

Abstract
Time-lapse X-ray crystallography allows visualization of intermediate structures during the DNA polymerase catalytic cycle. Employing time-lapse crystallography with human DNA polymerase β has recently allowed us to capture and solve novel intermediate structures that are not stable enough to be analyzed by traditional crystallography. The structures of these intermediates reveal exciting surprises about active site metal ions and enzyme conformational changes as the reaction proceeds from the ground state to product release. In this perspective, we provide an overview of recent advances in understanding the DNA polymerase nucleotidyl transferase reaction and highlight both the significance and mysteries of enzyme efficiency and specificity that remain to be solved.
Keywords: Base excision repair, DNA polymerase, DNA repair, fidelity, function, genome instability, structure, X-family, X-ray crystallography
1. Introduction
DNA polymerases play an essential role in DNA repair and replication. Accordingly, they are principal targets for chemotherapies and for understanding the efficiency and accuracy of DNA repair and replication. Additionally, DNA polymerases play a vital role in genetic engineering and the biotechnology industry. For these reasons, an enhanced view of their structure/function is a prerequisite for rational drug and protein design. Human cells have at least 16 DNA polymerases, and based on primary sequences have been categorized into four families (A-, B-, X-, and Y-families). DNA polymerases often include an accessory domain that complements its biological function (e.g., proofreading exonuclease or processivity).
DNA polymerase (pol) β (X-family) is the smallest human cellular polymerase and contributes two-enzymatic activities during base excision repair of simple DNA lesions. In addition to its metal-dependent DNA synthesis activity, a deoxyribose phosphate lyase activity is found on the amino-terminal lyase 8-kDa domain. The lyase activity is required to cleanse the 5′-end of a DNA gap to generate a substrate that can be ligated to complete repair. DNA polymerase β has been extensively characterized structurally, kinetically, computationally, and biologically [1, 2]. Consequently, pol β serves as a model DNA polymerase to explore the nucleotidyl transferase reaction as well as examine how polymerases gain specificity by choosing right from wrong nucleotide substrates. Here we review recent structural studies that provide a new framework to analyze the nucleotidyl transferase chemical reaction and substrate discrimination.
2. Structural Characterization of DNA Polymerase β
2.1. Approach
The first mammalian crystal structure of pol β was deposited in 1994; and to this day pol β remains a model DNA polymerase in deciphering the DNA polymerase chemical mechanism. As of November 2014, more than 240 X-ray crystallographic structures of pol β have been deposited in the protein data bank. These structural snapshots were obtained using multiple approaches ranging from modified protein or substrates to the recently developed time-lapse crystallography that employs natural substrates. In this section we will highlight several approaches utilized to capture key intermediate structures of pol β.
2.1.1. Modified substrates
The first approach to successfully capture a crystal structure of pol β bound to DNA utilized a modified DNA substrate to form an inactive pre-insertion complex. The ternary complex (+ddCTP) of rat pol β bound to DNA with an incoming nucleotide was determined to 2.9 Å using a dideoxy-terminated primer [3]. The dideoxy-terminated primer does not support catalysis due to the missing 3′-OH (Fig. 1A). Using this approach, a general pol β catalytic mechanism (see below) and key active site residues were identified. A subsequent study using human pol β with the preferred 1-nucleotide (1-nt) gapped DNA containing a dideoxy-terminated primer diffracted to ~3.0 Å. These structures identified key conformational changes that occur upon pol β binding to the DNA and an incoming dNTP [4]. The use of a dideoxy-terminated primer was instrumental in capturing these early pre-insertion structural snapshots of pol β and remained the standard until non-hydrolysable deoxynucleotide analogs became readily available.
Fig. 1.

Structural apporaches for capturing ternary pol β structural snapshots. A diagram of the pol β active site with either a (A) dideoxy-terminated primer or (B) a non-hydrolysable deoxynucleotide analog are shown. The active site aspartic acid residues, catalytic metal (Mc), nucleotide metal (Mn), and coordinating water molecules (blue) are indicated. The metal coordination is shown with dashes. In panel A, the position of the missing O3′ of the dideoxy-terminated primer is indicated in red. Additionally, the reactive oxygen group between Pα and Pβ of the triphosphate of a natural dNTP is indicated. The use of a dideoxy-terminated hinders catalytic metal binding and proper Asp256 coordination. In panel B, the 3′-OH for the primer terminus, proper orientation/coordination of Asp256, and Mc are shown. The bridging position of the non-hydrolysable deoxynucleotide analog is indicated with a red X. This position commonly contains either a non-reactive carbon or nitrogen group to prevent catalysis. (C) The time-lapse crystallography schematic for correct insertion is shown going from left to right. The global conformation of pol β is indicted at the top and the bottom corresponds to the ligands in the pol β active site at each step. The transfer to a solution containing MgCl2 starts the reaction (i.e., t = 0). The structural snapshots are captured by freezing the reaction at various time points (downward facing arrows). Correct insertion results in a closed ternary product complex.
A limitation of using a dideoxy-terminated primer is that the catalytic metal binding site is distorted by the absence of a key coordinating ligand (i.e., primer sugar O3′; Fig. 1A). To overcome this limitation, ternary complex pre-insertion structures with a non-hydrolysable deoxynucleotide analog (2′-deoxyuridine 5′-α,β-imido-triphosphate) were determined for pol β in 2006 [5]. This analog contains nitrogen at the bridging oxygen position between the α- and β-phosphates to prevent catalysis while allowing the utilization of a natural primer terminus with a 3′-OH (Fig. 1B). Soaking binary DNA pol β crystals in a solution containing the non-hydrolysable analog results in a ternary complex that routinely diffracts well past 2.0 Å [5]. The first structural snapshots using non-hydrolysable analogs provided direct evidence that the 3′-OH and the catalytic Mg2+ ion are required to achieve the proper geometry necessary for an in-line nucleophilic attack of O3′ (primer terminus) on the α-phosphate of the incoming nucleotide. The availability of non-hydrolysable nucleotide analogs has allowed for subsequent studies looking at polymerase fidelity and lesion bypass [6–9].
2.1.2. Site-directed mutagenesis
Prior to 2012 the main approach for capturing structural snapshots utilized wild-type pol β with modified DNA or nucleotides. A caveat with this approach is that it only captures a closed pre-insertion complex. To overcome this limitation and trap structural intermediates prior to the closed pre-insertion complex, selective mutations within the pol β active site have been utilized to alter the equilibrium between the open and closed states that occur upon substrate binding (see below). For example, Arg283 contacts the DNA minor groove in the active site and stabilizes the closed ternary complex. A R283K point mutant shifts the pol β equilibrium to favor the open conformation thereby providing an opportunity to capture structural intermediates prior to polymerase closure [6]. These snapshots revealed that Watson-Crick hydrogen bonding of the nascent base pair is assessed prior to enzyme closing around the nascent base pair. The closed/open equilibrium is also altered by another mutation, the E295K variant. Interestingly, this enzyme has been associated with some gastric carcinomas and has a dramatically lower polymerase activity [1, 7]. Structural studies with this protein provided insight into the structural rearrangements that occur during nucleotide insertion and provide structural evidence that alternate conformations modulate insertion efficiency and discrimination. Together, the R283K and E295K variant enzymes are examples of using site directed mutagenesis to alter the conformational equilibria in the active site so as to capture novel structural intermediates.
2.1.3. Time-lapse crystallography
In 1996 it was first reported that pol β undergoes catalysis within the crystal using Mn2+, while Ca2+ failed to support chemistry [10]. Around this same time, visualization of insertion within a crystal was also observed with Bacillus DNA polymerase I [11]. In both of these studies the ternary complex crystals were soaked in a solution that supported extension of the primer terminus to observe the subsequent product. Using a similar soaking method one can stage the reaction to capture key steps before, during, and after catalysis by time-lapse crystallography. This approach has been successfully utilized with both pol β [12] and the Y-family DNA polymerase η [13] to capture intermediate snapshots during catalysis. We refer to the technique as time-lapse crystallography to differentiate the approach from the Laue based time-resolved crystallography that can follow product formation on a much shorter time scale.
Time-lapse crystallography involves a series of staged soaks (Fig. 1C). For pol β, binary crystals of pol β bound to unmodified 1-nt gapped DNA are soaked in a cryo-solution containing CaCl2 and the incoming natural nucleotide. Since Ca2+ does not support catalysis, the resulting complex is in the closed ternary pre-insertion state with natural substrates. These crystals are then transferred to a solution containing MgCl2, but lacking CaCl2 and the incoming nucleotide. Varying the time in the MgCl2 soak prior to flash freezing at 100 K allows one to capture key structural snapshots during and after chemistry. Importantly, this technique identified key intermediate states that had previously been overlooked using alternative approaches. Studies of pol β with this approach have resulted in novel adjunct metal ions being identified and conformational changes that occur after chemistry in a substrate dependent phenomenon [12, 14]. These findings are discussed in detail below.
2.2. Domains and subdomains
Controlled proteolytic or chemical cleavage of pol β indicated that it is folded into distinct domains [15]. Subsequent crystallographic and activity measurements demonstrated that the enzyme was folded into two domains that each contributes a critical activity during the repair of simple base lesions in DNA. The amino-terminal 8-kDa lyase domain has a dRP lyase activity that removes the 5′-sugar-phosphate AP-site repair intermediate and a 31-kDa polymerase domain (Fig. 2) that has DNA synthesis activity. The polymerase domain has a modular organization with three functionally distinct subdomains. The catalytic subdomain coordinates divalent metal cations (Mg2+) that facilitate DNA synthesis (nucleotidyl transferase). The other subdomains are spatially situated on opposite sides of the catalytic subdomain (Fig. 2A). While the catalytic subdomain of X- and bacterial C-family DNA polymerases share structural homology, those from other families (e.g., A-family members) exhibit a similar but unique fold [16]. Based on structural homology of the catalytic domain of E. coli DNA polymerase I [17], structures of A-, B-, and Y-family polymerases have likened these enzymes to a right-hand with fingers, palm, and thumb subdomains. Since this nomenclature is opposite to that originally proposed for pol β [18], there is not a consistent usage of the hand-like nomenclature in the literature. Because the hand-like architectural analogy lacks functional insight, we have employed functionally based designations to the subdomains. Accordingly, the polymerase domain includes the C– (Catalytic), D– (DNA binding), and N– (Nascent base pair binding) subdomains and are equivalent to the palm, thumb, and fingers subdomains, respectively, of right-handed DNA polymerases (Fig. 2A).
Fig. 2.

Domain/subdomain organization of human DNA polymerase β. (A) A ribbon representation of pol β illustrating the polymerase (blue) and amino-terminal lyase (gray) domains. The polymerase domain is composed of three subdomains: DNA-binding, D; catalytic, C; and nascent base pair interacting, N. These correspond to the thumb, palm, and fingers subdomains of DNA polymerases, respectively, that utilize an architectural analogy to a right hand. The nascent base pair (yellow carbons) is illustrated in a stick representation while the remaining DNA is omitted for clarity. (B) The molecular surface of pol β bound to single-nucleotide gapped DNA exhibits a global doughnut-like structure where the lyase domain interacts with the N–subdomain of the polymerase domain. The viewpoint is the same as panel A. The molecular surface is clipped through the nascent base pair (illustrated in a stick representation) binding pocket. The backbone of the template and broken strands are shown in a ribbon representation; yellow and red, respectively. The 3′-ends of the gapped strand are indicated (the 3′-end label of the primer strand near the polymerase active site is white).
2.3. Conformational adjustments
High fidelity DNA polymerases and their substrates undergo several conformational transitions during catalytic cycling. These conformational changes play key roles in substrate discrimination facilitating correct substrate (dNTP) selection and are often referred to as ‘induced fit’ (see below). Pol β transitions from an extended to a doughnut-like conformation upon binding to single-nucleotide gapped DNA that creates a channel through which nucleotides enter the active site (Fig. 2B) [16].
Pol β rapidly repositions the N-subdomain (fingers) upon nucleotide binding to close around the nascent base pair (Fig. 3). As described above, reducing interactions of the N-subdomain with the nascent base pair through site-directed mutagenesis (i.e., R283K) destabilizes the closed conformation. Crystallographic structures of intermediate complexes with an incoming nucleotide bound to the open polymerase conformation revealed that the coding template base was able to Watson-Crick hydrogen bond with the correct incoming nucleotide [6]. In this open conformation, the nascent base pair is severely buckled, since the sugar/triphosphate moieties interact with protein side chains that have not moved to their closed positions. Unexpectedly, the negative charge on the incoming nucleotide triphosphate was neutralized through protein interactions rather than a magnesium ion. The structure of the metal-free complex indicates that the polymerase has a strong influence on metal coordination and triphosphate reorganization.
Fig. 3.

Conformational adjustments as DNA polymerase β transitions between open and closed conformations. (A) Ribbon representation of the pol β open DNA binary complex (gray) superimposed with the ternary (+dNTP) complex (blue). DNA is omitted for clarity, but the incoming nucleotide of the ternary complex is shown in a stick representation. The N-subdomain closes upon the nascent base pair upon formation of the ternary complex. This repositioning is indicated by the altered position of α-helix N (open conformation, orange; closed conformation, yellow). Concomitant with the closed conformation, the lyase domain also increases interactions with the N-subdomain. (B) The position of the N–subdomain can be monitored in the active site through a series of altered interactions between Asp192 (D192), that coordinate both active site metals (orange), and Arg283 (R283) that is situated in the N–subdomain. The arrows represent motions associated with the closing transition. N–subdomain closing is associated with Arg258 (R258) releasing Asp192 and forming hydrogen bonds with Glu295 (E295) and Tyr296 (Y296). Phe272 (F272) is repositioned in the closed complex to insulate Asp192 from Arg258. The DNA is omitted for clarity. (C) A view of the DNA major groove edge of the nascent base pair and primer terminus (O3′, stick representation) illustrating repositioning of the template strand and primer bases upon forming the closed ternary complex. The black arrow represent a shift in the template strand associated with the closing transition where the template strand moves toward the N–subdomain. Base pairs upstream of the primer terminus are illustrated in a wire representation. The polymerase is omitted for clarity. The incoming nucleotide (dUMPPNP) and its coding template base of the ternary complex are labeled N and n0, respectively. The green arrow illustrates the repositioned O3′ (labeled) attacking Pα of the incoming nucleotide. The catalytic and nucleotide magnesium ions are shown as orange spheres. Panels B and C were adapted from Beard et al. [29].
The N-subdomain repositioning upon nucleotide binding also results in many subtle conformational adjustments that lead to catalytic activation. One key aspect of this activation is that Asp192 is released from a salt bridge interaction with Arg258 permitting it to coordinate both the catalytic and nucleotide binding metals (Fig. 3B). Accordingly, enzyme mutations distant from the active site (e.g., Arg283 and Glu295) that destabilize the closed conformation dramatically decrease activity [1, 6, 7].
The templating (coding) base must be precisely positioned for the polymerase to faithfully determine nascent base pair geometry. Comparing the position of the templating base in the open DNA binary and ternary closed (+dNTP) complex indicates that the template base slides downstream to base pair with the incoming nucleotide (Fig. 3C) providing an opportunity to check for Watson-Crick geometry. Additionally, a key structural adjustment of the pre-catalytic complex resides in precise positioning of O3′ of the deoxyribose of the primer terminus. The catalytic metal coordinates a non-bridging oxygen of the α-phosphate of the incoming nucleotide as well as O3′ of the primer terminus. In the absence of the catalytic magnesium, crystallographic structures indicate that the sugar pucker is 2′-endo. Binding of the catalytic metal alters the sugar pucker (3′-endo) of the primer terminus repositioning O3′ for in-line attach on the α-phosphate of the incoming nucleotide [5, 19]. Computational studies also indicate that the path to its binding site can influence the open/closed subdomain transition prior to chemistry [20].
After chemistry, the ternary product complex remains in the closed conformation [12]. However, there are subtle active site changes. The formation of the phosphodiester bond alters the coordination state of the catalytic Mg2+ that facilitates its dissociation. In contrast, the nucleotide associated Mg2+ remains in the active site, coordinating active site aspartates (Asp190 and Asp192) and PPi (Fig. 4A). Surprisingly, crystals of the product complex after correct nucleotide insertion remain in a closed conformation with bound PPi even with extended incubations. Since catalytic cycling in solution occurs much more rapidly, other factors must influence the conformational state of the enzyme and facilitate opening. In contrast to the stable closed conformation observed after correct insertion, structures after nucleotide misinsertion indicate that the enzyme can rapidly open and release PPi and metals [12]. Misinsertion is also discouraged in the closed ternary complex through repositioning of the primer terminus (i.e., O3′) as the templating base vacates the coding position [21, 22].
Fig. 4.
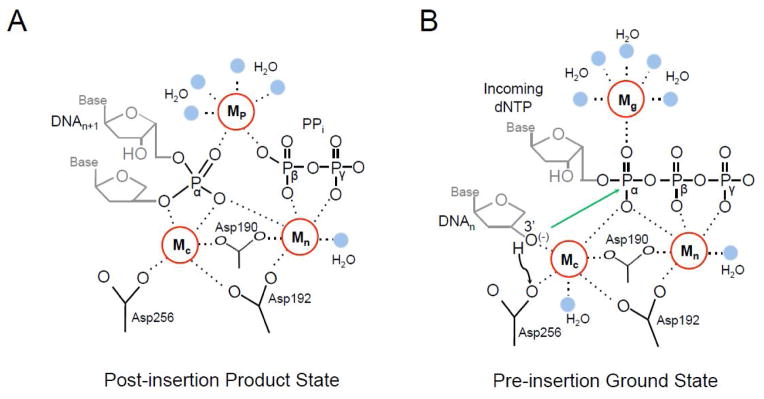
Adjunct metal binding sites in the pol β active site. (A) The post-insertion product state of the pol β active site is shown following insertion of the incoming dNTP (DNAn+1). The catalytic (Mc), nucleotide (Mn), and product (Mp) metal binding sites are shown as red circles with dashed lines to coordinating groups. Four Mp coordinating water molecules are in blue. Active site aspartic acid residues, the remaining bound pyrophosphate (PPi) group, and a coordinating water molecule (blue) are indicated. The groups corresponding to Pα, Pβ, and Pγ of the inserted dNTP triphosphate are labeled for clarity. The Mc binding site has only five coordinating ligands due to the missing 3′-OH. This results in the catalytic Mg2+ vacating the active site and being replaced by a Na2+ for pol β. (B) The pre-insertion state of the pol β active site is shown with an incoming dNTP and a primer terminus with a 3′-OH (i.e., 2′-deoxyribose). The black arrow corresponds to deprotonation of the 3′-OH to Asp256. The green arrow indicates the resulting O3′ anion attack on Pα. The catalytic (Mc), nucleotide (Mn), and ground (Mg) metal binding sites are shown as red circles with dashed lines to coordinating groups. Active site aspartic acid residues and coordinating waters (blue) are shown.
3. Nucleotide Insertion
3.1. Two-metal mechanism
In 1991 the two-metal mechanism for the nucleotidyl transferase reaction during DNA synthesis was proposed from crystallographic structures of substrates bound to the Escherichia coli Klenow fragment proofreading active site [23]. From these early structures, both activating/catalytic (Site A, Mc) and transition state stabilizing (Site B, Mn) metal binding sites were identified. These metal binding sites support catalysis using Mg2+ as the biological cofactor. For DNA synthesis, the Mc facilitates formation of the attacking oxyanion by lowering the pKa of the primer terminus O3′. Chemistry proceeds by an in-line nucleophilic attack of the oxyanion on the α-phosphate of the incoming nucleotide, leading to a penta-coordinated bipyramidal α-phosphate transition state that results in dNMP insertion and formation of pyrophosphate (PPi). The Mn coordinates the non-bridging oxygens of the incoming nucleotide triphosphate and would stabilize the developing negative charge during nucleophillic attack. Ternary complex structures of pol β are consistent with this proposed model [3–5].
For pol β the octahedral-coordinated catalytic metal binding site (Mc) is composed of O3′ at the primer terminus, the non-bridging oxygen of Pα, active site aspartic acid residues (Asp190, Asp192, and Asp256), and a water molecule (Fig. 1B). The octahedral-coordinated nucleotide metal binding site (Mn) is composed of the non-bridging oxygens of the incoming nucleotide triphosphate, active site aspartic acid residues (Asp190 and Asp192), and a water molecule (Fig. 1B). For pol β, Mc activates O3′ with the proton migrating to Asp256 that also coordinates Mc. Until the development of time-lapse crystallography the polymerase mechanism was believed to utilize only two metals (Mc and Mn) during catalysis [23]. Recently the role of metal ions during DNA polymerase catalysis has been analyzed with advances in structural and computational biology identifying adjunct metal ions [12–14].
3.2. Adjunct metal ions
The recent development of time-lapse crystallography has uncovered novel structural intermediates during DNA polymerase catalysis. An unexpected finding was the observation of additional transient divalent metal ions that are distinct from the catalytic and nucleotide metals [12–14]. The appearance of one transient divalent metal ion coincided with product formation after insertion of the incoming dNMP and generation of PPi. Importantly, this metal ion-binding site is dependent on having product present and therefore is termed the product metal (Mp) binding site. The Mp coordinates the non-bridging oxygens on the phosphates of the scissile bond from what previously was Pα and Pβ of the incoming nucleotide (Fig. 4B). The preferred metal ion for this binding site is Mg2+ or Mn2+, similar to the catalytic and nucleotide binding sites. Product formation also promotes the release of the catalytic Mg2+, likely arising from the loss of the coordinating O3′ following insertion of the incoming nucleotide. The role of the product metal binding site is an active area of research. It has been postulated to facilitate the forward reaction by stabilizing the transition state and may be involved in promoting the reverse reaction, pryophosphorolysis (discussed below).
Another adjunct divalent metal was recently identified during the insertion of the damaged nucleotide 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodGTP) opposite a templating cytosine [14]. This metal was observed coordinating Pα of the incoming nucleotide and five water molecules in the pre-insertion ground state (Fig. 4A). Since this metal was only observed in the pre-insertion complex it is referred to as the ground state metal (Mg) binding site. The role of the ground state metal in mediating insertion was further probed using computational analysis. Quantum mechanical analysis determined that a metal at the Mg site makes Pα more positive and the bridging oxygen between Pα and Pβ more negative. The more positive Pα with Mg present would facilitate the attack of O3′ anion at Pα (dNTP). The additional negative charge at the bridging oxygen between Pα and Pβ promotes bond breakage and the subsequent protonation of PPi which is rate limiting [24]. The role of the Mg binding site during catalysis with natural unmodified nucleotides is currently being investigated. Together, these adjunct metal ions within the polymerase active site facilitate chemistry by neutralizing the developing charge over the course of the reaction.
3.3. Substrate discrimination
To maintain the integrity of the genome, DNA polymerases must select the correct dNTP from a pool of chemically and structurally similar molecules. The ability of these enzymes to discriminate right from wrong is often quantified by the probability of a polymerase to produce a base substitution error (i.e., mismatch). The base substitution error frequency for DNA replication and repair polymerases is typically between 10−3 to > 10−6 [25]. These frequencies denote one error per thousand or million nucleotides synthesized, respectively. In general, the specificity constants (kcat/Km) for wrong nucleotides are much lower than for right nucleotides, so that polymerase fidelity is simply the ratio of specificity constants (right/wrong). Importantly, since fidelity is the simple ratio of specificity constants for insertion of right and wrong nucleotides, fidelity is altered by an unbalanced change in the specificity constants (i.e., the specificity constants do not change in parallel).
An induced-fit mechanism has been proposed to amplify DNA polymerase discrimination. It proposes that substrate-induced conformational adjustments alter the position of key catalytic groups. Good substrates align catalytic participants for optimal activity whereas poor substrates would deter catalysis through the misalignment of the reactive atoms. Crystallographic structures of polymerase binary DNA and ternary (+dNTP) complexes indicate that the N-subdomain closes around the nascent base pair (Fig. 3A). Concomitant with subdomain re-positioning, there are key conformational adjustments in protein side-chains and DNA positioning that lead to altered hydrogen bonding, DNA sugar puckering, and metal coordination. Using site-directed mutagenesis, substrate analogs, or time-lapse crystallography has revealed new catalytically relevant intermediate states that impact nucleotide insertion efficiency.
4. Computational Studies
4.1. Deprotonation of primer terminus
In the two-metal mechanism described above, three conserved acidic residues in the catalytic subdomain bind two divalent magnesium ions. One Mg2+ (Mn) coordinates Asp190 and Asp192 and the triphosphate moiety of the incoming nucleotide thereby facilitating nucleotide binding. The other Mg2+ (Mc) coordinates all three active site aspartates and O3′ of the primer terminus. It lowers the pKa of the primer terminus 3′-OH hastening attack on Pα of the incoming nucleotide. Asp256 serves as a general base upon O3′ activation [26]. Accordingly, the pKa of both the donor (primer sugar O3′) and the acceptor groups (OD2 of Asp256) are altered by Mc.
Site-directed mutagenesis of Asp256 indicates that this residue plays a fundamental role in nucleotidyl transfer [27]. Quantum calculations show a charge transfer into the catalytic metal and charge loss in OD2 of Asp256 that accompanies deprotonation of the primer terminus. Importantly, the charge on O3′ remains constant, fine-tuning and facilitating its attack on Pα (dNTP). The structure of mutant pol β, where glutamate is substituted for aspartate at residue 256 (D256E), indicates that a water molecule replaces OD2 of Asp256 and coordinates Mc. In contrast to the wild-type situation, quantum calculations show that this water is not involved in a charge transfer to the Mc. The critical role of Asp256 appears to be facilitated by a stabilizing salt-bridge interaction with the nearby Arg254.
4.2. Transition state development–a computational challenge
Understanding the chemistry of nucleotidyl transfer by a polymerase involves rationalizing the in-line attack of the O3′ oxyanion on Pα of the incoming nucleotide. In most cases, the attack occurs in a tightly confined and highly charged active site with phosphate-bound oxygen atoms near the O3′ oxyanion. As described above, computational analysis demonstrates a requisite charge neutralization of the phosphate-bound oxygen atoms and fine-tuning of the charge on O3′ that hastens nucleophilic attack on Pα [27]. Thus, there are critical active site events that accommodate the charge and steric requirements in forming and stabilizing the transition state. The corresponding charge and structural changes that promote transition state resolution into products are beginning to emerge from detailed structural and QM studies [28]. It remains a great challenge to identify which atoms to include in computationally intensive calculations. The recent discovery of transient adjunct metals ions [12–14], in addition to the general two metals necessary for catalysis, highlights the strong possibility that there are other atoms (i.e., water molecules) and features (conformational adjustments) that remain to be determined that could have an impact on both the forward and reverse reactions. For template-directed DNA polymerases, transition state development and stabilization must be linked to template base recognition for the incoming nucleoside triphosphate. Accordingly, it is generally acknowledged that an “induced fit” mechanism where the active site facilitates correct nucleotide insertion and discourages incorrect insertion is a key feature that contributes to high fidelity DNA synthesis. While the influence of DNA structure on polymerase function is well recognized, the subtle intricacies of their impact on the chemical reaction are unknown due to our lack of understanding of the ionic and conformational active site adjustments necessary to accommodate “aberrant” DNA structures.
5. Implications
The dynamic nature of the adjunct metals during nucleotide insertion is consistent with an induced-fit mechanism for nucleotide discrimination since the product-associated metal has only been observed during correct nucleotide insertion. It remains to be determined through computational studies what the affect of these metals may have on the transition state for the forward (DNA synthesis) and/or reverse (pyrophosphorolysis) reactions. Whether the observed metals represent an ordered movement of the metals within the active site or are in equilibrium from an external pool remains to be determined. Since replicative DNA polymerases have a basic side chain in the N-subdomain that could substitute for the adjunct metals [1, 12], the results imply a versatile role of this basic side chain that is multi-faceted. Further studies are needed to explore this possibility.
The unique global/local conformational changes that are inherent for each insertion event for right/wrong or modified nucleotides suggest that the induced fit mechanism may have wider implications by impacting downstream DNA repair events through substrate channeling. For example, the stable closed conformation observed after inserting the right nucleotide could provide a targeting mechanism for the next step in base excision repair (DNA nick ligation). In contrast, the rapid re-opening of pol β following incorrect insertion may provide a conformational signal discouraging ligation and highlighting the need of a proofreading enzyme. Accordingly, structures of molecular complexes in specific conformational states will provide critical clues about substrate channeling of repair intermediates. However, such studies remain to be conducted.
Acknowledgments
We thank K. Bebenek and J. K. Horton for critical reading of the manuscript. This research was supported by Research Project Numbers Z01-ES050158 and Z01-ES050159 in the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
Abbreviations
- 1-nt
1-nucleotide
- AP
apurinic/apyrimidinic
- BER
base excision repair
- dNTP
deoxynucleoside triphosphate
- dRP
deoxyribose phosphate
- Mc
catalytic metal
- Mg
ground state metal
- Mn
nucleotide or transition state metal
- Mp
product metal
- PPi
pyrophosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Biochemistry. 2014;53:2768–2780. doi: 10.1021/bi500139h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bienstock RJ, Beard WA, Wilson SH. Phylogenetic analysis and evolutionary origins of DNA polymerase X-family members. DNA Repair. 2014;22:77–88. doi: 10.1016/j.dnarep.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pelletier H, Sawaya MR, Kumar A, Wilson SH, Kraut J. Structures of ternary complexes of rat DNA polymerase β, a DNA template-primer, and ddCTP. Science. 1994;264:1891–1903. [PubMed] [Google Scholar]
- 4.Sawaya MR, Prasad P, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 5.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freudenthal BD, Beard WA, Wilson SH. Structures of dNTP intermediate states during DNA polymerase active site sssembly. Structure. 2012;20:1829– 1837. doi: 10.1016/j.str.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckenroth BE, Towle-Weicksel JB, Sweasy JB, Doublié S. The E295K cancer variant of human polymerase β favors the mismatch conformational pathway during nucleotide selection. J Biol Chem. 2013;288:34850–34860. doi: 10.1074/jbc.M113.510891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koag MC, Nam K, Lee S. The spontaneous replication error and the mismatch discrimination mechanisms of human DNA polymerase β. Nucleic Acid Res. 2014;42:11233–11245. doi: 10.1093/nar/gku789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koag MC, Lai L, Lee S. Structural basis for the inefficient nucleotide incorporation opposite cisplatin-DNA lesion by human DNA polymerase β. J Biol Chem. 2014;289:31341–31348. doi: 10.1074/jbc.M114.605451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelletier H, Sawaya MR, Wolfle W, Wilson SH, Kraut J. A structural basis for metal ion mutagenicity and nucleotide selectivity in human DNA polymerase β. Biochemistry. 1996;35:12762–12777. doi: 10.1021/bi9529566. [DOI] [PubMed] [Google Scholar]
- 11.Kiefer JR, Mao C, Braman JC, Beese LS. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 1998;391:304– 307. doi: 10.1038/34693. [DOI] [PubMed] [Google Scholar]
- 12.Freudenthal BD, Beard WA, Shock DD, Wilson SH. Observing a DNA polymerase choose right from wrong. Cell. 2013;154:157–168. doi: 10.1016/j.cell.2013.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura T, Zhao Y, Yamagata Y, Hua Y-j, Yang W. Watching DNA polymerase η make a phosphodiester bond. Nature. 2012;487:196–201. doi: 10.1038/nature11181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freudenthal BD, Beard WA, Perera L, Shock DD, Kim T, Schlick T, Wilson SH. Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature. 2015;517:635–639. doi: 10.1038/nature13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beard WA, Wilson SH. Purification and domain-mapping of mammalian DNA polymerase ®. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- 16.Wu S, Beard WA, Pedersen LG, Wilson SH. Structural comparison of DNA polymerase architecture suggests a nucleotide gateway to the polymerase active site. Chem Rev. 2014;114:2759–2774. doi: 10.1021/cr3005179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ollis DL, Brick P, Hamlin R, Xuong NG, Steitz TA. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 18.Sawaya MR, Pelletier H, Kumar A, Wilson SH, Kraut J. Crystal structure of rat DNA polymerase β: Evidence for a common polymerase mechanism. Science. 1994;264:1930–1935. doi: 10.1126/science.7516581. [DOI] [PubMed] [Google Scholar]
- 19.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Freudenthal BD, Beard WA, Wilson SH, Schlick T. Optimal and variant metal-ion routes in DNA polymerase β conformational pathways. J Am Chem Soc. 2014;136:3630–3639. doi: 10.1021/ja412701f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structures of DNA polymerase β with active site mismatches suggest a transient abasic site intermediate during misincorporation. Mol Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beard WA, Shock DD, Batra VK, Pedersen LC, Wilson SH. DNA polymerase β substrate specificity: Side chain modulation of the “A-rule”. J Biol Chem. 2009;284:31680–31689. doi: 10.1074/jbc.M109.029843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beese LS, Steitz TA. Structural basis for the 3′–5′ exonuclease activity of Escherichia coli DNA polymerase I: A two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oertell K, Chamberlain BT, Wu Y, Ferri E, Kashemirov BA, Beard WA, Wilson SH, McKenna CE, Goodman MF. Transition state in DNA polymerase β catalysis: Rate-limiting chemistry altered by base-pair configuration. Biochemistry. 2014;53:1842–1848. doi: 10.1021/bi500101z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beard WA, Shock DD, Vande Berg BJ, Wilson SH. Efficiency of correct nucleotide insertion governs DNA polymerase fidelity. J Biol Chem. 2002;277:47393–47398. doi: 10.1074/jbc.M210036200. [DOI] [PubMed] [Google Scholar]
- 26.Lin P, Pedersen LC, Batra VK, Beard WA, Wilson SH, Pedersen LG. Energy analysis of chemistry for correct insertion by DNA polymerase β. Proc Natl Acad Sci USA. 2006;103:13294–13299. doi: 10.1073/pnas.0606006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Batra VK, Perera L, Lin P, Shock DD, Beard WA, Pedersen LC, Pedersen LG, Wilson SH. Amino acid substitution in the active site of DNA polymerase β explains the energy barrier of the nucleotidyl transfer reaction. J Am Chem Soc. 2013;135:8078–8088. doi: 10.1021/ja403842j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perera L, Beard WA, Pedersen LG, Wilson SH. Applications of Quantum Mechanical/Molecular Mechanical Methods to the Chemical Insertion Step of DNA and RNA Polymerization. In: Christo ZC, editor. Adv Protein Chem Struct Biol. Academic Press; 2014. pp. 83–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beard WA, Shock DD, Batra VK, Prasad R, Wilson SH. Substrate-induced DNA polymerase β activation. J Biol Chem. 2014;289:31411–31422. doi: 10.1074/jbc.M114.607432. [DOI] [PMC free article] [PubMed] [Google Scholar]