

Abstract
DNA double-strand breaks (DSBs) in cells can undergo nucleolytic degradation to generate long 3′ single-stranded DNA tails. This process is termed DNA end resection, and its occurrence effectively commits to break repair via homologous recombination, which entails the acquisition of genetic information from an intact, homologous donor DNA sequence. Recent advances, prompted by the identification of the nucleases that catalyze resection, have revealed intricate layers of functional redundancy, interconnectedness, and regulation. Here, we review the current state of the field with an emphasis on the major questions that remain to be answered. Topics addressed will include how resection initiates via the introduction of an endonucleolytic incision close to the break end, the molecular mechanism of the conserved MRE11 complex in conjunction with Sae2/CtIP within such a model, the role of BRCA1 and 53BP1 in regulating resection initiation in mammalian cells, the influence of chromatin in the resection process, and potential roles of novel factors.
Keywords: Resection, Recombination, Double-Strand Breaks, Nucleases, Helicases
Introduction
Cells sustain double-strand breaks (DSBs) as a result of frequent injury to DNA replication forks, and also of exposure to environmental agents (e.g., high energy radiation) and endogenous reactive metabolites (e.g., radicals). The timely and efficient elimination of DSBs is of paramount importance to genome preservation and for disease avoidance, cancer in particular (1).
DSBs are eliminated via two conserved mechanisms: nonhomologous end joining (NHEJ) and homologous recombination (HR). In NHEJ, the DNA ends are brought in close proximity, processed if necessary, and then rejoined via the action of a specialized DNA ligase (2). NHEJ occurs during all phases of the cell cycle. Mechanistically, HR is more complex and involves the use of a homologous DNA template to guide accurate repair (3,4). Since the newly replicated sister chromatid most often serves as the repair template, cells must be in S or G2 phase for HR to be an efficient repair option (5,6). NHEJ must be avoided when single-ended DSBs arise as a result of replication fork collision with a DNA lesion, such as an interstrand crosslink. Inappropriate joining of such DSBs leads to dicentric chromosomes, initiating a catastrophic breakage-fusion-bridge cycle that generates complex chromosome rearrangements (Figure 1A) (7). This occurs frequently in the multigenic disorder Fanconi anemia (FA), in which patients harbor mutations that affect DNA damage checkpoint signaling and impair interstrand DNA crosslink repair (8,9). In normal cells, the DSB stemming from processing of a DNA crosslink is funneled into the HR pathway for repair, but in FA cells, NHEJ becomes the default repair mechanism (10). Accordingly, the large-scale genomic rearrangements observed in FA cells can be prevented by inactivating the Ku70-Ku80 heterodimer, a central player in NHEJ (10). Thus, the FA phenotype reflects the adverse consequence of engaging NHEJ within an aberrant context.
Figure 1.
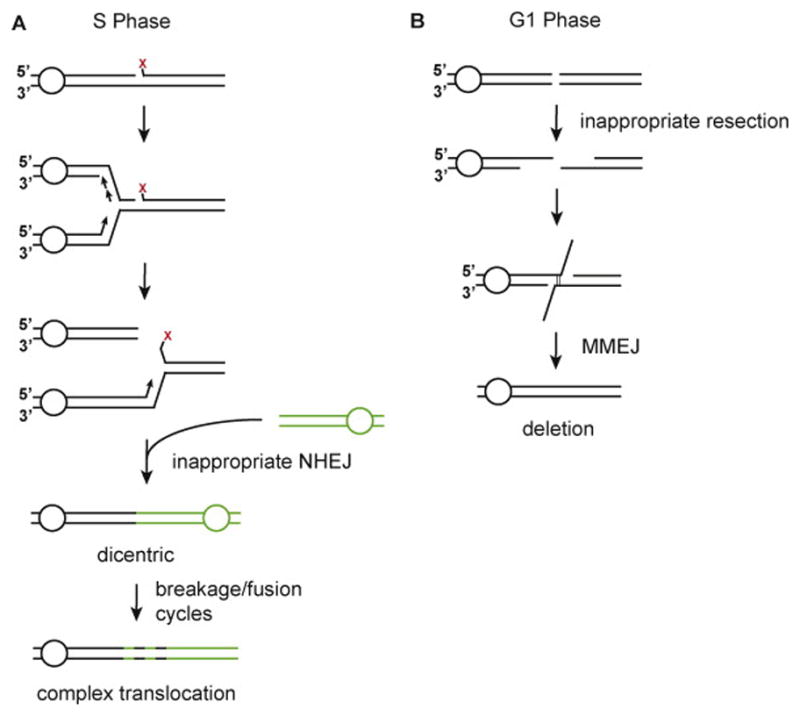
(A) Inappropriate NHEJ of replication-associated DSBs in S phase leads to dicentric chromosomes and complex chromosome rearrangements. (B) Extensive resection of DNA ends in G1 cells can lead to deletion-associated MMEJ events.
Any attempt to repair DSBs by HR in the G1 phase can also be catastrophic, as it may lead to deletions and translocations through an error-prone microhomology-based end joining mechanism (11). In order for HR to occur, the ends of the DSB must first undergo a significant amount of nucleolytic resection that entails the degradation of the 5′ DNA strand to generate 3′ ssDNA tails (12–15). This process precludes the pairing of short ssDNA overhangs that NHEJ relies on to guide break joining. Specifically, 5′ overhangs are ablated by resection, and 3′ overhangs become separated by large gaps, both of which effectively eliminate the possibility of repair by NHEJ (16). Cell death ordinarily ensues should DSB end resection be permitted to occur in the absence of a homologous DNA template, or when the HR machinery is defective (17). However, a small fraction of such cells can survive using the last-ditch mechanism known as alternative end joining or microhomology-mediated end joining (MMEJ) (Figure 1B) (11,18). MMEJ involves hybridization of short regions of homology between the resected overhangs and always leads to deletions. The microhomologies that are hallmarks of MMEJ are often found at translocation breakpoints in tumor cells, providing evidence that inappropriate resection in G1 can lead to tumorigenesis (19–21). The foregoing discussions highlight the importance of DSB repair pathway choice, as the inadvertant use of a repair pathway can results in the type of genomic rearrangements seen in tumor cells.
As resection occurs, the 3′ tails are promptly coated by the ubiquitous and abundant ssDNA binding protein RPA. DNA bound RPA must be replaced by the conserved recombinase protein Rad51, and this protein-protein exchange is facilitated by a class of HR factors known as recombination mediators (22). Once a small cluster of Rad51 protomers are loaded onto the ssDNA, they serve as the nucleus for the assembly of a right-handed Rad51 helical filament, with the concomitant displacement of RPA. The resulting Rad51-ssDNA nucleoprotein filament, commonly referred to as the presynaptic filament, engages a duplex DNA molecule, performs a search for DNA homology in the bound duplex, and catalyzes the intermediate steps of HR, including invasion of and DNA strand exchange with the duplex molecule. These steps are accompanied by DNA synthesis, followed by the resolution of the resulting DNA structures, and, finally, DNA ligation (3).
In E. coli, DSB end resection is catalyzed by the nuclease/helicase complex RecBCD (23,24), with a minor pathway being dependent on the RecQ helicase working in conjunction with Exonuclease I (25). The DNA resection process is much more complicated in eukaryotes, however, involving three distinct nucleases (Mre11, Exo1, and Dna2), a RecQ-related DNA helicase (Sgs1 in the budding yeast and either BLM or WRN in humans and other mammals), and a number of accessory factors (13,15). In mammals, an intricate, cell cycle-dependent control mechanism that involves the breast cancer tumor suppressor BRCA1 and the 53BP1 protein regulates the onset of resection (26). The past few years have witnessed major advances in understanding how the multiplicity of nucleases operate to resect DSB ends and the regulatory mechanisms that help impose cell cycle dependency of NHEJ versus HR at the level of end resection. Below, we review some of these advances and also focus on unresolved questions currently under active investigation.
Multiplicity of conserved nucleases in DNA end resection
Genetic studies by several laboratories in the budding yeast S. cerevisiae have played a pivotal role in identifying the three nucleolytic entitites that catalyze 5′ DNA strand resection during HR (27,28). We will first provide background information on these nucleases, namely, Mre11, Exo1, and Dna2, followed by an analysis of biochemical investigations revealing how they functionally co-operate with their cofactors and with one another in mediating long range DNA end resection in cells.
(A) The Mre11-Rad50-Xrs2 complex
The Mre11-Rad50-Xrs2 (MRX) complex (MRE11-RAD50-NBS1, or MRN, in humans) had long been known to mediate the processing of meiotic DSBs introduced by a type II topoisomerase-like protein called Spo11 (29–31). Spo11 remains covalently bound to the 5′ strands of the break ends and MRX, and presumably MRN, is needed to eliminate the Spo11 conjugate from the DNA ends. Other studies have established that the MRX complex is also important for processing mitotic DNA breaks that are capped by a trapped topoisomerase conjugate (32).
Unexpectedly, Mre11, the nuclease subunit of MRX, digests DNA exonucleolytically with a 3′ to 5′ polarity, which is the exact opposite of what is needed to generate 3′ ssDNA overhangs for HR promotion in cells (33). On the other hand, Mre11 also possesses an endonuclease activity on hairpin structures (33–35). Both of the 3′-5′ exonuclease and endonuclease activities of Mre11 are enhanced by Rad50 and Xrs2 (34,36,37). Genetic evidence has also implicated the MRX-associated protein Sae2 (CtIP in humans) in DNA end resection (27,38). Like mutants of the MRX complex, SAE2 deletion renders cells sensitive to DNA damaging agents, and diploid sae2Δ cells fail to process Spo11-induced DSBs and thus undergo cell cycle arrest early in meiosis (39,40). Emerging evidence indicates that MRX-Sae2, and by inference MRN-CtIP, initiates DNA end resection via the introduction of internal incisions proximal to DSB ends (see below) (41,42).
(B) Exo1 and Sgs1-Dna2
Even when the MRX-Sae2 pathway is impaired, mitotic cells are surprisingly adept at processing “clean” DSBs, such as those created by endonucleases, to yield ssDNA tails suitable for the assembly of the HR machinery (43,44). This finding revealed that at least one other nuclease is able to process clean DNA breaks in the absence of MRX-Sae2. Painstaking analyses have uncovered the conserved nucleases Exo1 and Dna2 as redundant entities that can process DNA breaks in parallel in the absence of MRX (27,28,45).
Exo1 and Dna2, already known to be involved in DNA mismatch repair and Okazaki fragment processing, respectively, were shown to act independently to process a DSB induced by the HO endonuclease (28). Exo1 digests duplex DNA exonucleolytically in the 5′ to 3′ direction, an attribute expected of a resection nuclease (46). Dna2 has endonuclease activity that can cleave both 5′ and 3′ single-stranded DNA overhangs adjoining a duplex region, such as those present in a Y structure stemming from the separation of strands in duplex DNA molecule by a helicase (47). Dna2 has a modest 5′ to 3′ helicase activity that is dispensable for DNA end resection (see below) (28,48,49). Instead, the separation of DNA strands in the Dna2-mediated resection reaction is carried out by the RecQ helicase Sgs1 (BLM in humans) (27,28,45,49). In cells, Sgs1 associates with a type IA topoisomerase, Top3 (Topo IIIα in humans), and an OB-fold containing protein, Rmi1, to form a trimeric complex called the STR complex (50–53). Both Rmi1 and Top3 are needed for the optimal activity of Sgs1-Dna2 in DNA end resection in vitro and in cells (49). There is compelling evidence that the human equivalent of the STR complex, referred to as BTR, which harbors BLM, Topo IIIα, RMI1 and another OB-fold protein, RMI2, also works in conjunction with DNA2 in DNA end resection (54,55). It should be noted that while the role of STR and BTR in the resolution of the double Holliday junction (dHJ), a late HR intermediate, is reliant on the catalytic activity of Top3/Topo IIIα (56,57), this activity is completely dispensable for the resection function of these protein complexes (49,54).
Biochemical reconstitution and insights into DNA end resection reactions
(A) Resection catalyzed by MRX-Sae2
As alluded to above, even though nuclease null mutants of Mre11 show a resection phenotype in both mitotic and meiotic cells, the exonuclease activity of Mre11 has a polarity opposite to what is needed to generate 3′ ssDNA tails in DSB end resection. It thus seems clear that Mre11 acts as an endonuclease in the resection process in cells (42). This premise is consistent with the observation that the MRX-Sae2 ensemble introduces internal incisions a short distance from Spo11-bound ends in meiosis (Figure 3) (58), and with results from the analysis of a mre11 mutant that is differentially inactivated for the protein’s exonuclease activity (59). The examination of small molecule inhibitors that target either the endo- or exonuclease activity of Mre11 has also provided supporting evidence in this regard (60).
Figure 3.
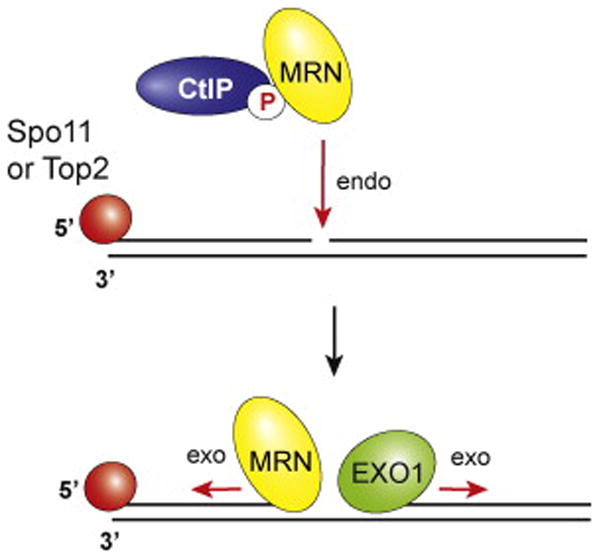
Model for nick-initiated resection by MRN-CtIP at protein-occluded DSBs. Sae2/CtIP activates the MRX/MRN endonuclease activity to generate a nick, followed by bidirectional resection catalyzed by MRN in the 3′ to 5′ direction and EXO1 in the 5′ to 3′ direction.
A breakthrough in understanding the mechanism by which the endonuclease activity of Mre11 acts to incise DNA proximal to DSB ends came from a recent biochemical study by Cannavo and Cejka (41). Using highly purified Saccharomyces cerevisiae proteins, these investigators showed that Sae2 activates a latent dsDNA-specific endonuclease activity of Mre11 within the context of the MRX complex. Importantly, the endonuclease activity cleaves the 5′-terminated dsDNA strand in a manner that is strongly enhanced by a protein block at the DNA end. This activity is also dependent on the ATPase activity of Rad50 as well as the physical interaction between MRX and Sae2. The results of Cannavo and Cejka (41) thus provide direct support to the premise, as first proposed by Garcia et al. (42), that MRX-Sae2 creates internal entry sites for the Mre11 exonuclease activity, Exo1, and possibly Sgs1-Dna2 for further processing of the DNA template in preparation for the HR reaction (see the section titled “Model for the conserved mechanism of DNA end resection” below and Figure 3). It can be expected that CtIP, the ortholog of Sae2 in humans, will likewise activate the endonuclease function of MRN to introduce DNA nicks into the 5′ terminated strand of a DSB. Recent structural work has revealed CtIP to be a flexible molecule with a conserved N-terminal alpha helical tetramerization domain that is essential for resection in vivo (61,62). Its S. pombe ortholog Ctp1 binds a variety of DNA structures and has the ability to bridge DNA ends, raising the possibility that it helps tether DSB ends during their nucleolytic processing (61). These results are consistent with the idea that CtIP/Ctp1 plays a structural role in coordinating and positioning the MRN nuclease.
(B) Exo1-mediated resection
Biochemical reconstitution studies have revealed several interesting aspects of the resection reaction mediated by Exo1, including crosstalk with the other resection pathways (Figure 2). Specifically, MRX/MRN was found to strongly upregulate the activity of Exo1 (49,63). Moreover, RPA stimulates end resection by preventing nonspecific binding of Exo1 to single-stranded DNA (49,63). It should be noted that Exo1 can access pre-existing nicks and gaps in duplex DNA (64), thus making it suited to mediating long range DNA resection at an incision or gap introduced via the action of MRX-Sae2. Interestingly, the activity of human EXO1 protein is enhanced by BLM in a manner that does not require the helicase activity of the latter (Figure 2) (65). Sgs1, on the other hand, does not appear to influence the activity of yeast Exo1 (49), suggesting that functional synergy is unique to the human proteins.
Figure 2.
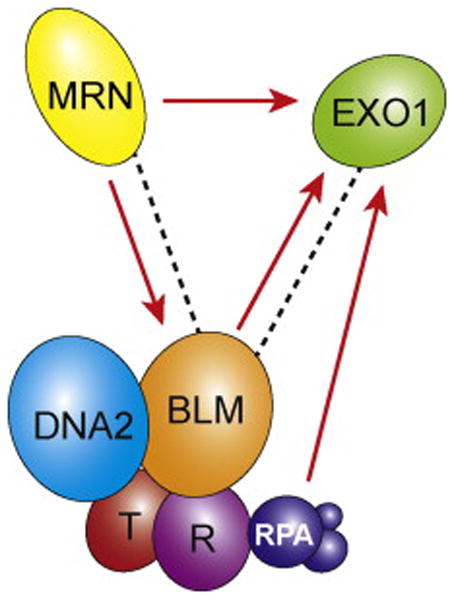
Crosstalk among the three nucleases in DNA end resection. Dotted lines indicate protein-protein interactions and red arrows indicate a stimulatory effect. BLM interacts directly with EXO1, and in yeast MRX interacts with Sgs1 (the BLM ortholog). RPA, BLM, and MRN are all capable of stimulating the nuclease activity of EXO1, and MRN(X) enhances the helicase activity of BLM/Sgs1.
(C) ATP-hydrolysis driven resection mediated by Sgs1/BLM and Dna2
DNA end resection that is dependent on the Sgs1-Dna2 protein pair has been reconstituted with highly purified proteins (49,66). The combination of RPA, Sgs1, and Dna2 is minimally sufficient for 5′ DNA strand resection to occur. In this reaction, Sgs1 helicase unwinds the DNA to produce an intermediate that is digested by Dna2. The DNA unwinding reaction is strongly stimulated by RPA in a species-specific manner (66). Consistent with genetic results, Dna2’s nuclease activity, but not its helicase activity, is needed for resection (49). Aside from sequestering ssDNA unwound by Sgs1, RPA also enhances the ability of Dna2 to incise the 5′ strand while attenuating degradation of the 3′ DNA strand (49,63,66). As such, RPA helps impose the strict 5′ polarity of DNA end resection in vitro and also in cells (67). Importantly, the MRX complex and the Top3-Rmi1 heterodimer individually enhance the ability of Sgs1 to unwind DNA, and, consequently, stimulate resection efficiency (Figure 2) (49,66). MRX and Top3 both recognize DNA ends, likely enabling these factors to properly target Sgs1 to DSBs (54,68). Findings similar to the above have been reported for reconstituted DNA end resection systems harboring the human orthologs of the yeast factors (63). Moreover, a recent study from our laboratory has provided evidence (i) for a subsidiary role of Topo IIIα in helping impose the 5′ polarity of DNA end resection, (ii) that DNA2 stimulates the helicase activity of BLM, and (iii) that both DNA2 and Topo IIIα-RMI1-RMI2 increase the processivity of the BLM helicase activity (54).
Multi-faceted role of RPA in DNA end resection
As discussed above, both the Exo1- and Sgs1-Dna2-mediated resection pathways are dependent on RPA for maximal efficiency, and in the case of the latter pathway, RPA also plays a major role in the imposition of DNA strand polarity during resection (49,63,66). In cells, depletion of RPA (accomplished using a degron system because the protein is essential for DNA replication) abolishes extensive DNA end resection, while still allowing short-range MRX-Sae2-dependent end trimming to occur (67). Interestingly, this study also revealed that the 3′ ssDNA tails derived from end resection become destabilized upon RPA depletion (67).
Model for the conserved mechanism of DNA end resection
In summary, the weight of the available evidence is in strong support of the model first proposed by Garcia et al. (42) and summarized in Figure 3. This model illustrates how MRX-Sae2/MRN-CtIP initiates resection in both vegetative and meiotic cells via endonucleolytic incision of the 5′-terminated strand adjacent to the DSB end. The model posits that, once internal scissions have been introduced, the exonuclease activities of Mre11 and Exo1, which have opposite polarities, generate a DNA gap. While Exo1 is well known to act at a nick in duplex DNA (indeed, this corresponds to its natural substrate in DNA mismatch repair) (69,70), it remains to be determined experimentally whether MRX and the STR-Dna2 ensemble are capable of processing a nicked DNA structure. Moreover, there is every reason to believe that MRN-CtIP acts in a similar fashion to generate a nicked DNA molecule to be processed further by the MRN exonuclease and EXO1, and perhaps the BTR-DNA2 ensemble as well. Although Sae2 and CtIP lack homology to known nucleases, it has been reported that they possess endonuclease activity in vitro (71–73). Other groups have purified nuclease-free Sae2 and CtIP, however, necessitating further investigation of the conflicting results (41,49,61,62).
DNA end resection within the context of chromatin
Multiple lines of evidence indicate that extensive changes in chromatin structure occur during DNA end resection, and the conserved, ATP hydrolysis-dependent chromatin remodelers Fun30, RSC, and INO80 have been implicated in this process (74–80). Mutant analyses have provided evidence for an overlapping role of these chromatin remodelers within the vincinity of a DSB, with RSC mutants showing the most severe phenotype in this regard (74). Importantly, Fun30 appears to be the major remodeler that promotes long-range resection (74). Fun30 likely remains associated with the resection machinery, as it interacts with multiple resection factors including Dna2, Exo1 and RPA (74,75). The human SMARCAD1 protein, homologous to Fun30, is also required for extensive resection (75).
In yeast, Rad9, the functional equivalent of mammalian 53BP1 (discussed below), associates with nucleosomes containing methylated H3K79 and also H2A phosphorylated at S129 (γ-H2A) (81–83). Deletion of RAD9 leads to faster resection (81,84) and largely suppresses the resection defect of fun30Δ (74). The latter observation suggests that the role of Fun30 is to help alleviate the Rad9-dependent chromatin barrier to resection.
The resection of a nucleosomal DNA substrate by Exo1 and the Sgs1-Dna2 ensemble has been reconstituted and examined in detail by Adkins et al. (85). The presence of nucleosomes impedes resection by both the Exo1- and Sgs1-Dna2-dependent mechanisms, although the former is more strongly affected in this regard (85). The presence of a nucleosome-free region allows the Sgs1-Dna2 ensemble to partially overcome the nucleosomal barrier downstream (85). The implication is that, once fully assembled on adjoining nucleosome free DNA, the Sgs1-Dna2 ensemble, at the expense of ATP hydrolysis, is able to negotiate through nucleosomes in its path. Interestingly, resection by Exo1 can be largely restored by removing the H2A-H2B dimer from nucleosomes, and biochemical and genetic evidence reveals that nucleosomes harboring H2AZ, an H2A variant that has been linked to DSB repair, are more accessible to Exo1 (85). Consistent with this, the SCRAP complex, which incorporates H2AZ into chromatin, has recently been shown to promote resection in human cells (86).
Much remains to be learned about the role of chromatin in regulating the extent and efficiency of DNA resection. For instance, in cells lacking the Sgs1-Dna2-dependent pathway, Exo1 is still able to conduct long range resection many kilobases away from the DSB end. It will be important to devise a reconstituted system encompassing the requisite chromatin remodelers, modified nucleosomes, and other cofactors to decipher the mechanism by which the chromatin barrier is overcome within the context of Exo1-mediated resection. Another key question is whether nucleosomes are evicted prior to the onset of resection or whether chromatin remodelers help the resection machinery navigate through chromatin, with nucleosome loss then occurring as a consequence of nucleolytic digestion. How histone modifications, such as H3K79 methylation and H2A S129 phosphorylation mentioned above, affect resection is another area that is ripe for exploration, as a number of these modifications have been implicated in the cell-cycle dependent regulation of resection as summarized below.
Resection regulation by cyclin-dependent kinase during the cell cycle
HR normally employs the sister chromatid as homologous information donor, and a dedicated mechanism exists to prevent it from occuring in G1 cells. Indeed, G1 cells do not engage the Rad53-dependent DNA damage checkpoint upon DSB induction (87), as activation of this checkpoint is dependent on ssDNA derived from DNA end resection (88). The first direct evidence that resection is regulated during the cell cycle came from Ira et al., who showed that holding yeast cells in G1 or inhibiting the cyclin-dependent kinase (CDK) blocks resection (89). Sae2/CtIP was subsequently identified as one of the critical CDK targets integral to the cell-cycle dependent control of resection (90,91). Specifically, S267 in Sae2 (S327 in human CtIP) is phosphorylated at the G1-S transitional boundary, an event that is important for the activation of DNA end resection (90,92). Work in S. pombe suggests that phosphorylation of Ctp1 (the Sae2 ortholog) is important for its interaction with the FHA domain of NBS1, an integral component of the MRN complex (93). NBS1 is also targeted by CDK at S432 (94). This phosphorylation event is important for resection, but, since it is not required for the interaction of CtIP with MRN, its functional impact remains to be delineated.
In addition to Sae2/CtIP, recent work has identified yeast Dna2 and human EXO1 as targets of CDK-dependent phosphorylation as well. Yeast Dna2 is phosphorylated by Cdk1 on T4, S17 and S237 to ensure its timely recruitment to DSBs (95). S17 phosphorylation is also important for the nuclear localization of DNA2 (96). Mutating these sites to alanine causes long-range resection to become almost completely dependent on Exo1 (95). Four CDK-dependent phosphorylation sites - S639, T732, S815 and T824 - have been identified in human EXO1 (97). As in the case of yeast Dna2, phosphorylation promotes the recruitment of human EXO1 to DSB sites (97). Additionally, EXO1 co-immunoprecipitates from cells with BRCA1, known to be involved in the channeling of DSBs into HR, in a phospho-dependent manner (97). Whether this interaction is direct or not remains to be determined.
Regulation of resection in mammalian cells by 53BP1 and BRCA1
The tumor suppressor BRCA1 forms an obligate dimer with BARD1 and the resulting protein complex possesses an E3 ubiquitin ligase activity (98,99). BRCA1 has a well known function in HR (100–103) and it has recently been linked to the promotion of DNA resection (104–107). Recent studies have also identified 53BP1, a phosphorylation target of the ATM kinase, as a negative regulator of DNA end resection. 53BP1 helps prevent resection in G1 (104), but is removed in a BRCA1-dependent manner in S phase, thus allowing resection to occur (106). As such, 53BP1 and BRCA1 fulfill opposing roles in DSB repair, with the former being a NHEJ facilitator and the latter acting to commit breaks to repair via HR.
The recruitment of 53BP1 to DSB sites involves multiple protein-protein interactions and its post-translational modifications (26). These include interactions with the tandem BRCT domains of MDC1 (108,109), histone H4 that is dimethylated on lysine 20 (H4K20me2) (110), and histone H2A ubiquitinated on K15 (111). Each of these interactions entails a distinct domain on 53BP1, and several deubiquinating enzymes (DUBs) and proteins compete with 53BP1 for binding to the aforementioned partners, further adding to the complexity of regulation (112–114).
53BP1 mediates its inhibition on resection through two effector proteins: RIF1 and PTIP, both of which interact with 53BP1 in a manner that requires the ATM-dependent phosphorylation of 53BP1 (115–120). How these effectors block resection to lead to NHEJ is not yet known. They might inhibit the resection nucleases, either by preventing them from accessing the DNA ends and/or via direct interactions with the nucleases or their cofactors to attenuate their catalytic activity. In support of this idea, RIF1 has been shown to interact with BLM, but, curiously, RIF1 also appears to be required for BLM recruitment to DSBs (118,121,122). Further investigation will be needed to understand the basis for the negative influence that RIF1 exerts on resection. Determining whether RIF1 and PTIP are functionally redundant or have specific roles at DSBs with different end structures, as suggested by their differential penotypes in V(D)J and class switch recombination (115–117,119,123), will also be an important goal.
The mechanism by which BRCA1 helps mediate the removal of 53BP1 in S phase remains obscure. What we do know is that DNA damage recruitment of 53BP1 occurs in S phase cells when BRCA1 is depleted, and loss of 53BP1 leads to the recruitment of BRCA1 in the G1 phase (117). Thus, the molecular platform needed for the DNA damage recruitment of 53BP1 and BRCA1 in DNA end resection and DSB repair pathway choice is intact throughout the cell cycle, but somehow becomes differentially masked as the two proteins and their associated effectors antagonize one another. CtIP may co-operate with BRCA1 in 53BP1 removal (26). Specifically, CtIP, upon its CDK-dependent phosphorylation in G2 (124), forms a complex with BRCA1 and MRN (125,126), and CtIP phosphorylation is necessary for preventing the DSB recruitment of 53BP1 (117). However, the importance of the CtIP-BRCA1 interaction is subject to debate, as one publication showed resection defects in CtIP mutants defective for BRCA1 binding (92) but others have not observed such an effect (127–129). Recently, a study involving the use of a high resolution technique for measuring resection tract lengths in cells has provided evidence that while the BRCA1-CtIP interaction is not essential for resection, it helps ensure the timeliness of the process (130).
There is a paucity of mechanistc information on how the ubiquitin E3 ligase activity of BRCA1 contributes to its DNA end resection role. Specifically, while phosphorylated CtIP is ubiquitinated by BRCA1-BARD1 (131), the functional relevance of this modification is unknown. Recently, lysines 127 and 129 of histone H2A have been identified as ubiquitination targets of BRCA1, but how this affects the DNA damage response also remains to be established (132). EXO1 is a potential BRCA1 target given the aforementioned association between the two proteins (97). Examination of the I26A RING domain mutant of BRCA1 has suggested that its E3 ligase activity is dispensable for tumor suppression and DNA end resection (133,134). Further investigation will be important here, however, as it has been reported that the BRCA1-I26A mutant retains residual catalytic activity (135). It is nonetheless possible that 53BP1 displacement is the primary function of BRCA1 in DNA end resection, with other ubiquitin ligases, such as RNF8 and RNF168, providing the enzymatic activity relevant to resection regulation. In this regard, it has been shown that histone H2A is monoubiquitinated by RNF168 on K13 and K15, modifications that are necessary for recruiting the RAP80 complex, which in turn brings BRCA1 to the DSB site via its interaction with the associated protein Abraxas (136).
Other proteins implicated in DNA end resection
(A) Role for the WRN helicase in resection
Aside from BLM, four additional RecQ family helicases - RECQ1, WRN, RECQ4/RTS, and RECQ5 - exist in mammals (137). Interestingly, emerging evidence implicates WRN in DNA resection independently of BLM. Studies done using Xenopus egg extracts first revealed a role of WRN in DNA unwinding during end resection (138). More recently, human WRN has been shown to interact physically with DNA2, and the two proteins work in a co-operative fashion to mediate DNA resection in vitro (55). Experimentation involving the use of an HR reporter that measures gene conversion frequency has provided support for redundant cellular roles of BLM and WRN in HR (55). Intertesingly, WRN has also been suggested to function in MMEJ (139).
(B) PCNA and the 9-1-1 complex
EXO1 contains a canonical PIP box in its C-terminus which confers the ability to interact with the DNA polymerase processivity clamp PCNA (140). This interaction is critical for the retention of EXO1 at DSB sites and for its processivity as an exonuclease (141). The 9-1-1 complex, a DNA damage checkpoint-specific PCNA-related molecule, has also been identified as a resection accessory factor (142,143). Specifically, the 9-1-1 complex is required for extensive resection in yeast cells, and its human orthologue stimulates the nuclease activity of EXO1 and DNA2 in vitro (142,143). Why two distinct DNA clamps are needed for resection is unclear, and future work should address potential redundancy between them and to further delineate their biochemical roles.
(C) The SSB/SOSS complexes
Over many years, RPA was thought to be the sole functional equivalent of bacterial single-strand DNA binding protein (SSB) in eukaryotes. In 2008, however, two human protein complexes harboring either the SSB1 or SSB2 protein, both related to archaeal SSB, were identified (144). These ssDNA binding heterotrimeric complexes, referred to as SOSS1 and SOSS2, share two common subunits, INTS3 and SSBIP1 (144–146). The SSB1 complex is of higher cellular abundance and appears to play a more prominent role in the DNA damage response. It is stabilized upon DNA damage by ATM-dependent phosphorylation and localizes rapidly to DSB sites (144). SSB1 interacts directly with NBS1 and is required for efficient DSB recruitment of the MRN complex (147). Importantly, SSB1 alone has been shown to stimulate the nuclease activity of MRN (148), and the full SSB1 complex also enhances the activity of EXO1 (149). Taken together, the available results suggest that the SSB1 complex functions as a cofactor for EXO1 and MRN, while RPA acts specifically with DNA2. Unlike RPA, however, SSB1 does not localize to stalled replication forks (144). The loss of SSB1 is lethal in mice due to skeletal defects, but surprisingly, has no obvious effect on the DNA damage response (150,151). Much remains to be learned about the biological roles and mechanisms of action of the two SSB complexes, e.g. whether these complexes function redundantly of one another in DNA end resection, and if they would also enhance the BLM-DNA2 ensemble.
(D) The hnRNPUL proteins
Ribonuclear hnRNP proteins are involved in transcription, translation and the nuclear export of RNAs (152,153), but hnRNPUL1 and 2 have also been implicated in DNA end resection. These proteins localize to sites of DNA damage in an MRN-dependent fashion (154), and they physically interact directly with BLM and are also required for BLM recruitment to damage sites, suggesting a role in long-range end resection (154). These results are intriguing in view of the finding that small RNAs are generated by DROSHA and DICER at DSB sites and are required for the formation of MRN-dependent DNA damage response foci that harbor numerous DNA checkpoint and repair proteins (155).
Conclusions
A great deal of progress has been made in understanding the genetic requirements of the conserved pathways of DNA end resection in yeast and other eukaryotes in just the past few years. Importantly, the in vitro reconstitution of DNA end resection reactions with yeast and human proteins has yielded a great deal of mechanistic insights into these pathways and how pathway crosstalk occurs. However, we are only beginning to understand the layers of positive and negative controls, e.g. the underpinnings of the BRCA1-53BP1 regulatory network, that affect DNA end resection and hence the choice among NHEJ, MMEJ, and HR for DNA lesion removal. Given the involvment of DSB repair mechanisms in tumor biology, ongoing studies in the above areas have strong relevance to human health and disease.
Acknowledgments
The studies in our laboratory have been supported by research grants and a program project grant (SBDR) from the National Institutes of Health. We apologize to colleagues whose work is not cited owing to space limitation.
Abbreviations
- DSB
Double-strand break
- HR
Homologous recombination
- NHEJ
Nonhomologous end joining
- MMEJ
Microhomolgy-mediated end joining
- MRN
MRE11-RAD50-NBS1 complex
- RPA
Replication protein A
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Malkova A, Haber JE. Mutations arising during repair of chromosome breaks. Annu Rev Genet. 2012;46:455–473. doi: 10.1146/annurev-genet-110711-155547. [DOI] [PubMed] [Google Scholar]
- 2.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harbor perspectives in biology. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 4.Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor perspectives in biology. 2014;6:a016428. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 6.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 7.Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunological reviews. 2003;194:77–95. doi: 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 8.Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annual review of biophysics. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- 10.Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010;329:219–223. doi: 10.1126/science.1192277. [DOI] [PubMed] [Google Scholar]
- 11.McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends in genetics: TIG. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blackwood JK, Rzechorzek NJ, Bray SM, Maman JD, Pellegrini L, Robinson NP. End-resection at DNA double-strand breaks in the three domains of life. Biochemical Society transactions. 2013;41:314–320. doi: 10.1042/BST20120307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009;8:983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mimitou EP, Symington LS. DNA end resection--unraveling the tail. DNA Repair (Amst) 2011;10:344–348. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niu H, Raynard S, Sung P. Multiplicity of DNA end resection machineries in chromosome break repair. Genes Dev. 2009;23:1481–1486. doi: 10.1101/gad.1824209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daley JM, Laan RL, Suresh A, Wilson TE. DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J Biol Chem. 2005;280:29030–29037. doi: 10.1074/jbc.M505277200. [DOI] [PubMed] [Google Scholar]
- 17.Vaze MB, Pellicioli A, Lee SE, Ira G, Liberi G, Arbel-Eden A, Foiani M, Haber JE. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell. 2002;10:373–385. doi: 10.1016/s1097-2765(02)00593-2. [DOI] [PubMed] [Google Scholar]
- 18.Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol Cell Biol. 2003;23:8820–8828. doi: 10.1128/MCB.23.23.8820-8828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Current opinion in genetics & development. 2012;22:211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. J Biol Chem. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 23.Smith GR. How RecBCD enzyme and Chi promote DNA break repair and recombination: a molecular biologist’s view. Microbiology and molecular biology reviews: MMBR. 2012;76:217–228. doi: 10.1128/MMBR.05026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiology and molecular biology reviews: MMBR. 2008;72:642–671. doi: 10.1128/MMBR.00020-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Handa N, Morimatsu K, Lovett ST, Kowalczykowski SC. Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 2009;23:1234–1245. doi: 10.1101/gad.1780709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daley JM, Sung P. 53BP1, BRCA1, and the Choice between Recombination and End Joining at DNA Double-Strand Breaks. Mol Cell Biol. 2014;34:1380–1388. doi: 10.1128/MCB.01639-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furuse M, Nagase Y, Tsubouchi H, Murakami-Murofushi K, Shibata T, Ohta K. Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J. 1998;17:6412–6425. doi: 10.1093/emboj/17.21.6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nairz K, Klein F. mre11S--a yeast mutation that blocks double-strand-break processing and permits nonhomologous synapsis in meiosis. Genes Dev. 1997;11:2272–2290. doi: 10.1101/gad.11.17.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartsuiker E, Neale MJ, Carr AM. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol Cell. 2009;33:117–123. doi: 10.1016/j.molcel.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 34.Trujillo KM, Yuan SS, Lee EY, Sung P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J Biol Chem. 1998;273:21447–21450. doi: 10.1074/jbc.273.34.21447. [DOI] [PubMed] [Google Scholar]
- 35.Trujillo KM, Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J Biol Chem. 2001;276:35458–35464. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- 36.Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trujillo KM, Roh DH, Chen L, Van Komen S, Tomkinson A, Sung P. Yeast xrs2 binds DNA and helps target rad50 and mre11 to DNA ends. J Biol Chem. 2003;278:48957–48964. doi: 10.1074/jbc.M309877200. [DOI] [PubMed] [Google Scholar]
- 38.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKee AH, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prinz S, Amon A, Klein F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cannavo E, Cejka P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature. 2014;514:122–125. doi: 10.1038/nature13771. [DOI] [PubMed] [Google Scholar]
- 42.Garcia V, Phelps SE, Gray S, Neale MJ. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature. 2011;479:241–244. doi: 10.1038/nature10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 44.Nakada D, Hirano Y, Sugimoto K. Requirement of the Mre11 complex and exonuclease 1 for activation of the Mec1 signaling pathway. Mol Cell Biol. 2004;24:10016–10025. doi: 10.1128/MCB.24.22.10016-10025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee BI, Wilson DM., 3rd The RAD2 domain of human exonuclease 1 exhibits 5′ to 3′ exonuclease and flap structure-specific endonuclease activities. J Biol Chem. 1999;274:37763–37769. doi: 10.1074/jbc.274.53.37763. [DOI] [PubMed] [Google Scholar]
- 47.Budd ME, Campbell JL. A yeast gene required for DNA replication encodes a protein with homology to DNA helicases. Proc Natl Acad Sci U S A. 1995;92:7642–7646. doi: 10.1073/pnas.92.17.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Budd ME, Choe WC, Campbell JL. DNA2 encodes a DNA helicase essential for replication of eukaryotic chromosomes. J Biol Chem. 1995;270:26766–26769. doi: 10.1074/jbc.270.45.26766. [DOI] [PubMed] [Google Scholar]
- 49.Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, Prakash R, Seong C, Liu D, Lu L, et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature. 2010;467:108–111. doi: 10.1038/nature09318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang M, Bellaoui M, Zhang C, Desai R, Morozov P, Delgado-Cruzata L, Rothstein R, Freyer GA, Boone C, Brown GW. RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J. 2005;24:2024–2033. doi: 10.1038/sj.emboj.7600684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu D, Guo R, Sobeck A, Bachrati CZ, Yang J, Enomoto T, Brown GW, Hoatlin ME, Hickson ID, Wang W. RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 2008;22:2843–2855. doi: 10.1101/gad.1708608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin J, Sobeck A, Xu C, Meetei AR, Hoatlin M, Li L, Wang W. BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. EMBO J. 2005;24:1465–1476. doi: 10.1038/sj.emboj.7600622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh TR, Ali AM, Busygina V, Raynard S, Fan Q, Du CH, Andreassen PR, Sung P, Meetei AR. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008;22:2856–2868. doi: 10.1101/gad.1725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Daley JM, Chiba T, Xue X, Niu H, Sung P. Multifaceted role of the Topo IIIalpha-RMI1-RMI2 complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 2014;42:11083–11091. doi: 10.1093/nar/gku803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sturzenegger A, Burdova K, Kanagaraj R, Levikova M, Pinto C, Cejka P, Janscak P. DNA2 cooperates with the WRN and BLM RecQ helicases to mediate long-range DNA end resection in human cells. J Biol Chem. 2014;289:27314–27326. doi: 10.1074/jbc.M114.578823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bussen W, Raynard S, Busygina V, Singh AK, Sung P. Holliday junction processing activity of the BLM-Topo IIIalpha-BLAP75 complex. J Biol Chem. 2007;282:31484–31492. doi: 10.1074/jbc.M706116200. [DOI] [PubMed] [Google Scholar]
- 57.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 58.Neale MJ, Pan J, Keeney S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature. 2005;436:1053–1057. doi: 10.1038/nature03872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell. 2014;53:7–18. doi: 10.1016/j.molcel.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andres SN, Appel CD, Westmoreland JW, Williams JS, Nguyen Y, Robertson PD, Resnick MA, Williams RS. Tetrameric Ctp1 coordinates DNA binding and DNA bridging in DNA double-strand-break repair. Nat Struct Mol Biol. 2015;22:158–166. doi: 10.1038/nsmb.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davies OR, Forment JV, Sun M, Belotserkovskaya R, Coates J, Galanty Y, Demir M, Morton CR, Rzechorzek NJ, Jackson SP, et al. CtIP tetramer assembly is required for DNA-end resection and repair. Nat Struct Mol Biol. 2015;22:150–157. doi: 10.1038/nsmb.2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szankasi P, Smith GR. A DNA exonuclease induced during meiosis of Schizosaccharomyces pombe. J Biol Chem. 1992;267:3014–3023. [PubMed] [Google Scholar]
- 65.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467:112–116. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen H, Lisby M, Symington LS. RPA coordinates DNA end resection and prevents formation of DNA hairpins. Mol Cell. 2013;50:589–600. doi: 10.1016/j.molcel.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 70.Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J Biol Chem. 2005;280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP, et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell. 2014;54:1022–1033. doi: 10.1016/j.molcel.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H, Li Y, Truong LN, Shi LZ, Hwang PY, He J, Do J, Cho MJ, Li H, Negrete A, et al. CtIP maintains stability at common fragile sites and inverted repeats by end resection-independent endonuclease activity. Mol Cell. 2014;54:1012–1021. doi: 10.1016/j.molcel.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28:638–651. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature. 2012;489:576–580. doi: 10.1038/nature11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012;489:581–584. doi: 10.1038/nature11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van Attikum H, Fritsch O, Gasser SM. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J. 2007;26:4113–4125. doi: 10.1038/sj.emboj.7601835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell. 2004;16:979–990. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 78.van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–788. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 79.Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 2004;119:767–775. doi: 10.1016/j.cell.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 80.Seeber A, Hauer M, Gasser SM. Nucleosome remodelers in double-strand break repair. Current opinion in genetics & development. 2013;23:174–184. doi: 10.1016/j.gde.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 81.Lazzaro F, Sapountzi V, Granata M, Pellicioli A, Vaze M, Haber JE, Plevani P, Lydall D, Muzi-Falconi M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008;27:1502–1512. doi: 10.1038/emboj.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hammet A, Magill C, Heierhorst J, Jackson SP. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 2007;8:851–857. doi: 10.1038/sj.embor.7401036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol. 2005;25:8430–8443. doi: 10.1128/MCB.25.19.8430-8443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lydall D, Weinert T. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- 85.Adkins NL, Niu H, Sung P, Peterson CL. Nucleosome dynamics regulates DNA processing. Nat Struct Mol Biol. 2013;20:836–842. doi: 10.1038/nsmb.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dong S, Han J, Chen H, Liu T, Huen MS, Yang Y, Guo C, Huang J. The human SRCAP chromatin remodeling complex promotes DNA-end resection. Curr Biol. 2014;24:2097–2110. doi: 10.1016/j.cub.2014.07.081. [DOI] [PubMed] [Google Scholar]
- 87.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 88.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 89.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, Li J, Carr AM, Jackson SP, Smerdon SJ. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell. 2009;139:100–111. doi: 10.1016/j.cell.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Falck J, Forment JV, Coates J, Mistrik M, Lukas J, Bartek J, Jackson SP. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012;13:561–568. doi: 10.1038/embor.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen X, Niu H, Chung WH, Zhu Z, Papusha A, Shim EY, Lee SE, Sung P, Ira G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct Mol Biol. 2011;18:1015–1019. doi: 10.1038/nsmb.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kosugi S, Hasebe M, Tomita M, Yanagawa H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A. 2009;106:10171–10176. doi: 10.1073/pnas.0900604106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, Burma S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nature communications. 2014;5:3561. doi: 10.1038/ncomms4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 99.Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276:14537–14540. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 100.Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 101.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 102.Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- 103.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 104.Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35:534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eliezer Y, Argaman L, Rhie A, Doherty AJ, Goldberg M. The direct interaction between 53BP1 and MDC1 is required for the recruitment of 53BP1 to sites of damage. J Biol Chem. 2009;284:426–435. doi: 10.1074/jbc.M807375200. [DOI] [PubMed] [Google Scholar]
- 109.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 110.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, Dantuma NP. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat Struct Mol Biol. 2011;18:1345–1350. doi: 10.1038/nsmb.2188. [DOI] [PubMed] [Google Scholar]
- 113.Butler LR, Densham RM, Jia J, Garvin AJ, Stone HR, Shah V, Weekes D, Festy F, Beesley J, Morris JR. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012;31:3918–3934. doi: 10.1038/emboj.2012.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell reports. 2012;2:1233–1243. doi: 10.1016/j.celrep.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 115.Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–1280. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 118.Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288:11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Grabarz A, Guirouilh-Barbat J, Barascu A, Pennarun G, Genet D, Rass E, Germann SM, Bertrand P, Hickson ID, Lopez BS. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell reports. 2013;5:21–28. doi: 10.1016/j.celrep.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 122.Xu D, Muniandy P, Leo E, Yin J, Thangavel S, Shen X, Ii M, Agama K, Guo R, Fox D, 3rd, et al. Rif1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 2010;29:3140–3155. doi: 10.1038/emboj.2010.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Callen E, Faryabi RB, Luckey M, Hao B, Daniel JA, Yang W, Sun HW, Dressler G, Peng W, Chi H, et al. The DNA damage- and transcription-associated protein paxip1 controls thymocyte development and emigration. Immunity. 2012;37:971–985. doi: 10.1016/j.immuni.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 126.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nakamura K, Kogame T, Oshiumi H, Shinohara A, Sumitomo Y, Agama K, Pommier Y, Tsutsui KM, Tsutsui K, Hartsuiker E, et al. Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet. 2010;6:e1000828. doi: 10.1371/journal.pgen.1000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Reczek CR, Szabolcs M, Stark JM, Ludwig T, Baer R. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. The Journal of cell biology. 2013;201:693–707. doi: 10.1083/jcb.201302145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Polato F, Callen E, Wong N, Faryabi R, Bunting S, Chen HT, Kozak M, Kruhlak MJ, Reczek CR, Lee WH, et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. The Journal of experimental medicine. 2014;211:1027–1036. doi: 10.1084/jem.20131939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cruz-Garcia A, Lopez-Saavedra A, Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell reports. 2014;9:451–459. doi: 10.1016/j.celrep.2014.08.076. [DOI] [PubMed] [Google Scholar]
- 131.Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20:1721–1726. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kalb R, Mallery DL, Larkin C, Huang JT, Hiom K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell reports. 2014;8:999–1005. doi: 10.1016/j.celrep.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kakarougkas A, Ismail A, Katsuki Y, Freire R, Shibata A, Jeggo PA. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS, deRooij DG, Hirsch S, Ravi K, Hicks JB, et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science. 2011;334:525–528. doi: 10.1126/science.1209909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Joukov V, Groen AC, Prokhorova T, Gerson R, White E, Rodriguez A, Walter JC, Livingston DM. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell. 2006;127:539–552. doi: 10.1016/j.cell.2006.08.053. [DOI] [PubMed] [Google Scholar]
- 136.Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150:1182–1195. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 137.Wu L, Davies SL, Hickson ID. Roles of RecQ family helicases in the maintenance of genome stability. Cold Spring Harbor symposia on quantitative biology. 2000;65:573–581. doi: 10.1101/sqb.2000.65.573. [DOI] [PubMed] [Google Scholar]
- 138.Toczylowski T, Yan H. Mechanistic analysis of a DNA end processing pathway mediated by the Xenopus Werner syndrome protein. J Biol Chem. 2006;281:33198–33205. doi: 10.1074/jbc.M605044200. [DOI] [PubMed] [Google Scholar]
- 139.Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, Cooper PK, Chen DJ, Tainer JA. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 140.Liberti SE, Andersen SD, Wang J, May A, Miron S, Perderiset M, Keijzers G, Nielsen FC, Charbonnier JB, Bohr VA, et al. Bidirectional routing of DNA mismatch repair protein human exonuclease 1 to replication foci and DNA double strand breaks. DNA Repair (Amst) 2011;10:73–86. doi: 10.1016/j.dnarep.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen X, Paudyal SC, Chin RI, You Z. PCNA promotes processive DNA end resection by Exo1. Nucleic Acids Res. 2013;41:9325–9338. doi: 10.1093/nar/gkt672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ngo GH, Balakrishnan L, Dubarry M, Campbell JL, Lydall D. The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 2014;42:10516–10528. doi: 10.1093/nar/gku746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tsai FL, Kai M. The checkpoint clamp protein Rad9 facilitates DNA-end resection and prevents alternative non-homologous end joining. Cell Cycle. 2014;13:3460–3464. doi: 10.4161/15384101.2014.958386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Richard DJ, Bolderson E, Cubeddu L, Wadsworth RI, Savage K, Sharma GG, Nicolette ML, Tsvetanov S, McIlwraith MJ, Pandita RK, et al. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature. 2008;453:677–681. doi: 10.1038/nature06883. [DOI] [PubMed] [Google Scholar]
- 145.Huang J, Gong Z, Ghosal G, Chen J. SOSS complexes participate in the maintenance of genomic stability. Mol Cell. 2009;35:384–393. doi: 10.1016/j.molcel.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li Y, Bolderson E, Kumar R, Muniandy PA, Xue Y, Richard DJ, Seidman M, Pandita TK, Khanna KK, Wang W. HSSB1 and hSSB2 form similar multiprotein complexes that participate in DNA damage response. J Biol Chem. 2009;284:23525–23531. doi: 10.1074/jbc.C109.039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Richard DJ, Savage K, Bolderson E, Cubeddu L, So S, Ghita M, Chen DJ, White MF, Richard K, Prise KM, et al. hSSB1 rapidly binds at the sites of DNA double-strand breaks and is required for the efficient recruitment of the MRN complex. Nucleic Acids Res. 2011;39:1692–1702. doi: 10.1093/nar/gkq1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Richard DJ, Cubeddu L, Urquhart AJ, Bain A, Bolderson E, Menon D, White MF, Khanna KK. hSSB1 interacts directly with the MRN complex stimulating its recruitment to DNA double-strand breaks and its endo-nuclease activity. Nucleic Acids Res. 2011;39:3643–3651. doi: 10.1093/nar/gkq1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang SH, Zhou R, Campbell J, Chen J, Ha T, Paull TT. The SOSS1 single-stranded DNA binding complex promotes DNA end resection in concert with Exo1. EMBO J. 2013;32:126–139. doi: 10.1038/emboj.2012.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shi W, Bain AL, Schwer B, Al-Ejeh F, Smith C, Wong L, Chai H, Miranda MS, Ho U, Kawaguchi M, et al. Essential developmental, genomic stability, and tumour suppressor functions of the mouse orthologue of hSSB1/NABP2. PLoS Genet. 2013;9:e1003298. doi: 10.1371/journal.pgen.1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Feldhahn N, Ferretti E, Robbiani DF, Callen E, Deroubaix S, Selleri L, Nussenzweig A, Nussenzweig MC. The hSSB1 orthologue Obfc2b is essential for skeletogenesis but dispensable for the DNA damage response in vivo. EMBO J. 2012;31:4045–4056. doi: 10.1038/emboj.2012.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Krecic AM, Swanson MS. hnRNP complexes: composition, structure, and function. Current opinion in cell biology. 1999;11:363–371. doi: 10.1016/S0955-0674(99)80051-9. [DOI] [PubMed] [Google Scholar]
- 153.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nature reviews Molecular cell biology. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 154.Polo SE, Blackford AN, Chapman JR, Baskcomb L, Gravel S, Rusch A, Thomas A, Blundred R, Smith P, Kzhyshkowska J, et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol Cell. 2012;45:505–516. doi: 10.1016/j.molcel.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d’Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–235. doi: 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]