.63 (m, 2 H), 7.75–7.72 (m, 2 H), 7.53 (d, J=16.3 Hz, 1 H), 7.27 ppm (d, J=16.3 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.2, 156.8, 150.4, 141.3, 138.7, 121.9, 117.6 ppm; ESI-MS m/z: 207 [M+H]+; HRMS m/z calcd for C9H6N2O2S [M+H]+ 207.0228, found 207.0255.
(E)-5-(2-(2-Methoxypyridin-3-yl)vinyl)-1,3,4-oxathiazol-2-one (50). According to Method D, 3-iodo-2-methoxy-pyridine (235 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After microwave irradiation, the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through silica gel (CH3CN). Evaporation of the solvent gave the crude corresponding acrylate, which was purified by silica column chromatography (pentane/EtOAc 1:1 → 0:1, then EtOAc/MeOH 9:1) to afford 184 mg of a 1:1 mixture of (E)-3-(2-methoxypyridin-3-yl)acrylamide and starting acrylamide as a pale yellow oil. TLC: Mizoroki–Heck product Rf=0.68, acrylamide Rf=0.60 (EtOAc/MeOH 9:1); ESI-MS m/z: 179 [M+H]+. According to Method A, the obtained Mizoroki–Heck reaction mixture (184 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.34 mL) to give, after purification by trituration with Et2O followed by precipitation from CH3CN/H2O, 91 mg (39 % isolated yield after two steps) of the title compound as a pale yellow solid. TLC: Rf=0.80 (EtOAc); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.23 (dd, J=4.9, 1.8 Hz, 1 H), 8.17 (dd, J=7.4, 1.8 Hz, 1 H), 7.56 (d, J=16.5 Hz, 1 H), 7.09 (dd, J=7.4, 4.9 Hz, 1 H), 7.06 (d, J=16.5 Hz, 1 H), 3.98 ppm (s, 3 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.6, 161.1, 157.4, 148.7, 138.2, 135.1, 117.8, 117.1, 115.4, 53.9 ppm; ESI-MS m/z: 237 [M+H]+; HRMS m/z calcd for C10H8N2O3S [M+H]+ 237.0334, found 237.0338.
(E)-5-(2-(Thiophen-2-yl)vinyl)-1,3,4-oxathiazol-2-one (51). According to Method C, 2-iodothiophene (210 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After 15 min of microwave irradiation (T=120 °C), the reaction mixture was irradiated for additional 30 min at the same temperature to afford full conversion. The resulting reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through silica gel (CH3CN→EtOAc→EtOAc/MeOH 10:1). Evaporation of the solvent gave the crude corresponding acrylate, which was purified by precipitation from CH3OH/H2O to afford 70 mg (46 % isolated yield) of (E)-3-(thiophen-2-yl)acrylamide as an off-white solid. TLC: Rf=0.52 (EtOAc); LC purity (254 nm): 95 %; 1H NMR (400 MHz, CDCl3): δ=7.71 (d, J=15.3 Hz, 1 H), 7.28 (bd, J=5.1 Hz, 1 H), 7.17 (bd, J=3.5 Hz, 1 H), 6.98 (dd, J=5.1, 3.5 Hz, 1 H), 6.19 (d, J=15.3 Hz, 1 H), 5.36 ppm (bs, 2 H); ESI-MS m/z: 154 [M+H]+. According to Method B, (E)-3-(thiophen-2-yl)acrylamide (70 mg, 0,46 mmol) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.40 mL) to give, after purification by silica column (pentane/EtOAc 100:0 → 1:1) followed by preparative HPLC purification, 46 mg (45 % isolated yield) of the title compound as a white powder. TLC: Rf=0.63 (pentane/EtOAc 20:1); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=7.76 (bd, J=5.1 Hz, 1 H), 7.72 (d, J=16.1 Hz, 1 H), 7.59 (bd, J=3.7 Hz, 1 H), 7.17 (dd, J=5.1, 3.7 Hz, 1 H), 6.61 ppm (d, J=16.1 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.4, 157.0, 138.9, 134.3, 131.8, 130.2, 128.7, 111.3 ppm; ESI-MS m/z: 212 [M+H]+; HRMS m/z calcd for C8H5NO2S2 [M+H]+ 211.9840, found 211.9837.
(E)-5-(2-(Thiophen-3-yl)vinyl)-1,3,4-oxathiazol-2-one (52). According to Method C, 3-iodothiophene (210 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After 15 min of microwave irradiation (T=120 °C), the reaction mixture was irradiated for additional 30 min at the same temperature to afford full conversion. The resulting reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through silica gel (CH3CN→EtOAc→EtOAc/MeOH 10:1). Evaporation of the solvent gave the crude corresponding acrylate, that was purified by precipitation from EtOAc/pentane followed by silica column purification (pentane/EtOAc 1:1 → 0:1) to afford 115 mg (75 % isolated yield) of (E)-3-(thiophen-3-yl)acrylamide as a white solid. TLC: Rf=0.46 (EtOAc); LC purity (254 nm): 95 %; 1H NMR (400 MHz, CDCl3): δ=7.64 (d, J=15.4 Hz, 1 H), 7.47 (bd, J=2.9 Hz, 1 H), 7.33 (dd, J=5.1, 2.9 Hz, 1 H), 7.28 (bd, J=5.1 Hz, 1 H), 6.30 ppm (d, J=15.4 Hz, 1 H), 5.53 (bs, 2 H); ESI-MS m/z: 154 [M+H]+. According to Method B, (E)-3-(thiophen-3-yl)acrylamide (115 mg, 0,75 mmol) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.40 mL) to give, after purification by silica column (pentane/EtOAc 100:0 → 1:1) followed by preparative HPLC purification, 84 mg (53 % isolated yield) of the title compound as a white powder. TLC: Rf=0.55 (pentane/EtOAc=20:1); LC purity (254 nm): 95 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.02–8.00 (m, 1 H), 7.66–7.61 (m, 2 H), 7.55 (d, J=16.2 Hz, 1 H), 6.82 ppm (d, J=16.2 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.5, 157.6, 137.5, 135.4, 129.6, 127.9, 125.7, 112.6 ppm; ESI-MS: m/z 212 [M+H]+; HRMS m/z calcd for C8H5NO2S2 [M+H]+ 211.9840, found 211.9841.
(E)-5-(2-(2-Oxo-1,3,4-oxathiazol-5-yl)vinyl)furan-2-carbaldehyde (53). According to Method C, 5-iodo-2-furaldehyde (222 mg, 1.0 mmol) was used as starting iodide in the Mizoroki–Heck reaction. After 15 min of microwave irradiation (T=120 °C), the reaction mixture was irradiated for additional 30 min at the same temperature to afford full conversion. The resulting reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through silica gel (CH3CN→EtOAc→EtOAc/MeOH 10:1). Evaporation of the solvent gave a crude mixture that was taken in CH3OH/H2O and then filtered to remove the precipitated homocoupling byproduct. Evaporation of the solvent gave 262 mg of a crude mixture, which was purified by flash silica column (pentane/EtOAc 1:1 → 0:1) to give 88 mg of a mixture containing the desired (E)-3-(5-formylfuran-2-yl)acrylamide as a yellow solid. TLC: Mizoroki–Heck compound Rf=0.44 (EtOAc); LC purity (254 nm): 30 %; ESI-MS m/z: 166 [M+H]+. According to Method B, the obtained Mizoroki–Heck reaction mixture (88 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.40 mL) to give, after purification by silica column (pentane/EtOAc 100:0 → 1:1) followed by preparative HPLC purification, 10 mg (5 % isolated yield after two steps) of the title compound as a white powder. TLC: Rf=0.70 (pentane/EtOAc=20 :1); LC purity (254 nm): 90 %; 1H NMR (400 MHz, [D6]DMSO): δ=9.66 (s, 1 H), 7.63 (d, J=3.7 Hz, 1 H), 7.47 (d, J=16.2 Hz, 1 H), 7.24 (d, J=3.7 Hz, 1 H), 6.83 ppm (d, J=16.2 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=178.7, 173.1, 156.4, 154.5, 152.8, 127.1, 124.3, 116.8, 115.0 ppm; ESI-MS m/z: 224 [M+H]+; HRMS m/z calcd for C9H5NO4S [M+H]+ 224.0018, found 224.0015.
(E)-5-(2-(Pyridin-2-yl)vinyl)-1,3,4-oxathiazol-2-one (54). According to Method E, 2-bromopyridine (237 mg, 1.5 mmol) was used as starting bromide in the Mizoroki–Heck reaction. After 15 min of microwave irradiation (T=140 °C), the reaction mixture was irradiated for additional 60 min at 160 °C to afford full conversion. The resulting reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite (EtOAc/MeOH 1:1). Evaporation of the solvent under reduced pressure gave a crude mixture containing DMF, which was taken in EtOAc (20 mL). The organic phase was washed with H2O (2×40 mL), the H2O phases were extracted with EtOAc (2×100 mL), and the collected organic phases were dried over Na2SO4. Evaporation of solvent gave 126 mg of a mixture containing the desired (E)-3-(pyridin-2-yl)acrylamide as a yellow oil. TLC: Mizoroki–Heck compound Rf=0.07 (EtOAc/MeOH 10:1); LC purity (254 nm)=70 %; ESI-MS m/z: 149 [M+H]+. According to Method A, the obtained Mizoroki–Heck reaction mixture (126 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.50 mL) to give, after purification by preparative HPLC, 8 mg (3 % isolated yield after two steps) of the title compound as a brown powder. LC purity (254 nm): 92 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.69–8.65 (m, 1 H), 7.89 (td, J=7.8, 2.0 Hz, 1 H), 7.78 (dm, J=8.0 Hz, 1 H), 7.58 (d, J=16.0 Hz, 1 H), 7.43 (ddd, J=7.8, 4.7, 1.2 Hz, 1 H), 7.20 ppm (d, J=16.0 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.3, 156.9, 152.0, 150.1, 140.3, 137.4, 125.0, 124.8, 116.0 ppm; ESI-MS m/z: 207 [M+H]+; HRMS m/z calcd for C9H6N2O2S [M+H]+ 207.0228, found 207.0234.
(E)-5-(2-(Pyridin-3-yl)vinyl)-1,3,4-oxathiazol-2-one (55). According to Method E, 3-bromopyridine (237 mg, 1.5 mmol) was used as starting bromide in the Mizoroki–Heck reaction. After 15 min of microwave irradiation (T=140 °C), the reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite (EtOAc/MeOH 1:1). Evaporation of the solvent under reduced pressure gave a crude mixture containing DMF, which was taken in EtOAc (20 mL). The organic phase was washed with H2O (2×40 mL), the H2O phases were extracted with EtOAc (2×100 mL), and the collected organic phases were dried over Na2SO4. Evaporation of solvent gave 102 mg of a mixture containing the desired (E)-3-(pyridin-3-yl)acrylamide as a white solid. TLC: Mizoroki–Heck compound Rf=0.26 (EtOAc/MeOH 10:1); LC purity (254 nm): 90 %; ESI-MS m/z: 149 [M+H]+. According to Method A, the obtained Mizoroki–Heck reaction mixture (102 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.50 mL) by microwave irradiation, followed by thermal heating at 70 °C for 2 d to give, after purification by preparative HPLC, 45 mg (15 % isolated yield after two steps) of the title compound as a white powder. LC purity (254 nm): 99 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.92 (bd, J=2.1 Hz, 1 H), 8.59 (dd, J=4.9, 1.8 Hz, 1 H), 8.24 (dm, J=8.0 Hz, 1 H), 7.58 (d, J=16.4 Hz, 1 H), 7.47 (bdd, J=7.5, 4.3 Hz, 1 H), 7.16 ppm (d, J=16.4 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=173.4, 157.0, 150.9, 149.8, 138.1, 134.4, 130.1, 123.9, 115.1 ppm; ESI-MS m/z: 207 [M+H]+; HRMS m/z calcd for C9H6N2O2S [M+H]+ 207.0228, found 207.0229.
(E)-5-(2-(2-Oxo-2 H-[1,2,4]thiadiazolo[2,3-a]pyridin-8-yl)vinyl)-1,3,4-oxathiazol-2-one (56). According to Method E, 2-amino-3-bromopyridine (260 mg, 1.5 mmol) was used as starting bromide in the Mizoroki–Heck reaction. After 15 min of microwave irradiation (T=140 °C), the reaction mixture was irradiated for additional 15 min at 160 °C to afford full conversion. The resulting reaction mixture was cooled down to rt, and the formed Pd black was removed by filtration through Celite (EtOAc/MeOH 1:1). Evaporation of the solvent under reduced pressure gave a crude mixture containing DMF, which was taken in EtOAc (20 mL). The organic phase was washed with H2O (2×40 mL), the H2O phases were extracted with EtOAc (2×100 mL), and the collected organic phases were dried over Na2SO4. Evaporation of solvent gave 94 mg of a mixture containing the desired (E)-3-(2-aminopyridin-3-yl)acrylamide as a yellow solid. TLC: Mizoroki–Heck compound Rf=0.22 (EtOAc/MeOH 10:1); LC purity (254 nm)=90 %; ESI-MS m/z: 164 [M+H]+. According to Method B, the obtained Mizoroki–Heck reaction mixture (94 mg) was reacted with an excess of chlorocarbonyl sulfenyl chloride (0.50 mL), to give, after purification by trituration in CH3CN/H2O, 16 mg (4 % isolated yield after two steps) of the title compound as a pale yellow solid. LC purity (254 nm): 97 %; 1H NMR (400 MHz, [D6]DMSO): δ=8.02 (dd, J=7.0, 1.3 Hz, 1 H), 7.95 (dm, J=7.0 Hz, 1 H), 7.72 (d, J=16.0 Hz, 1 H), 7.62 (dd, J=16.2, 0.6 Hz, 1 H), 6.89 ppm (t, J=7.0 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=174.3, 173.3, 157.1, 148.0, 137.1, 134.8, 126.1, 125.0, 117.4, 111.6 ppm; ESI-MS m/z: 280 [M+H]+; HRMS m/z calcd for C10H5N3O3S2 [M+H]+ 279.9851, found 279.9843. gHSQC and gHMQC experiments showed no correlations between proton HE and carbon C10, suggesting the following structure (Figure 3) as the most probable one.
Figure 3.
gHSQC and gHMQC experiment results of compound 56.
5-Phenethyl-1,3,4-oxathiazol-2-one (57). (2E)-3-(1H-Indol-3-yl)acrylic acid (378 mg, 2.0 mmol) and 1,1′-carbonyldiimidazole (656 mg, 2.0 mmol) were dissolved in anhydrous DMF (10 mL), and the mixture was stirred at rt. After 30 min, NH4HCO3 (639 mg, 4.0 mmol) was added, and the resulting suspension was stirred at rt o/n. EtOAc (100 mL) was added, and the organic phase was washed with H2O (2×50 mL) and brine (50 mL). Evaporation of solvent gave 397 mg of the crude corresponding amide, purified by silica column chromatography (pentane/EtOAc 1:1 → 0:1) to afford 116 mg (31 % isolated yield) of (E)-3-(1H-indol-3-yl)acrylamide as a yellow sticky oil. TLC: Rf=0.24 (EtOAc); LC purity (254 nm): 95 %; ESI-MS m/z: 187 [M+H]+. According to Method B, (E)-3-(1H-indol-3-yl)acrylamide (58 mg, 0.31 mmol) was reacted with chlorocarbonyl sulfenyl chloride to give, after preparative HPLC purification, 9 mg (12 % isolated yield) of the title compound as a pale yellow powder. LC purity (254 nm): 97 %; 1H NMR (400 MHz, [D6]DMSO): δ=11.84 (bs, 1 H), 8.00 (s, 1 H), 7.94 (dm, J=7.2 Hz, 1 H), 7.75 (d, J=16.2 Hz, 1 H), 7.48 (dm, J=8.1 Hz, 1 H), 7.26–7.15 (m, 2 H), 6.66 ppm (d, J=16.2 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=174.7, 159.3, 138.1, 136.7, 132.4, 125.4, 123.4, 121.8, 120.5, 113.1, 112.5, 107.0 ppm; ESI-MS m/z: 245 [M+H]+; HRMS m/z calcd for C12H8N2O2S [M+H]+ 245.0385, found 245.0384.
Proteasome cloning, expression, and purification. The sequences for the open-gate alpha8a (Rv2109c residues 9–240) and a slightly truncated beta (Rv2110c) (residues 1–285) subunits of the M. tuberculosis proteasome were amplified by PCR from total DNA of Mtb strain H37Rv28 using the polymerase Pfu Ultra (Agilent Technologies, La Jolla, USA). The C-terminus of the beta subunit was modified to incorporate a 6-histidine tag after the terminal glycine residue.8a The genes were inserted into the vector pRSF Duet-1, which is a vector for coexpressing two proteins, both controlled by T7 promoters, from Novagen (MerckMillipore, Darmstadt, Germany), and cloned in E. coli Top10 cells (Invitrogen, Carlsbad, USA).
Expression and purification were performed as previously described.8a Briefly, 6 L cultures of E. coli BL21(DE3) cells in Luria Bertani broth with 30 μg/mL kanamycin were grown for 20–22 h at 37 °C after induction with 0.4 mm isopropyl beta-D1 thiogalactopyranoside. The cells were harvested by centrifugation, resuspended in lysis buffer (50 mm Na3PO4, pH 8, 300 mm NaCl, 10 mm imidazole with 0.25 mg mL−1 lysozyme, 0.02 mg mL−1 RNase, and 0.01 mg mL−1 DNase), incubated 30 min at rt and lysed by passage through a Constant Cell Disruption System (Constant Systems Ltd, Daventry, UK) at 2 kBar. Full proteasome molecules were isolated from the cleared lysate with 1 mL Ni-Sepharose (GE-Healthcare, Uppsala, Sweden), and eluted with 250 mm imidazole. The buffer was exchanged on an Econo-Pac 10DG desalting column (Bio-Rad, Hercules, USA) to 10 mm Tris pH 7.5, 100 mm NaCl before a final purification step by size-exclusion chromatography on a HiLoad 16/60 Superdex 200 column (GE Healthcare, Little Chalfront, UK) equilibrated with the same buffer. The fractions containing the proteasome as analyzed by SDS-PAGE were pooled and concentrated to 1 mg mL−1 (measured by A280, extinction coefficient 33 810 m−1 cm−1) in a Vivaspin 20, molecular weight cut-off 100 kDa (Sartorius Stedin Biotech, Goettingen, Germany).
Proteasome assay. The protein production and assay were based on that of Lin et al.8b Both Mtb and human proteasomes were assayed in 20 mm Hepes, 0.5 mm EDTA pH 7.5, with 25 μm suc-LLVY-AMC (Bachem AG, Switzerland) as substrate, in a final volume of 100 μL on a white 96-well assay plate (Cliniplate, Thermo Fisher Scientific OY, Finland). The increase in fluorescence caused by cleavage of AMC (7-amino-4-methylcoumarin) was measured using a Fluoroskan Ascent fluorescent plate reader (Thermo Fisher Scientific OY, Finland) equipped with a 390 nm filter for excitation and 460 nm filter for emission. Human 20S proteasome (Boston Biochemicals, USA) was assayed at 1 nm concentration in the presence of a tenfold excess of human PA28 activator (Boston Biochemicals), as recommended by the manufacturer. The Mtb proteasome was assayed at a concentration of 2 nm. The assay was performed as follows. Threefold dilutions of inhibitor (10 mm stock solution in DMSO) from an initial concentration of 100 μm were performed on the assay plate in buffer containing the above concentrations of enzyme; the final volume in the well after dilution was 96 μL. The plate was sealed and preincubated at rt for 3 h for Mtb proteasome or 1 h for human proteasome. Next, substrate (5 μL of 500 μm in a buffer made from a 10 mm stock solution in DMSO) was added, and the fluorescence read in the plate reader immediately (t=0), then after 3 h for the Mtb proteasome and after 1 h for the human proteasome. The t=0 value was subtracted from the final value, and plotted against the logarithm of the inhibitor concentration; the data were fitted to the standard IC50 equation using GraphPad Prism 5 (GraphPad Software Inc., USA).
Minimum inhibitory concentration (MIC). The MIC were determined as previously described29 in Middlebrook 7H9 medium containing 10 % v/v OADC (oleic acid, bovine serum albumin, d-glucose, catalase; Becton Dickinson, USA) and 0.05 % w/v Tween 80. Briefly, compounds were tested in 10-point, two-fold serial dilutions with the highest concentration of 20 μm against Mtb H37Rv expressing a fluorescent protein (DsRed). Growth was measured using OD590 and RFU after 5 d. Curves were fitted using the Gompertz function, with MIC defined as the minimum concentration required to inhibit growth. Data are reported for optical density at 590 nm (OD590) and relative fluorescence units (RFU).
Kill kinetics against nonreplicating bacteria. Mtb was grown aerobically in Middlebrook 7H9 medium containing 10 % v/v OADC and 0.05 % w/v Tween 80, washed, and resuspended in PBS with 0.05 % w/v tyloxapol for 14 d. Compounds were added, and viability was determined by plating serial dilutions to determine colony forming units (CFU) on Middlebrook 7H10 agar plus 10 % v/v OADC. Colonies were counted after four weeks.
Cytotoxicity assay. Cytotoxicity was monitored against Vero African green monkey kidney epithelial cells (ATCC CCL-81) grown in DMEM, High Glucose, GlutaMAX™ (Invitrogen), supplemented with 10 % FBS, and penicillin–streptomycin solution (Gibco). The assay was run in 96-well plates using a 10-point, serial dilution curve. Cell viability was measured after 48 h using Cell Titer Glo (Promega), and growth inhibition curves were fitted using the Levenberg–Marquardt algorithm. The TC50 was defined as the compound concentration that gave 50 % inhibition of growth.
Determination of solubility in PBS (pH 7.4). HPLC analyses were performed using a Shimadzu 10 A system with a SCL-10Avp controller, 2 LC-10AD pumps with a 0.5 mL gradient mixer at the high pressure side, a SIL-10 A (rebuilt to also run 96-well plates), a CMA 260 Degasser, and a Waters PDA 996 Photodiode Array Detector. A Zorbax Eclipse XDB-C8, 5 μm, 2.1×50 mm column was utilized from Dalco Chromtech AB (Sollentuna, Sweden) with CH3CN/H2O in 0.05 % TFA as mobile phase and 1 mL min−1. The chromatograms were registered at the wavelengths 230, 254, and 280 nm. All solutions containing the sample compound were prepared from 10 mm DMSO stock solutions stored in glass vials at ambient temperature. High standards (HS) were made by diluting the DMSO stock solution 200 times with CH3CN+PBS pH 7.4 (1+1), obtaining a 50 μm solution. Low standards (LS) were prepared by diluting HS 20 times with CH3CN+PBS pH 7.4 (1+1), obtaining a 2.5 μm solution. The HS solution was chromatographed a second time after 24 h to determine the chemical stability. It was very important to exchange the septa after the first injection to avoid evaporation of the CH3CN. The solubility solutions were prepared by diluting the DMSO stock solution 100 times with PBS pH 7.4, obtaining a nominal concentration of 100 μm. The solutions were left for 18 h at ambient temperature before filtration. When filtering the sample solution, the first 0.1 mL was discarded, and the rest was collected. The solubility was determined using a two-point calibration curve forced through the origin. To evaluate the quality of the standard curve, the quotient of the area of HS to the LS was also calculated. The optimal quotient should equal the value of the concentration ratio, that is, 20.0. When evaluating the solubility, the wavelength used was that which gives the area quotient closest to 20. The stability (given in percent remaining) was obtained by multiplying the area after the 24 h run by 100 and dividing it by the area of the first HS injection.
Abbreviations used. MW, microwaves; Mtb, Mycobacterium tuberculosis; MDR, multidrug resistant; XDR, extensively drug resistant; TDR, totally drug resistant; MIC, minimum inhibitory concentration; CFU, colony forming units
Notes. The authors declare no competing financial interest.
Acknowledgments
The authors would like to acknowledge VINNOVA–the Swedish Governmental Agency for Innovation Systems for financial support and Medivir AB for technical support. The research was also supported in part by funding from the Bill and Melinda Gates Foundation, under grant OPP1024038, and by National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health under award numbers R56AI095652 and R01AI095652. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors also thank Stephanie Florio, Juliane Ollinger, and Julie Early at the Infectious Disease Research Institute (Seattle, USA) for technical assistance and helpful discussion.
References
- 1.2014. Global Tubercolosis Report, World Health Organization, Geneva.
- 2a.Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C. Clin. Infect. Dis. 2012;54:579–581. doi: 10.1093/cid/cir889. [DOI] [PubMed] [Google Scholar]
- 2b.Migliori GB, De Iaco G, Besozzi G, Centis R, Cirillo DM. Euro Surveill. 2007;12:3194. doi: 10.2807/esw.12.20.03194-en. , pii= [DOI] [PubMed] [Google Scholar]
- 2c.Velayati AA, Masjedi MR, Farnia P, Tabarsi P, Ghanavi J, Ziazarifi AH, Hoffner SE. Chest. 2009;136:420–425. doi: 10.1378/chest.08-2427. [DOI] [PubMed] [Google Scholar]
- 3.Society AmericanThoracic, Prevention CentersforDiseaseControland, America InfectionsDiseasesSocietyof. Am. J. Resp. Crit. Care Med. 2003;167:603–662. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 4a.Mikusova K, Slayden RA, Besra GS, Brennan PJ. Antimicrob. Agents Chemother. 1995;39:2484–2489. doi: 10.1128/aac.39.11.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b.Rozwarski DA, Grant GA, Barton DHR, Jacobs WR, Sacchettini JC. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 4c.Sacchettini JC, Rubin EJ, Freundlich JS. Nat. Rev. Microbiol. 2008;6:41–52. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- 5.Mcclure WR, Cech CL. J. Biol. Chem. 1978;253:8949–8956. [PubMed] [Google Scholar]
- 6a.Reinstein E, Ciechanover A. Ann. Inter. Med. 2006;145:676–684. doi: 10.7326/0003-4819-145-9-200611070-00010. [DOI] [PubMed] [Google Scholar]
- 6b.Burns KE, Liu WT, Boshoff HIM, Dorrestein PC, Barry CE. J. Biol. Chem. 2009;284:3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Adams J. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 7b.Coux O, Tanaka K, Goldberg AL. Annu. Rev. Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 7c.Lin G, Li DY, de Carvalho LPS, Deng HT, Tao H, Vogt G, Wu KY, Schneider J, Chidawanyika T, Warren JD, Li HL, Nathan C. Nature. 2009;461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d.Kisselev AF, Goldberg AL. Chem. Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 7e.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 8a.Lin G, Hu GQ, Tsu C, Kunes YZ, Li HL, Dick L, Parsons T, Li P, Chen ZQ, Zwickl P, Weich N, Nathan C. Mol. Microbiol. 2006;59:1405–1416. doi: 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- 8b.Hu GQ, Lin G, Wang M, Dick L, Xu RM, Nathan C, Li HL. Mol. Microbiol. 2006;59:1417–1428. doi: 10.1111/j.1365-2958.2005.05036.x. [DOI] [PubMed] [Google Scholar]
- 9.Lin G, Tsu C, Dick L, Zhou XK, Nathan C. J. Biol. Chem. 2008;283:34423–34431. doi: 10.1074/jbc.M805324200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groll M, Berkers CR, Ploegh HL, Ovaa H. Structure. 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 11a.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma Y-T, Plamondon L, Stein RL. Bioorg. Med. Chem. Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 11b.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- 12.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 13.Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. Nat. Med. 2007;13:1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14a.Dahlmann B, Kopp F, Kuehn L, Niedel B, Pfeifer G, Hegerl R, Baumeister W. Febs Lett. 1989;251:125–131. doi: 10.1016/0014-5793(89)81441-3. [DOI] [PubMed] [Google Scholar]
- 14b.Tamura T, Nagy I, Lupas A, Lottspeich F, Cejka Z, Schoofs G, Tanaka K, Demot R, Baumeister W. Curr. Biol. 1995;5:766–774. doi: 10.1016/s0960-9822(95)00153-9. [DOI] [PubMed] [Google Scholar]
- 15a.Mak PA, Rao SPS, Tan MP, Lin XH, Chyba J, Tay J, Ng SH, Tan BH, Cherian J, Duraiswamy J, Bifani P, Lim V, Lee BH, Ma NL, Beer D, Thayalan P, Kuhen K, Chatterjee A, Supek F, Glynne R, Zheng J, Boshoff HI, Barry CE, Dick T, Pethe K, Camacho LR. ACS Chem. Biol. 2012;7:1190–1197. doi: 10.1021/cb2004884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b.Gomez JE, McKinney JD. Tuberculosis. 2004;84:29–44. doi: 10.1016/j.tube.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 16a.Darby CM, Nathan CF. J. Antimicrob. Chemother. 2010;65:1424–1427. doi: 10.1093/jac/dkq145. [DOI] [PubMed] [Google Scholar]
- 16b.Tabarrini O, Sabatini S, Massari S, Pieroni M, Franzblau SG, Cecchetti V. Chem. Biol. Drug Des. 2012;80:781–786. doi: 10.1111/cbdd.12022. [DOI] [PubMed] [Google Scholar]
- 16c.Silveira GP, Ferreira M, Fernandes L, Moraski GC, Cho SY, Hwang C, Franzblau SG, Sa MM. Bioorg. Med. Chem. Lett. 2012;22:6486–6489. doi: 10.1016/j.bmcl.2012.08.048. [DOI] [PubMed] [Google Scholar]
- 16d.de Carvalho LPS, Lin G, Jiang XJ, Nathan C. J. Med. Chem. 2009;52:5789–5792. doi: 10.1021/jm9010719. [DOI] [PubMed] [Google Scholar]
- 16e.Heifets L, Simon J, Pham V. Ann. Clin. Microbiol. Antimicrob. 2005;4:6. doi: 10.1186/1476-0711-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16f.Lee SH, Kim S, Yun MH, Lee YS, Cho SN, Oh T, Kim P. Bioorg. Med. Chem. Lett. 2011;21:1515–1518. doi: 10.1016/j.bmcl.2010.12.128. [DOI] [PubMed] [Google Scholar]
- 16g.Yang J, Pi W, Xiong L, Ang W, Yang T, He J, Liu Y, Chang Y, Ye W, Wang Z, Luo Y, Wei Y. Bioorg. Med. Chem. Lett. 2013;23:1424–1427. doi: 10.1016/j.bmcl.2012.12.065. [DOI] [PubMed] [Google Scholar]
- 17.Gezginci MH, Martin AR, Franzblau SG. J. Med. Chem. 2001;44:1560–1563. doi: 10.1021/jm000350w. [DOI] [PubMed] [Google Scholar]
- 18.Gryder BE, Guerrant W, Chen CH, Oyelere AK. Medchemcomm. 2011;2:1083–1086. [Google Scholar]
- 19.Nathan C, Lin G. 2011. , (Cornell Research Foundation, Inc., Ithaca, NY, USA), US Pat. Appl. Pub. No. US 2011/0118274 A1.
- 20a.Datta G, Vallin KA, Larhed M. Mol. Diversity. 2003;7:107–114. doi: 10.1023/b:modi.0000006798.53091.a2. [DOI] [PubMed] [Google Scholar]
- 20b.Datta GK, von Schenck H, Hallberg A, Larhed M. J. Org. Chem. 2006;71:3896–3903. doi: 10.1021/jo0602367. [DOI] [PubMed] [Google Scholar]
- 20c.Larhed M, Hallberg A. J. Org. Chem. 1996;61:9582–9584. doi: 10.1021/jo952112s. [DOI] [PubMed] [Google Scholar]
- 21.Nilsson P, Gold H, Larhed M, Hallberg A. Synthesis. 2002:1611–1614. [Google Scholar]
- 22.Goldberg AL. J. Cell Biol. 2012;199:583–588. doi: 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bassett I, Lun S, Bishai W, Guo H, Kirman J, Altaf M, OToole R. J. Microbiol. 2013;51:651–658. doi: 10.1007/s12275-013-3099-4. [DOI] [PubMed] [Google Scholar]
- 24.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Mol. Microbiol. 2002;43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 25.Tsutsui Y, Yamada N, Kubo D, Tanikawa T, Ushiki Y, Hattori N, Yamamoto K, Ono N, Yamaguchi T, Ushiyama F. 2013. , (Toyama Chemical Co. Ltd. And Taisho Pharmaceutical Co. Ltd., Tokyo, Japan), Int. PCT Pub. No. WO/2013/031694.
- 26.Cosford NDP, McDonald IA, Bleicher LS, Cube RV, Schweiger EJ, Vernier J-M, Hess SD, Varney MA, Munoz B. 2001. , (Merck & Co., Inc., Rahway, NJ, USA), Int. PCT Pub. No. WO/2001/016121.
- 27.Paton RM, Stobie I, Mortier RM. Phosphorus Sulfur Relat. Elem. 1983;15:137–142. [Google Scholar]
- 28.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry 3rd CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 29.Ollinger J, Bailey MA, Moraski GC, Casey A, Florio S, Alling T, Miller MJ, Parish T. PLoS ONE. 2013;8:60531. doi: 10.1371/journal.pone.0060531. , e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. The background of the Table of Contents graphic is a public domain image originally contributed by the CDC/Melissa Brower to the Public Health Image Library (PHIL ID#16881)