

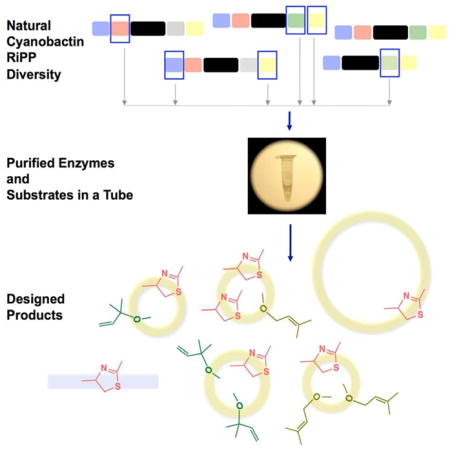
SUMMARY
Macrocyclases and other posttranslational enzymes afford derived peptides with improved properties for pharmaceutical and biotechnological applications. Here, we asked whether multiple posttranslational modifications could be simultaneously controlled and matched to rationally generate new peptide derivatives. We reconstituted the cyanobactin peptide natural products in vitro with up to five different posttranslational enzymes in a single tube. By manipulating the order of addition and identity of enzymes and exploiting their broad-substrate tolerance, we engineered the production of highly unnatural derivatives, including an N-C peptide macrocycle of 22 amino acids in length. In addition to engineering, this work better defines the macrocyclization mechanism, provides the first biochemical demonstration of Ser/Thr posttranslational prenylation, and is the first example of reconstitution of a native, multistep RiPP pathway with multiple enzymes in one pot. Overall, this work demonstrates how the modularity of posttranslational modification enzymes can be used to design and synthesize desirable peptide motifs.
Keywords: Cyclic peptide, Cyanobactin, Posttranslational modification, RiPP
Graphical abstract
INTRODUCTION
Cyclic peptides are of particular interest among the various bioactive macrocyclic scaffolds known so far. Their stability, coupled with their ability to access hard to reach targets such as protein-protein interactions, has led to significant enthusiasm concerning their therapeutic potential (White and Yudin, 2011). Most FDA-approved cyclic peptide drugs, such as cyclosporine and daptomycin, are synthesized by non-ribosomal multienzyme assembly lines (Sieber and Marahiel, 2003), but an increasing number of related cyclic compounds are also ribosomally synthesized (Arnison et al., 2013). The chemical synthesis of macrocyclic scaffolds has been well explored, but many drawbacks exist including those of specific sequence requirements, length restrictions, poor yield and high cost (Yu and Sun, 2013). In contrast, enzyme-catalyzed peptide macrocyclization offers many advantages over the conventional synthetic chemistry methods.
Well-studied enzymatic N-C macrocyclizations include those mediated by subtiligases, sortases and inteins (Zhang et al., 2014; Scott et al., 1999). In contrast to these unnatural processes, PatG-like enzymes from cyanobactin pathways are serine proteases that natively form macrocycles (Lee et al., 2009; McIntosh et al., 2010a; Agarwal et al., 2012; Koehnke et al., 2012). The macrocyclases creating the cyclotide and amanitin natural products are also being explored (Craik and Malik, 2013; Luo et al., 2012; Luo et al., 2014; Barber et al., 2013; Nguyen et al., 2014). In addition, a wide array of enzymes cyclizing products via side-chains (rather than as main chain N-C macrocycles) is also known (Arnison et al., 2013). Beyond simple synthesis of macrocycles, it is desirable to introduce tailoring reactions, which greatly modify the pharmacological properties of the peptides. In light of this, the aim of this study was to exploit a series of cyanobactin tailoring enzymes to produce highly modified cyclic peptides in vitro, using hybrid biosynthetic machineries comprising mixtures of enzymes from different pathways.
The cyanobactins are a class of natural products first isolated from marine ascidian animals (Ireland and Scheuer, 1980), where they are produced by symbiotic cyanobacteria (Schmidt et al., 2005). Cyanobactins are found in approximately 10–30% of all cyanobacteria on Earth (Leikoski et al., 2013), creating an enormous wealth of enzymes and substrates that can be exploited in synthetic biology. They are ribosomally synthesized and posttranslationally modified peptides (RiPPs) that are mostly cyclic, although some linear derivatives have been reported recently (Leikoski et al., 2013; Donia and Schmidt, 2011). The biosynthetic pathway involves a linear precursor peptide substrate, containing a core sequence that matures into the final natural product. The enzymes that modify the core sequence include at least two proteases, PatA and PatG and their homologs, which proteolyze the N- and C-termini of the core sequence, respectively (Lee et al., 2009). PatG also carries out N-C macrocyclization of the core in tandem with proteolysis (Lee et al., 2009; Schmidt et al., 2005). Often, multiple core peptides are encoded in a single precursor, such that multiple cyclic peptides are derived from single substrates. Further modifications such as heterocyclization, oxidation, prenylation and/or methylation maybe carried out by additional enzymes (Figure 1).
Figure 1. Simplified schematic of cyanobactin biosynthesis.
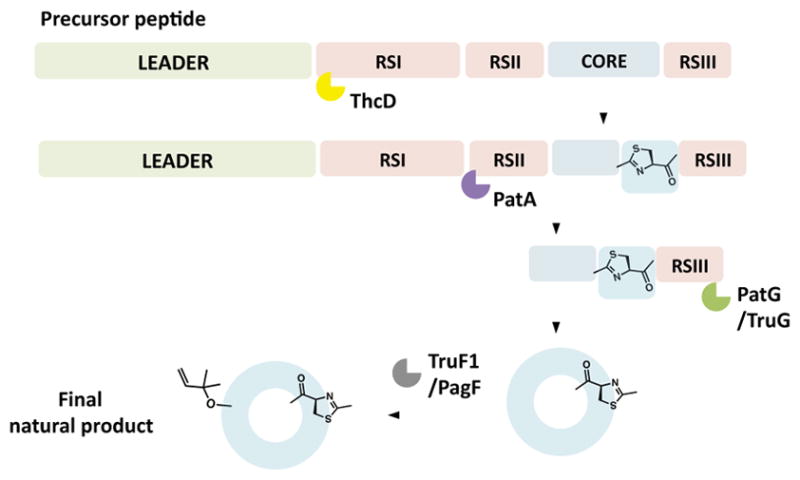
A schematic of cyanobactin biosynthetic machinery showing the leader peptide, core peptide, recognition sequences (RSs) and the posttranslational enzymes used in this study. ThcD is a heterocyclase that uses RSI to modify Cys residues to thiazoline. This is followed by the action of proteases PatA and PatG (or TruG) that use RSII and RSIII to cleave off the N- and C-termini of the core peptides respectively. PatG (or TruG) also carries out macrocyclization in tandem with proteolysis. The prenyltransferases TruF1 and PagF add isoprene units to the cyclic peptide scaffold and do not require RSs.
In nature, cyanobactins exist as large combinatorial libraries, leading to hundreds of known natural products and probably thousands of unknown compounds. This diversity arises in part from hypervariable core sequences, which are tolerated by exceptionally broad-substrate biosynthetic enzymes (Donia et al., 2006; Sardar et al., 2014). Broad-substrate tolerance is possible because the precursor contains specific recognition sequences (RSs) directing enzymes to otherwise hypervariable cores. RS conservation therefore at least in part explains the observed promiscuity of the posttranslational enzymes (Donia et al., 2006; Sardar et al., 2014). As elucidated from earlier work, RSI recruits a heterocyclase enzyme (PatD homolog) that acts on cysteine, serine, or threonine in a regio- and chemoselective manner. RSII and RSIII recruit proteases related to PatA (N-terminal protease) and PatG (C-terminal protease/macrocyclase), respectively. Proteolysis cleaves RSs from the core peptide, enabling “scarless” formation of the desired cyclic peptide with the recognition elements removed. The only exception in RS requirements lies in the final enzymatic step of prenylation, which does not seem to need RSs and instead recognizes cyclic peptides with diverse sequences (McIntosh et al., 2011) (Figure S1).
Our previous in vitro effort toward cyclic peptide production used a combination of PatA and PatG to generate eptidemnamide (an analog of the anticoagulant eptifibatide) (Lee et al., 2009). Subsequent work focused only on PatG activity using short synthetic substrates as starting material (McIntosh et al., 2010a, Agarwal et al., 2012; Koehnke et al., 2012). These substrates carried a proline residue at the C-terminus of the core since a heterocycle at this position was observed to be required for PatG activity (McIntosh et al., 2010a), although natural PatG substrates carry thiazole at this position. We have also synthesized small libraries in vitro using discrete substrates for the prenyltransferase enzymes (McIntosh et al., 2011). Despite having the ability to reconstitute individual enzymatic steps in vitro, the combination of multiple enzymatic steps to generate modified cyclic peptides has been much simpler in E. coli in vivo (Ruffner et al., 2014; Tianero et al., 2011). Using this methodology, the single tru pathway has been used to encode libraries that potentially synthesize millions of “unnatural natural products”. While in vivo libraries are extremely powerful in drug discovery, not all types of compounds are readily synthesized. For example, compounds toxic to E. coli are unlikely to be produced, and substrates that are degraded faster than they are modified by enzymes would not be observable.
Recent advances in heterocyclization chemistry using PatD and homologs (Sardar et al., 2014; Koehnke et al., 2013; Goto et al., 2014) enabled us to make short heterocycle containing peptides, which provided a simple method to generate native-like substrates for downstream enzymes in vitro. This in turn allowed us to optimize the conditions necessary to exploit the chemistry of a combination of these enzymes. Here, our efforts involved the coupling of PatD-like enzymes along with a mixture of other posttranslational enzymes to produce modified cyclic peptides. We based this work on the hypothesis that the RS elements act independently both from each other and from the core peptides. Therefore, appropriately embedded RS elements would universally and predictively recruit posttranslational modification enzymes, even when mixed from different biosynthetic pathways. We tested this hypothesis by designing and synthesizing a series of modified cyclic peptides, both natural and unnatural, using enzymes, precursors, and RS elements from the trunkamide, prenylagaramide, cyanothecamide, patellamide, and aestuaramide cyanobactin families.
RESULTS
Sequential enzymatic reactions reconstitute full pathway activity
Synthetic substrate 1, which carries the trunkamide core sequence TSIAPFC (Figure 2), was selected for initial studies because it exhibited promising biological activity (Salvatella et al., 2002). Additionally, we have developed substantial technology around this sequence (Ruffner et al., 2014; Tianero et al., 2011). In place of the complete precursor peptide, substrate 1 was previously engineered to contain solely RS elements I, II and III (Sardar et al., 2014), such that 1 represents a minimal substrate. Precursor 1 was only 25 amino acids in length, enabling chemical synthesis, whereas the native substrates are >50–80 amino acids and require more cumbersome methods. The enzymes used included: heterocyclase ThcD from the recently characterized cyanothecamide thc pathway (Donia and Schmidt, 2011; Sardar et al., 2014), N-terminal protease PatA (Lee et al., 2009), the C-terminal protease/macrocyclase PatG and TruG (Lee et al., 2009, McIntosh et al., 2010a; Agarwal et al., 2012; Koehnke et al., 2012) and the prenyltransferase TruF1 (McIntosh et al., 2011), from the well-characterized patellamide pat and trunkamide tru pathways. Although ThcD is not from the tru pathway, it exhibits the same chemoselectivity as TruD (Sardar et al., 2014). Additionally, PatA and TruA are virtually sequence identical (Lee et al., 2009).
Figure 2. In vitro synthesis of trunkamide derivatives.

(A) Schematic representation of the step-by-step reaction starting from the linear unmodified substrate 1 carrying the trunkamide core sequence TSIAPFC along with the necessary RSs. Action of ThcD heterocyclizes the Cys in the core to lead to 2, followed by proteolysis by N-terminal protease PatA to release the intermediate 4, which is further processed by the C-terminal protease/macrocyclase PatG to lead to the cyclic product 5. The addition of the prenyltransferase TruF1 further prenylates 5 to 6. (B) Mass spectra showing the masses of each reaction intermediate: 1, 1272.8 Da [M+2H]2+ and 849.0 Da [M+3H]3+; 2, 1264.2 Da [M+2H]2+ and 843.1 [M+3H]3+; 4, 1084.8 Da [M+H]+ and 543.1 Da [M+2H]2+; 5, 702.3 Da [M+H]+; and 6, 770.4 Da [M+H]+. See Fig. S2 for structures and higher resolution MS of each species. (C) Mass spectrum of the [M+H]+ of the product 5 resulting from a one-pot addition of enzymes ThcD, PatA and PatG. The structure of 5 is given with the thiazoline modification circled in yellow. See Figure S2-E for high-resolution mass spectrum of 5 with a mass error of < 2.5 ppm.
We investigated the action of enzymes on substrate 1 by sequentially adding the enzymes into the same tube along with the necessary enzyme cofactors at each step (see Methods). Since heterocyclization requires RSI, the ThcD reaction was performed first, leading to the thiazoline-containing product 2 (Sardar et al., 2014). Subsequent addition of PatA excised both RSI and the PatA recognition sequence RSII, leading to products 3 and 4. Since 4 carries thiazoline, it is a substrate for PatG. Addition of PatG macrocyclase domain to the reaction mixture cleaved the RSIII element in tandem with cyclization to provide the cyclic peptide 5. The addition of prenyltransferase TruF1 provided the monoprenylated product 6 (Figures 2 and S2-A). In our hands, only about 30% of 5 (as judged from area of integrated chromatogram) was converted to the prenylated product 6, and the doubly prenylated product was not detected (Figure S2-A). The same products 5 and 6 were observed with full-length protein TruG in place of the macrocyclase/protease domain of PatG (Figure S2-B). The structure of 5 was further confirmed by NMR characterization (Figure S2-C).
The substrate 1 was quantitatively consumed in the reaction, and less than 5% was converted to side products as detected by LC-MS (Figure S2-D). In the subtiligase and sortase systems, a yield of 67% and 85% respectively has been reported (Zhang et al., 2014), whereas with PatG, as shown here, it is possible to achieve nearly complete conversion to cyclic product in the reaction mixture.
Biochemical characterization of each posttranslational enzyme from previous studies has shown that each enzyme is fully functional independently, and no protein-protein interactions were detected between enzymes (McIntosh et al., 2010a; McIntosh et al., 2010b; McIntosh et al., 2010c). Here, we reinvestigated the possible requirement for protein interactions. Reactions were carried out where intermediates from each step were purified by HPLC and used as a substrate for the next modification step. These pure intermediates (2 and 4), free from enzymes and cofactors from the previous modification reaction, were fully competent substrates for the next enzymatic reaction in that they could be completely processed to the respective products (Figure S2-E).
Previous studies reported extremely slow reaction rates for PatG such that in many cases reaction was complete by 24 h only with a 50% catalyst load (McIntosh et al., 2010a; McIntosh et al., 2010c; Agarwal et al., 2012), although the rate could be modified by adjusting buffer conditions (Koehnke et al., 2012). In contrast, in this study PatG was comparatively faster in the formation of product 5 such that complete conversion to product was observed by 24 h using a 1:5 enzyme to substrate ratio. To test if presence of a thiazoline heterocycle instead of a proline could be responsible for this acceleration, we synthesized a variant of substrate 1 containing the same core sequence with a single point mutation of TSIAPFC to TSIAPFP (1a). Reactions were carried out with PatG to give the expected cyclic product 5a. A time-course comparison of amount of proline containing product 5a formed versus the thiazoline containing product 5 showed that formation of 5a was significantly slower (up to 4-fold) indicating that presence of the thiazoline at the C-terminal end improved PatG activity (Figure S2-F).
One-pot synthesis of cyanobactins
When all enzymes, cofactors and the substrate 1 were added together in one pot, rather than sequentially, the cyclic product 5 was readily synthesized (Figures 2 and S2-G). As observed for the sequential reaction, an essentially quantitative conversion to 5 was measured (Figure S2-H). The reaction was sensitive to the concentration of protease PatA, which could produce the non-heterocyclized N-terminally cleaved product 7 as a side-product (Figure S2-I). Absent heterocyclization, 7 is not a substrate for the macrocyclization reaction. When prenyltransferase TruF1 was added, the monoprenylated product 6 was produced (Figure S2-J), although as in the sequential reactions the complete conversion to 6 was not observed. Analysis of the one-pot reaction showed that PatG step is the limiting step in that it is much slower in comparison to either the ThcD or PatA step (Figure S2-K). To our knowledge, this is the first demonstration of reconstituting a complete, multistep RiPP biosynthetic pathway with multiple enzymes in a single pot.
Unprecedented macrocycle size plasticity
For several years, the ability to perform one-pot reactions eluded us and has proved challenging in other hands as well, where proteases other than PatA were required (Houssen et al., 2014). Here, we showed that this was due to differential redox sensitivity of various enzymes in the pathway. Neither ThcD nor PatG require the addition of reducing agent for activity, but PatA showed a significant decrease in activity if dithiothreitol (DTT) was used in the assay. DTT has usually been added to these reactions because of the prevalence of disulfide bonds in the precursor peptide. Hence, both the sequential and one-pot reactions described above to yield 5 were carried out in absence of DTT.
To our surprise, in one-pot reactions with 1 where PatA was inhibited by DTT, a macrocyclic product (8, m/z = 1072 Da, [M+2H]2+) was observed, resulting from macrocyclization of 2 (Figure S3-A). This product could also be recapitulated in reactions containing solely ThcD and PatG. PatG macrocyclase domain could also be replaced with full-length TruG, again affording the same product 8 (Figure S3-B). Complete conversion of 1 to the product 8 was observed, whether the reactions were performed in a single one-pot step (Figure S3-C) or via stepwise addition of enzymes (Figure 3). As with the cyclic peptide 5, the linear derivative of 8 (8 + H2O) was present only in barely detectable trace amounts.
Figure 3. Synthesis of 22-ring size macrocycle.

Using ThcD and PatG only, macrocycle 8 comprising 22 residues was synthesized from 1. The structure of 8 is given with the thiazoline modification shown in yellow along with the high-resolution FT-ICR mass spectrum showing the [M+2H]2+ with an error <1.5 ppm.
The structure of 8 was anticipated by high-resolution FT-ICR MS/MS of 8 in comparison to 2 and further confirmed by purification and NMR analysis (Figure S3-D,E and Table S1). Ring opening of the macrocycle as detailed in the next section provides further evidence confirming the structure of 8. Purified 8 was quantified by NMR using boc-tyrosine as a standard (Tianero et al., 2011). This in turn was used to draw a standard curve of 8, which was used to compute the yield of the reaction as 81% (Figure S3-F). A time-course of the formation of 8 in comparison to 5 in one-pot reactions is given in Figure S3-G. Since this unusually long substrate 2 was macrocyclized, we also examined a short substrate 9, comprising only 5 amino acids in the core peptide. Substrate 9 was also processed to the cyclic product 11, although with a lower yield (<40%) (Figure S3-H).
Here, we show that pat and tru enzymes can synthesize macrocycles in a size range of 5–22 amino acids in length. Previously, natural macrocycles from these pathways were known to comprise 6–8 amino acids, while artificial substrates had been 6–11 amino acids (McIntosh et al., 2010a). Beyond the pat and tru pathways, cyanobactins exist varying from sizes of as small as 3 residues (the linear products, viridisamides) (Leikoski et al., 2013) to up to 20 residues in cyclic products (Leikoski et al., 2012). A 43-residue macrocycle could be a possible product in Calothrix sp. PCC 7103 (Figure S3-I). However, the 22-residue 8 is the largest confirmed product of a cyanobactin pathway.
Enzymatic reactions in designed unnatural synthesis
RiPPs carry enormous potential for designer synthesis of derivatives. As an initial examination of this process, we noted that the large macrocycle 8 carries the PatA recognition sequence, RSII. Recognition motifs within cyclic peptide scaffolds have been shown to hold potential in stabilizing these scaffolds for enhanced protein-protein interactions (Davidson et al., 2009; de Vega et al., 2007). Thus, we expected that the macrocyclic product 8 might be proteolysed by PatA, which in turn will produce a derivative with the thiazoline residue in an abnormal position. The HPLC-purified product 8 (Figure S4-A) was used in assays with PatA, and the expected linear product 13 was indeed observed (Figure S4-B). A complete conversion to the linear product 13 was observed, as judged by disappearance of 8 in the mass spectrum. High-resolution FT-ICR MS/MS confirmed the sequence of the product 13 (Figure S4-B and Table S2). Additionally, PatA cleavage of both the cyclic substrate 8 and the linear substrate 2 was comparable in that both reactions were complete by 30 min (Figure S4-C). Thus, proteolysis within the cyclic peptide motif shifts the position of the heterocycle (Figure 4). This holds potential for engineering thiazoline containing peptides, wherein heterocycles can be introduced readily in any desired position within a linear peptide.
Figure 4. Manipulation of reaction order alters position of heterocycle.
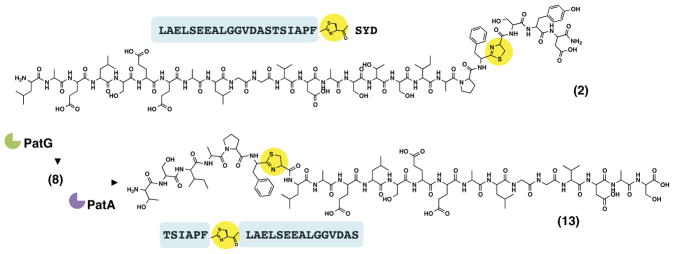
In native cyanobactin biosynthesis the order of enzymatic reactions starts with heterocyclization followed by N- and C-terminal proteolysis/macrocyclization, such that in this study ThcD would act first followed by PatA and PatG. Here, we changed the order of the reaction such that proteolysis by PatA was carried out after PatG macrocyclization of 2 to 8. This provided the linear product 13, which has its thiazoline ring (shown in yellow) shifted toward the N-terminus from the original C-terminal end position in the precursor 2.
Cyclic peptides from full-length artificial precursor peptides
Using the tru pathway in E. coli, we have synthesized many peptides with highly artificial sequences (Ruffner et al., 2014; Tianero et al., 2011). However, many sequences were not accepted. Here, we asked whether these in vitro failures were due to the cyanobactin enzymes, or due to some limiting factor in E. coli.
We previously sought to use 14 in vivo, but no products were detected. Peptide 14 encodes a precursor peptide sequence derived from the natural pag pathway (Donia and Schmidt, 2011), but where the native INPYLYP has been replaced with INPYLYC. Peptide 14 was a substrate for heterocyclization in vitro but not macrocyclization in vivo (Sardar et al., 2014).
Many tru precursor peptides contain exceptionally strong intra- or intermolecular disulfide bonds (McIntosh et al., 2010b; McIntosh et al., 2010c), necessitating use of the PatA-inhibiting DTT. Therefore 14 and ThcD were allowed to react in the presence of DTT, affording 14a, which was purified by HPLC. Addition of PatA led to production of 14b, which was then HPLC purified and used with PatG, affording the expected cyclic product 14c (cyc-INPYLYC*) (Figure 5). Minor amounts of the linearized precursor (14c + H2O) were also produced. However, the yield of 14c was quite low in comparison to other reactions performed in this study. The limiting factor was observed to be macrocyclization by PatG (Figure S5). Therefore, at least in this case the factor limiting in vivo production appears to be the macrocyclization reaction, since in vivo linear intermediates would likely be nonspecifically proteolyzed in the 5-day fermentation time course.
Figure 5. Synthesis of thiazoline containing pag derivative from full-length linear precursor.

ThcD modification on 14 adds a thiazoline to the core to yield 14a, which is followed by PatA to yield 14b that undergoes PatG processing to lead to 14c. The structure of 14c is shown along with its mass spectrum. The FT-ICR mass spectrum is given in Figure S5 with a mass error <4.5 ppm. Structures and mass spectrum of intermediates are also given in Figure S5. It is to be noted that natural prenylagaramide (pag) pathway products lack thiazolines unlike the unnatural derivative 14c.
Synthesis of multiple modified cyclic peptides in a single reaction batch from a precursor peptide carrying multiple cores
Some RiPP pathways contain multiple core sequences in a single precursor peptide. Apart from cyanobactins where multiple core sequences are very common, this feature has been observed only rarely as in the cyclotide and orbitide family of plant RiPPs (Arnison et al., 2013; Condie et al., 2011; Jennings et al., 2001), and also the ustiloxins fungal cyclic peptides that have been recently reported to be ribosomally synthesized (Umemura et al., 2014; Tsukui et al., 2015). We therefore asked whether a precursor peptide (15) containing multiple cores could be processed in vitro. Precursor 15 is a hybrid containing core sequence TFPVPTVC from the tru pathway and core sequence ACMPCYP from the lyn pathway (McIntosh et al., 2013). Using the stepwise methods applied to substrate 14, the expected products cyc-TFPVPTVC* 15c and cyc-AC*MC*YP 15d (where C* is thiazoline) were observed (Figures 6 and S6). As found with substrate 14, the PatG reaction was slow, and intermediates 15a–b were not fully cyclized within 24 h. The linear derivatives of 15c and 15d (15c–d + H2O) were not detectable in the reaction mixture. Cyclic peptide 15d previously was not synthesized in vivo (Sardar et al., 2014), again likely because of the slow reaction with PatG. Overall, it is clear that application of the in vitro system complements the in vivo method when macrocyclization becomes rate limiting.
Figure 6. Multiple cyclic peptides produced in a single reaction batch from a hybrid precursor peptide carrying multiple core sequences derived from the tru and lyn pathways.

As detailed for substrate 14, processing of 15 by ThcD, PatA and PatG led to the cyclic peptides 15c and 15d. The schematics of the reaction series are given along with the mass spectra of the products. See Figure S6 for mass spectra and structures of intermediates.
Combining posttranslational modifications from multiple pathways
RiPP prenylation has been found in the cyanobactins and the quorum sensing ComX derivatives, where there are many otherwise unprecedented posttranslational modifications (Arnison et al. 2013; McIntosh et al., 2011; Okada et al., 2005). We aimed to cross prenyltransferases between pathways, swapping TruF1, from the trunkamide tru pathway, and PagF, from the prenylagaramide pag pathway. Based upon the natural products of these pathways, one would expect that TruF1 would reverse prenylate serine and threonine, while PagF would forward prenylate tyrosine.
The cyclic peptides 15c and 15d were prenylated in a single batch reaction containing both TruF1 and PagF, which led to prenylated products cyc-TFPVPTVC* and cyc-AC*MPC*YP (where the underlined threonine or tyrosine residue represents prenylation and C* represents thiazoline). In case of 15c both singly and doubly prenylated products were observed on the two threonine residues. For 15d carrying one tyrosine residue, the expected single prenylation event was observed. Additionally, cyclic peptide 14c that carries two tyrosine residues was doubly prenylated to yield 14d. In both cases the prenylation reaction was complete in that the cyclic peptide substrate was not detectable after 18 h of reaction. The natural product analog of 14d (prenylagaramide B) does not carry a thiazoline modification and is only singly prenylated, whereas in vitro exploitation of these enzymes allows us to make this unnatural derivative. These results show that it is possible to cross multiple posttranslational modifications to create unusual products representing hybrids of natural biosynthetic pathways (Figures 7 and S7).
Figure 7. Designed cross-pathway products.

(A) Prenylation of cyclic peptides 15c–d by a combination of the prenyltransferases TruF1 and PagF to yield the products 15e–f, and of 14c by PagF to yield the product 14d. It is to be noted that 15e is a natural product (patellin 6 from the tru pathway) whereas 15f is a derivative of the lyn pathway lacking thiazoline oxidation. The product 14d is an unusual derivative of the pag pathway carrying thiazoline and double prenylation that is natively absent in the parent pag natural product (prenylagaramide B) (B) The mass spectrum of the product 14d. See Figure S7 for the mass spectra of 15e–f products.
DISCUSSION
Macrocycles are an underrepresented drug class, and their potential is gradually being recognized. They exhibit unique stability, membrane permeability and ability to access difficult targets such as protein-protein interactions. The cyanobactin class of macrocyclic natural products provides access to an unprecedented diversity of broad substrate enzymes that can be used for synthetic biology. The natural tru pathway has previously been shown to be capable of producing millions of macrocyclic derivatives using just proteinogenic amino acids in vivo in an E. coli expression platform. Non-proteinogenic amino acids have also been incorporated, greatly adding to this already exceptional chemical diversity (Tianero et al., 2011).
Here, we define practical methods to make derivatives using mixtures of cyanobactin enzymes in vitro. Both natural and unnatural derivatives of the patellamide, trunkamide, aestuaramide and prenylagaramide pathways were successfully made. Moreover, the reactions using 14 and 15 showed that these are competent substrates, and that a former lack of detection in E. coli expression experiments (Sardar et al., 2014) was not due to incompetency of the tru pathway enzymes to accept these substrates. Thus, in total the enormous chemical potential of cyanobactin pathways determined through in vivo work greatly underestimates the potential products that can be synthesized by this highly plastic biosynthetic pathway.
This work is complementary to a recent report wherein simple derivatives of the patellamide core sequence were synthesized in vitro (Houssen et al., 2014). Here, this work synthesizes highly distant sequences using the same set of enzymes, including a hybrid of modifications from different pathways. In addition, this work exploits the prenylation posttranslational modifications, macrocycle size plasticity and use of a one-pot methodology. A combination of these technologies will find use in the generation of important cyclic peptides other than cyanobactins. For example, the Suga group produced heterocyclic precursors to the telomestatins using an in vitro translation system (Goto et al., 2014). Perhaps a combination with the methods developed here, more advanced telomestatin analogs could be synthesized.
The cyanobactin macrocyclase PatG is a serine protease (Lee et al., 2009; McIntosh et al., 2010a; Agarwal et al., 2012; Koehnke et al., 2012); its mechanism is similar to that of other serine proteases in first creating a covalently tethered ester intermediate, which is then displaced by a nucleophile. In the case of PatG, that nucleophile is the N-terminus, leading to macrocycle formation. In contrast, most serine proteases use water as the nucleophile. A remaining question was how this selectivity for macrocyclization instead of hydrolysis is achieved. One proposal is that a capping helix that binds to RSIII also excludes water from the active site (Koehnke et al., 2012). However, nucleophiles readily enter the PatG active site (McIntosh et al., 2010a; Agarwal et al., 2012). For example, transamination can occur via both side-chain and main-chain amines (McIntosh et al., 2010a), as well as from short dipeptides in the reaction buffer (Agarwal et al., 2012). A peptide with a glycolate terminus (containing OH in place of NH2 at the terminus) is hydrolyzed efficiently by water (McIntosh et al., 2010a). PatG could function by exploiting native substrate pre-organization that would hold the amine terminus in proximity to the ester intermediate. However, PatG has been used to cyclize hundreds of vastly different substrates (Lee et al., 2009; McIntosh et al., 2010a; Agarwal et al., 2012; Koehnke et al., 2012; Schmidt et al., 2005; Donia et al., 2006; Ruffner et al., 2014; Tianero et al., 2011; Houssen et al., 2014). Here, PatG efficiently macrocyclizes a 22-amino acid precursor containing much of the leader peptide. This peptide necessarily adopts a much different shape than the small, natural substrates, which is at odds with the idea of substrate pre-organization. Therefore, PatG itself likely scaffolds peptides for macrocyclization, preferring nitrogen to oxygen nucleophiles. The role of protein-substrate interactions in promoting cyclization versus hydrolysis is reminiscent of that found in nonribosomal peptide thioesterases and should be further explored (Jiang et al., 2011).
EXPERIMENTAL PROCEDURES
Genes and Cloning
The genes for the enzymes ThcD, PatA, PatG protease/macrocyclase domain and TruG along with those for the substrates 14 and 15 were made previously (Lee et al., 2009; Sardar et al., 2014). The codon-optimised version of TruF1 was obtained from GenScript. All enzymes and peptides were expressed as histidine-tagged constructs in pET-28b (+) expression vectors.
Protein Expression, Purification and/or Synthesis
Peptides 1 and 9 were obtained from the Peptide Core facility at the University of Utah. Precursor peptides 14 and 15 were made as described previously, where R2D-BL21 cells were electroporated with the respective plasmid, a seed culture grown overnight in 2xYT media, an aliquot of which was inoculated the next day in 1L 2xYT cultures. The cultures were grown at 37°C until an OD600 of about 0.4 was achieved, followed by 1 mM IPTG induction for 3 h at the same temperature. The enzymes PatA, PatG, TruG, TruF1 and PagF were expressed such that all of the conditions were maintained the same as above except that 0.1 mM IPTG induction was carried out overnight at a lower temperature of 18°C. The cells were harvested and purified by Ni-NTA affinity chromatography by denaturing (for 14 and 15) or native (for all modification enzymes) conditions, according to the manufacturer’s protocol (Qiagen). All peptides were dialyzed, aliquoted, flash-frozen and stored at −80°C till used. The enzymes were additionally stored in 10% (v/v) glycerol.
Assay conditions for sequential enzyme addition
In all assays a substrate concentration of 50 – 100 μM was maintained in 50 mM Tris buffer of pH 7.5. The heterocyclization reactions used 2 μM of the enzyme in reaction mixtures containing 1 mM ATP and 5 mM MgCl2. The N-terminal proteolysis reactions with PatA used 2–5 μM of the enzyme in reaction mixtures carrying 10 mM CaCl2, and C-terminal proteolysis/macrocyclization with PatG or TruG used 10–20 μM of the enzymes with 5 mM MgCl2. Prenylation with TruF1 and PagF used enzyme at 10 μM in optimised reaction mixtures containing 10 mM DMAPP. A temperature of 37°C with variable time points was used for all reactions. Reactions with substrate 1 were carried out for 2 h with ThcD, 2 h with PatA and overnight with PatG/TruG and TruF1/PagF. With substrate 14 and 15, each step was carried out for 18 h (sometimes longer in reactions with PatG).
Assay conditions for one-pot reactions
All reagents and enzymes were maintained at the same concentrations as above such as 50 mM Tris buffer pH 7.5, 5 mM MgCl2, 10 mM CaCl2, 1 mM ATP, 2 μM ThcD, 2 μM PatA and 10 μM PatG at 37°C. If prenyltransferase was used, 10 μM of the enzyme was used with 10 mM DMAPP. Reactions were carried out for 18 h in most cases.
HPLC purification of reaction intermediates
Reaction mixtures were centrifuged and the supernatant was used for purification of intermediates by RP-HPLC on a LaChrom Elite System (Hitachi). A semi-preparative Supelco Discovery HS C18 column (25 cm × 10 mm, 5 μm) was used for purification of the ThcD and PatA products of substrate 1 (intermediates 2 and 4), and also for ThcD products of full-length substrates 14 and 15, and PatA product of substrate 14a (intermediate 14b). A gradient of 99% H2O/1% ACN to 1% H2O/99% ACN was used over 20 min at a flow rate of 3.0 ml min−1 with 250 μL injection volumes. No TFA was used in the mobile phase because of the possibility of thiazoline ring opening. For PatA product intermediates of substrate 15 (intermediates 15a–b) an analytical Agilent Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm) was used. The same gradient as above was maintained, with the exception of adding 0.05% formic acid to the aqueous mobile phase. Reactions were dried down to 100 μL for injection and a flow rate of 0.7 ml min−1 was used.
Characterization and yield of compounds
The reaction leading to 8 were performed using the same reagent concentrations described above, but were scaled up to 2 mL to guarantee sufficient amounts of 1 (2 mg) for characterization. The product was purified as described above to afford ~1 mg quantities of pure 8, which was characterized by NMR and HR-MS. 1H NMR and 1H-1H COSY spectra were obtained using a Varian INOVA 500 (1H 500 MHz) NMR spectrometer with a 3 mm Nalorac MDBG probe. HSQC spectra were recorded on a Varian INOVA 600 (1H 600 MHz and 13C 150 MHz) NMR equipped with a 5 mm 1H[13C,15N] triple resonance cold probe with a z-axis gradient, utilizing residual solvent signals for referencing. The amount of isolated 8 was measured using NMR standards consisting of varying concentrations of boc-tyrosine. Both sample and standard were dissolved in 3:2 D2O/ACN-d3. To determine the yield of 8 in enzyme reactions, the isolated, quantified standard was used to generate a standard curve with HPLC on an analytical C18 column (Kinetex, 250 × 4.5 mm, 5μ) over a gradient of 100% H2O-0.5% formic acid/0% ACN-0.5% formic to 0% H2O-0.5% formic acid/100% ACN-0.5% formic acid for 40 min at a flow rate of 1.0 ml min−1.
PatA proteolysis reaction on macrocycle 8
HPLC purified 8 was lyophilized and an aqueous stock solution of 1 mM was made. PatA cleavage of 8 was carried out using 7 μM of the enzyme, 200 μM of the purified 8, 4 mM MgCl2 and 10 mM CaCl2 at 37°C for 18 h. A reaction without PatA was used as a negative control.
Liquid chromatography-mass spectrometric based product characterization
All product characterization was based on liquid chromatography-mass spectrometric analyses. Low-resolution LC/ESI-MS was done using a Micromass Quattro-II (Waters) instrument on an analytical Agilent Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm) with a linear gradient of 1% – 99% B over 20 min, where the mobile phase consisted of solvent A (H2O with 0.05% formic acid) and solvent B (ACN). High-resolution mass spectra were recorded using the same LC conditions on a Micromass Q-TOF Micro mass spectrometer (Waters) in positive ion mode. FT-ICR/MS was performed using a LTQ FT Ultra Hybrid Mass Spectrometer (Thermo Scientific) at the University of Utah Mass Spectrometry Core Facility. See Tables S3 and S4 for a complete list of all substrates, intermediates and products made in this study along with their expected masses.
Supplementary Material
SIGNIFICANCE.
Posttranslationally modified peptides are found in all forms of life and are increasingly important as drug leads and biochemical tools. Nature provides an enormous diversity of peptide products and posttranslational modifications. Here, we demonstrate how enzymes from multiple pathways can be combined in vitro to produce new natural products with tailored posttranslational modifications. Because cyanobactin enzymes are exceptionally promiscuous, these combined enzymes could be applied to highly unnatural substrates to design the synthesis of new compounds. Such a strategy holds promise in applying the many RiPP enzymes to synthesizing drug-like compounds. Finally, application of these enzymes provided new insights into important and novel posttranslational modifications.
Highlights.
The use of cyanobactin enzymes in vitro to synthesize modified cyclic peptides
Manipulation of cyanobactin enzyme requirements for one pot synthesis
In vitro synthesis of cyanobactins inaccessible through expression in E. coli
TruF1 was shown to be a Ser/Thr prenyltransferase in vitro
Acknowledgments
This work was funded by NIH GM102602. We thank Krishna Parsawar at the University of Utah Mass Spectrometry Core Facility for FT-ICR MS/MS data collection and Dr. Chris Ireland and Tom Smith for use of Micromass Q-TOF mass spectrometer.
Footnotes
Supplemental information includes 8 figures, 3 tables and Supplemental Experimental Procedures, which can be found with this article online.
AUTHOR CONTRUBUTIONS
D. S. and Z.L. carried out the experimental work. D. S. and E. W. S. designed the experiments. D. S. and E. W. S. wrote the manuscript.
The authors have declared conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal V, Pierce E, McIntosh JA, Schmidt EW, Nair SK. Structures of cyanobactin maturation enzymes define a family of transamidating proteases. Chem Biol. 2012;19:1411–1422. doi: 10.1016/j.chembiol.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy GL, et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013;30:108–160. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber CJ, Pujara PT, Reed DW, Chiwocha S, Zhang H, Covello PS. The two-step biosynthesis of cyclic peptides from linear precursors in a member of the plant family Caryophyllaceae involves cyclization by a serine protease-like enzyme. J Biol Chem. 2013;288:12500–12510. doi: 10.1074/jbc.M112.437947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condie JA, Nowak G, Reed DW, Balsevich JJ, Reaney MJ, Arnison PG, Covello PS. The biosynthesis of Caryophyllaceae-like cyclic peptides in Saponaria vaccaria L. from DNA-encoded precursors. Plant J. 2011;67:682–690. doi: 10.1111/j.1365-313X.2011.04626.x. [DOI] [PubMed] [Google Scholar]
- Craik DJ, Malik U. Cyclotide biosynthesis. Curr Opin Chem Biol. 2013;17:546–554. doi: 10.1016/j.cbpa.2013.05.033. [DOI] [PubMed] [Google Scholar]
- Davidson A, Leeper TC, Athanassiou Z, Patora-Komisarska K, Karn J, Robinson JA, Varani G. Simultaneous recognition of HIV-1 TAR RNA bulge and loop sequences by cyclic peptide mimics of Tat protein. Proc Natl Acad Sci U S A. 2009;106:11931–11936. doi: 10.1073/pnas.0900629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vega MJP, Martin-Martinez M, Muniz-Gonzalez R. Modulation of protein-protein interactions by stabilizing/mimicking protein secondary structure elements. Curr Top Med Chem. 2007;7:33–62. doi: 10.2174/156802607779318325. [DOI] [PubMed] [Google Scholar]
- Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat Chem Biol. 2006;2:729–735. doi: 10.1038/nchembio829. [DOI] [PubMed] [Google Scholar]
- Donia MS, Schmidt EW. Linking chemistry and genetics in the growing cyanobactin natural products family. Chem Biol. 2011;18:508–519. doi: 10.1016/j.chembiol.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Ito Y, Kato Y, Tsunoda S, Suga H. One-pot synthesis of azoline-containing peptides in a cell-free translation system integrated with a posttranslational cyclodehydratase. Chem Biol. 2014;21:766–774. doi: 10.1016/j.chembiol.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Houssen WE, Bent AF, McEwan AR, Pieiller N, Tabudravu J, Koehnke J, Mann G, Adaba RI, Thomas L, Hawas UW, Liu H, Schwarz-Linek U, Smith MCM, Naismith JH, Jaspars M. An efficient method for the in vitro production of azol(in)e-based cyclic peptides. Angew Chem Int Ed. 2014;53:14171–14174. doi: 10.1002/anie.201408082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houssen WE, Wright SH, Kalverda AP, Thompson GS, Kelly SM, Jaspars M. Solution structure of the leader sequence of the patellamide precursor peptide, PatE1-34. Chembiochem. 2010;11:1867–73. doi: 10.1002/cbic.201000305. [DOI] [PubMed] [Google Scholar]
- Houssen WE, Koehnke J, Zollman D, Vendome J, Raab A, Smith MCM, Naismith JH, Jaspars M. The discovery of new cyanobactins from Cyanothece PCC 7425 defines a new signature for processing of patellamides. ChemBioChem. 2012;13:2683–2689. doi: 10.1002/cbic.201200661. [DOI] [PubMed] [Google Scholar]
- Ireland CM, Scheuer PJ. Ulicyclamide and Ulithiacyclamide, two new small peptides from a marine tunicate. J Am Chem Soc. 1980;102:5688–5691. [Google Scholar]
- Jennings C, West J, Waine C, Craik D, Anderson M. Biosynthesis and insecticidal properties of plant cyclotides: the cyclic knotted proteins from Oldenlandia affinis. Proc Natl Acad Sci U S A. 2001;98:10614–10619. doi: 10.1073/pnas.191366898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Morley KL, Schrag JD, Kazlauskas RJ. Different active-site loop orientation in serine hydrolases versus acyltransferases. Chembiochem. 2011;12:768–776. doi: 10.1002/cbic.201000693. [DOI] [PubMed] [Google Scholar]
- Koehnke J, Bent A, Houssen WE, Zollman D, Morawiz F, Shirran S, Vendome J, Nneoyiegbe AF, Trembleau L, Botting CH, et al. The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat Struct Mol Biol. 2012;19:767–772. doi: 10.1038/nsmb.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J, Bent AF, Zollman D, Smith K, Houssen WE, Zhu X, Mann G, Lebl T, Scharff R, Shirran S, et al. The cyanobactin heterocyclase enzyme: a processive adenylase that operates with a defined order of reaction. Angew Chem Int Edit. 2013;52:13991–13996. doi: 10.1002/anie.201306302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Hathaway BJ, Schmidt EW. Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J Am Chem Soc. 2009;131:2122–2124. doi: 10.1021/ja8092168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikoski N, Fewer DP, Jokela J, Alakoski P, Wahlsten M, Sivonen K. Analysis of an inactive cyanobactin biosynthetic gene cluster leads to discovery of new natural products from strains of the genus Microcystis. PLoS One. 2012;7:1–9. doi: 10.1371/journal.pone.0043002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikoski N, Liu L, Jokela J, Wahlsten M, Gugger M, Calteau A, Permi P, Kerfield CA, Sivonen K, Fewer DP. Genome mining expands the chemical diversity of the cyanobactin family to include highly modified linear peptides. Chem Biol. 2013;20:1033–1043. doi: 10.1016/j.chembiol.2013.06.015. [DOI] [PubMed] [Google Scholar]
- Luo H, Hallen-Adams HE, Scott-Craig JS, Walton JD. Ribosomal biosynthesis of alpha-amanitin in Galerina marginata. Fungal Genet Biol. 2012;49:123–129. doi: 10.1016/j.fgb.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Hong SY, Sgambelluri RM, Angelos E, Li X, Walton JD. Peptide macrocyclization by a prolyl-oligopeptidase involved in α-amanitin biosynthesis. Chem Biol. 2014;21:1–8. doi: 10.1016/j.chembiol.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Robertson CR, Agarwal V, Nair SK, Bulaj GW, Schmidt EW. Circular logic: nonribosomal peptide-like macrocyclization with a ribosomal peptide catalyst. J Am Chem Soc. 2010a;132:15499–15501. doi: 10.1021/ja1067806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Schmidt EW. Insights into heterocyclization from two highly similar enzymes. J Am Chem Soc. 2010b;132:4089–4091. doi: 10.1021/ja9107116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Schmidt EW. Marine molecular machines: heterocyclization in cyanobactin biosynthesis. ChemBioChem. 2010c;11:1413–1421. doi: 10.1002/cbic.201000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Donia MS, Nair SK, Schmidt EW. Enzymatic basis of ribosomal peptide prenylation in cyanobacteria. J Am Chem Soc. 2011;133:13698–13705. doi: 10.1021/ja205458h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh JA, Lin Z, Tianero MDB, Schmidt EW. Aestuaramides, a natural library of cyanobactin cyclic peptides resulting from isoprene-derived Claisen rearrangements. ACS Chem Biol. 2013;8:887–883. doi: 10.1021/cb300614c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne BF, Long PF, Starcevic A, Hranueli D, Jaspars M. Spontaneity in the patellamide biosynthetic pathway. Org Biomol Chem. 2006;4:631–638. doi: 10.1039/b515938e. [DOI] [PubMed] [Google Scholar]
- Nguyen GKT, Wang S, Qiu Y, Hemu X, Yilong L, Tam JP. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat Chem Biol. 2014;10:732–738. doi: 10.1038/nchembio.1586. [DOI] [PubMed] [Google Scholar]
- Okada M, Sato I, Cho SJ, Iwata H, Nishio T, Dubnao D, Sakagami Y. Structure of the Bacillus subtilis quorum-sensing peptide pheromone ComX. Nat Chem Biol. 2005;1:23–24. doi: 10.1038/nchembio709. [DOI] [PubMed] [Google Scholar]
- Ruffner DE, Schmidt EW, Heemstra JR. Assessing the combinatorial potential of the RiPP cyanobactin tru pathway. ACS Synth Biol. 2014 doi: 10.1021/sb500267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatella X, Caba JM, Albericio F, Giralt E. Solution structure of the antitumor candidate trunkamide A by 2D NMR and restrained simulated annealing methods. J Org Chem. 2002;68:211–215. doi: 10.1021/jo026464s. [DOI] [PubMed] [Google Scholar]
- Sardar D, Pierce E, McIntosh JA, Schmidt EW. Recognition sequences and substrate evolution in cyanobactin biosynthesis. ACS Synth Biol. 2014 doi: 10.1021/sb500019b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EW, Nelson JT, Rasko DA, Sudek S, Elsen JA, Haygood MG, Ravel J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc Natl Acad Sci USA. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Production of cyclic peptides and proteins in vivo. Proc Natl Acad Sci USA. 1999;96:13638–13643. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber SA, Marahiel MA. Learning from nature’s drug factories: nonribosomal synthesis of macrocyclic peptides. J Bacteriol. 2003;185:7036–7043. doi: 10.1128/JB.185.24.7036-7043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tianero MDB, Donia MS, Young TS, Schultz PG, Schmidt EW. Ribosomal route to small-molecule diversity. J Am Chem Soc. 2011;134:418–425. doi: 10.1021/ja208278k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukui T, Nagano N, Umemura M, Kumagai T, Terai G, Machida M, Asai K. Ustiloxins, fungal cyclic peptides, are ribosomally synthesized in Ustilaginoidea virens. Bioinformatics. 2015;31(7):981–985. doi: 10.1093/bioinformatics/btu753. [DOI] [PubMed] [Google Scholar]
- Umemura M, Nagano N, Koike H, Kawano J, Ishii T, Miyamura Y, Kikuchi M, Tamano K, Yu J, Shin-ya K, Machida M. Characterization of the biosynthetic gene cluster for the ribosomally synthesized cyclic peptide ustiloxin B in Aspergillus flavus. Fungal Genet Biol. 2014;68:23–30. doi: 10.1016/j.fgb.2014.04.011. [DOI] [PubMed] [Google Scholar]
- White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nat Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- Yu X, Sun D. Macrocyclic drugs and synthetic methodologies toward macrocycles. Molecules. 2013;18:6230–6268. doi: 10.3390/molecules18066230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Dai P, Spokoyny AM, Pentelute BL. Enzyme-catalyzed macrocyclization of long unprotected peptides. Org Lett. 2014;16:3652–3655. doi: 10.1021/ol501609y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.