

Abstract
Apolipoprotein A-I is the major protein in high-density lipoprotein (HDL) and plays an important role during the process of reverse cholesterol transport (RCT). Knowledge of the high-resolution structure of full-length apoA-I is vital for a molecular understanding of the function of HDL at the various steps of the RCT pathway. Due to the flexible nature of apoA-I and aggregation properties, the structure of full-length lipid-free apoA-I has evaded description for over three decades. Sequence analysis of apoA-I suggested that the amphipathic α-helix is the structural motif of exchangeable apolipoprotein, and NMR, X-ray and MD simulation studies have confirmed this. Different laboratories have used different methods to probe the secondary structure distribution and organization of both the lipid-free and lipid-bound apoA-I structure. Mutation analysis, synthetic peptide models, surface chemistry and crystal structures have converged on the lipid-free apoA-I domain structure and function: the N-terminal domain [1–184] forms a helix bundle while the C-terminal domain [185–243] mostly lacks defined structure and is responsible for initiating lipid-binding, aggregation and is also involved in cholesterol efflux. The first 43 residues of apoA-I are essential to stabilize the lipid-free structure. In addition, the crystal structure of C-terminally truncated apoA-I suggests a monomer-dimer conversation mechanism mediated through helix 5 reorganization and dimerization during the formation of HDL. Based on previous research, we have proposed a structural model for full-length monomeric apoA-I in solution and updated the HDL formation mechanism through three intermediate states. Mapping the known natural mutations on the full-length monomeric apoA-I model provides insight into atherosclerosis development through disruption of the N-terminal helix bundle or deletion of the C-terminal lipid-binding domain.
Keywords: Apolipoproteins, Lipoproteins, Lipids, Structure, Atherosclerosis
Introduction
Diseases of lipid metabolism, in particular cardiovascular disease, remain the number one cause of health problems and death, especially CHD caused by atherosclerosis (1). One of the main risk factors for atherosclerosis is high blood cholesterol. Plasma levels of high-density lipoprotein (HDL) are negatively correlated with the incidence of atherosclerosis and the mechanism of the anti-atherogenic effects of HDL are mainly related to its involvement in the pathways of reverse cholesterol transport (RCT). It has been recently demonstrated that it does not simply lower HDL cholesterol levels in plasma that correlate with the anti-atherogenic role of HDL but the cholesterol efflux ability of the HDL that determines the role in RCT (2) underscore the need for molecular understanding of the function of HDL at the various steps of the RCT pathway.
Apolipoprotein A-I (apoA-I), the major protein component of HDL, plays vital roles throughout the RCT process as follows: formation and stabilization of the HDL particle structure, interacting with the ABCA-I transporter (3), activating lecithin cholesterol acyl transferase (LCAT) (4) and acting as a ligand for the hepatic scavenger receptor (SRB1) (5).
Plasma apoA-I (243 amino acids, 28kd) exists in lipid-free, lipid-poor and lipid-bound states and, as a consequence, has a flexible and adaptable structure similar to the molten globular state (6). This flexible nature has hindered high-resolution structural studies. Until now, the high-resolution structure of full-length lipid-free apoA-I remains enigmatic. A lipid-free apoA-I structure in full-length is crucial to understanding HDL formation and atherosclerosis development.
Structural Motif of Exchangeable Apolipoproteins
Amphipathic α-helices that exist in the exchangeable apolipoproteins (apoA-I, apoA-IV and apoE) were first proposed as a unique structural and functional motif involved in lipid interaction by Segrest (7, 8). The class A amphipathic helix is a major lipid-binding motif and is characterized by basic residues near the hydrophobic/hydrophilic interface with acidic residues at the center of the polar face. The hydrophobic interface provides the lipid-binding surface. Based on the sequence analysis of apoA-I, apoA-IV and apoE-3, consensus sequence units A (PLAEELRARLR) and B (AQLEELRERLG) were classified in early studies (9). A helix-wheel representation of the AB repeat shows the typical class A amphipathic helix (Figure 1). In addition, salt bridges between acidic and basic residues at positions (i, i+3/i, i+4) may provide additional stabilization of the helical structure. X-ray, NMR and MD simulations have confirmed the formation of helix-loop-helix structure by the ABAB repeat sequence peptide (10–12).
Figure 1.

Helix wheel diagrams of the consensus sequence AB model. Consensus sequence AB shows typical class A amphipathic helix.
Secondary Structure
Figure 2 summarizes the helical distribution of apoA-I probed by different methods and the details are as the follows. Mature apoA-I contains 243 residues that are encoded by two exons. The first 43 residues are endcoded by exon-3 and the 44–243 region is encoded by exon-4 (13). Segrest first proposed a G* helix in the exon-3 encoded region followed by ten continuous tandem repeat helices (22 or 11 residues) in the exon 4 encoded region separated by prolines: H1 [44–65], H2 [66–87], H3 [88–98], H4 [99–120], H5 [21–142], H6 [143–164], H7 [165–187], H8 [188–208], H9 [209–219], and H10 [220–243] (Figure 2) (7). The 22 amino acid repeat was further classified into the two 11-mer repeats sequence units A and B with different homology to consensus sequence units (9). The exon-4 encoded region is thus divided into a series of putative helical segments with different homologies: H1 (BB), H2 (AA), H3 (B), H4 (AB), H5 (AB), H 6(AB), H7 (AB), H8 (BB), H9 (A), H10 (BB) (Figure 2). This classification results in five AB (22 residues) repeating motifs in the center of the apoA-I sequence: AB1 (H2/H3), AB2 (H4), AB3 (H5), AB4 (H6) and AB5 (H7).
Figure 2.

Illustration of helix distribution of lipid-free, lipid-bound apoA-I derived from different methods and crystal structures of truncated apoA-I. Exon-3-encoded region (residues 1–43) is green. Exon-4-encoded region [44–243] is ice-blue. Consensus sequence peptide (CSP) A and B homology sequences are in purple and cyan, respectively. Prolines are labeled in yellow. Five AB repeats are labeled with black dotted lines with arrows. Helical structure is labeled with a rectangle while beta strand is labeled with a red arrow. Bimodal structure is labeled with a rectangle with mesh.
Nolte and Atkinson proposed a secondary structure model of lipid-free apoA-I based on circular dichroism data and computer modeling (Figure 2) (9). N-terminal residues 1–59 contained the most ambiguously defined structure composed mostly of random coil or β-structure with a short region of amphipathic alpha-helix in residues 9–13. The central region of apoA-I [60–184] was suggested to be composed solely of amphipathic α-helix. C-terminal residues 185–243 were predicted as helices from residues 187–223 and 231–243. Residues 224–230 are very hydrophobic and assigned to a random coil conformation. Mutation and deletion studies in our laboratory further refined the secondary structure model. Double substitution (G185P, G186P) increased the protein stability without altering the secondary structure, suggesting G185 and G186 are located in a loop/disordered region (14). Deletion of residues 136–143 led to stabilization without altering the number of residues in helical conformation, suggesting that this region is unstructured in lipid-free apoA-I (15). The quadruple substitution E125K/E128K/K133E/E139K led to ~17 additional residues in helical conformation consistent with a disordered structure in the segment of residues 123–142 that becomes helical as a result of the quadruple mutation or upon lipid binding (15). Deletion of residues 121–142 led to a loss of ~8 residues in helical conformation compared to WT apoA-I (16). Considering that the region [136–143] is unstructured in lipid-free apoA-I suggests that these eight helical residues are in the region [123–130], whereas P121 and L122 may form a kink between H4 and H5. Thus, the segment of residues 131–143 is disordered and may function as a “hinge domain.” Furthermore, single and double terminal truncations (Δ[1–41], Δ[1–59], Δ[198–243], Δ[209–243], Δ[1–41 and 185–243], and Δ[1–59 and 185–243]) suggested the close proximity of the N- and C-termini in the lipid-free apoA-I tertiary conformation (17). With these mutation studies, our laboratory suggested an updated secondary structure model of lipid-free apoA-I (Figure 2).
Other investigators have used different methods to probe the secondary structure distribution of lipid-free apoA-I or lipid-bound apoA-I, which have suggested different helix distributions. Site-directed spin-label electron paramagnetic resonance spectroscopy (SDSL-EPR) studies of the C-terminal fragment [163–243] of apoA-I (18) suggested a β-strand in this region from residues 208–219 as shown in Figure 2 that can bind lipid and change into helical structure. Recent hydrogen-deuterium exchange experiments on plasma apoA-I in solution suggested that residues 7–44, 54–65, 70–78, 81–115, 147–178 form α-helices, whereas residues 116–146 and 179–243 lack defined structure (19). An EPR study of full-length apoA-I suggested different helical distribution compared to the HX experiments (20). In order to gain insight into the lipid-bound apoA-I structure, NMR assignment data for intact A-I in SDS have been reported and are shown in Figure 2 (21). EPR studies of the N-terminal [6–98] (22), central [99–163] and C-terminal [164–238] fragments on nascent rHDL also combine to suggest a secondary structure assignment for full-length lipid-bound apoA-I (23). The helical distribution of apoA-I on 9.6 nm rHDL has been proposed by HX and indicates continuous helical structure except for the central region 125–158 that functions to give a bimodal structure (Figure 2) (23, 24).
Folding Domains and Domain Function
Studies of Δ[1–43] and Δ[187–243] compared to plasma apoA-I suggested that Δ[187–243] has a similar tertiary structure to plasma apoA-I (25). However, Δ[1–43] exhibits a different conformation in comparison to plasma apoA-I, suggesting that the N-terminus [1–43] is important to stabilize the lipid-free apoA-I structure in solution.
Point mutations and the 11-residue helix deletions in the region 165–243 of apoA-I suggested that residues 165–175 corresponding to H7A are critical for proper folding of apoA-I in its lipid-free form (14). Eleven residue helix deletions and reverse charged mutations (E125K/E128K/K133E/E139K) of the central region of apoA-I [123–165] suggested that the 144–165, H6 region forms a helical structure whereas 123–142, H5, had a disordered structure (15). Deletion studies of residues 121–142 further confirmed that the 131–143 segment is disordered and may function as a “hinge domain” as discussed previously (16). Thus, the central region H5 [121–143] may function as a hinge region to connect the two terminal regions, N [1–120] and C [145–243].
Two truncation mutants [44–186]A-I (26) and [1–192]A-I (27) were shown to be a mixture of monomer and dimer in solution with decreased phospholipid binding ability, which suggested that the C-terminal [193–243] region is responsible for the self-association of A-I and the initiation of phospholipid binding. Cross-linking experiments confirmed that the C-terminal domain of apoA-I participates in the self-association of the protein as demonstrated by the inability of the C-terminal deletion mutants Δ[185–243] and Δ[209–243] to form higher order aggregates in solution (28). Studies of the peptide representing the C-terminal 46-residues [198–243]apoA-I suggested that it is responsible for self-association and has high lipid affinity (29). Thus, results from different laboratories converge on the concept that the C-terminus, possibly residues 185–243, is responsible for the aggregation of lipid-free apoA-I. Single or multiple point mutations of hydrophobic residues in this region significantly decreased dimyristoyl-phophatidylcholine (DMPC) clearance and 8-anilino-1-naphthalenesulfonate (ANS) binding also suggests that the C-terminal domain is responsible for lipid binding and aggregation (30–32). Deletion of residues 191–220 showed intermediate lipid binding between wild type and Δ[223–243], suggesting that residues 191–220 play a role in enhancing the ability of apoA-I to bind to and solubilized lipids by forming helical structure upon lipid interaction (33). Segment deletion (34), replacement with the sequence of the C-terminal segment (35) and reversal of the sequence (36) suggest that residues 143–164, H6 are involved in LCAT activation. Mutations of R149, R153, and R160 in this region lead to loss of LCAT activity possibly through the disruption of charge interactions between LCAT and apoA-I (37). Mutation E110A/E111A (H4) has also been shown to affect LCAT activation (38).
In contrast to the activation of LCAT, the interactions between A-I and SR-BI or ABCA1 are less well understood. A binding and cross-linking study suggested the class A amphipathic helix as a recognition motif by SR-BI (39). A natural mutant, apoA-I Nichinan, Glu235del, showed decreased cellular cholesterol efflux and thus the C-terminus of apoA-I was suggested to be involved in the interaction with ABCA1 (40). Single or multiple mutations of hydrophobic residues in the C-terminal region [185–243] significantly decrease cholesterol efflux activity (30–32).
The phospholipid-binding affinities of the different helix repeats have been studied using synthetic peptide models by a number of groups including our laboratory. Forty-four residue consensus sequence peptides (ABAB), real sequence peptides, RSP-1 (H4–5) and RSP-2 (H5–H6) were studied in our laboratory. RSP-1 exhibited higher lipid affinity than RSP-2 in terms of helical structure induced by detergent, suggesting that H4 has higher lipid-binding affinity than H6 (41). Twenty-two residue peptides (42) followed by 33 residue (G*/[1–33]A-I, H2–3, H3–4, H8–9 and H9–10), 44 residue (H1–2, H4–5, H5–6, H6–7 and H7–8) and 55 residue peptides (H2–3–4) (43) have also been studied. These studies suggest that only the helices corresponding to amino acids 44–87 (H1) and 220–241 (H9 and H10) had significant high affinity for lipid. In contrast, H4–H5 and H5–H6 had low lipid binding affinity. The lipid affinity of H6–7 was suggested to be moderate. In addition, little difference was seen in the α-helical content or lipid affinity of the larger vs. the 22-mer peptides, indicating that the individual domains do not fold/bind lipid cooperatively as was proposed earlier (43). Comparison of the [198–243]apoA-I and [1–43]apoA-I peptides suggested that the C-terminal peptide has higher lipid affinity that was confirmed by surface monolayer experiments (44, 45). Comparison of the surface behavior of Δ[1–59][185–243]apoA-I to Δ[185–243]apoA-I showed that Δ[1–59][185–243]apoA-I desorbs at lower pressures compared with wild type apoA-I and Δ[185–243]apoA-I indicating that it is missing a strong lipid association motif (H1) (46). In summary, it is generally accepted that the N-terminus (residues 1–43) is important to stabilize lipid-free apoA-I conformation; H5 in the central region may function as a hinge segment between the N and C terminal domains; the region of H4 and H6 is required for LACT activation; and the extreme C-terminal domain (H9, H10) possesses the highest lipid binding affinity and thus may initiate the lipid binding of apoA-I and be responsible for the aggregation properties of apoA-I. Additionally the region may be involved in cholesterol efflux as shown in Figure 2. Phillips’ group first suggested a two-folding domain structure of lipid-free apoA-I through analyzing a series of deletion mutants (47) and suggested that the lipid-free apoA-I molecule is organized into a two-domain structure similar to apoE; the N-terminal domain [1–189] forms a helix bundle, whereas the C-terminal domain [190–243] forms a less organized structure. ApoA-I initially binds to a lipid surface through amphipathic helices in the C-terminal domain followed by opening of the helix bundle in the N-terminal domain. ApoE behaves similarly; this mechanism may represent a general feature for lipid interaction of other exchangeable apolipoproteins, such as apoA-IV. The two-domain structure of apoE has been verified by the full-length apoE3 NMR structure (48). In addition, a mouse apoA-I NMR structure deposited in the Protein Data Bank has shown the residues corresponding to human apoA-I 1–184 forms the N-terminal helix bundle (PDB: 2LEM). This is consistent with our model in which residues 1–184 forms a N-terminal helix bundle, whereas residues 185–243 form a C-terminal domain with little defined structure (49).
Crystal Structures of apoA-I
High resolution structural information is critical to explore the biological function of the apolipoprotein. However, multiple conformations and the tendency to aggregate in lipid-free solution due to large hydrophobic surfaces have prevented the growth of crystals apolipoproteins in general. There are only two legitimate crystal structures of apoA-I; both are truncated forms and represent a lipid-bound and intermediate state.
N-terminal Truncated ApoA-I
The low resolution (4Å) crystal structure of a N-terminal truncated apoA-I, Δ[1–43] (PDB: 1AV1) was determined by Borhani et al. (50) (Figure 3A). In this structure, two apoA-l molecules with elongated α-helical structure are aligned antiparallel to form a horseshoe-like shape (Figure 3A). Two dimers are aligned antiparallel to form a tetramer with their hydrophobic surfaces facing each other and well shielded from the aqueous solution (Figure 3A). Each dimer was suggested to be stabilized by salt bridges. In the crystal structure, Δ[1–43]apoA-I has an α-helical content >90% and was thus proposed to be a lipid-bound state of apoA-I during the process of HDL formation. In fact, the crystal was grown in the presence of high salt, which is a highly hydrophilic environment, possibly enhancing the hydrophobic interaction between monomers and thus simulating a lipid-bound conformation. The hydrophobic surface between the dimers is presumably the lipid-binding interface. Notably, the pseudo-continuous α-helix has regular kinks containing proline residues. However, the poor quality of the diffraction data (resolution at 4Å, completeness 85% Rmerge = 16.6%) and the final model (Rwork = 38.2% and Rfree = 42.8%) preclude any detailed interpretation of this structure.
Figure 3.

Comparison of the two crystal structures of truncated apoA-I. A, Δ(1–43)apoA-I: N-terminal truncated apoA-I dimer. The small picture shows the tetramer structure formed by two dimers. B, Δ[185–243]apoA-I: C-terminal truncated apoA-I dimer. Each region is colored to correspond to Figure 2.
C-terminal truncated apoA-I
Following deletion of the C-terminal domain [185–243] of apoA-I that is responsible for aggregation, we successfully crystallized and solved the high-resolution structure of apoA-I at a resolution of 2.2Å for the first time (49). As shown in Figure 3B, two monomers of apoA-I form a half-circle dimer. The backbone of the dimer consists of two elongated antiparallel proline-kinked helices. The N-terminal domain of each molecule forms a four-helix bundle with the helical C-terminal region of the symmetry-related partner. The central region (H5) forms a flexible segment with two antiparallel helices connecting the bundles at each end. The structure substantiates many features of the secondary structure predictions of the type A and type B helical repeats with different homologies. The structure also shows the molecular details of the stabilization of lipid-free apoA-I by the N-terminal exon-3 encoded residues and suggests the role of dimerization in the assembly of HDL. In addition, the structure suggests how the central domain (H5) may function as a hinge region to facilitate a monomer to dimer conversion. With a semicircular backbone formed from antiparallel helical repeats, the structure allows us to model the formation of discoidal HDL particles with different geometries.
Comparison of the Two Crystal Structures
The prior crystal structure of Δ[1–43]apoA-I shows the formation of a dimer of dimers because it lacks the first 43 residues that are responsible for stabilizing the lipid-free structure and shielding the hydrophobic surface of the helices resulting in a dimer-dimer interaction to cover the hydrophobic surface. Although the resolution of the Δ[1–43]apoA-I crystal structure is low, dimerization of the five AB-repeating motifs is clearly a common feature with both structures exhibiting the same five AB repeat antiparallel interacting-motifs (Figure 3). H5 is in the center of the five AB repeat dimerization motifs. In the Δ[1–43]apoA-I crystal structure, H1 and the first A unit of H2 forms a dimer interaction with the C-terminal H8 and H9. This implies potential dimerization ability and a possible dimerization interface in full-length apoA-I. Following lipid binding and unfolding of the N-terminal helix bundle, interaction of the exposed H1 and the first A unit of H2 with the C-terminal H8 and H9 to form the dimer results in a “double belt” model as seen for Δ[1–43]apoA-I. Thus, the Δ[185–243]apoA-I structure may represent the intermediate state during the process of HDL formation.
Lipid-free ApoA-I in Solution and HDL Formation Through Dimerization
Previously we proposed the monomer conformation of Δ[185–243]apoA-I with the helix 5 region changed from helical structure into a loop conformation leading the H6 and H7 helices folding back to form the helical bundle with the N-terminal region based on the crystal dimer of Δ[185–243]apoA-I (Figure 4). This domain swap mechanism, also found in the apoA-IV crystal structure (51), suggests a common mechanism of lipid binding to form lipoprotein particles through dimerization. With the knowledge of C-terminal domain formation by the loop region and interaction between the N and C terminal domains, we proposed a full-length lipid-free apoA-I monomer model in solution with the C-terminal domain folded back. This result is consistent with Segrest’s simulation models (52). Based on the full-length apoA-I model, we updated our HDL formation mechanism. In the initial state, the monomeric apoA-I forms a helix bundle with the C- terminal domain presenting exposed hydrophobic surface with little defined structure. In the intermediate state, when the monomeric apoA-I is close to the lipid surface of the cell membrane, the C-terminal domain, especially the H10 region will bind the lipid to form helical structure. This binding facilitates the anchoring of apoA-I to the lipid bilayer that can bring two monomeric apoA-I in close juxtaposition for dimerization to occur through helix formation in the H5 hinge region. It is possible that apoA-I interaction with the ABCA1 transporter plays a role in the juxtaposition of two apoA-I molecules for dimerization. Furthermore, interaction with the lipid surface may play a role in the H5 region transitioning from loop to the helix facilitating the dimerization. The dimeric organization in our crystal structure thus represents the intermediate state. In the final state, lipids will fill the double loop and transform the remainder of C terminal domain from unstructured into helical conformation and result in the formation and release of a discoidal, nascent HDL from the cell membrane (Figure 5).
Figure 4.
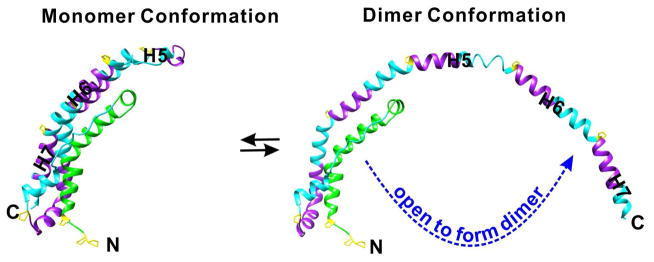
Monomer dimer conversation of Δ[185–243] in solution governed by the H5 region. The H5 region functions as a hinge through folding and unfolding to control the dimerization of Δ[185–243] monomer through a domain swap of the H6/H7 helices.
Figure 5.

Proposed full-length apoA-I monomeric model and HDL formation mechanism through three states.
Disease-related Mutations
With the full-length apoA-I model based on the high-resolution crystal structure in hand, the detailed molecular information aids in understanding disease related mutations to better clarify the HDL formation mechanism and atherosclerosis development.
A novel apoA-I truncation (apoA-I Mytilene) due to a heterozygous nonsense mutation (Gln216) has been reported to be associated with markedly decreased levels of HDL-C and decreased total cholesterol efflux associated with premature CHD and is consistent with the model that the C-terminal domain is responsible for the initiation of the lipid binding and cholesterol efflux (53). Partial deletion of the C-terminal domain as shown in Figure 6 inhibits the lipid binding and interaction with ABCA1 resulting in low HDL and decreased cholesterol efflux. A naturally occurring human apoA-I mutation, apoA-I[Lys107del], associated with hypertriglyceridemia (HTG), showed enhanced binding to TG-rich particles, lower stability, and greater exposure of hydrophobic surfaces compared to wild-type apoA-I. The location lys107 on the apoA-I monomer model (Figure 6A) suggests that deletion of Lys107 disrupts helix registration and disturbs a stabilizing salt bridge network in the N-terminal helical bundle and might lead to the opening of the helix bundle. Mutation studies of lys107del and lys107A confirmed that the registry shift and ensuing disruption of the inter-helical salt bridges in apoA-I[Lys107del] result in destabilization of the helical bundle structure and greater exposure of hydrophobic surfaces. Opening of the N-terminal helical bundle in the apoA-I[Lys107del] variant facilitated its binding to TG-rich lipoproteins and thus, may reduce their lipolysis and contribute to the development of HTG in carriers of the mutation (54).
Figure 6.

Mapping the natural mutations on full-length apoA-I monomeric model. A, K107 and R151 are in the salt-bridge network that stabilize the N-terminal helix bundle. B, R173 is in the salt-bridge network that stabilized the N-terminal helix bundle. C, L159 is located in the hydrophobic center inside of the N-terminal helix bundle. Orange color shows hydrophobic surface, whereas blue shows hydrophilic surface.
Previously we have mapped the HDL deficiencies mutations apoA-I Milano (R173C) and apoA-I Paris (R151C) on the crystal structure dimer and found that both are in the center of the salt bridge networks that hold the dimer together (49). Mutation of these residues is likely to cause the disruption of the inter-helical salt bridges that determine the H5/H5 registration. In the Milano and Paris double-belt model, different helix registration is proposed due to the formation of the disulfide bond between the cysteine residues with fewer or no inter-helical salt bridges (55). This decrease in inter-helical salt bridge interaction may cause instability in the antiparallel double helix backbone, thus leading to the monomeric form of the protein. We mapped the mutations on the monomeric apoA-I model (Figures 6A and 6B), and they are both located in the center of salt bridge networks that stabilized the N-terminal helix bundle. Introduction of a negatively charged residue will disrupt the salt bridge network and thus lead to instability of the helix bundle in addition to shifting the dimerization registration. In summary, these mutations disrupt the helix bundle domain in the apoA-I molecule and modify the lipid-binding characteristics to higher dynamics due to different helix registration (56, 57).
Family members with the apoA-I Fin (L159R) mutation have been reported with reduced plasma HDL cholesterol (20%) and apoA-I (25%) compared with unaffected family members. Proteolytic degradation of apoA-I Fin in plasma is thought to be the reason for the low apoA-I concentrations (55). As shown in Figure 6C, Leu159 is situated in the middle of the N-terminal bundle in the hydrophobic core. Mutation of this Leu into a strongly charged Arg will disrupt the hydrophobic core and lead to the disruption of the N-terminal helix bundle and its stabilizing role in the structure may render the lipid-free apoA-I accessible to the protease thus leading to degradation.
In summary, mapping the naturally occurred mutations on the full-length monomeric apoA-I model provides insight into atherosclerosis development. Mutations (L159R, K107del) in the N-terminal domain causing the destabilization of the helical bundle that lead to opening up of the helix bundle resulting in degradation of the protein or binding to TG-rich particles, thus leading to HTG. Cysteine mutations (Milano and Paris) in the N-terminal domain will disrupt the helix bundle as well as modifying the lipid-binding characteristics due to different helix registration. Truncation mutations like Mytilene at the C-terminal domain will result in decreased HDL and cholesterol efflux due to the lack of interaction with lipids and ABCA-1.
Conclusion
The full-length lipid-free apoA-I monomeric model based on high-resolution crystal structure of C-terminal truncated apoA-I provides molecular details to study the function of apoA-I during HDL formation and reverse cholesterol transport. The structure verified previous functional studies of apoA-I and the low-resolution crystal structure. In addition, it helps us to propose the formation of HDL through dimerization governed by the H5 region through three states. However, the C-terminal domain that plays a vital role during this process is still missing. The high-resolution structure of full-length apoA-I will result in a better understanding of the HDL formation and its function at various steps of the RCT pathways, which may lead to strategies for the prevention of atherosclerosis development.
Acknowledgments
Work was funded by NIH/NHLBI Grants 1R01HL116518-01A1 and 5P01HL026335-30 to D.A.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2010. Natl Vital Stat Rep. 2013;61:1–117. [PubMed] [Google Scholar]
- 2.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 3.Lee JY, Parks JS. ATP-binding cassette transporter AI and its role in HDL formation. Curr Opin Lipidol. 2005;16:19–25. doi: 10.1097/00041433-200502000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Fielding CJ, Shore VG, Fielding PE. A protein cofactor of lecithin:cholesterol acyltransferase. Biochem Biophys Res Commun. 1972;46:1493–1498. doi: 10.1016/0006-291x(72)90776-0. [DOI] [PubMed] [Google Scholar]
- 5.Acton S, Rigotti A, Landschulz KT, et al. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 6.Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc Natl Acad Sci USA. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segrest JP, Jackson RL, Morrisett JD, et al. A molecular theory of lipid-protein interactions in the plasma lipoproteins. FEBS Lett. 1974;38:247–258. doi: 10.1016/0014-5793(74)80064-5. [DOI] [PubMed] [Google Scholar]
- 8.Segrest JP, Jones MK, De Loof H, et al. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 9.Nolte RT, Atkinson D. Conformational analysis of apolipoprotein A-I and E-3 based on primary sequence and circular dichroism. Biophys J. 1992;63:1221–1239. doi: 10.1016/S0006-3495(92)81698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wen M. PhD thesis. Boston University School of Medicine; Boston, MA: 2013. Structural studies of a consensus sequence peptide (CSP) ABAB of apolipoproteins through NMR spectroscopy. [Google Scholar]
- 11.Luo D. PhD thesis. Boston University School of Medicine; Boston, MA: 2009. Structure, stability and lipid binding of consensus sequences CSP-BABA and CSP-ABBA of exchangeable apolipoproteins. [Google Scholar]
- 12.Yang C. PhD thesis. Boston University School of Medicine; Boston, MA: 2003. Conformational studies of a consensus sequence peptide (CSP) and a real sequence peptide (RSP) of apolipoproteins by circular dichroism spectroscopy and X-ray crystallography. [Google Scholar]
- 13.Marcel YL, Kiss RS. Structure-function relationships of apolipoprotein A-I: a flexible protein with dynamic lipid associations. Curr Opin Lipidol. 2003;14:151–157. doi: 10.1097/00041433-200304000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Gorshkova IN, Liadaki K, Gursky O, et al. Probing the lipid-free structure and stability of apolipoprotein A-I by mutation. Biochemistry. 2000;39:15910–15919. doi: 10.1021/bi0014406. [DOI] [PubMed] [Google Scholar]
- 15.Gorshkova IN, Liu T, Zannis VI, et al. Lipid-free structure and stability of apolipoprotein A-I: probing the central region by mutation. Biochemistry. 2002;41:10529–10539. doi: 10.1021/bi025807d. [DOI] [PubMed] [Google Scholar]
- 16.Gorshkova IN, Liu T, Kan HY, et al. Structure and stability of apolipoprotein a-I in solution and in discoidal high-density lipoprotein probed by double charge ablation and deletion mutation. Biochemistry. 2006;45:1242–1254. doi: 10.1021/bi051669r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang Y, Gursky O, Atkinson D. Structural studies of N- and C-terminally truncated human apolipoprotein A-I. Biochemistry. 2003;42:6881–6890. doi: 10.1021/bi034152t. [DOI] [PubMed] [Google Scholar]
- 18.Oda MN, Forte TM, Ryan RO, et al. The C-terminal domain of apolipoprotein A-I contains a lipid-sensitive conformational trigger. Nat Struct Biol. 2003;10:455–460. doi: 10.1038/nsb931. [DOI] [PubMed] [Google Scholar]
- 19.Chetty PS, Mayne L, Lund-Katz S, et al. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lagerstedt JO, Budamagunta MS, Liu GS, et al. The “beta-clasp” model of apolipoprotein A-I--a lipid-free solution structure determined by electron paramagnetic resonance spectroscopy. Biochim Biophys Acta. 2012;1821:448–455. doi: 10.1016/j.bbalip.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okon M, Frank PG, Marcel YL, et al. Heteronuclear NMR studies of human serum apolipoprotein A-I. Part I. Secondary structure in lipid-mimetic solution. FEBS Lett. 2002;517:139–143. doi: 10.1016/s0014-5793(02)02600-5. [DOI] [PubMed] [Google Scholar]
- 22.Lagerstedt JO, Cavigiolio G, Budamagunta MS, et al. Structure of apolipoprotein A-I N terminus on nascent high density lipoproteins. J Biol Chem. 2011;286:2966–2975. doi: 10.1074/jbc.M110.163097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda MN, Budamagunta MS, Borja MS, et al. The secondary structure of apolipoprotein A-I on 9. 6-nm reconstituted high-density lipoprotein determined by EPR spectroscopy. FEBS J. 2013;280:3416–3424. doi: 10.1111/febs.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sevugan Chetty P, Mayne L, Kan ZY, et al. Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2012;109:11687–11692. doi: 10.1073/pnas.1209305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers DP, Roberts LM, Lebowitz J, et al. Structural analysis of apolipoprotein A-I: effects of amino- and carboxy-terminal deletions on the lipid-free structure. Biochemistry. 1998;37:945–955. doi: 10.1021/bi9713512. [DOI] [PubMed] [Google Scholar]
- 26.Beckstead JA, Block BL, Bielicki JK, et al. Combined N- and C-terminal truncation of human apolipoprotein A-I yields a folded, functional central domain. Biochemistry. 2005;44:4591–4599. doi: 10.1021/bi0477135. [DOI] [PubMed] [Google Scholar]
- 27.Ji Y, Jonas A. Properties of an N-terminal proteolytic fragment of apolipoprotein AI in solution and in reconstituted high density lipoproteins. J Biol Chem. 1995;270:11290–11297. doi: 10.1074/jbc.270.19.11290. [DOI] [PubMed] [Google Scholar]
- 28.Laccotripe M, Makrides SC, Jonas A, et al. The carboxyl-terminal hydrophobic residues of apolipoprotein A-I affect its rate of phospholipid binding and its association with high density lipoprotein. J Biol Chem. 1997;272:17511–17522. doi: 10.1074/jbc.272.28.17511. [DOI] [PubMed] [Google Scholar]
- 29.Zhu HL, Atkinson D. Conformation and lipid binding of a C-terminal (198–243) peptide of human apolipoprotein A-I. Biochemistry. 2007;46:1624–1634. doi: 10.1021/bi061721z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyssenko NN, Hata M, Dhanasekaran P, et al. Influence of C-terminal alpha-helix hydrophobicity and aromatic amino acid content on apolipoprotein A-I functionality. Biochim Biophys Acta. 2012;1821:456–463. doi: 10.1016/j.bbalip.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka M, Dhanasekaran P, Nguyen D, et al. Contributions of the N. C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry. 2006;45:10351–10358. doi: 10.1021/bi060726t. [DOI] [PubMed] [Google Scholar]
- 32.Kono M, Tanaka T, Tanaka M, et al. Disruption of the C-terminal helix by single amino acid deletion is directly responsible for impaired cholesterol efflux ability of apolipoprotein A-I Nichinan. J Lipid Res. 2010;51:809–818. doi: 10.1194/jlr.M002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagao K, Hata M, Tanaka K, et al. The roles of C-terminal helices of human apolipoprotein A-I in formation of high-density lipoprotein particles. Biochim Biophys Acta. 2014;1841:80–87. doi: 10.1016/j.bbalip.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sorci-Thomas M, Kearns MW, Lee JP. Apolipoprotein A-I domains involved in lecithin-cholesterol acyltransferase activation. Structure:function relationships. J Biol Chem. 1993;268:21403–21409. [PubMed] [Google Scholar]
- 35.Sorci-Thomas MG, Curtiss L, Parks JS, et al. Alteration in apolipoprotein A-I 22-mer repeat order results in a decrease in lecithin:cholesterol acyltransferase reactivity. J Biol Chem. 1997;272:7278–7284. doi: 10.1074/jbc.272.11.7278. [DOI] [PubMed] [Google Scholar]
- 36.Sorci-Thomas MG, Curtiss L, Parks JS, et al. The hydrophobic face orientation of apolipoprotein A-I amphipathic helix domain 143–164 regulates lecithin:cholesterol acyltransferase activation. J Biol Chem. 1998;273:11776–11782. doi: 10.1074/jbc.273.19.11776. [DOI] [PubMed] [Google Scholar]
- 37.Roosbeek S, Vanloo B, Duverger N, et al. Three arginine residues in apolipoprotein A-I are critical for activation of lecithin:cholesterol acyltransferase. J Lipid Res. 2001;42:31–40. [PubMed] [Google Scholar]
- 38.Chroni A, Kan HY, Kypreos KE, et al. Substitutions of glutamate 110 and 111 in the middle helix 4 of human apolipoprotein A-I (apoA-I) by alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in apoA-I-deficient mice. Biochemistry. 2004;43:10442–10457. doi: 10.1021/bi049782p. [DOI] [PubMed] [Google Scholar]
- 39.Williams DL, de La Llera-Moya M, Thuahnai ST, et al. Binding and cross-linking studies show that scavenger receptor BI interacts with multiple sites in apolipoprotein A-I and identify the class A amphipathic alpha-helix as a recognition motif. J Biol Chem. 2000;275:18897–18904. doi: 10.1074/jbc.M002411200. [DOI] [PubMed] [Google Scholar]
- 40.Huang W, Sasaki J, Matsunaga A, et al. A single amino acid deletion in the carboxy terminal of apolipoprotein A-I impairs lipid binding and cellular interaction. Arterioscler Thromb Vasc Biol. 2000;20:210–216. doi: 10.1161/01.atv.20.1.210. [DOI] [PubMed] [Google Scholar]
- 41.Loren J, Wally DA. PhD thesis. Boston University School of Medicine; 2005. Conformation and lipid binding properties of peptide models of exchangable apolipoproteins. [Google Scholar]
- 42.Palgunachari MN, Mishra VK, Lund-Katz S, et al. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity. Implications for HDL assembly. Arterioscler Thromb Vasc Biol. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- 43.Mishra VK, Palgunachari MN, Datta G, et al. Studies of synthetic peptides of human apolipoprotein A-I containing tandem amphipathic alpha-helixes. Biochemistry. 1998;37:10313–10324. doi: 10.1021/bi980042o. [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Hua N, Atkinson D, et al. The N-terminal (1–44) and C-terminal (198–243) peptides of apolipoprotein A-I behave differently at the triolein/water interface. Biochemistry. 2007;46:12140–12151. doi: 10.1021/bi7010114. [DOI] [PubMed] [Google Scholar]
- 45.Zhu HL, Atkinson D. Conformation and lipid binding of the N-terminal (1–44) domain of human apolipoprotein A-I. Biochemistry. 2004;43:13156–13164. doi: 10.1021/bi0487894. [DOI] [PubMed] [Google Scholar]
- 46.Wang L, Mei X, Atkinson D, et al. Surface behavior of apolipoprotein A-I and its deletion mutants at model lipoprotein interfaces. J Lipid Res. 2014;55:478–492. doi: 10.1194/jlr.M044743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saito H, Dhanasekaran P, Nguyen D, et al. Domain structure and lipid interaction in human apolipoproteins A-I and E, a general model. J Biol Chem. 2003;278:23227–23232. doi: 10.1074/jbc.M303365200. [DOI] [PubMed] [Google Scholar]
- 48.Chen J, Li Q, Wang J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc Natl Acad Sci USA. 2011;108:14813–14818. doi: 10.1073/pnas.1106420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286:38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borhani DW, Rogers DP, Engler JA, et al. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci USA. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deng X, Morris J, Dressmen J, et al. The structure of dimeric apolipoprotein A-IV and its mechanism of self-association. Structure. 2012;20:767–779. doi: 10.1016/j.str.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Segrest JP, Jones MK, Shao B, et al. An experimentally robust model of monomeric apolipoprotein A-I created from a chimera of two X-ray structures and molecular dynamics simulations. Biochemistry. 2014;53:7625–7640. doi: 10.1021/bi501111j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anthanont P, Polisecki E, Asztalos BF, et al. A novel ApoA-I truncation (ApoA-IMytilene) associated with decreased ApoA-I production. Atherosclerosis. 2014;235:470–476. doi: 10.1016/j.atherosclerosis.2014.05.935. [DOI] [PubMed] [Google Scholar]
- 54.Gorshkova IN, Mei X, Atkinson D. Binding of human apoA-I[K107del] variant to TG-rich particles: implications for mechanisms underlying hypertriglyceridemia. J Lipid Res. 2014;55:1876–1885. doi: 10.1194/jlr.M047241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klon AE, Segrest JP, Harvey SC. Comparative models for human apolipoprotein A-I bound to lipid in discoidal high-density lipoprotein particles. Biochemistry. 2002;41:10895–10905. doi: 10.1021/bi020315m. [DOI] [PubMed] [Google Scholar]
- 56.Alexander ET, Tanaka M, Kono M, et al. Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009;50:1409–1419. doi: 10.1194/jlr.M800578-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gursky O, Jones MK, Mei X, et al. Structural basis for distinct functions of the naturally occurring Cys mutants of human apolipoprotein A-I. J Lipid Res. 2013;54:3244–3257. doi: 10.1194/jlr.R037911. [DOI] [PMC free article] [PubMed] [Google Scholar]