

Abstract
Structure-prone DNA repeats are common components of genomic DNA in all kingdoms of life. In humans, these repeats are linked to genomic instabilities that result in various hereditary disorders, including many cancers. It has long been known that DNA repeats are not only highly polymorphic in length but can also cause chromosomal fragility and stimulate gross chromosomal rearrangements, i.e. deletions, duplications, inversions, translocations and more complex shuffles. More recently, it has become clear that inherently unstable DNA repeats dramatically elevate mutation rates in surrounding DNA segments and that these mutations can occur upto ten kilobases away from the repetitive tract, a phenomenon we call repeat-induced mutagenesis (RIM). This review describes experimental data that led to the discovery and characterization of RIM and discusses the molecular mechanisms that could account for this phenomenon.
Keywords: DNA repeats, expansions, contractions, mutagenesis, replication, transcription, repair, hereditary disease, cancer, chromosomal fragility
1. Introduction
About half of the human genome is made up of repetitive DNA elements i.e. multiple copies of identical sequences, often categorized based on their location and length of the repeating unit (interspersed vs tandem repeats, microsatellites vs minisatellites etc.) [1,2]. While the polymorphic nature of these repeats is believed to contribute to genetic variability, their instability is known to cause various human diseases. One striking example is the expansion of short tandem DNA repeats, a phenomenon responsible for ~40 human hereditary neurological, neurodegenerative and developmental disorders such as Huntington’s disease, myotonic dystrophy type 1, Friedreich’s ataxia, fragile X syndrome, amyotrophic lateral sclerosis and others (reviewed in [3–5]). Molecular mechanisms underlying DNA repeats instability have been extensively studied in various experimental systems, including bacteria, yeast, mice and cultured human cells [6–8]. An unexpected outcome of these studies has been the discovery that besides being inherently unstable, DNA repeats can also induce mutations in flanking DNA sequences, a phenomenon called repeat-induced mutagenesis (RIM) [9]. Here, we review the historical backdrop as well as recent experimental data demonstrating RIM and discuss the molecular pathways through which it compromises genomic integrity.
2. Historical backdrop of repeat-induced mutagenesis
The discovery of repeat-induced mutagenesis was totally serendipitous in nature. The story goes back to the 80s during which many alternative DNA structures, including left-handed Z-DNA, cruciform DNA, three-stranded H-DNA and four-stranded G-quadruplex DNA were discovered (reviewed in [10]). The first of multi-stranded DNA structures to be discovered was H-DNA – an intramolecular DNA triplex formed by homopurine-homopyrimidine mirror repeats under the influence of negative super coiling [11]. This discovery was almost instantly followed by the realization that intermolecular triplexes could form between a triplex-forming oligonucleotide (TFO) and its homopurine-homopyrimidine target in duplex DNA [12–14]. Researchers found that targeting a homopurine-homopyrimidine sequence within the promoter region of the c-myc proto-oncogene with a TFO repressed its transcription, both in vitro as well as in cultured HeLa cells [15,16]. Because such sequence elements are found commonly in the human genome and often located in the regulatory portions of various genes, it was speculated that TFOs could be used as prospective antigene tools to gain control of gene expression at the transcriptional level [17]. Subsequent demonstrations of TFO-mediated gene modulation by various groups invariably helped antigene technology gain momentum as an attractive therapeutic strategy against viral infections as well as cancer [18]. Investigations into the mechanisms behind TFO-mediated modulation of gene expression revealed two major causes – (1) direct blockage of transcriptional initiation and/or elongation at the site of triplex formation [19–22] and (2) induction of localized mutations by TFOs through site-specific DNA damage (see [23–25] and references therein).
The latter was a totally unexpected outcome, nonetheless exploited by researchers, who began deliberately conjugating DNA damaging agents, such as psoralen or bleomycin, to the TFOs in the hope to develop a powerful yet facile method for site-specific genome modification [23,25,26]. In experiments with a supF plasmid reporter system carried out in cultured primate cells, it was found that a 30-nucleotide long TFO increased the rate of localized mutations 10-fold above control. Psoralen-conjugates of this TFO increased the mutation rate upto 100-fold above control upon activation by irradiation [27,28]. Similar TFO-mediated mutagenesis was reported in cultured human cells [29] but was absent however in xerodermapigmentosa group A (XPA) cells deficient in nucleotide excision repair or in Cockayne’s syndrome group B (CSB) cells deficient in transcription-coupled repair. These results implied that transcription-coupled and/or nucleotide excision repair pathways are essential for the TFO-induced mutagenesis. Researchers soon discovered that TFOs could also induce point mutations, small insertions and deletions at or around their chromosomal targets in mammalian cells [30,31]. Remarkably, in all cases, these mutations were found to lie either within the TFO target site or less than 50 base pairs upstream of it. Using the same supF reporter system, it was further demonstrated that the H-DNA-forming region from the human c-myc promoter could induce a greater than 20-fold increase in mutation frequency by itself, i.e. without the presence of any TFO [32]. Additionally, frequent double-strand breaks (DSB) were found to occur around the H-DNA site in this system, implying that naturally occurring triplex-forming DNA sequences could act as an endogenous source of genomic instability [24]. Sequences capable of forming other unusual DNA structures, such as Z-DNA [33] or G-quadruplex DNA [34,35], also appeared to be mutagenic. It was suggested therefore that unusual DNA structures, which have the ability to stall the DNA replication or transcription machinery, might result in DNA breakage and subsequently lead to mutagenesis (reviewed in [36]).
3. Recent insights into repeat-induced mutagenesis
3.1 Mutations induced by Friedreich’s at axia GAA repeats
Friedreich’s ataxia (FRDA) is an autosomal recessive neurodegenerative disorder caused by the expansion of (GAA) n repeats in the first intron of the frataxin (FXN) gene [37]. Studies from various labs have revealed that the GAA repeat element is capable of forming an intramolecular triplex and blocking transcriptional elongation in vitro as well as in vivo[38,39]. Expanded (GAA)n repeats also trigger heterochromatinization of surrounding DNA, leading to the inhibition of FXN gene expression (reviewed in [40]). While studying GAA repeat expansions in serially passaged lymphoblastoid cell lines from FRDA patients, researchers inadvertently observed a 3-fold increase in mutagenesis within a 135 bp region immediately upstream of the expanded (GAA)n repeat tract [41]. Our lab recently developed a tractable genetic system to study large-scale repeat expansions in yeast [42]. Various lengths of GAA repeats were cloned into the intron of the artificially split URA3 selectable marker. Large-scale expansions of the repeats would turn off URA3 splicing, allowing for selection of such events on media containing 5-fluoroorotic acid (5-FOA). However, we observed three different types of events among the 5-FOA resistant clones: (1) large-scale expansions of GAA repeats, (2) mutations in the body of the URA3 gene and (3) chromosomal rearrangements. Relative to a control construct without repeats, the rate of mutation was elevated 10-fold in the presence of a (GAA)78 run and 100-fold in the presence of (GAA)150 run. Thus, doubling the size of the repeat increased mutation rate by an order of magnitude. Interestingly, these mutations consisted largely of point substitutions and were found at significant distances, i.e. up to 1 kb away, both upstream as well as down stream of the repeat tract.
More recently, we investigated the role of various DNA polymerases in GAA-mediated RIM [9]. In eukaryotes, faithful duplication of the genome requires the action of several DNA polymerases. These include three distinct and specialized replicative DNA polymerases – leading and lagging strand polymerases Pol ε (epsilon) and Pol δ (delta) respectively as well as polymerase-primase Polα (alpha) (reviewed in [43,44]). In addition to the replicative DNA polymerases, various translesion (TLS) polymerases also carry out specialized roles, and these include DNA polymerases Pol ζ (zeta) and Rev1 (for a detailed description, see [45] and references therein). Using the above yeast system, we found that defects in the leading or lagging strand polymerase (Pol ε and Pol δ), but not polymerase-primase (Pol α), drastically elevated the rate of RIM relative to a wild-type background. Consistent with prior observations, the repeat-induced mutations in DNA polymerase mutants could be mapped as far as 1 kb away and on either side of the repetitive tract. The increase in rate of RIM in yeast strains containing defective-replisomes seemed to depend, at least partially, on the presence of TLS polymerases Pol ζ or Rev1. Majority of the point substitutions observed in these cases were C-to-G transversions, which is a characteristic signature of these two TLS polymerases [46,47]. Recent studies have found that TLS polymerases are recruited to the replication fork by tethering to proliferating cell nuclear antigen (PCNA) [46,48]. The signaling cascade is initiated by post-translational modification of a lysine residue (K164) on PCNA (reviewed in [49]). We observed that blocking modification of this residue through a mutation in PCNA (K164R) also had the same effect as the absence of Polζ or Rev1. Note, however, that we did not observe a Polζ-dependence of RIM in the wild-type background.
Petes and co-workers studied GAA-mediated RIM in a slightly different yeast system, in which a URA3 reporter was placed more than 1 kb away from a very long (GAA)230 repeat [50,51]. Both deletions and mutations were observed in the body of the URA3 reporter, but an over whelming majority of these events were point substitutions. While the results in this system are largely consistent with our observations described above, there are also some important differences. First, almost all mutational events in [52] were accompanied by changes in the length of the GAA. Second, in many instances, the URA3 gene was found to contain more than one mutation. Third, and the most significant difference was that these mutations were exquisitely Pol ζ-dependent, since deletion of its catalytic subunit (rev3) ablated nearly all of the mutagenesis in the wild-type background. These differences could be explained by the fact that different tract lengths were used in the two systems– (GAA)100 in our case and (GAA)230 in theirs. However, strain or locus-specific differences cannot be ruled out. Lobachev and colleagues also designed a yeast system to study RIM, in which the URA3 reporter was positioned at short (0.4-to-0.6 kb), medium (8 kb) or long (30 kb) distances on either side ofa very long (GAA)230 repeat [53]. The presence of a long GAA tract caused substantial chromosomal fragility, resulting in double strand breaks (DSB) at the junction of the repeats. This increase in chromosomal fragility went hand in hand with a significant increase in GAA-mediated mutagenesis and was observed upto 8 kb away from the repeat tract. However, this effect was only seen in strains containing defective DNA Pol δ. Consistent with our observations, GAA-mediated mutagenesis in replisome-defective strains was found to be partially dependent on TLS polymerase Pol ζ.
3.2 Mutations induced by other repeats
Recent investigations have uncovered a relationship between RIM-associated phenomena and several other classes of repeats, including inverted repeats and telomere-like repeats. Inverted repeats (IR), known to fold into hairpin or cruciform structures, are a potent source of chromosomal rearrangements and genomic instability [54–57]. Alu retrotransposons, a type of IR consisting of ~320 bp-long repeat element, are found littered throughout the human genome (reviewed in [58]). Alu-mediated recombination is known to cause as many as 33 inherited disorders such as insulin-resistant diabetes type II, Tay-Sachs disease, familial hypercholesterolaemia, α-thalassaemia and others as well as 16 different types of cancer, including Ewing sarcoma, breast cancer, acute myelogenous leukemia etc. (reviewed in [59]). Lobachev and his colleagues used their yeast experimental (described above) to assess the mutagenic potential of long inverted repeats, including the 320 bp Alu palindrome [53]. Similarly to GAA-mediated RIM, the mutations in IR-mediated RIM were (1) increased upto 30-fold in strains with defective DNA Pol δ but not in the wild-type strain, (2) partially dependent on the TLS polymerase Pol ζ and (3) detected as far as 8 kb away from the IR site. Additionally, these inverted repeats were also found to cause substantial chromosomal fragility, resulting in DSBs.
Telomeric DNA repeats help in maintaining chromosomal stability and genomic integrity by protecting the ends of chromosomes from fusion and degradation (reviewed in [60]). While telomeric repeats are usually located at chromosomal termini, interstitial telomeric sequences (ITS) are found at internal sites on the chromosomes of many organisms (reviewed in [61–63]). ITSs co-localize to sites of several human chromosomal aberrations and rearrangements implicated in chromosomal fragility, various cancers, Prader-Willi syndrome etc. (reviewed in [61,62]). While investigating the mechanisms behind ITS instability in our yeast experimental system, we found that even short telomere-like (TGTGTGGG)n repeats caused a 25-to-75 fold increase in the mutation rate on either side the repeat tract [64]. Similarly to other structure-prone repeats, telomeric repeats are known to be potent blockers of replication fork progression [65] and promote DSB formation [64].
4. Molecular models of repeat-induced mutagenesis
Structure-prone DNA repeats frequently co-localize to break points of various chromosomal aberrations, including deletions, translocations, duplications, inversions and complex chromosomal rearrangements linked to human disease (reviewed in [36,66–69]. It is becoming increasingly clear that formation of some sort of a DNA double-strand break (DSB) intermediate is at the heart of all these aberrations. In several model systems, increased chromosomal fragility (i.e. DSB) goes hand in hand with an increase in RIM [53]. Increased local mutations have also been observed in the vicinity of complex human chromosomal rearrangements [70]. We believe, therefore, that RIM is the hidden side of repeat instability, resulting from the repair of a DSB event. In light of these findings, we discuss how and when these DSBs could occur and in what way their repair could result in RIM.
4.1 Mutagenesis induced by short structure-prone DNA repeats
The molecular mechanisms of RIM could be some what different between short repeats and longer structure-prone repeats. Short repeats are not known to stall replication fork progression in vivo [57,71–73], and unless they are present on strongly super coiled DNA, formation of alternative DNA structures by short repetitive tracts is not energetically favorable [74]. At the same time, short repetitive tracts, including the triplex-forming GAA repeats and the H-DNA region from the c-myc promoter, are known tostall RNA polymerase in vitro as well as in vivo [75–77]. Could transcriptional stalling by short repeats result in DNA breakage? Transcription-coupled repair (TCR), a sub pathway of nucleotide excision repair (NER), is activated when the transcriptional machinery encounters a damaged DNA template, such as an abasic site or a DNA adduct [78]. An alternative form of TCR, called “gratuitous” TCR, may be invoked when an RNA polymerase pauses at sites of unusual structure formation [79]. A wide body of evidence suggests that various short repeats can indeed stall translocating RNA polymerase in vivo by forming transient structures on the non-template strand or extended DNA-RNA hybrids (R-loops), leading to the initiation of gratuitous TCR [80–83]. Endonuclease components of the TCR machinery can induce single strand breaks (SSBs) in the template strand and it was recently shown that SSBs within R-loops could also be converted to double strand breaks (DSBs) ([84], reviewed in [85]).
A DSB formed during the transcription of a repeat locus in the G1 phase of the cell cycle could be repaired by various mechanisms. Of particular interest to us is repair via the single-strand annealing pathway (SSA) (Fig. 1A) where the 5′ ends of the break undergo resection, followed by re-annnealing of the 3′ repetitive ends and extension through gap filling [86,87]. DNA synthesis during gap filling can be carried out by various DNA repair polymerases, including TLS polymerase DNA Pol ζ, and give rise to point mutations on either side of the break. Several other scenarios can also be envisioned. Unrepaired SSBs could lead to the formation of a one-ended DSB during DNA replication, which can be repaired via the break-induced replication (BIR) pathway discussed below. Another mechanism is the so-called alternative end-joining pathway, which can lead to the accumulation of small deletions and templated insertions around the break site (reviewed in [88]). Interestingly, all the above-predicted outcomes, including deletions, insertions and point mutations, have been observed in various RIM studies [9,51,53]. The varying occurrence of these outcomes in different experimental systems is probably due to the delicate balance of factors affecting multiple DNA repair pathways.
Fig. 1. Molecular mechanisms leading to repeat-induced mutagenesis.
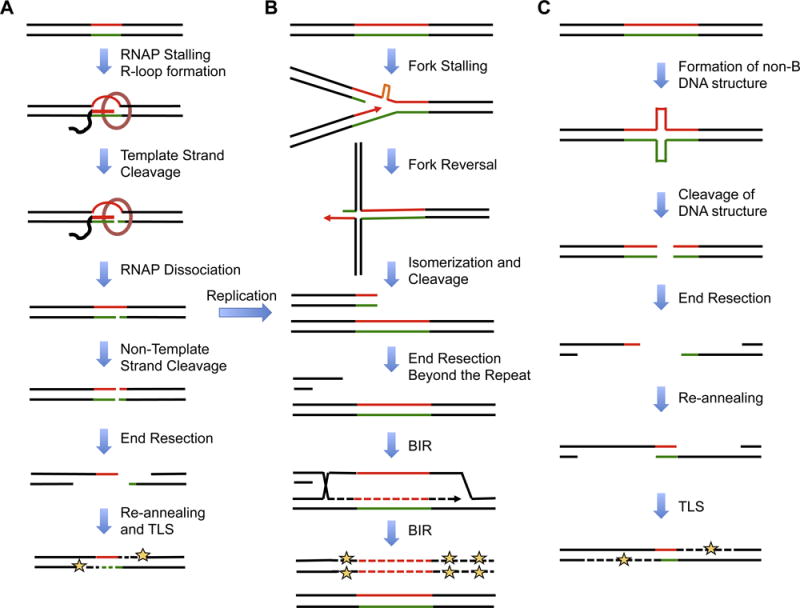
(A) Short repeats block transcription by forming alternative DNA structures or R-loops. This initiates ‘gratuitous’ transcription-coupled repair (TCR) and generates single strand breaks (SSBs) that can be converted to double strand breaks (DSBs). To repair the break, the ends undergo resection and single-strand annealing followed by gap-filling through TLSDNA polymerases that gives rise to mutations on either side of the repeat tract. (B) Long repeats block replication fork progression and result in reversed forks that are similar to Holliday junctions. Upon isomerization and resolution, these structures generate a one-ended DSB, which can be repaired by break-induced replication (BIR). During BIR, the end undergoes resection followed by invasion and copying of several kilobases of DNA from the donor. Due to the conservative mode of replication in BIR, DNA synthesis is highly error-prone and gives rise to mutations at large distances from the initial break site. Unrepaired SSBs generated by gratuitous TCR can also give rise to a one-ended DSB upon replication. (C) Long structure-prone repeats can form unusual DNA structures even in the G1 phase of the cell cycle. Cleavage of these structures leads to the formation of two-ended DSBs that can anneal ‘out-of-register’ and undergo homologous recombination (HR) repair. Gap-filling of the resected ends by error-prone DNA polymerases can give rise to mutations on either side and at large distances from the break site. In the case of (A) and (C), mutations will be incorporated in the next round of replication (not shown). Repetitive strands are shown in red and green. Dashed lines indicate DNA synthesized during repair and yellow star indicates mutations.
4.2 Mutagenesis induced by long structure-prone DNA repeats
Long structure-prone repeats are known to pose various challenges during DNA replication, recombination and repair. Numerous studies have shown that long repeats are potent blockers of DNA polymerases in vitro and replication fork progression in vivo (reviewed in [89]) Stalled replication forks often lead to fork reversal [90], the resulting structure of which (Fig. 1B) is practically indistinguishable from a Holliday junction [91]. Upon isomerization and enzymatic resolution, a reversed fork could be converted into a one-ended DSB (Fig. 1B), and breaks with only one free end are believed to be repaired by break-induced replication (BIR) [92]. In BIR, the 5′ end of the break undergoes resection, followed by invasion of the 3′ end into a homologous region on the sister chromatid and extension by DNA synthesis. BIR can involve copying of several kilobases of DNA, often times extending all the way to the telomere of the donor chromosome [93]. The nascent DNA is synthesized by DNA Pol δ and is exquisitely dependent on its Pol32 subunit [94]. Despite being independent of error-prone TLS polymerases, BIR appears to bea highly mutagenic form of DSB repair [95–97]. Remarkably, this is due to BIR involving a “conservative” mode of replication [98,99], for which the nascent leading strand serves as a template for the nascent lagging strand (Fig. 1B), thus precluding the role of mismatch repair. Since the mutational signature of BIR can be observed many kilobases away from the site of DSB, it could explain the large-scale effects of RIM and its independence from translesion polymerase observed in our studies [9].
Apart from forming alternative DNA structures during DNA replication in the S phase, long DNA repeats can also do so in the G1 phase of the cell cycle (Fig. 1C). In this scenario, limited DNA supercoiling required for theses structural transitions can easily be generated during transcriptional elongation [100]. Alternative DNA structures are known to be cleaved by various proteins in vivo, resulting in the formation of two-ended DSBs (Fig. 1C). For example, a DNA triplex formed by the long GAA repeats in yeast is converted into a DSB by the endonuclease activity of MutLα [101]. The Mre11p-Rad50p-Xrs2p (MRX) complex in yeast processes the ends of two-ended DSBs, giving rise to 3′-overhangs [54]. These over hangscan quicklyre-anneal, owing to the repetitive nature of their ends, followed by gap-filing carried out by DNA repair polymerases, including translesion DNA Pol ζ (Fig. 1C). This can explain the occurrence of point mutations on either side of the repeat tract. Importantly, re-annealing of repetitive tracts would most likely occur out of register, causing a change in the repeat’s length together with RIM, as was indeed observed in [51].
4. Future directions
While Fig. 1 summarizes current ideas on genetic transactions leading to RIM, detailed molecular mechanisms of this phenomenon still remain to be unraveled. In the short run, genetic and biochemical studies are needed to understand the role of Rad26p in TCR-mediated RIM, Pol32p subunit of DNA Polδ and DNA-helicase Pif1p in BIR mediated RIM as well as recombination proteins Rad51p and Rad52p in SSA-mediated RIM. Experimental systems designed to study repeat instability in a controlled transcription environment could be used to distinguish the relative contributions of transcription and replication in RIM [102]. One common and important feature of the replication-dependent and replication-independent pathways leading to RIM is the presence of single-stranded DNA (ssDNA) at the DSB site. Sensing of ssDNA by cell-cycle check point proteins should trigger a DNA damage response by the ATR signaling pathway (Mec1 pathway in S. cerevisiae) (reviewed in [103]). Furthermore, if the ssDNA is converted to a DSB, the ATM signaling pathway becomes rapidly activated (Tel1 protein in S. cerevisiae). ssDNA sensing and coating by RPA (ssDNA-binding protein) is necessary to activate various checkpoint pathways. However, alternative DNA structures are not known to bind RPA, and could thus escape detection by the checkpoint machinery. Future studies on how the Mec1p and Tel1p kinases of the ATR/ATM pathway affect RIM could help elucidate these mechanisms.
While mutagenesis mediated by short repeats is well documented in mammalian cells [55,67], very little is known about RIM caused by long structure-prone repeats in higher eukaryotes, including humans. Recent studies have observed elevated mutation levels around human chromosomal loci that have undergone complex, repeat-associated genome rearrangements [70] but almost nothing is known about their basis. Thus, developing tractable genetic systems capable of investigating both the mutability (repeat instability) as well as the mutagenicity (RIM) of long repeats is of high priority.
Finally, it would be of great interest to compare mutation rates in DNA segments located adjacent or apart from structure-prone DNA repeats using computational genomics approaches.
Acknowledgments
We thank Tom Petes, Kirill Lobachev, Polina Shcherbakova, Anna Malkova, Anna Aksenova, Jim Haber, Lorraine Symington and Hanna Klein for many fruitful discussions of the mechanisms for repeat-induced mutagenesis. This study was supported by NIH grants GM105473 and GM60987, and by generous contribution from the White family to S.M.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hardman N. Structure and function of repetitive DNA in eukaryotes. Biochem J. 1986;234:1–11. doi: 10.1042/bj2340001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 3.Pearson CE, Edamura KN, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 4.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 5.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nature Publishing Group. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wells RD, Dere R, Hebert ML, Napierala M, Son LS. Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucleic Acids Research. 2005;33:3785–3798. doi: 10.1093/nar/gki697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovtun IV, McMurray CT. Features of trinucleotide repeat instability in vivo. Cell Res. 2008;18:198–213. doi: 10.1038/cr.2008.5. [DOI] [PubMed] [Google Scholar]
- 8.Orr HT, Zoghbi HY. Trinucleotide Repeat Disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 9.Shah KA, Shishkin AA, Voineagu I, Pavlov YI, Shcherbakova PV, Mirkin SM. Role of DNA Polymerases in Repeat-Mediated Genome Instability. Cell Reports. 2012:1–13. doi: 10.1016/j.celrep.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirkin SM. Discovery of alternative DNA structures: a heroic decade (1979–1989) Front Biosci (Landmark Ed) 2008;13:1064–1071. doi: 10.2741/2744. [DOI] [PubMed] [Google Scholar]
- 11.Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, Frank-Kamenetskii MD. DNA H form requires a homopurine-homopyrimidine mirror repeat. Nature. 1987;330:495–497. doi: 10.1038/330495a0. [DOI] [PubMed] [Google Scholar]
- 12.Moser HE, Dervan PB. Sequence-specific cleavage of double helical DNA by triple helix formation. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 13.Lyamichev VI, Mirkin SM, Frank-Kamenetskii MD, Cantor CR. A stable complex between homopyrimidine oligomers and the homologous regions of duplex DNAs. Nucleic Acids Research. 1988;16:2165–2178. doi: 10.1093/nar/16.5.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Praseuth D, Perrouault L, Le Doan T, Chassignol M, Thuong N, Hélène C. Sequence-specific binding and photocrosslinking of alpha and beta oligodeoxynucleotides to the major groove of DNA via triple-helix formation. Proceedings of the National Academy of Sciences. 1988;85:1349–1353. doi: 10.2307/31225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooney M, Czernuszewicz G, Postel EH, Flint SJ, Hogan ME. Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science. 1988;241:456–459. doi: 10.1126/science.3293213. [DOI] [PubMed] [Google Scholar]
- 16.Postel EH, Flint SJ, Kessler DJ, Hogan ME. Evidence that a triplex-forming oligodeoxyribonucleotide binds to the c-myc promoter in HeLa cells, thereby reducing c-myc mRNA levels. Proceedings of the National Academy of Sciences. 1991;88:8227–8231. doi: 10.1073/pnas.88.18.8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hélène C, Thuong NT, Harel-Bellan A. Control of gene expression by triple helix-forming oligonucleotides. The antigene strategy. Ann N Y Acad Sci. 1992;660:27–36. doi: 10.1111/j.1749-6632.1992.tb21054.x. [DOI] [PubMed] [Google Scholar]
- 18.Chubb JM, Hogan ME. Human therapeutics based on triple helix technology. Trends in Biotechnology. 1992;10:132–136. doi: 10.1016/0167-7799(92)90195-2. [DOI] [PubMed] [Google Scholar]
- 19.Young SL, Krawczyk SH, Matteucci MD, Toole JJ. Triple helix formation inhibits transcription elongation in vitro. Proceedings of the National Academy of Sciences. 1991;88:10023–10026. doi: 10.1073/pnas.88.22.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duval-Valentin G, Thuong NT, Hélène C. Specific inhibition of transcription by triple helix-forming oligonucleotides. Proceedings of the National Academy of Sciences. 1992;89:504–508. doi: 10.2307/2358569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xodo L, Alunni-Fabbroni M, Manzini G, Quadrifoglio F. Pyrimidine phosphorothioate oligonucleotides form triple-stranded helices and promote transcription inhibition. Nucleic Acids Research. 1994;22:3322–3330. doi: 10.1093/nar/22.16.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovacs A, Kandala JC, Weber KT, Guntaka RV. Triple helix-forming oligonucleotide corresponding to the polypyrimidine sequence in the rat alpha 1(I) collagen promoter specifically inhibits factor binding and transcription. Journal of Biological Chemistry. 1996;271:1805–1812. doi: 10.1074/jbc.271.3.1805. [DOI] [PubMed] [Google Scholar]
- 23.Havre PA, Gunther EJ, Gasparro FP, Glazer PM. Targeted mutagenesis of DNA using triple helix-forming oligonucleotides linked to psoralen. Proceedings of the National Academy of Sciences. 1993;90:7879–7883. doi: 10.1073/pnas.90.16.7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vasquez KM, Wilson JH. Triplex-directed modification of genes and gene activity. Trends Biochem Sci. 1998;23:4–9. doi: 10.1016/S0968-0004(97)01158-4. [DOI] [PubMed] [Google Scholar]
- 25.Havre PA, Glazer PM. Targeted mutagenesis of simian virus 40 DNA mediated by a triple helix-forming oligonucleotide. Journal of Virology. 1993;67:7324–7331. doi: 10.1128/jvi.67.12.7324-7331.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faruqi AF, Egholm M, Glazer PM. Peptide nucleic acid-targeted mutagenesis of a chromosomal gene in mouse cells. Proceedings of the National Academy of Sciences. 1998;95:1398–1403. doi: 10.2307/44294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang G, Levy DD, Seidman MM, Glazer PM. Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Molecular and Cellular Biology. 1995;15:1759–1768. doi: 10.1128/mcb.15.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majumdar A, Khorlin A, Dyatkina N, Lin FL, Powell J, Liu J, et al. Targeted gene knockout mediated by triple helix forming oligonucleotides. Nat Genet. 1998;20:212–214. doi: 10.1038/2530. [DOI] [PubMed] [Google Scholar]
- 29.Wang G, Seidman MM, Glazer PM. Mutagenesis in Mammalian Cells Induced by Triple Helix Formation and Transcription-Coupled Repair. Science. 1996;271:802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- 30.Vasquez KM, Wang G, Havre PA, Glazer PM. Chromosomal mutations induced by triplex-forming oligonucleotides in mammalian cells. Nucleic Acids Research. 1999;27:1176–1181. doi: 10.1093/nar/27.4.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vasquez KM, Narayanan L, Glazer PM. Specific mutations induced by triplex-forming oligonucleotides in mice. Science. 2000;290:530–533. doi: 10.1126/science.290.5491.530. [DOI] [PubMed] [Google Scholar]
- 32.Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proceedings of the National Academy of Sciences. 2004;101:13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang G, Christensen LA, Vasquez KM. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proceedings of the National Academy of Sciences. 2006;103:2677–2682. doi: 10.2307/30049472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat Struct Mol Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 35.Lopes J, Piazza A, Bermejo R, Kriegsman B, Colosio A, Teulade-Fichou MP, et al. G-quadruplex-induced instability during leading-strand replication. Embo J. 2011;30:4033–4046. doi: 10.1038/emboj.2011.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G, Vasquez KM. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair. 2014;19:143–151. doi: 10.1016/j.dnarep.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 38.Bidichandani SI, Ashizawa T, Patel PI. The GAA triplet-repeat expansion in Friedreich ataxia interferes with transcription and may be associated with an unusual DNA structure. Am J Hum Genet. 1998;62:111–121. doi: 10.1086/301680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohshima K, Montermini L, Wells RD, Pandolfo M. Inhibitory effects of expanded GAA. TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivo. Journal of Biological Chemistry. 1998;273:14588–14595. doi: 10.1074/jbc.273.23.14588. [DOI] [PubMed] [Google Scholar]
- 40.Kumari D, Usdin K. Chromatin remodeling in the noncoding repeat expansion diseases. Journal of Biological Chemistry. 2009;284:7413–7417. doi: 10.1074/jbc.R800026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bidichandani SI, Purandare SM, Taylor EE, Gumin G, Machkhas H, Harati Y, et al. Somatic sequence variation at the Friedreich ataxia locus includes complete contraction of the expanded GAA triplet repeat, significant length variation in serially passaged lymphoblasts and enhanced mutagenesis in the flanking sequence. Hum Mol Genet. 1999;8:2425–2436. doi: 10.1093/hmg/8.13.2425. [DOI] [PubMed] [Google Scholar]
- 42.Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, et al. Large-Scale Expansions of Friedreich’s Ataxia GAA Repeats in Yeast. Molecular Cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol. 2008;18:521–527. doi: 10.1016/j.tcb.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pavlov YI, Shcherbakova PV. DNA polymerases at the eukaryotic fork-20 years later. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2010;685:45–53. doi: 10.1016/j.mrfmmm.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhuang Z, Johnson RE, Haracska L, Prakash L, Prakash S, Benkovic SJ. Regulation of polymerase exchange between Poleta and Poldelta by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc Natl Acad Sci USA. 2008;105:5361–5366. doi: 10.1073/pnas.0801310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Northam MR, Moore EA, Mertz TM, Binz SK, Stith CM, Stepchenkova EI, et al. DNA polymerases ζ and Rev1 mediate error-prone bypass of non-B DNA structures. Nucleic Acids Research. 2014;42:290–306. doi: 10.1093/nar/gkt830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulrich HD. Timing and spacing of ubiquitin-dependent DNA damage bypass. FEBS Lett. 2011;585:2861–2867. doi: 10.1016/j.febslet.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 49.Maga G. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. Journal of Cell Science. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 50.Tang W, Dominska M, Greenwell PW, Harvanek Z, Lobachev KS, Kim HM, et al. Friedreich’s ataxia (GAA)n•(TTC)n repeats strongly stimulate mitotic crossovers in Saccharomyces cerevisae. PLoS Genet. 2011;7:e1001270–e1001270. doi: 10.1371/journal.pgen.1001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang W, Dominska M, Gawel M, Greenwell PW, Petes TD. Genomic deletions and point mutations induced in Saccharomyces cerevisiae by the trinucleotide repeats (GAA·TTC) associated with Friedreich’s ataxia. DNA Repair. 2013;12:10–17. doi: 10.1016/j.dnarep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang W, Dominska M, Gawel M, Greenwell PW, Petes TD. Genomic deletions and point mutations induced in Saccharomyces cerevisiae by the trinucleotide repeats (GAA·TTC) associated with Friedreich’s ataxia. DNA Repair. 2013;12:10–17. doi: 10.1016/j.dnarep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saini N, Zhang Y, Nishida Y, Sheng Z, Choudhury S, Mieczkowski P, et al. Fragile DNA Motifs Trigger Mutagenesis at Distant Chromosomal Loci in Saccharomyces cerevisiae. PLoS Genet. 2013;9:e1003551. doi: 10.1371/journal.pgen.1003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/S0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 55.Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2006;598:103–119. doi: 10.1016/j.mrfmmm.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 56.Kurahashi H, Inagaki H, Ohye T, Kogo H, Kato T, Emanuel BS. Palindrome-mediated chromosomal translocations in humans. DNA Repair. 2006;5:1136–1145. doi: 10.1016/j.dnarep.2006.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA. 2008;105:9936–9941. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Batzer MA, Deininger PL. ALU REPEATS AND HUMAN GENOMIC DIVERSITY. Nat Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 59.Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 60.Wellinger RJ, Zakian VA. Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics. 2012;191:1073–1105. doi: 10.1534/genetics.111.137851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruiz-Herrera A, Nergadze SG, Santagostino M, Giulotto E. Telomeric repeats far from the ends: mechanisms of origin and role in evolution. Cytogenet Genome Res. 2008;122:219–228. doi: 10.1159/000167807. [DOI] [PubMed] [Google Scholar]
- 62.Lin KW, Yan J. Endings in the middle: current knowledge of interstitial telomeric sequences. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2008;658:95–110. doi: 10.1016/j.mrrev.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 63.Bolzán AD. Chromosomal aberrations involving telomeres and interstitial telomeric sequences. Mutagenesis. 2012;27:1–15. doi: 10.1093/mutage/ger052. [DOI] [PubMed] [Google Scholar]
- 64.Aksenova AY, Greenwell PW, Dominska M, Shishkin AA, Kim JC, Petes TD, et al. Genome rearrangements caused by interstitial telomeric sequences in yeast. Proceedings of the National Academy of Sciences. 2013;110:19866–19871. doi: 10.1073/pnas.1319313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anand RP, Shah KA, Niu H, Sung P, Mirkin SM, Freudenreich CH. Overcoming natural replication barriers: differential helicase requirements. Nucleic Acids Research. 2012;40:1091–1105. doi: 10.1093/nar/gkr836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bacolla A, Wells RD. Non-B DNA conformations, genomic rearrangements, and human disease. Journal of Biological Chemistry. 2004;279:47411–47414. doi: 10.1074/jbc.R400028200. [DOI] [PubMed] [Google Scholar]
- 67.Bacolla A, Wells RD. Non-B DNA conformations as determinants of mutagenesis and human disease. Mol Carcinog. 2009;48:273–285. doi: 10.1002/mc.20507. [DOI] [PubMed] [Google Scholar]
- 68.Zhang F, Carvalho CMB, Lupski JR. Complex human chromosomal and genomic rearrangements. Trends in Genetics. 2009;25:298–307. doi: 10.1016/j.tig.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lupski JR. Hotspots of homologous recombination in the human genome: not all homologous sequences are equal. Genome Biol. 2004;5:242–242. doi: 10.1186/gb-2004-5-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carvalho CMB, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, et al. Replicative mechanisms for CNV formation are error prone. Nat Genet. 2013;45:1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pelletier R, Krasilnikova MM, Samadashwily GM, Lahue R, Mirkin SM. Replication and expansion of trinucleotide repeats in yeast. Molecular and Cellular Biology. 2003;23:1349–1357. doi: 10.1128/MCB.23.4.1349-1357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Krasilnikova MM, Mirkin SM. Replication stalling at Friedreich’s ataxia (GAA)n repeats in vivo. Molecular and Cellular Biology. 2004;24:2286–2295. doi: 10.1128/MCB.24.6.2286-2295.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol. 2009;16:226–228. doi: 10.1038/nsmb.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mirkin SM. DNA Topology: Fundamentals. John Wiley & Sons, Ltd; 2001. [DOI] [Google Scholar]
- 75.Krasilnikova MM, Samadashwily GM, Krasilnikov AS, Mirkin SM. Transcription through a simple DNA repeat blocks replication elongation. Embo J. 1998;17:5095–5102. doi: 10.1093/emboj/17.17.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krasilnikova MM, Kireeva ML, Petrovic V, Knijnikova N, Kashlev M, Mirkin SM. Effects of Friedreich’s ataxia (GAA)n*(TTC)n repeats on RNA synthesis and stability. Nucleic Acids Research. 2007;35:1075–1084. doi: 10.1093/nar/gkl1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. Journal of Biological Chemistry. 2007;282:32433–32441. doi: 10.1074/jbc.M704618200. [DOI] [PubMed] [Google Scholar]
- 78.Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 51(1987):241–249. doi: 10.1016/0092-8674(87)90151-6. doi: http://dx.doi.org/10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 79.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 80.Lin Y, Wilson JH. Transcription-induced CAG repeat contraction in human cells is mediated in part by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2007;27:6209–6217. doi: 10.1128/MCB.00739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, Hanawalt PC. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proceedings of the National Academy of Sciences. 2010;107:12816–12821. doi: 10.1073/pnas.1007580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salinas-Rios V, Belotserkovskii BP, Hanawalt PC. DNA slip-outs cause RNA polymerase II arrest in vitro: potential implications for genetic instability. Nucleic Acids Research. 2011;39:7444–7454. doi: 10.1093/nar/gkr429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Belotserkovskii BP, Neil AJ, Saleh SS, Shin JHS, Mirkin SM, Hanawalt PC. Transcription blockage by homopurine DNA sequences: role of sequence composition and single-strand breaks. Nucleic Acids Research. 2013;41:1817–1828. doi: 10.1093/nar/gks1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sollier J, Stork CT, García-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Molecular Cell. 2014;56:777–785. doi: 10.1016/j.molcel.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aguilera A, García-Muse T. R loops: from transcription byproducts to threats to genome stability. Molecular Cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 86.Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carter SD, Vigasová D, Chen J, Chovanec M, Aström SU. Nej1 recruits the Srs2 helicase to DNA double-strand breaks and supports repair by a single-strand annealing-like mechanism. Proceedings of the National Academy of Sciences. 2009;106:12037–12042. doi: 10.1073/pnas.0903869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends in Genetics. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiology and Molecular Biology Reviews. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Research. 2009;37:3475–3492. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol. 2010;11:683–687. doi: 10.1038/nrm2974. [DOI] [PubMed] [Google Scholar]
- 92.Lydeard JR, Lipkin-Moore Z, Sheu YJ, Stillman B, Burgers PM, Haber JE. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes & Development. 2010;24:1133–1144. doi: 10.1101/gad.1922610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 94.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 95.Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, et al. Break-induced replication is highly inaccurate. PLoS Biol. 2011;9:e1000594–e1000594. doi: 10.1371/journal.pbio.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Malkova A, Haber JE. Mutations arising during repair of chromosome breaks. Genetics. 2012;46:455–473. doi: 10.1146/annurev-genet-110711-155547. [DOI] [PubMed] [Google Scholar]
- 97.Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, et al. Break-Induced Replication Is a Source of Mutation Clusters Underlying Kataegis. Cell Reports. 2014;7:1640–1648. doi: 10.1016/j.celrep.2014.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, et al. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature. 2013;502:389–392. doi: 10.1038/nature12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, et al. Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013;502:393–396. doi: 10.1038/nature12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Krasilnikov AS, Podtelezhnikov A, Vologodskii A, Mirkin SM. Large-scale effects of transcriptional DNA supercoiling in vivo. Journal of Molecular Biology. 1999;292:1149–1160. doi: 10.1006/jmbi.1999.3117. [DOI] [PubMed] [Google Scholar]
- 101.Kim HM, Narayanan V, Mieczkowski PA, Petes TD, Krasilnikova MM, Mirkin SM, et al. Chromosome fragility at GAA tracts in yeast depends on repeat orientation and requires mismatch repair. Embo J. 2008;27:2896–2906. doi: 10.1038/emboj.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shah KA, McGinty RJ, Egorova VI, Mirkin SM. Coupling Transcriptional State to Large-Scale Repeat Expansions in Yeast. Cell Reports. 2014;9:1594–1602. doi: 10.1016/j.celrep.2014.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Voineagu I, Freudenreich CH, Mirkin SM. Checkpoint responses to unusual structures formed by DNA repeats. Mol Carcinog. 2009;48:309–318. doi: 10.1002/mc.20512. [DOI] [PMC free article] [PubMed] [Google Scholar]