

Abstract
Billions of base pairs of DNA must be replicated trillions of times in a human lifetime. Complete and accurate replication once and only once per cell division cycle is essential to maintain genome integrity and prevent disease. Impediments to replication fork progression including difficult to replicate DNA sequences, conflicts with transcription, and DNA damage further add to the genome maintenance challenge. These obstacles frequently cause fork stalling, but only rarely cause a failure to complete replication. Robust mechanisms ensure that stalled forks remain stable and capable of either resuming DNA synthesis or being rescued by converging forks. However, when failures do happen the fork collapses leading to genome rearrangements, cell death and disease. Despite intense interest, the mechanisms to repair damaged replication forks, stabilize them, and ensure successful replication remain only partly understood. Different models of fork collapse have been proposed with varying descriptions of what happens to the DNA and replisome. Here, I will define fork collapse and describe what is known about how the replication checkpoint prevents it to maintain genome stability.
Keywords: fork collapse, replication checkpoint, ATR, replisome, iPOND
Introduction
The diploid human genome consists of over 6 billion base pairs of DNA that must be copied completely and accurately each cell division cycle. Given the size of the genome, multiple copy machines (replisomes) work simultaneously, each initiated from an origin of replication. Initiation is regulated to ensure only a single round of DNA replication per cell division cycle.
In a human cell, approximately 30,000–50,000 origins are used creating nearly 100,000 replication forks [1]. Although the average fork moves at about 1–2kb/minute, replication requires approximately 8 hours due to a controlled replication-timing program that ensures a balance between the number of active forks, available replisome proteins, and abundance of nucleotide precursors.
Many sources of replication stress can challenge the replisome [2]. These include DNA damage since thousands of DNA lesions happen per cell per day. Even very efficient repair mechanisms sometimes fail to provide a clean template for DNA synthesis. Environmental genotoxins add to this damage burden. Other challenges to replication fork movement include difficult to replicate sequences, DNA-protein crosslinks and even collisions with transcription complexes [3]. Finally, abnormal situations that disrupt origin timing programs such as oncogene activation or chemotherapeutic interventions can greatly increase the replication stress burden [4].
While the basic mechanism of DNA synthesis has been known for decades, we are still discovering how the replisome works at the molecular level and the regulatory mechanisms that ensure success in the context of replication stresses. One of these mechanisms is the replication stress response, which includes activation of the replication checkpoint kinase ATR and its downstream effector kinase CHK1. These kinases are conserved in all eukaryotic organisms (Mec1 and Rad53/Chk1 in budding yeast; Rad3 and Cds1 in fission yeast). I point the reader to reviews of how ATR is activated, and how it controls the replication checkpoint [5–7].
Here, I will focus on what happens when the replication checkpoint fails and forks collapse. I will describe the molecular changes to the DNA and replisome that accompany fork collapse, and discuss how the ATR-dependent checkpoint prevents it.
What is Fork Collapse?
In most cases, replication stress only yields a transient pausing of the replisome. In fact, most types of lagging-strand template DNA damage will not impede the fork at all since a new Okazaki fragment will simply bypass the lesion. Leading strand damage is more likely to stall the replisome and uncouple helicase and polymerase activities [8]. However, even in these cases, repriming that allows continued fork movement may be possible [9, 10].
Some types of DNA damage and replication stress may cause more persistent fork stalling. For example, inter-strand crosslinks can interfere with helicase activities creating a more lasting challenge to the fork. There may also be cases in which multiple clustered DNA lesions, aberrant replication programs, or other impediments to DNA synthesis yield persistent fork stalling. Replication can complete even in these cases because another fork, initiated from an adjacent origin, can complete synthesis of the replicon. However, in rare cases when two converging forks both stall without an intervening origin or when a fork stalls persistently in origin poor regions of the genome, there is a need to restart DNA synthesis from the stalled fork. The replication checkpoint ensures these stalled forks remain stable and promotes their restart.
Failures to stabilize stalled replication forks will cause their collapse. Fork collapse has been used to describe several potentially different processes including replisome protein dissociation and the formation of double-strand breaks at stalled forks [2, 11]. While these descriptions of the molecular events are essential to understand the process of fork collapse, fork inactivation without one or both of them may happen. Thus, I will define a collapsed fork more generally as a replication fork that has lost the capacity to perform DNA synthesis. This is an operational definition that can reflect more than one molecular event.
Experimentally, fork collapse can be visualized using a technique called DNA fiber labeling or molecular combing [12, 13]. This method monitors incorporation of nucleoside analogs at a single molecule level. Thus, the rate of fork elongation, inter-origin distances, frequency of origin firing, and frequency of fork collapse can be determined. Typically the method does not distinguish where the forks are in the genome, although it can be combined with DNA probes to provide site specificity [14].
Alternative methods of measuring fork collapse are possible in systems with highly efficient origins of replication that can be induced to fire synchronously. For example, in budding yeast, a Meselson-Stahl type of density substitution experiment yielded the first strong evidence that the replication checkpoint prevents fork collapse [15]. It is also possible to monitor the appearance of abnormal DNA structures during DNA replication in budding yeast using two dimensional gels of replication intermediates [16]. Finally, budding yeast replication is compatible with chromatin immunoprecipitation (ChIP) to measure the abundance of replisome proteins at sites of DNA synthesis [17, 18].
These methods don’t work well in most eukaryotic organisms because they lack highly defined, efficient, synchronously firing origins; therefore, it is not possible to track a replication fork as it moves through genomic sequence. Instead, more indirect methods to monitor fork collapse have been utilized. Assays for double-strand breaks and excess ssDNA are fairly easy and can be indicative of fork collapse [19–21]. Electron microscopy is a powerful method of visualizing aberrant replication structures although only a few labs have this capability [22–24]. Finally, immunofluorescence microscopy and chromatin fractionation have been used to examine changes in the spatial patterns of replication factories and the overall abundance of replisome proteins on chromatin respectively.
The frequency of fork collapse is rare in normally dividing cells and even in cells treated with DNA damaging agents or replication inhibitors. However, fork collapse is much more common in cells lacking the ATR-dependent replication checkpoint. For example, about 40% of forks become inactivated in checkpoint-deficient budding yeast cells treated with a DNA damaging agent prior to completing 20kb of DNA synthesis [15].
The best estimates of fork collapse frequency in the absence of added genotoxic stress come from bacteria and are based on the phenotypes of mutants in fork repair pathways [25]. These analyses indicate that at least 15% of cells undergo replication-dependent recombination events during normal growth. Recombination is likely a major mechanism to recover from a fork collapse event. However, this is an underestimation of true fork collapse rates since the assay only scores crossover recombination products. The poor growth of E. coli PriA mutants suggests fork collapse may be as frequent as once per cell per generation since this enzyme is required for restarting replication by helicase re-loading [26].
The frequency of fork collapse in human cells is unknown. However, if we assume that recombination during replication is a measure of fork collapse, then its frequency must be greater than once per cell division cycle since crossover recombination products generated during replication are detected even though they are actively selected against by enzymes like the BLM-TOP-RMI1 dissolvase [27].
Consequences of fork collapse
Fork collapse generates the risk of incomplete DNA replication. While it may be possible to recover collapsed forks through recombination and break-induced replication [28, 29], the trade-off for completing DNA synthesis will be increased genetic instability with deletions and chromosomal translocations due to incorrect repair and more error-prone replication. Thus, checkpoint mutants have a greatly elevated rate of gross chromosomal instability [30].
In many organisms including budding yeast, mice, and humans, the replication checkpoint kinases are essential for cell viability. Preventing fork collapse or recovering stalled forks is thought to be the essential function of the replication checkpoint in these organisms. Mutations in the ATR HEAT repeats generates a kinase that cannot support cell viability or prevent fork collapse, but retains sufficient activity to signal a G2 checkpoint [31]. Thus, preventing cell division is not sufficient to rescue cell viability in the absence of fork stabilization. Consistent with this interpretation, preventing cell division also does not rescue the viability of checkpoint-deficient yeast [15, 32]. Finally, a partially inactive Mec1 mutant (mec1-100) cannot inhibit origin firing but does inhibit fork collapse [33, 34]. The mec1-100 strain is much less sensitive to replication stress than the mec1Δ yeast again suggesting fork collapse is the primary cause of cell lethality associated with checkpoint inactivation.
In budding yeast, the lethality caused by mec1Δ can be rescued by increasing intracellular dNTP pools [32, 35]. Ribonucleotide reductase is regulated downstream of Mec1. Increasing dNTP levels may decrease the frequency of fork stalling and promote repair thereby reducing the chance of fork collapse. Atr and Chk1 knockout mice do not survive the earliest embryonic stages [36, 37], and the chromosomes from these checkpoint-deficient embryos are shattered. Likewise, human cells cannot complete even a single cell division cycle without ATR [38]. When replication stress is combined with an ATR-selective inhibitor, human cells that are in S-phase lose the ability to complete DNA replication within 45 minutes [21]. Removing the stress and ATR inhibition does not restore cell viability suggesting the essential function of the checkpoint is to prevent fork collapse and not to promote fork restart. Similarly, reintroduction of the checkpoint in budding yeast mutants after a short treatment with replication stress agents is insufficient to allow completion of DNA synthesis or rescue viability [34, 39].
How does the replication checkpoint prevent fork collapse?
ATR and CHK1 phosphorylate hundreds of proteins suggesting there are multiple mechanisms by which the replication checkpoint prevents fork collapse. I will discuss three: regulation of origin firing to prevent RPA exhaustion, stabilization of the replisome, and regulation of fork repair enzymes (Figure 1).
Figure 1.
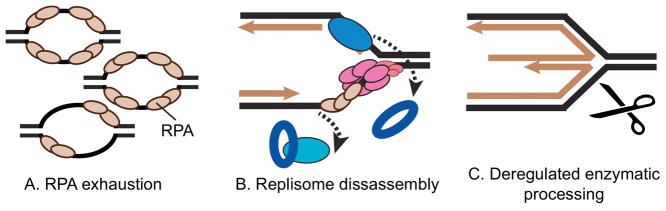
Three models of how the replication checkpoint prevents fork collapse. See text for details.
Origin firing as the target of the replication checkpoint needed to stabilize stalled forks
One possibility is that the replication checkpoint prevents fork collapse as an indirect consequence of regulating the replication-timing program. Origin firing is blocked in replication checkpoint-proficient yeast even after prolonged HU treatment [40]. Based on the replication patterns seen by immunofluorescence imaging and fiber labeling experiments, the same is true human cells [41, 42]. In contrast, human cells treated with checkpoint inhibitors and HU transition to replication patterns consistent with later stages of S-phase [41]. Similar changes can be observed in S. pombe replication patterns [43]. Thus, the deregulated origin firing in the checkpoint mutants may cause redistribution of essential replisome components to new forks thereby inactivating the old ones prior to completing replicon synthesis.
More direct evidence for this model has come recently from studies of the kinetics of fork collapse in human cells. When treated with high doses of HU, replication checkpoint-deficient cells accumulate increasing amounts of ssDNA [21, 44]. This excess ssDNA is at least partly due to excess origin firing, and it eventually causes exhaustion of the ssDNA binding protein RPA [44]. When the RPA levels are depleted, the exposed ssDNA is no longer protected and the forks collapse. Overexpression of RPA is sufficient to delay fork collapse in cells treated with HU and an ATR inhibitor [44].
While this mechanism elegantly explains the extensive fork collapse that happens in HU-treated and checkpoint-inhibited cells, it is likely an incomplete explanation of how the checkpoint prevents fork collapse. First, the persistence of stalled forks and the amount of replication stress will determine whether RPA exhaustion happens. All forks in HU-treated cells would be stalled simultaneously and continuously. This experimental situation would be rare in vivo where stabilization of even small numbers of stalled forks is likely to be important. Second, origin regulation can be separated from the fork stabilization functions of the replication checkpoint in budding yeast [34]. The same is true in vertebrate systems, since preventing origin firing in Xenopus in vitro replication systems only reduced the number of replication-associated double-strand breaks caused by ATR inhibition by 25% [45]. Likewise, we have observed continued fork collapse in HU-treated human cells even when new origin firing is blocked (Dungrawala and Cortez, unpublished data). Finally, as described below, there are multiple ATR substrates at the fork that must be phosphorylated to prevent aberrant fork processing events associated with fork collapse [21, 46, 47].
Replisome stabilization by the replication checkpoint
Historically, prevention of fork collapse was thought to work through stabilization of the replisome machinery itself [23, 39, 48]. A fork collapse model emerged suggesting that replisome disassembly precedes DNA processing. Evidence for checkpoint-dependent replisome stabilization comes from both indirect and direct experimental approaches. These data include decreases in the abundance of some replisome components at stalled forks in checkpoint-deficient yeast as measured by ChIP [17, 49–51], loss of early PCNA staining patterns in human and fission yeast cells [43, 52], and decreases in the amount of some replisome proteins bound to chromatin in both Xenopus in vitro replication systems and human cells [45, 53, 54].
However, there are some experimental observations that suggest that replisome stabilization is not a key mechanism to prevent fork collapse. Most importantly, not all ChIP studies have found replisome protein dissociation at stalled forks in checkpoint-deficient yeast. The Labib group in particular found that the replisome remains associated with stalled forks using genome wide ChIP analyses and replisome proteins continue to interact in a complex [18]. Furthermore, a recent paper examining strand-specific protein localization in budding yeast suggested that the checkpoint actually promotes PCNA unloading from the lagging strand instead of preventing its dissociation [55].
Table 1 summarizes the evidence for and against replisome stabilization as a key function of the replication checkpoint. How can the data for and against the replisome stabilization model be reconciled? The Labib group suggests that the focus on a very early firing origin in the earlier yeast ChIP studies might have been misleading [18]. The behavior of the replisome at very early firing origins may be different than other origins because there are sufficient nucleotides to allow some DNA synthesis at these locations. While these experimental differences may explain at least some of the disparate yeast data, what about vertebrate systems?
Table 1.
Published evidence for and against a function for the replication checkpoint in regulating replisome stability
| Evidence the replication checkpoint DOES regulate replisome stability | Evidence the replication checkpoint DOES NOT regulate replisome stability |
|---|---|
| ChIP data in S. cerevisiae | |
| DNA polymerase subunits and RPA are reduced in abundance near an early firing origin in mec1Δ cells treated with HU. In contrast, the MCM helicase but not the polymerases are dissociated from the forks in rad53Δ cells [17, 49]. | The replisome including polymerase and helicase subunits remains stable and associated with forks throughout the genome in HU-treated mec1Δ or rad53Δ budding yeast cells [18]. |
| Polα and ε subunits are decreased in abundance near the early origin ARS305 in rad53 mutants and Polδ is lost at later times after HU treatment [50]. | Polα and Cdc45 are retained near the early ARS305 origin in a rad53 mutant strain treated with HU even though they also start associating with a late origin[98]. Similarly, inactivation of the checkpoint using dpb11-1 or rad53 mutants did not cause loss of Polε from ARS305 in HU-treated cells [99]. |
| Cdc45 is much less abundant near the early origin ARS305 in HU-treated mec1Δtel1Δ cells than in controls [51]. | Replisome subunits remain associated with stalled forks induced by a protein-based replication fork barrier even when the checkpoint is inactivated by mec1 or rad53 deletion [100] |
| Examination of PCNA abundance on leading vs. lagging strand in HU treated cells showed that the checkpoint actually promotes PCNA unloading from the lagging strand [55]. | |
| Other yeast data | |
| When monitored by 2D gel electrophoresis of replication intermediates, the aberrant fork structures in rad53 mutant cells look the same as the structures caused by inactivation of replisome proteins [39]. | S. pombe cds1Δ cells treated with HU retain the ability to synthesize DNA, and the ssDNA accumulated in these circumstances is dependent on MCM activity suggesting the helicase remains associated with the fork [101]. |
| Checkpoint-deficient fission yeast show decreased PCNA staining in early replication patterns and increased PCNA staining in late patterns [43] | MMS-treated S. cerevisiae rad53Δexo1Δ double mutants retain functional forks that are capable of continuing DNA synthesis indicating the replisome may remain stable [102]. |
| Xenopus extract replication data | |
| In Xenopus extracts, Polε dissociates from chromatin when replication proceeds in the presence of CPT. The ATM/ATR kinases are needed for polymerase reloading in this circumstance [45]. | Polε did not dissociate from the replicating Xenopus chromatin in response to aphidicolin treatment in checkpoint-deficient extracts [45]. |
| When fork collapse was triggered by direct induction of a replication-associated double-strand break using a nuclease in the Xenopus extracts, the GINS subunits of the replicative helicase were lost from chromatin[53]. Polε was also lost in this system but MCM2-7, CDC45, and POLα were not. | |
| Human and mouse cell data | |
| The total amount of PCNA, POLE, POLD2, and CDC45 associated with chromatin is reduced in ATR-deficient cells treated with aphidicolin compared to control cells [54]. The level of MCM3 on chromatin was not changed. | |
| Checkpoint-deficient human cells show decreased PCNA staining in early replication patterns and increased PCNA staining in late patterns [52] | |
Unfortunately, the direct ChIP methods are largely ineffective at monitoring the replisome in vertebrates, and all of the data from mammalian cells on replisome stabilization are indirect. The studies examining replication factories by PCNA staining convincingly demonstrate that the replication checkpoint regulates replication timing [52]; however, they are largely incapable of addressing the question of replisome stability and fork collapse. Consider the following scenario: as DNA elongation proceeds, there are likely several PCNA molecules on the lagging strand template but fewer (perhaps one) on the leading strand template. PCNA loading and unloading on the lagging strand is at equilibrium. Treatment with HU changes this equilibrium since the rate of loading is greatly decreased (since new Okazaki fragments would start much less frequently) while the rate of unloading may be unaffected. Thus, the amount of PCNA at forks in HU-treated cells would be reduced (Figure 2). In the absence of the checkpoint, new PCNA molecules will associate with aberrantly fired origins explaining the altered replication patterns visualized by IF. Since the PCNA staining patterns examined by IF imaging is qualitative, it may appear that PCNA is disassociating from the existing stalled forks and re-associating with new forks even if the absolute number of PCNA molecules per stalled fork is the same in wild-type vs. checkpoint-deficient cells.
Figure 2.
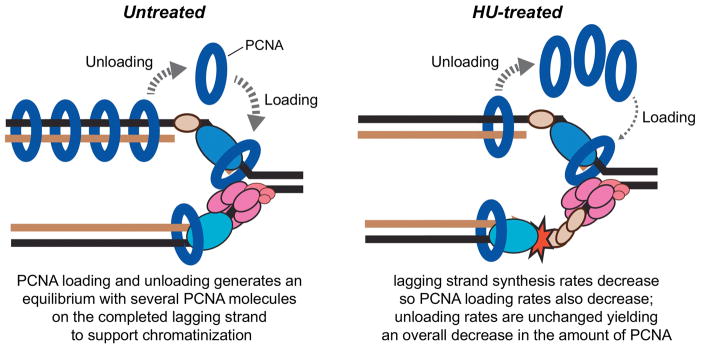
Fork stalling causes a decrease in PCNA levels associated with replication forks. After an Okazaki fragment is completed and chromatin is deposited, PCNA is unloaded from the lagging strand. When a fork is elongating normally, PCNA is rapidly placed back onto the lagging strand to support generation of a new Okazaki fragment and continued DNA synthesis. When the fork stalls due to nucleotide depletion, PCNA is no longer loaded since new Okazaki fragments are not being initiated rapidly. The PCNA that was already on the lagging strand is unloaded so the total amount of PCNA at the fork is less. This model predicts that the amount of PCNA at forks DNA should be higher in normal replicating cells than in HU-treated cells. This is exactly what is observed when the amount of PCNA associated with the nascent DNA is examined by iPOND [56].
Experimental evidence for this explanation comes from measuring the amount of PCNA associated with newly synthesized DNA using iPOND (isolation of proteins on nascent DNA) [56]. This methodology demonstrated that the amount of PCNA associated with nascent DNA reaches equilibrium even as more total DNA is labeled with EdU since the rate of unloading and loading at the moving fork balance. In response to HU, a new equilibrium is reached rapidly with less overall PCNA associated with the much slower moving forks [56].
Importantly, iPOND also provides a way to directly assess replisome stability in checkpoint-deficient cells. In fact, by combining iPOND with highly quantitative mass spectrometry, all replisome subunits can be examined simultaneously. In time-course experiments, we found no evidence that ATR signaling regulates replisome stability in response to replication stress (Dungrawala and Cortez, unpublished data). Thus, replisome stabilization does not appear to be a major function of the replication checkpoint to prevent fork collapse in either yeast or human cells.
Regulation of fork repair activities to prevent fork collapse
In addition to regulating origin firing and potentially controlling replisome stability, the replication checkpoint prevents fork collapse by regulating fork repair enzymes directly. ATR targets include RecQ helicases like WRN and BLM, nucleases like DNA2 and EXO1, and DNA translocases like SMARCAL1. While these enzymes have multiple repair activities, one thing they have in common is the ability to regulate a process called fork regression or fork reversal.
Fork regression is the movement of the fork backwards opposite the direction needed for DNA synthesis. The newly synthesized DNA strands are annealed during this process forming a Holliday junction. Fork regression provides three possible advantages to the cell. First, it may be a mechanism to limit ssDNA at the fork and thereby promote stabilization. Second, if the fork stalled due to a DNA lesion, it places that lesion back into the context of double-stranded DNA where excision repair mechanisms can operate. Third, it provides an opportunity for lesion bypass through a template switching mechanism.
There is considerable evidence that fork regression is a fork repair mechanism in prokaryotic cells. However, electron microscopy and two-dimension gel analyses of replication intermediates indicated that regressed forks only formed in replication checkpoint mutants in S. cerevisiae [23, 39]. Thus, a model emerged that the replication checkpoint prevents fork collapse by preventing fork regression.
More recent data suggest that regulated fork regression is actually a fork stabilization/repair mechanism at least in higher eukaryotes, and that only un-regulated regression is a problem. The best data supporting this model comes from Lopes and colleagues who have documented fork reversal in checkpoint-proficient human and mouse cells in response to many types of replication stress including difficult to replicate sequences and oncogene-induced replication stress [24, 57, 58]. The electron microscopy studies suggest that up to 30% of all forks reverse in some circumstances [24, 57, 58]. However, this measurement must be considered an estimate since detection by electron microscopy involves a purification step that may not equally enrich all types of replication intermediates. Nonetheless, such a high rate of fork reversal indicates it is a programmed repair mechanism that can prevent fork collapse.
While topological stress due to topisomerase inhibition could drive fork regression, other types of replication stress such as HU or DNA lesions would require an enzymatic reaction. The DNA changes needed for fork regression are actually not energetically difficult. In fact, early observations of abundant nascent-nascent DNA hybrids consistent with reversed forks was due to spontaneous branch migration after DNA isolation [59]. In vitro studies of fork remodeling require modifications to the DNA such as mismatches at the fork junction to prevent spontaneous branch migration. Even with these modifications, there are many enzymes that can catalyze fork regression of naked DNA substrates including BLM, WRN, HLTF, FANCM, FBH1, ZRANB3, and SMARCAL1 [60–69].
In a living cell, fork-bound proteins are an obstacle to branch migration necessitating a motor enzyme for regression. The most obvious blocking protein is the RPA bound to the ssDNA at a stalled fork. Thus, it is essential to evaluate fork regression enzymes using biochemical reactions containing RPA and preferably, the entire replisome. The only system where this has been possible is T4 phage replication. Single-molecule studies of the T4 replication reaction in the context of a DNA lesion demonstrated that the UvsW translocase catalyzed fork regression allowing time for lesion bypass synthesis [70].
The SMARCAL1 enzyme fulfills the requirements for a human fork regression enzyme and importantly has structural homology to UvsW suggesting evolutionary conservation of the mechanism [71]. First, as a DNA translocase, SMARCAL1 catalyzes fork regression in a concerted reaction that does not require the generation of additional ssDNA intermediate like a helicase might [61]. Second, SMARCAL1 is recruited to stalled replication forks through an interaction with RPA, which becomes more abundant when the fork stalls [72–76]. Third, SMARCAL1 fork regression activity is stimulated when RPA is bound to the leading-strand template but inhibited by RPA bound to the lagging-strand template [62, 77]. Thus, its substrate preference is for a stalled fork structure that would form when the leading strand polymerase is block. RPA actually stimulates SMARCAL1 regression activity on this substrate.
In addition to catalyzing fork regression, SMARCAL1 can also catalyze the opposite reaction, fork restoration [62]. Fork restoration re-generates a replication fork structure that can resume DNA synthesis. Again, this restoration reaction is regulated by RPA. In this case, RPA inhibits SMARCAL1-catalyzed restoration if bound to a longer lagging nascent-strand and activates SMARCAL1 restoration activity if bound to a leading nascent-strand. This is exactly the substrate preference that would be required to restore a normal replication fork with gaps on the lagging template-strand. However, the product of the fork regression reaction due to a leading strand blockage would typically be a regressed fork in which the lagging strand would be longer than the leading in the nascent-nascent duplex. Thus, an exonuclease would be needed to generate the preferred SMARCAL1 fork restoration substrate. DNA2 appears to fulfill this requirement as it was recently shown to drive restart of reversed forks [78].
As yet there is no definitive evidence that SMARCAL1 catalyzes either fork regression or restoration in cells. New genetic evidence suggests that the homologous recombination protein RAD51 is essential for fork regression [79], and the FBH1 helicase also plays a role [69]. Another enzyme, RECQ1 participates in restoring replication forks that form due to low dose CPT treatment [80]. The ZRANB3 enzyme can also perform fork regression and restoration reactions in vitro [62, 68], but RPA acts as a block to its translocase activity [62]. WRN, and BLM associate with RPA, but it is not clear that this binding promotes their ability to catalyze fork regression. Thus, clarifying which enzymes catalyze fork regression and restoration in vivo remains an important research goal. Furthermore, defining the exact substrates and fork remodeling reactions for each of the candidates will be essential.
The best evidence that SMARCAL1 does remodel replication forks in cells comes from studying its regulation by ATR. As would be expected for a repair or fork stabilization function, silencing SMARCAL1 by RNA interference in human cells or deletion in mouse cells yields increased replication-associated DNA damage and collapsed forks [72, 76, 81]. Fork collapse in this circumstance depends on the action of the MUS81 structure-specific endonuclease [82]. Importantly, too much SMARCAL1 activity can also cause fork collapse [72]. In this case, MUS81 is not absolutely required due to redundancy with additional nucleases scaffolded by SLX4 [21]. This is reminiscent of ATR-deficient cells in which double-strand breaks are generated at stalled forks even in MUS81-deficient cells by another SLX4-dependent nuclease [21, 54]. Furthermore, both ATR inhibition and SMARCAL1 overexpression yield a pan-nuclear γH2AX pattern providing another genetic link between ATR inhibition and too much SMARCAL1 activity [21, 72].
These genetic observations suggest the following hypothesis: In the presence of active ATR, SMARCAL1 is regulated by RPA to catalyze fork remodeling reactions that repair or stabilize stalled forks and prevent fork collapse. When ATR is inactivated, SMARCAL1 activity is no longer regulated properly and the structures it generates are substrates for SLX4-dependent nucleases (Figure 3). Consistent with this model, SMARCAL1 is a direct substrate of ATR [72, 74]; ATR-dependent phosphorylation reduces SMARCAL1 activity in vitro and in cells [21]; and silencing or depleting SMARCAL1 in human or Xenopus systems reduces the amount of nascent ssDNA observed in response to ATR inhibition [21]. Thus, ATR-dependent SMARCAL1 phosphorylation helps to prevent fork collapse by ensuring the right amount of stalled fork remodeling or coordinating it with other fork repair mechanisms.
Figure 3.

The checkpoint regulates fork regression and enzymatic cleavage at stalled replication forks. Checkpoint-deficient cells have excessive or persistent fork regression creating substrates for structure-specific nucleases. Nuclease action generates double-strand breaks and excessive ssDNA when the checkpoint is inactive.
Other fork repair enzymes directly targeted by ATR include WRN and BLM. BLM is required for fork restart after aphidicolin-induced stalling, and mutation of an ATR phosphorylation site on BLM generates an enzyme that is unable to support fork restart [47, 83]. WRN phosphorylation helps recruit WRN to stalled forks, and WRN-deficient cells accumulate double-strand breaks [46]. As is the case for SMARCAL1, the double-strand breaks in WRN-deficient cells are caused by the action of MUS81 [82, 84]. Interestingly, SMARCAL1 and WRN form a complex that is bridged by RPA [82]. It is not yet known whether the complex is important for fork stabilization and repair, but given the similarities in phenotypes caused by deficiencies in each individual protein, it is tempting to speculate that they cooperate in a fork repair pathway.
There are multiple additional ATR pathway substrates at forks including endonucleases, RPA, and the MCM proteins [85–90]. In S. pombe, direct phosphorylation of Dna2 by Cds1 regulates its association with stalled forks and Dna2 action prevents fork collapse [86]. In S. cerevisiae, Exo1 processes collapsed replication forks and reduces fork reversal rates in checkpoint-deficient cells [85]. Phosphorylation of RPA is complex and likely controls multiple fork stabilization, repair, and restart activities including the action of recombination proteins [91]. MCM phosphorylation by ATR promotes the firing of additional nearby origins to rescue the stalled fork [89]. Fork rescue explains why many more origins are licensed than actually utilized in any single cell division cycle [92–94]. Fork cleavage by MUS81 and other SLX4-scaffolded nucleases can cause fork collapse but also promote fork recovery through recombination processes. Their regulation is essential to ensure that they act appropriately to promote replication completion. I point the reader to a recent review for further discussion of these nucleases [95].
The ability of fork regression and fork cleavage to be both beneficial and detrimental to the completion of DNA synthesis depending on context is another illustration of a more universal paradigm in DNA repair. Repair mechanisms can sometimes cause more problems than they fix. In the case of fork repair, sometimes the best solution is to do nothing since replication can normally be completed using other forks or bypass mechanisms. By regulating regression and nuclease activities, the checkpoint may ensure that double-strand breaks are only generated when no other mechanism can finish replication.
Summary
I have focused this discussion on how the replication checkpoint prevents fork collapse. There are many replication fork protection mechanisms that can operate with or independently of the checkpoint pathway (the Timeless-Tipin complex for example [96]), and I have not discussed even all the known mechanisms by which ATR signaling ensures complete replication (see for example its regulation of tethering chromatin to the nuclear pore [97]). However, I have tried to emphasize the following concepts: (1) Fork collapse can include replisome dissociation and generation of double-strand breaks, but should more generally be considered the inactivation of a replication fork prior to completion of replicon synthesis. (2) The replication checkpoint prevents fork collapse through multiple mechanisms, and this is likely the essential function of the checkpoint to maintain cell viability. (3) The mechanisms by which the replication checkpoint prevents fork collapse include regulation of origin firing so as to prevent depletion of RPA and direct regulation of DNA processing enzymes to control fork regression, cleavage, and other enzymatic events.
There are many unanswered questions. For example, whether a key function of the checkpoint is to prevent replisome disassembly remains controversial. A future focus on helicase dynamics is probably appropriate since its association with the fork is likely to be the key determinant of whether the fork can easily continue DNA synthesis. Fork reversal and fork cleavage can be both beneficial and pathological. Regulating these DNA changes appears to be an essential aspect of the checkpoint although exactly which enzymes do which reactions remains speculative. Finally, we will need to convert our understanding of fork collapse into a better ability to intervene in diseases like cancer that are associated with higher levels of replication stress and genome instability as well as the rare developmental disorders caused by mutations in fork stabilization and repair proteins.
Acknowledgments
Research on the replication checkpoint and fork collapse in the Cortez lab is supported by NIH grant R01CA102729.
abbreviations
- ATR
ATM and Rad3-related
- RPA
replication protein A
- iPOND
isolation of proteins on nascent DNA
- ChIP
chromatin immunoprecipitation
- HU
hydroxyurea
- ssDNA
single-stranded DNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mechali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev Mol Cell Biol. 2010;11:728–738. doi: 10.1038/nrm2976. [DOI] [PubMed] [Google Scholar]
- 2.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Molecular cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 4.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 5.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nam EA, Cortez D. ATR signalling: more than meeting at the fork. The Biochemical journal. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Contreras AJ, Fernandez-Capetillo O. The ATR barrier to replication-born DNA damage. DNA Repair (Amst) 2010;9:1249–1255. doi: 10.1016/j.dnarep.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, Mendez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol. 2013;20:1383–1389. doi: 10.1038/nsmb.2719. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ, Blanco L. PrimPol, an archaic primase/polymerase operating in human cells. Molecular cell. 2013;52:541–553. doi: 10.1016/j.molcel.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carr AM, Paek AL, Weinert T. DNA replication: failures and inverted fusions. Seminars in cell & developmental biology. 2011;22:866–874. doi: 10.1016/j.semcdb.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Alignment and sensitive detection of DNA by a moving interface. Science. 1994;265:2096–2098. doi: 10.1126/science.7522347. [DOI] [PubMed] [Google Scholar]
- 13.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norio P, Schildkraut CL. Visualization of DNA replication on individual Epstein-Barr virus episomes. Science. 2001;294:2361–2364. doi: 10.1126/science.1064603. [DOI] [PubMed] [Google Scholar]
- 15.Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 16.Brewer BJ, Fangman WL. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- 17.Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. The EMBO journal. 2003;22:4325–4336. doi: 10.1093/emboj/cdg391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Piccoli G, Katou Y, Itoh T, Nakato R, Shirahige K, Labib K. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Molecular cell. 2012;45:696–704. doi: 10.1016/j.molcel.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Olive PL. Cell proliferation as a requirement for development of the contact effect in Chinese hamster V79 spheroids. Radiat Res. 1989;117:79–92. [PubMed] [Google Scholar]
- 20.Ager DD, Dewey WC, Gardiner K, Harvey W, Johnson RT, Waldren CA. Measurement of radiation-induced DNA double-strand breaks by pulsed-field gel electrophoresis. Radiat Res. 1990;122:181–187. [PubMed] [Google Scholar]
- 21.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordeiro-Stone M, Makhov AM, Zaritskaya LS, Griffith JD. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. Journal of molecular biology. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 23.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 24.Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 25.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 26.Gabbai CB, Marians KJ. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 2010;9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bizard AH, Hickson ID. The dissolution of double Holliday junctions. Cold Spring Harbor perspectives in biology. 2014;6:a016477. doi: 10.1101/cshperspect.a016477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anand RP, Lovett ST, Haber JE. Break-induced DNA replication. Cold Spring Harbor perspectives in biology. 2013;5:a010397. doi: 10.1101/cshperspect.a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Current opinion in genetics & development. 2013;23:271–279. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 31.Nam EA, Zhao R, Cortez D. Analysis of mutations that dissociate G(2) and essential S phase functions of human ataxia telangiectasia-mutated and Rad3-related (ATR) protein kinase. J Biol Chem. 2011;286:37320–37327. doi: 10.1074/jbc.M111.276113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 1998;12:2956–2970. doi: 10.1101/gad.12.18.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paciotti V, Clerici M, Scotti M, Lucchini G, Longhese MP. Characterization of mec1 kinase-deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Molecular and cellular biology. 2001;21:3913–3925. doi: 10.1128/MCB.21.12.3913-3925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tercero JA, Longhese MP, Diffley JF. A central role for DNA replication forks in checkpoint activation and response. Molecular cell. 2003;11:1323–1336. doi: 10.1016/s1097-2765(03)00169-2. [DOI] [PubMed] [Google Scholar]
- 35.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Molecular cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 36.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 38.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 39.Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 40.Alvino GM, Collingwood D, Murphy JM, Delrow J, Brewer BJ, Raghuraman MK. Replication in hydroxyurea: it’s a matter of time. Molecular and cellular biology. 2007;27:6396–6406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merrick CJ, Jackson D, Diffley JF. Visualization of altered replication dynamics after DNA damage in human cells. J Biol Chem. 2004;279:20067–20075. doi: 10.1074/jbc.M400022200. [DOI] [PubMed] [Google Scholar]
- 43.Meister P, Taddei A, Ponti A, Baldacci G, Gasser SM. Replication foci dynamics: replication patterns are modulated by S-phase checkpoint kinases in fission yeast. The EMBO journal. 2007;26:1315–1326. doi: 10.1038/sj.emboj.7601538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 45.Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. The EMBO journal. 2006;25:1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, Pichierri P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. The EMBO journal. 2010;29:3156–3169. doi: 10.1038/emboj.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Molecular and cellular biology. 2004;24:1279–1291. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 49.Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005;19:3055–3069. doi: 10.1101/gad.361805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lucca C, Vanoli F, Cotta-Ramusino C, Pellicioli A, Liberi G, Haber J, Foiani M. Checkpoint-mediated control of replisome-fork association and signalling in response to replication pausing. Oncogene. 2004;23:1206–1213. doi: 10.1038/sj.onc.1207199. [DOI] [PubMed] [Google Scholar]
- 51.Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–1083. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- 52.Dimitrova DS, Gilbert DM. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol. 2000;2:686–694. doi: 10.1038/35036309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hashimoto Y, Puddu F, Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol. 2012;19:17–24. doi: 10.1038/nsmb.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ragland RL, Patel S, Rivard RS, Smith K, Peters AA, Bielinsky AK, Brown EJ. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev. 2013;27:2259–2273. doi: 10.1101/gad.223180.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu C, Gan H, Han J, Zhou ZX, Jia S, Chabes A, Farrugia G, Ordog T, Zhang Z. Strand-Specific Analysis Shows Protein Binding at Replication Forks and PCNA Unloading from Lagging Strands when Forks Stall. Molecular cell. 2014;56:551–563. doi: 10.1016/j.molcel.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Follonier C, Oehler J, Herrador R, Lopes M. Friedreich’s ataxia-associated GAA repeats induce replication-fork reversal and unusual molecular junctions. Nat Struct Mol Biol. 2013;20:486–494. doi: 10.1038/nsmb.2520. [DOI] [PubMed] [Google Scholar]
- 58.Neelsen KJ, Zanini IM, Herrador R, Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol. 2013;200:699–708. doi: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tatsumi K, Strauss B. Production of DNA bifilarly substituted with bromodeoxyuridine in the first round of synthesis: branch migration during isolation of cellular DNA. Nucleic acids research. 1978;5:331–347. doi: 10.1093/nar/5.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Molecular cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 61.Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, Eichman BF, Cortez D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Betous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3:1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Machwe A, Karale R, Xu X, Liu Y, Orren DK. The Werner and Bloom syndrome proteins help resolve replication blockage by converting (regressed) holliday junctions to functional replication forks. Biochemistry. 2011;50:6774–6788. doi: 10.1021/bi2001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc Natl Acad Sci U S A. 2008;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Opresko PL, Sowd G, Wang H. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PloS one. 2009;4:e4825. doi: 10.1371/journal.pone.0004825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Achar YJ, Balogh D, Haracska L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1101951108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Machwe A, Xiao L, Groden J, Orren DK. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- 68.Ciccia A, Nimonkar AV, Hu Y, Hajdu I, Achar YJ, Izhar L, Petit SA, Adamson B, Yoon JC, Kowalczykowski SC, Livingston DM, Haracska L, Elledge SJ. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Molecular cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fugger K, Mistrik M, Neelsen KJ, Yao Q, Zellweger R, Kousholt AN, Haahr P, Chu WK, Bartek J, Lopes M, Hickson ID, Sorensen CS. FBH1 Catalyzes Regression of Stalled Replication Forks. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.028. [DOI] [PubMed] [Google Scholar]
- 70.Manosas M, Perumal SK, Croquette V, Benkovic SJ. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science. 2012;338:1217–1220. doi: 10.1126/science.1225437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mason AC, Rambo RP, Greer B, Pritchett M, Tainer JA, Cortez D, Eichman BF. A structure-specific nucleic acid-binding domain conserved among DNA repair proteins. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1324143111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bhat KP, Betous R, Cortez D. High-affinity DNA-binding Domains of Replication Protein A (RPA) Direct SMARCAL1-dependent Replication Fork Remodeling. J Biol Chem. 2015;290:4110–4117. doi: 10.1074/jbc.M114.627083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, Cejka P, Stewart S, Lopes M, Vindigni A. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol. 2015;208:545–562. doi: 10.1083/jcb.201406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ, Jr, Lopes M, Vindigni A. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013;20:347–354. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bansbach CE, Boerkoel CF, Cortez D. SMARCAL1 and replication stress: An explanation for SIOD? Nucleus. 2010;1:245–248. doi: 10.4161/nucl.1.3.11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Betous R, Glick GG, Zhao R, Cortez D. Identification and Characterization of SMARCAL1 Protein Complexes. PLoS One. 2013;8:e63149. doi: 10.1371/journal.pone.0063149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol. 2007;14:677–679. doi: 10.1038/nsmb1267. [DOI] [PubMed] [Google Scholar]
- 84.Franchitto A, Pirzio LM, Prosperi E, Sapora O, Bignami M, Pichierri P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J Cell Biol. 2008;183:241–252. doi: 10.1083/jcb.200803173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Molecular cell. 2005;17:153–159. doi: 10.1016/j.molcel.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 86.Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W, Sun F, Ji J, Murray JM, Carr AM, Kong D. The intra-s phase checkpoint targets dna2 to prevent stalled replication forks from reversing. Cell. 2012;149:1221–1232. doi: 10.1016/j.cell.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 87.Binz SK, Sheehan AM, Wold MS. Replication Protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst) 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 88.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci U S A. 2004;101:10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trenz K, Errico A, Costanzo V. Plx1 is required for chromosomal DNA replication under stressful conditions. The EMBO journal. 2008;27:876–885. doi: 10.1038/emboj.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoo HY, Shevchenko A, Dunphy WG. Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J Biol Chem. 2004;279:53353–53364. doi: 10.1074/jbc.M408026200. [DOI] [PubMed] [Google Scholar]
- 91.Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, Borowiec JA. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol. 2014;206:493–507. doi: 10.1083/jcb.201404111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–3341. doi: 10.1101/gad.457807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ibarra A, Schwob E, Mendez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proceedings of the National Academy of Sciences. 2008;105:8956–8961. doi: 10.1073/pnas.0803978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends in biochemical sciences. 2014;39:409–419. doi: 10.1016/j.tibs.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 96.Leman AR, Noguchi E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle. 2012;11:3945–3955. doi: 10.4161/cc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bermejo R, Kumar A, Foiani M. Preserving the genome by regulating chromatin association with the nuclear envelope. Trends Cell Biol. 2012;22:465–473. doi: 10.1016/j.tcb.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 98.Aparicio OM, Stout AM, Bell SP. Differential assembly of Cdc45p and DNA polymerases at early and late origins of DNA replication. Proc Natl Acad Sci U S A. 1999;96:9130–9135. doi: 10.1073/pnas.96.16.9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Masumoto H, Sugino A, Araki H. Dpb11 controls the association between DNA polymerases alpha and epsilon and the autonomously replicating sequence region of budding yeast. Molecular and cellular biology. 2000;20:2809–2817. doi: 10.1128/mcb.20.8.2809-2817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–1919. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sabatinos SA, Green MD, Forsburg SL. Continued DNA synthesis in replication checkpoint mutants leads to fork collapse. Molecular and cellular biology. 2012;32:4986–4997. doi: 10.1128/MCB.01060-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Segurado M, Diffley JFX. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008;22:1816–1827. doi: 10.1101/gad.477208. [DOI] [PMC free article] [PubMed] [Google Scholar]