

Abstract
Del(20q), a common cytogenetic abnormality in myeloid neoplasms, is rare in chronic lymphocytic leukemia. We report 64 patients with chronic lymphocytic leukemia and del(20q), as the sole abnormality in 40, a stemline abnormality in 21, and a secondary abnormality in 3 cases. FISH analysis revealed an additional high-risk abnormality, del(11q) or del(17p), in 27/64 (42%) cases. In most cases, the leukemic cells showed atypical cytologic features, unmutated IGHV genes and ZAP70 positivity. The del(20q) was detected only after chemotherapy in all 27 cases with initial karyotypes available. With a median follow-up of 90 months, 30 patients (47%) died, most as a direct consequence of chronic lymphocytic leukemia. Eight patients developed a therapy-related myeloid neoplasm, seven with a complex karyotype. Combined morphologic and FISH analysis for del(20q) performed in 12 cases without morphologic evidence of a myeloid neoplasm localized the del(20q) to the chronic lymphocytic leukemia cells in 5 (42%) cases, and to myeloid/erythroid cells in 7 (58)% cases. The del(20q) was detected in myeloid cells in all 4 cases of myelodysplastic syndrome. In aggregate, these data indicate that chronic lymphocytic leukemia with del(20q) acquired after therapy is heterogeneous. In cases with morphologic evidence of dysplasia, the del(20q) likely resides in the myeloid lineage. However, in cases without morphologic evidence of dysplasia, the del(20q) may represent clonal evolution and disease progression. Combining morphologic analysis with FISH for del(20q) or performing FISH on immunomagnetically-selected subpopulations to localize the cell population with this abnormality may help guide patient management.
Keywords: chronic lymphocytic leukemia, del(20q), disease progression, unmutated IGHV genes, combined morphologic and FISH analysis
Introduction
Interstitial deletion of the long arm of chromosome 20, del(20q), is a common recurrent cytogenetic abnormality in myeloid malignancies, including myeloproliferative neoplasms, myelodysplastic syndromes, and acute myeloid leukemias, reported in approximately 10%, 4%, and 2% of cases, respectively (1–3). In myeloproliferative neoplasms, the presence of del(20q) appears to have no adverse effect on patient survival (4, 5). Similarly, del(20q) as the sole cytogenetic abnormality in patients with myelodysplastic syndromes is associated with good survival and a low risk of leukemic transformation (6, 7). In contrast, del(20q) has been associated with a poor response to treatment and reduced survival in acute myeloid leukemia (4).
In patients with chronic lymphocytic leukemia, the common recurrent cytogenetic abnormalities identified by fluorescence in situ hybridization (FISH) analysis in about 80% of patients include del(11)(q22.3), del(13)(q14.3), +12, and del(17)(p13.1) (8). Each of these cytogenetic subtypes is associated with distinct clinical, prognostic, and pathologic features (8). Deletion 20q is unusual in lymphoproliferative disorders including chronic lymphocytic leukemia. The clinical features of chronic lymphocytic leukemia with del(20q) have been described in detail in only a single case report (9). Deletion 20q in chronic lymphocytic leukemia without clinical information is reported in seven other publications as single cases (10–16).
We report the clinicopathologic, morphologic, immunophenotypic, and molecular genetic features of 64 cases of chronic lymphocytic leukemia with del(20q), the largest series to date. We performed combined morphologic and FISH analysis for del(20q) in a subset of cases. Our results indicate that chronic lymphocytic leukemia with del(20q) is heterogeneous. In a small subset of patients, we identified the del(20q) in myeloid or erythroid cells, where it may represent an age- or therapy-related myeloid neoplasm. In the majority of the patients, we identified the del(20q) in chronic lymphocytic leukemia cells, where it is likely a manifestation of disease progression. These two groups require different therapeutic approaches.
Materials and Methods
Case selection
We searched the files of our Clinical Cytogenetics Laboratory for cases of chronic lymphocytic leukemia with del(20q) between 1/1//1991 and 5/31/2014. The cases were reviewed, and the diagnoses of chronic lymphocytic leukemia and myeloid neoplasms were characterized using the morphologic and immunophenotypic criteria as specified in the World Health Organization classification (17, 18). The clinical data were obtained by review of medical records.
Morphologic examination
We reviewed H&E-stained bone marrow core biopsy and clot specimens, as well as Wright-Giemsa-stained aspirate smears and touch imprints. The bone marrow cellularity and pattern of lymphocytic infiltration were assessed in the core biopsy specimens; the pattern was classified as nodular, interstitial, diffuse, or a combination of these patterns. We performed 500-cell differential counts on aspirate smears or touch imprints. We paid particular attention to the cytologic features of the lymphocytes with respect to atypical morphologic features, including indented or clefted nuclei, plasmacytoid features, and the presence of prolymphocytes. The percentages of plasmacytoid lymphocytes, defined as cells with eccentrically placed nuclei, moderately abundant cytoplasm, and/or cartwheel-like chromatin, and lymphocytes with indented nuclei were recorded. Dysplasia in myeloid cells, erythrocytes, and megakaryocytes was assessed based on the criteria as specified in the World Health Organization classification (18).
Immunophenotypic analysis
Flow cytometry immunophenotyping was performed on bone marrow aspirates using a FACScan instrument (Becton Dickinson, San Jose, CA) as described previously (19). The lymphocyte population was gated using right-angle side scatter and CD45 expression. The panel of monoclonal antibodies included reagents specific for CD3, CD5, CD10, CD11c, CD19, CD20, CD22, CD23, CD38, CD79b, FMC-7, surface immunoglobulin light chains, and IgM/IgD.
We calculated chronic lymphocytic leukemia scores according to the system proposed by Matutes and colleagues (20) and subsequently modified by Moreau and colleagues (21). Scores were based on five variables: expression of dim surface immunoglobulin (1 point), CD5 (1 point) and CD23 (1 point); dim or absent CD22/CD79b (1 point); and absent FMC-7 (1 point). Cases with a score of 4 or 5 were considered to have a typical immunophenotype; cases with a score of less than 4 were considered to have an atypical immunophenotype.
Immunohistochemical stains were performed on formalin-fixed, paraffin-embedded bone marrow core biopsies or clot sections. Staining for ZAP70 (dilution 1:500, Upstate Cell Signaling Systems, Lake Placid, NY) was performed as described previously (22). Cases with nuclear staining in ≥20% of the neoplastic cells were considered positive. Nuclear staining of admixed non-neoplastic T-cells served as an internal control. We also performed immunostain for cyclin D1 (SP4, Lab Vision/NeoMarkers, Fremont, CA) in a subset of cases.
Conventional cytogenetic and fluorescence in situ hybridization analyses
Conventional cytogenetic analysis was performed on metaphase cells from bone marrow aspirates or peripheral blood cultured for 24 hours without mitogens and for 72 hours with lipopolysaccharide, using standard techniques. Giemsa-banded metaphases were analyzed, and results were reported using the International System for Human Cytogenetic Nomenclature.
FISH analysis for the common chronic lymphocytic leukemia-associated cytogenetic abnormalities was performed in 46 cases on interphase nuclei from cultures of bone marrow or peripheral blood using a panel of probes designed to detect deletions of 11q22.3 (ATM), 13q14.3, 13q34, and 17p13 (TP53), and trisomy 12, according to the manufacturer’s instructions (Vysis/Abbott, Des Plaines, IL). FISH to detect del(20)(q12) was performed using the LSI D20S108 probe (Vysis/Abbott).
Combined morphologic and fluorescence in situ hybridization analysis
Combined morphologic and FISH analysis was performed as described previously (23). Briefly, Wright-Giemsa-stained bone marrow aspirate smears were reviewed to select target cell populations, and images were captured using bright-field microscopy. The smears were then destained using 1% acid alcohol (1% HCl in 70% ethanol), treated with protease II (Vysis/Abbott), and hybridized with a fluorescently-labeled LSI D20S108 probe (Vysis/Abbott). The target cell populations were then matched under fluorescent microscope, and photographed.
Somatic mutation status of the IGHV genes
Sequence analysis of the immunoglobulin heavy chain variable region (IGHV) gene was performed in 33 cases using total RNA extracted from bone marrow aspirates or peripheral blood as described previously (24). Patients’ sequences were aligned to the germline sequences in the V-BASE 2 database (25). The IGHV mutation status was designated as “unmutated” if there were fewer than 2% mutations (≥98% homology to germline sequences) or as “mutated” if there were 2% or more mutations (<98% homology) compared with the germline sequences (26).
Results
Clinical findings
We identified 64 cases of chronic lymphocytic leukemia with del(20q), 53 men and 11 women, representing <1% of all chronic lymphocytic leukemia cases. At initial diagnosis of chronic lymphocytic leukemia, the median age was 57 years (range, 31–77). The patients presented with absolute lymphocytosis (n=35), anemia (n=1), or thrombocytopenia (n=1) detected on routine physical examination, lymphadenopathy (n=21), or absolute lymphocytosis found during evaluation for B-symptoms (n=3) or chest pain (n=2); clinical presentation information was unavailable for one patient.
The clinical and laboratory data when patients presented to our institution are summarized in Table 1. Forty-two patients had received treatment for chronic lymphocytic leukemia and 22 were treatment-naive. Twelve patients (19%) reported B-symptoms, 56 (88%) had generalized lymphadenopathy, and 25 (39%) had splenomegaly. Forty-eight patients (75%) had absolute lymphocytosis (range, 5.0–324.3x109/L; median, 25.4x109/L; reference range, 1.0–4.8×109/L), 43 (67%) were anemic (hemoglobin range, 4.1–13.9 g/dL; median, 11.8 g/dL; reference range, 14.0–18.0 g/dL for men and 12.0–16.0 g/dL for women), and 36 (56%) were thrombocytopenic (range, 4–139×109/L; median, 82×109/L; reference range, 140–440×109/L). Serum lactate dehydrogenase was elevated in 30 patients (47%) (range, 621–3357 U/L; median, 752 U/L; reference range, 313–618 U/L), as was serum β2-microglobulin (≥4 mg/L) in 26 patients (40%) (range, 4.1–10.3 mg/L; median, 5.9 mg/L; reference range, 0.7–1.8 mg/L). The patients were in Rai stages 0 (n=2; 3%), I (n=21; 33%), II (n=9; 14%), III (n=4; 6%) and IV (n=28; 44%).
Table 1.
Clinical findings*
| Number | Median | Range | |
|---|---|---|---|
| Gender | |||
| Male | 53 | ||
| Female | 11 | ||
| B-symptoms | 12 | ||
| Lymphadenopathy | 56 | ||
| Splenomegaly | 25 | ||
| Lymphocytosis (x109/L) | 48 | 25.4 | 5.0–324.3 |
| Anemia (Hgb, g/dL) | 43 | 11.8 | 4.1–13.9 |
| Thrombocytopenia (x109/L) | 36 | 82 | 4–139 |
| LDH (U/L) | 30 | 752 | 621–3357 |
| β2M (mg/L) | 26 | 5.9 | 4.1–10.3 |
| Rai stage | |||
| 0 | 2 | ||
| I | 21 | ||
| II | 9 | ||
| III | 4 | ||
| IV | 28 | ||
At presentation to our institution
Abbreviations: Hgb, hemoglobin; LDH, lactate dehydrogenase; β2M, β-2 microglobulin
Sixty-two patients received treatment for chronic lymphocytic leukemia at our institution; 58 received fludarabine, cyclophosphamide and rituximab (FCR)-based therapy, 2 received fludarabine and cyclophosphamide, 1 received bendamustine and rituximab, and 1 received rituximab. Fourteen patients subsequently underwent bone marrow transplantation. Two patients remain untreated.
Of the nine patients (14%) who subsequently developed diffuse large B-cell lymphoma (Richter’s transformation), seven died, five after therapy with cyclophosphamide, vincristine, doxorubicin and dexamethasone (hyper-CVAD), and two after therapy with cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) and bone marrow transplantation. Two patients remain alive, one with persistent chronic lymphocytic leukemia following treatment with ABT-199, a BCL2 inhibitor, and one in clinical remission following treatment with hyper-CVAD and bone marrow transplantation.
Ten patients (16%) received a diagnosis of a myeloid neoplasm. Three patients had a myeloid neoplasm at presentation to our institution: one had a 10-year history of JAK2 V617F-positive polycythemia vera and received hydroxyurea before developing chronic lymphocytic leukemia; one had a 2-year history of chronic lymphocytic leukemia treated with FCR, and presented with both chronic lymphocytic leukemia and myelodysplastic syndromes; and one treatment-naïve patient developed myelodysplastic syndrome two months after developing chronic lymphocytic leukemia. After presenting to our institution, seven additional patients developed a myeloid neoplasm following therapy for chronic lymphocytic leukemia; one patient developed JAK2 V617F-positive primary myelofibrosis 139 months after treatment and six developed myelodysplastic syndromes after treatment (median, 67 months; range, 28–167). Thus, the myeloid neoplasms in eight patients, seven with myelodysplastic syndrome and one with myeloproliferative neoplasm, are considered therapy-related.
With a median follow-up of 90 months from the initial diagnosis of chronic lymphocytic leukemia (range, 12–252), 30 patients (47%) died of chronic lymphocytic leukemia (n=21, including 7 with development of diffuse large B-cell lymphoma), graft vs. host disease (n=5), secondary acute myeloid leukemia (n=2), or lung cancer with persistent chronic lymphocytic leukemia (n=2). Thirty-four patients (53%) remain alive, 19 with persistent chronic lymphocytic leukemia and 15 in clinical remission.
Morphologic and immunophenotypic findings
At presentation to our institution, all patients showed bone marrow involvement by chronic lymphocytic leukemia. The morphologic features of a representative case are illustrated in Figure 1A. The core biopsies showed a median cellularity of 60% (range, 20–95%). Lymphoid cells infiltrated the bone marrow in an interstitial (n=24), diffuse (n=10), nodular (n=8), or mixed (n=22) pattern, and involved 5–100% of the total cellular elements (median, 80%). The aspirate smears showed predominantly small lymphoid cells, but 43 cases (67%) showed atypical cytologic features. Thirty-one cases contained a subset (≥15%) of lymphoid cells with indented and irregular nuclear contours, and 28 showed a subset (≥15%) of plasmacytoid lymphocytes. Fifteen patients had increased (≥5%) prolymphocytes (range, 5–15%; median, 7%) (Figure 1B). Three patients also showed a myeloid neoplasm in addition to chronic lymphocytic leukemia, two with myelodysplastic syndromes (Figure 2) and one with polycythemia vera.
Figure 1. Morphologic findings of chronic lymphocytic leukemia with del(20q).
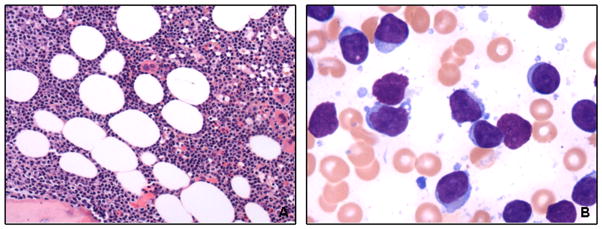
(A) Bone marrow biopsy from a representative case shows chronic lymphocytic leukemia cells extensively infiltrating the marrow (H&E, 200x). (B) The aspirate smear shows atypical chronic lymphocytic leukemia cells with irregular nuclear contours and plasmacytoid differentiation (Wright-Giemsa, 1 000x).
Figure 2. Morphologic findings of a case of chronic lymphocytic leukemia with myelodysplastic syndrome.

Bone marrow biopsy and aspirate smear demonstrate dysplastic features in (A) megakaryocytes (H&E, 400x), (B) erythrocytes (Wright-Giemsa, 1 000x), and (C) granulocytes (Wright-Giemsa, 1 000x), as well as (D) residual chronic lymphocytic leukemia (H&E, 200x).
Despite the presence of atypical cytologic features in 43 cases, the leukemic cells demonstrated typical immunophenotypes in 62 cases (97%), with a chronic lymphocytic leukemia score of 4 or 5 (17, 20, 21). Flow cytometry immunophenotyping demonstrated that the neoplastic cells in all 64 cases were positive for CD5, CD19, and dim surface monotypic immunoglobulin light chain (40 kappa, 23 lambda, 1 lacking expression of surface light chains). In addition, the neoplastic cells were positive for CD23 in 62/64 (97%), dim to moderate CD20 in 60/64 (94%), CD11c in 37/54 (69%), CD38 in 41/60 (68%), CD79b (dim) in 22/55 (40%), CD22 (dim) in 20/54 (37%), and FMC-7 (dim) in 4/64 (6%) cases. In all cases assessed, the cells were negative for CD3 (n=64) and CD10 (n=45). Two cases showed atypical immunophenotypes, with a chronic lymphocytic leukemia score of 3; one showed moderate expression of CD20 and FMC-7, and the other lacked CD23 and expressed FMC-7. Immunostain demonstrated that 30/36 cases (83%) expressed ZAP70, with >90% of the leukemic cells positive. Immunostain for cyclin D1 performed in 17 cases, including the two cases with atypical immunophenotypes, was negative.
Cytogenetic and molecular diagnostic findings
Overall, at the time the del(20q) was identified, 46 cases showed simple abnormal karyotypes (1–2 abnormalities) and 18 showed complex abnormal karyotypes (≥3 abnormalities). The del(20q) was the sole abnormality in 40 cases. In two cases (one simple and one complex) it was the sole abnormality in the stemline, i.e., the most basic clone of the tumor cell population, and in eight cases (one simple and seven complex) it was one of two or more stemline abnormalities. In twelve cases (three simple and nine complex) the del(20q) was the sole abnormality in one clone in cases that showed clonal divergence, i.e., two or more cytogenetically-unrelated clones. It was a secondary abnormality in only three cases (two simple and one complex).
The del(20q) was detected only after chemotherapy for chronic lymphocytic leukemia in all 27 cases with karyotypes at initial diagnosis available for comparison, with a median interval from the time of initial diagnosis to the detection of del(20q) of 65 months (range, 2–212). The results of conventional cytogenetic analysis performed at the time of initial chronic lymphocytic leukemia diagnosis were unavailable for 37 cases.
FISH analysis to assess the common recurrent chronic lymphocytic leukemia abnormalities performed in 46 cases at presentation to our institution demonstrated at least one abnormality in 37 (80%) (Table 2). Deletions of 11q22.3, 13q14.3, 17p13.1, and trisomy 12 were detected in 16 (35%), 20 (43%), 11 (24%), and 15 (33%) cases, respectively. Twenty-five cases (54%) had one FISH abnormality, seventeen cases (37%) had two abnormalities, three cases (7%) had three abnormalities, and one case (2%) had four abnormalities. Two cases with del(13)(q14.3) also showed del(13)(q34), suggesting monosomy 13. Interphase FISH analysis for del(20)(q12) performed as part of the initial clinical evaluation at our institution was positive in all 27 cases tested.
Table 2.
Cytogenetic and molecular genetic features
| Number | |
|---|---|
| del(20q)* | |
| sole abnormality | 40 |
| sole abnormality with clonal divergence | 12 |
| stemline abnormality | 9 |
| secondary abnormality | 3 |
| FISH | |
| del(13)(q14.3) | 20/46 |
| del(11)(q22.3) | 16/46 |
| trisomy 12 | 15/46 |
| del(13)(q34) | 2/46 |
| del(17)(p13) | 11/46 |
| Unmutated IGHV genes | 28/33 |
On conventional karyotypic analysis
Of the eight patients who developed a therapy-related myeloid neoplasm, the del(20q) was identified in three before the diagnosis of therapy-related myelodysplastic syndrome (12, 17, and 27 months), in one before the diagnosis of primary fibrosis (160 months), in three at the time of diagnosis of therapy-related myelodysplastic syndrome, and in one after the diagnosis of myelodysplastic syndrome (15 months). In order to determine the lineage of the cells that harbored the del(20q), we performed combined morphologic and FISH analysis in 16 cases with aspirate smears available for study, 13 previously-untested cases and three case previously assessed by interphase FISH. We identified the del(20)(q12) in myeloid and/or erythroid cells in 11 cases (69%): all four cases tested with therapy-related myelodysplastic syndrome (Figure 3A and 3B), one case with slight dysplasia that did not meet the morphologic criteria for myelodysplastic syndrome, and six cases without myeloid dysplasia. We identified the del(20)(q12) in lymphoid cells in five cases (31%), none with morphologic evidence of a myeloid neoplasm (Figures 3C and 3D). Thus, in 11 cases without evidence of myelodysplasia, combined morphologic and FISH analysis localized the del(20q) to the chronic lymphocytic leukemia cells in about half of cases, and to the myeloid/erythroid cells in the other half. Upon review, we could identify no karyotypic abnormalities that would have allowed us to distinguish between these cases.
Figure 3. Combined morphologic and FISH analysis.

(A and B) The del(20q) is identified in myeloid and erythroid cells in a case of myelodysplastic syndrome (100x). (C and D) The del(20q) is identified in lymphoid cells in a case without morphologic evidence of a myeloid neoplasm (100x).
Sequence analysis to evaluate the somatic mutation status of the IGHV genes performed in 33 cases demonstrated that 28 cases were unmutated and 5 were mutated.
Discussion
Because del(20q) is a common cytogenetic abnormality in myeloid neoplasms of older adults (1–3) and unusual in lymphoproliferative disorders, it would be reasonable to assume that the presence of a clone with del(20q) in a chronic lymphocytic leukemia patient represents an evolving myeloid neoplasm, either age-related or secondary to chemotherapy. However, we observed that in a subset of chronic lymphocytic leukemia patients with del(20q), the abnormality was present in a fraction of interphase nuclei by FISH, i.e., non-dividing cells, that far exceeded the fraction of myeloid cells in the bone marrow aspirates, and was more consistent with the extent of involvement by chronic lymphocytic leukemia. In addition, most of these cases lacked morphologic features of myelodysplasia either at the time the del(20q) was detected or at follow-up. Based on these observations, we hypothesized that, in at least a subset of cases, the del(20q) resided in the chronic lymphocytic leukemia, rather than in the myeloid cells. We performed combined morphologic and FISH analysis to determine the lineage of the cells that contained the cytogenetic abnormality. Although our sample size is small, in approximately half of the patients we identified the del(20q) in the chronic lymphocytic leukemia cells; in the other half we identified it in the myeloid and/or erythroid cells. Thus, chronic lymphocytic leukemia with del(20q) is a heterogeneous group.
There are few reports in the literature of chronic lymphocytic leukemia with del(20q). We found a single well-documented case report. The patient, an 80-year-old woman, presented with generalized lymphadenopathy, anemia, and thrombocytopenia. Small lymphoid cells, some with plasmacytoid features, infiltrated the bone marrow. Cytogenetic analysis revealed del(20)(q11.2) as the sole cytogenetic abnormality. The patient received supportive care, and died 12 months later from bilateral bronchopneumonia and ischemic heart disease (9). The cytogenetic features of a handful of other cases have been reported in the literature (10–16).
A number of features suggest that del(20q) in chronic lymphocytic leukemia is associated with disease progression and poor prognosis. In general, the neoplastic cells showed one or more markers of poor prognosis, including atypical cytologic features, unmutated IGHV gene, ZAP70 expression, and one of the high-risk recurrent cytogenetic abnormalities (8, 27–32). With a median follow-up of less than 8 years, almost all patients required therapy for chronic lymphocytic leukemia and almost half died, most as a consequence of chronic lymphocytic leukemia. Most of these cases lacked morphologic features of myelodysplasia either at the time del(20q) was detected or at follow-up. Only a small subset of the patients developed a myeloid neoplasm.
The presence of del(20q) as either the sole or a stemline karyotypic abnormality in more than 95% of our cases raises the possibility that it is a critical genetic event. It also suggests the presence of a gene (or genes) on the long arm of chromosome 20 that functions as a tumor suppressor. In myeloid neoplasms, potential candidate genes that map to this region include Dido (33), L3MBTL1 (34), and MYBL2 (35). There are few genetic studies of del(20q) in lymphoproliferative lesions. However, loss of heterozygosity (LOH) in the region of 20q11.2 to 20q13.3 has been described in chronic lymphocytic leukemia patients, raising the possibility of a tumor suppressor gene in this region (36, 37). In a study of 19 patients with large cell transformation, Lee et al. described LOH in a region that corresponds to 20q12 within large cells in over half of the cases studied (36). Recently, Clifford, et al. has identified SAMHD1, located at chromosome 20q11.23, as a tumor suppressor in chronic lymphocytic leukemia. The protein encoded by SAMHD1 normally degrades intracellular deoxynucleoside triphosphates, and is involved in the response to DNA damage and in cell proliferation and survival. They found that SAMHD1 is recurrently mutated in 11% of recurrent/refractory chronic lymphocytic leukemia cases. Further, these mutations were associated with copy-neutral LOH or loss of the other allele (38).
In conclusion, our results show that chronic lymphocytic leukemia cases with del(20q) that acquired after therapy is heterogeneous. In cases with morphologic evidence of myelodysplasia, the del(20q) likely resides in the myeloid cells. However, in cases that lack morphologic evidence of myelodysplastic syndrome, del(20q) may represent clonal evolution and chronic lymphocytic leukemia progression. In the absence of morphologic evidence of a myelodysplastic syndrome, either combining morphologic analysis with FISH for del(20q), or performing FISH on immunomagnetically-selected subpopulations in order to localize the cell population in which this abnormality resides may help guide patient management.
Supplementary Material
Acknowledgments
The authors thank Dr. Nyla Heerema for critically reviewing the manuscript.
Footnotes
Disclosure/Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Heim S, Mitelman F. Cytogenetic analysis in the diagnosis of acute leukemia. Cancer. 1992;70:1701–9. doi: 10.1002/1097-0142(19920915)70:4+<1701::aid-cncr2820701609>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 2.Fenaux P, Morel P, Lai JL. Cytogenetics of myelodysplastic syndromes. Semin Hematol. 1996;33:127–38. [PubMed] [Google Scholar]
- 3.Bench AJ, Aldred MA, Humphray SJ, et al. A detailed physical and transcriptional map of the region of chromosome 20 that is deleted in myeloproliferative disorders and refinement of the common deleted region. Genomics. 1998;49:351–62. doi: 10.1006/geno.1998.5231. [DOI] [PubMed] [Google Scholar]
- 4.Campbell LJ, Garson OM. The prognostic significance of deletion of the long arm of chromosome 20 in myeloid disorders. Leukemia. 1994;8:67–71. [PubMed] [Google Scholar]
- 5.Hussein K, Van Dyke DL, Tefferi A. Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol. 2009;82:329–38. doi: 10.1111/j.1600-0609.2009.01224.x. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88. [PubMed] [Google Scholar]
- 7.Braun T, de Botton S, Taksin AL, et al. Characteristics and outcome of myelodysplastic syndromes (MDS) with isolated 20q deletion: a report on 62 cases. Leuk Res. 2011;35:863–7. doi: 10.1016/j.leukres.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 9.Cotter PD, Tumewu P, Browett PJ. Deletion of the long arm of chromosome 20 in a patient with small cell lymphocytic lymphoma. Cancer Genet Cytogenet. 1993;70:142–3. doi: 10.1016/0165-4608(93)90186-p. [DOI] [PubMed] [Google Scholar]
- 10.Barbieri D, Michaux JL, Bosly A, et al. Cytogenetic evaluation of bone marrow involvement in non-Hodgkin’s lymphomas. A survey of 94 cases. Haematologica. 1984;69:285–96. [PubMed] [Google Scholar]
- 11.Fegan C, Robinson H, Thompson P, Whittaker JA, White D. Karyotypic evolution in CLL: identification of a new sub-group of patients with deletions of 11q and advanced or progressive disease. Leukemia. 1995;9:2003–8. [PubMed] [Google Scholar]
- 12.Espinet B, Solé F, Woessner S, Florensa L, Lloveras E, Besses C. Cytogenetic and in situ hybridization findings in 27 patients with atypical B-cell chronic lymphocytic leukaemia. Haematologica. 1999;84:569–71. [PubMed] [Google Scholar]
- 13.Wiktor A, Van Dyke DL. Combined cytogenetic testing and fluorescence in situ hybridization analysis in the study of chronic lymphocytic leukemia and multiple myeloma. Cancer Genet Cytogenet. 2004;153:73–6. doi: 10.1016/j.cancergencyto.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Brezinová J, Zemanová Z, Ransdorfová S, et al. Prognostic significance of del(20q) in patients with hematological malignancies. Cancer Genet Cytogenet. 2005;160:188–92. doi: 10.1016/j.cancergencyto.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 15.Han JY, Theil KS, Hoeltge G. Frequencies and characterization of cytogenetically unrelated clones in various hematologic malignancies: seven years of experiences in a single institution. Cancer Genet Cytogenet. 2006;164:128132. doi: 10.1016/j.cancergencyto.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 16.Reddy KS. Chronic lymphocytic leukaemia profiled for prognosis using a fluorescence in situ hybridisation panel. Br J Haematol. 2006;132:705–22. doi: 10.1111/j.1365-2141.2005.05919.x. [DOI] [PubMed] [Google Scholar]
- 17.Müller-Hermelink HK, Montserrat E, Catovsky D, Campo E, Harris NL, Stein H. Chronic lymphocytic leukaemia/small lymphocytic lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC; Lyon: 2008. pp. 180–2. [Google Scholar]
- 18.Brunning RD, Porwit A, Orazi A, et al. Myelodysplastic syndromes/neoplasms, overview. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC; Lyon: 2008. pp. 88–93. [Google Scholar]
- 19.Yin CC, Lin P, Carney DA, et al. Chronic lymphocytic leukemia/small lymphocytic lymphoma associated with IgM paraprotein. Am J Clin Pathol. 2005;123:594–602. doi: 10.1309/FDGW-B5C2-MYRY-XH2E. [DOI] [PubMed] [Google Scholar]
- 20.Matutes E, Owusu-Ankomah K, Morilla R, et al. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia. 1994;8:1640–5. [PubMed] [Google Scholar]
- 21.Moreau EJ, Matutes E, A’Hern RP, et al. Improvement of the chronic lymphocytic leukemia scoring system with the monoclonal antibody SN8 (CD79b) Am J Clin Pathol. 1997;108:378–82. doi: 10.1093/ajcp/108.4.378. [DOI] [PubMed] [Google Scholar]
- 22.Admirand JH, Rassidakis GZ, Abruzzo LV, Valbuena JR, Jones D, Medeiros LJ. Immunohistochemical detection of ZAP-70 in 341 cases of non-Hodgkin and Hodgkin lymphoma. Mod Pathol. 2004;17:954–61. doi: 10.1038/modpathol.3800145. [DOI] [PubMed] [Google Scholar]
- 23.Wang SA, Hutchinson L, Tang G, Chen SS, Miron PM, Huh YO, et al. Systemic mastocytosis with associated clonal hematological non-mast cell lineage disease: clinical significance and comparison of chromosomal abnormalities in SM and AHNMD components. Am J Hematol. 2013;88:219–24. doi: 10.1002/ajh.23380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin CC, Medeiros LJ, Cromwell CC, et al. Sequence analysis proves clonal identity in five patients with typical and blastoid mantle cell lymphoma. Mod Pathol. 2007;20:1–7. doi: 10.1038/modpathol.3800716. [DOI] [PubMed] [Google Scholar]
- 25.Retter I, Althaus HH, Münch R, Muller W. VBASE2, an integrative V gene database. Nucleic Acids Res. 2005;33:D671–4. doi: 10.1093/nar/gki088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fais F, Ghiotto F, Hashimoto S, et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J Clin Invest. 1998;102:1515–25. doi: 10.1172/JCI3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oscier DG, Matutes E, Copplestone A, et al. Atypical lymphocyte morphology: an adverse prognostic factor for disease progression in stage A SLL independent of trisomy 12. Br J Haematol. 1997;98:934–9. doi: 10.1046/j.1365-2141.1997.3263141.x. [DOI] [PubMed] [Google Scholar]
- 28.Criel A, Michaux L, De Wolf-Peeters C. The concept of typical and atypical chronic lymphocytic leukaemia. Leuk Lymphoma. 1999;33:33–45. doi: 10.3109/10428199909093723. [DOI] [PubMed] [Google Scholar]
- 29.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–7. [PubMed] [Google Scholar]
- 30.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated IgVH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–54. [PubMed] [Google Scholar]
- 31.Crespo M, Bosch F, Villamor N, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. 2003;348:1764–75. doi: 10.1056/NEJMoa023143. [DOI] [PubMed] [Google Scholar]
- 32.Wiestner A, Rosenwald A, Barry TS, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood. 2003;101:4944–51. doi: 10.1182/blood-2002-10-3306. [DOI] [PubMed] [Google Scholar]
- 33.Futterer A, Campanero MR, Leonardo E, et al. Dido gene expression alterations are implicated in the induction of hematological myeloid neoplasms. J Clin Invest. 2005;115:2351–62. doi: 10.1172/JCI24177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurvich N, Perna F, Farina A, et al. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc Natl Acad Sci U S A. 2010;107:22552–7. doi: 10.1073/pnas.1017092108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinrichs S, Conover LF, Bueso-Ramos CE. MYBL2 is a sub-haploinsufficient tumor suppressor gene in myeloid malignancy. Elife. 2013;2:e00825. doi: 10.7554/eLife.00825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JN, Giles F, Huh YO, et al. Molecular differences between small and large cells in patients with chronic lymphocytic leukemia. Eur J Haematol. 2003;71:235–42. doi: 10.1034/j.1600-0609.2003.00089.x. [DOI] [PubMed] [Google Scholar]
- 37.Pei J, Robu V, Feder M, et al. Copy neutral loss of heterozygosity in 20q in chronic lymphocytic leukemia/small lymphocytic lymphoma. Cancer Genet. 2014;207:98–102. doi: 10.1016/j.cancergen.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clifford R, Louis T, Robbe P, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123:1021–31. doi: 10.1182/blood-2013-04-490847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.