

Abstract
DNA repair normally protects the genome against mutations that threaten genome integrity and thus cell viability. However, growing evidence suggests that in the case of the Repeat Expansion Diseases, disorders that result from an increase in the size of a disease-specific microsatellite, the disease-causing mutation is actually the result of aberrant DNA repair. A variety of proteins from different DNA repair pathways have thus far been implicated in this process. This review will summarize recent findings from patients and from mouse models of these diseases that shed light on how these pathways may interact to cause repeat expansion.
Keywords: Repeat Expansion Diseases, Mismatch Repair, Base excision repair, transcription coupled repair, global genome repair, oxidative damage
1. Introduction
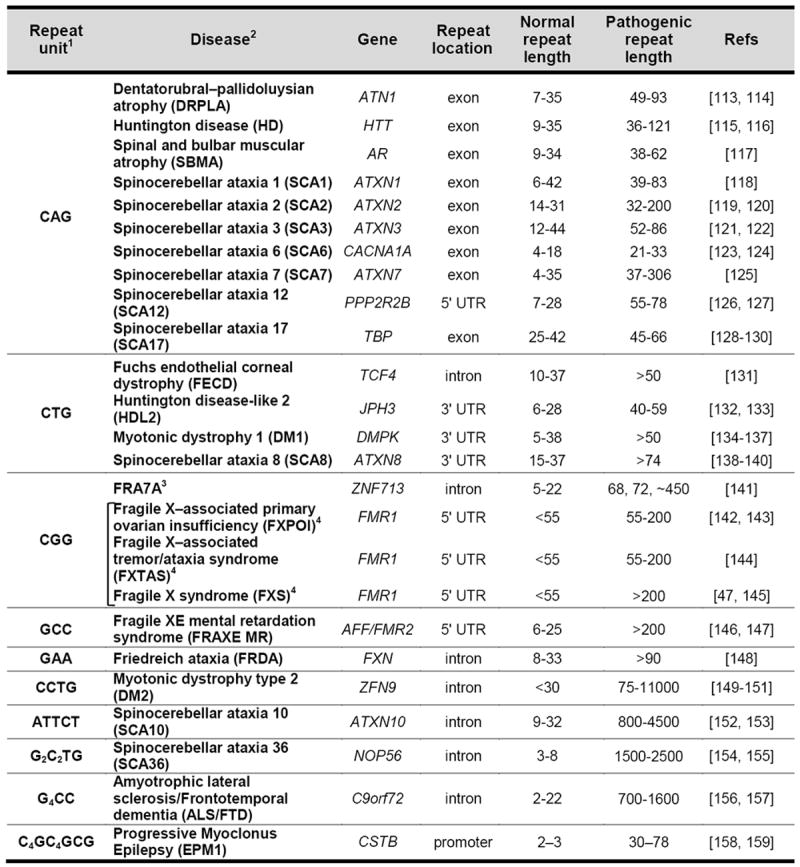
DNA damage repair is essential for human survival. However, it is becoming increasingly apparent that some repair processes act as double-edged swords, protecting the genome against some sorts of mutations whilst increasing the risk of others. The Repeat Expansion Diseases may represent one such example of a class of human genetic disorders that arise because of DNA repair gone wrong. These diseases comprise the 20+ human genetic conditions that arise from an increase (expansion) in the number of repeats in a particularly unstable tandem repeat array (Table 1). The disease-associated tandem repeats that have been identified thus far involve units of 3-12 bases. The consequences of the expansion depend on some combination of the location of the repeat within the affected gene, the size of the repeat unit, the number of repeats present in the allele and the sequence of the repeat (Discussed in [1] and chapters therein). The mechanism of expansion is unknown. However, as will be discussed below, growing evidence supports the idea that disease critical expansions result from the error-prone processing of these repeats by one or more DNA repair pathways that normally protect the genome against DNA damage.
Table1.
|
The disorders are organized based on the sequence of the repeat unit in the coding sequence of the affected gene and on its location within that gene. However, in some cases an antisense transcript is produced that may contribute to disease pathology. In addition, Repeat-associated Non-ATG (RAN) translation can occur in some of these genes. Thus while the repeat may nominally be a non-coding region of the affected gene, pathology may nevertheless arise from the production of a toxic polypeptide.
This table does not include a disorders like SCA31 that result from the “insertion” of a complex microsatellite [160]and the 9+ disorders that resultfrom the presence of a frequently interrupted and often stable microsatellite that encodes a polyalanine tract [161].
A firm link has yet to be established between the repeat expansion mutation and symptoms of autism spectrum disorder reported in 2 families [141].
These three diseases are all members of the Fragile X-related or FMR1-related disorders. Alleles with 55-200 repeats are referred to as Premutation (PM) alleles while alleles with >200 repeats are referred to as Full mutation (FM) alleles.
The repeat instability in these disorders is likely to be very different from the generalized microsatellite instability (MSI) seen in many forms of cancer since MSI involves the gain or loss of a few repeat units (reviewed in [2]), while in the Repeat Expansion Diseases the repeat tract shows a strong expansion bias, in some cases adding hundreds, if not thousands of repeats in the space of a single generation. In addition, as will be discussed in more detail later, mutations that increase MSI actually decrease repeat expansion.
2. Disease associated repeats form unusual DNA structures
All of the disease-associated repeats form unusual secondary structures. Most repeats form intrastrand structures like hairpins/stem-loops, G-quadruplexes and i-motif tetraplexes, whilst others form intramolecular triplexesandor become unpaired under moderate superhelical stress (See [3-7] for recent reviews). Many of these structures contain single-stranded regions, mismatched bases or unusual hydrogen bonding interactions such as Hoogsteen and other non-Watson-Crick base pairs. In principle, these structures could form any time that the DNA is unpaired e.g., during DNA replication, repair or transcription and there is evidence that some of these structures do indeed form in vivo [8, 9]. Interruptions to the purity of the repeat are commonly seen in different diseases and these interruptions frequently reduce both the stability of these secondary structures (e.g., [10, 11]) and the risk of expansion (e.g., [12-15]). Current thinking is that these structures are somehow responsible for the tendency of the repeats to expand.
Many of the repeats also form persistent co-transcriptional RNA:DNA hybrids (or R-loops) [8, 16-19]. Bidirectional transcription through these repeats can result in the generation of double R-loops [20]. The single-stranded regions of the R-loops may themselves be prone to damage that could trigger expansions. They are also likely to increase the chances that secondary structures are formed by the unpaired DNA strand.
Work in vitro, in E. coli, yeast, flies and various tissue culture model systems has provided important clues as to the many ways that expansions can be generated in different contexts and there are a number of comprehensive reviews that summarize this work in some detail [3-7]. Our current thinking about how these repeats expand in the Repeat Expansion Diseases is informed by this work. However, because of space limitations this review will focus on what we have learnt from the natural history of the expansion process in affected humans and from different mouse and human disease-derived models. Furthermore, while expansions and contractions may represent different outcomes of the same underlying process, evidence suggests that genetic and environmental factors that affect expansions don’t necessarily affect contractions the same way (e.g., [21-23]). Thus, it may be that expansions and contractions involve different mechanisms. Given the limited space available, this review will focus on data that specifically addresses the expansion process.
3. Expansion in mammalian cells does not require chromosomal replication
In affected humans expansions are often seen in tissues with a low proliferative capacity including brain, liver and muscle (e.g., [24-27]). Expansions have been observed in non-dividing cells in affected humans including neurons of patients with Huntington disease (HD) [28, 29], Dentatorubral–pallidoluysian atrophy (DRPLA) [30] and Friedreich ataxia (FRDA) [31]. Expansions are also seen in the oocytes of women with myotonic dystrophy type 1 (DM1) [32, 33] and a maternal age effect is seen on the transmission of expanded alleles in DM1, Fragile X syndrome (FXS) and FRDA [13, 34, 35]. Since neurons are post-mitotic and oocytes are suspended in dictyate arrest from late in fetal life, this would support the idea that expansion is independent of chromosomal replication. Furthermore, all mouse models for the Repeat Expansion Diseases studied to date also show expansions in tissues with a low proliferative capacity (e.g., [22, 36-38]). Expansion is seen in terminally differentiated neurons in different mouse models of HD [29, 39] and paternally transmitted expansions have been shown to occur in post-meiotic haploid cells in a mouse model of HD [40]. In embryonic fibroblasts from a DM1 mouse model, chemical or genetic cell arrest of the cell cycle at a variety of stages did not block expansions and in some cases actually increased the expansion frequency [41].
Thus, while some expansions may arise from problems associated with chromosomal replication, data from affected humans and of mouse models of the Repeat Expansion Diseases demonstrate that expansions in disease-relevant cells like neurons and gametes involves a process that is independent of genome duplication. Since expansion mechanisms involving chromosomal replication have been thoroughly covered elsewhere [3-6], this review will focus on mechanisms that can occur in non-dividing cells.
4. Expansion is facilitated by transcription or by transcriptionally competent chromatin
While depletion of proteins that cause transcriptional silencing or treatment with small molecule inhibitors of such proteins do affect repeat expansion in different ways [42-45], the reported effect is not thought to be mediated by a change in the chromatin or transcriptional activity of the repeat-containing sequence [42, 45]. Nonetheless, there is evidence from a number of different systems suggesting that transcription through the repeat is important for expansion. For example, a clear requirement for transcription or at least transcriptionally competent chromatin can be seen in FXS one of the Fragile X-related disorders (FXDs). In this disorder the repeat is stabilized when the gene in which the repeat is located undergoes repeat-mediated gene silencing [46-49]. Furthermore, in a mouse model of the FXDs where the repeat falls below the threshold for repeat-mediated silencing, expansions in females only occur when the repeat is on the active X chromosome [50]. A re-examination of data from women who carry similar sized alleles [51] demonstrates that the same is true in humans.
However, there is not a simple relationship between the amount of transcription and the extent of expansion in either mice or humans (e.g., [36, 37, 52]). It could be that expansion requires an open chromatin configuration rather than transcription per se or that transcription is not rate limiting for expansion. It has also been suggested that the expansion frequency is related to the rate of transcription elongation rather than to the absolute levels of transcription [53]. This idea is based on a comparison of the repeat instability in the striatum and cerebellum of HD mouse models. While these two tissues show similar steady state levels of transcription, transcription elongation rates are higher in the striatum, which also shows higher levels of expansion [53]. However, the difference in the expansion rates of these tissues has also been correlated with differences in the levels of expression of some of the proteins involved in the expansion process [54, 55]. It remains to be seen whether either of these correlations hold up when additional tissues are analyzed.
5. A diverse collection of proteins involved in DNA repair are important for expansion
A number of different proteins have been implicated in repeat expansion (Table 2). These proteins include general DNA processing enzymes that are involved in a wide variety of different biological processesas well as proteins central to specific DNA repair and recombination pathways.
Table 2.
DNA repair proteins directly implicated in repeat expansion
| Protein | Model system | Cells affected | References |
|---|---|---|---|
| General factors | |||
| LIG1 | HD mouse | maternal germ line | [56] |
| MMR proteins | |||
| MSH2 | DM1, FXD and HD mice; tissue culture model of CTG/CAG and GAA/TTC-repeats | germ line and somatic cells | [22, 40, 45, 60, 66, 71, 162] |
| MSH3 | DM1and HD mice, tissue culture model of CTG/CAG and GAA/TTC-repeats | germ line and somatic cells | [45, 65, 67, 68, 71, 163] |
| MSH6 | FRDA mouse; FRDA iPSCs | somatic cells, iPSCs | [69, 70] |
| MLH1 | HD and FRDA mice | germ line* and somatic cells** | [73, 74] |
| PMS2 | DM1 mouse | somatic cells** | [72] |
| MLH3 | HD mouse | somatic cells** | [73] |
| BER proteins | |||
| OGG1 | HD mouse | somatic cells | [164] |
| NEIL1 | HD mouse | somatic cells | [84] |
| NER proteins | |||
| CSB | FXD mouse | maternal germ line, some somatic cells | [90] |
| XPA | SCA1 mouse | neural cells | [88] |
| Recombination proteins | |||
| RAD52 | DM1 mouse | germ line | [60] |
in the FRDA mouse model loss of one Mlh1 allele led to significant decrease in germ line expansions
homozygous null mice are sterile so the effect of this mutation on germ line expansion was not examined in either the HD or the DM1 mouse models.
5.1. General DNA processing proteins
DNA ligase 1 (LIG1) is involved in sealing nicks generated during lagging strand DNA synthesis, as well as during DNA repair and recombination. A Lig1 hypomorphic mutation had no effect on repeat expansion in the FXD mouse [21]. However, the same mutation reduced maternally transmitted expansions in an HD mouse [56]. This may reflect a maternal-specific expansion process. However, since heterozygosity for this allele had the effect as homozygosity for this allele, this result is difficult to interpret. It has been suggested to reflect a potential dominant negative effect of the hypomorphic allele [56]. However, given that a Lig1 null mutation is embryonic lethal, it might be premature to exclude a more general role for this protein in repeat expansion.
There is also currently no genetic evidence for a role for Flap endonuclease 1 (FEN1) in repeat expansion in mammals. FEN1 often works upstream of LIG1 to generate the correct substrates for ligation in a variety of different DNA processing pathways. Despite the fact that work in vitro has shown that hairpins formed by some of the repeats block FEN1 processing [57], heterozygosity for a Fen1 null mutation did not reduce expansions in DM1, HD or FXD mouse models [21, 58, 59]. However, absence of FEN1 is also embryonic lethal and since it is possible that Fen1 heterozygous mice are not haplo insufficient, it is probably also too early to exclude a role for FEN1 in the expansion process.
5.2. Double-strand break repair (DSBR) proteins
Loss of RAD52, a protein involved in homologous recombination (HR), reduced the size of germ line but not somatic expansions in a mouse model of DM1 [60]. However, neither the loss of this protein nor the loss of RAD54, another HR protein, affected the expansion frequency [60]. Furthermore, expansions are seen in haploid sperm of HD mice in which there is no sister chromosome or sister chromatid with which to recombine [40]. Thus an HR-based mechanism for expansion is unlikely. However, since RAD52 also cooperates with 8-oxoguanine DNA glycosylase (OGG1) in the repair of oxidative lesions via base excision repair (BER) [61], this protein may affect the expansion size independent of its role in DSBR. The absence of DNA-dependent protein kinase (DNA-PK), a protein involved in non-homologous end joining (NHEJ), had no effect on the expansion frequency in this model [60].
5.3. Mismatch Repair (MMR) proteins
While the loss of MMR proteins increases MSI, the opposite is true in mouse and human cell models of repeat expansion where some of these proteins are actually required for expansion to occur. MutSα, a heterodimer of the MutS homolog 2 (MSH2) and the MutS homolog 6 (MSH6), and MutSβ, a heterodimer of MSH2 and the MutS homolog 3 (MSH3), are the complexes responsible for lesion recognition in the MMR pathway [62]. The substrates with which they interact are partially overlapping, with MutSα recognizing single base mismatches and small insertions and deletions (INDELs), and MutSβ recognizing larger INDELs. While there is some variability between different mouse models (e.g., [63, 64]), a case can be made for MutSβ being required for expansions in most mouse models [22, 45, 65-68]. However, MSH6, and thus MutSα has been suggested to promote somatic expansions in an FRDA mouse model [69]. The effect of knockdown of MSH6 in induced pluripotent stem cells derived from FRDA patients [70] and overexpression of MutSβ in a human tissue culture model [71] also supports a role for these complexes in generating expansions in humans.
The requirement for MMR proteins extends to complexes that act downstream of MutS in the MMR pathway such as MutLα, a heterodimer of MutL homolog 1 (MLH1) and postmeiotic segregation increased 2 (PMS2), and MutLγ, a heterodimer of MLH1 and the MutL homolog 3 (MLH3) protein. MutLα has been implicated in at least 50% of somatic expansions in a DM1 mouse model [72] but seems to be excluded from a role in expansion in the FRDA mouse [69]. MutLγ is required for all somatic expansions in an HD mouse model [73] and perhaps, by inference, in a FRDA mouse model as well [69, 74]. The importance of MutLγ is consistent with the observation that MutSβ is required for expansion in most mouse models and the fact that MutLγ is thought to interact with MutSβ but not MutSα [75].
While it was once thought that MMR was confined to S phase, recent work in vitro has shown that extrahelical CAG-repeats can activate the latent endonuclease activity of MutLα. This activation occurs in the absence of the strand discontinuities that arise during genomic replication that normally serve this purpose [76]. This activation allows loading of proliferating cell nuclear antigen (PCNA) thus enabling successful MMR to occur outside of S phase. However, whether other repeats are able to activate MutLα or whether the repeats are able to activate MutLγ is unknown.
While the requirement for MutS and MutL proteins makes a strong case for an MMR-based mechanism, work in Cynthia McMurray’s laboratory has shown that binding of MutSβ to the CAG-hairpin changes the properties of mismatch recognition [77]. While this result has been challenged [78], the McMurray group have gone on to show using single-molecule fluorescence resonance energy transfer (smFRET) that a subset of the hairpins form a unique DNA junction that traps MutSβ on the template [79]. It has been suggested that this prevents classical MMR and may divert the hairpin substrate into another DNA repair pathway that ultimately gives rise to expansions. Expansion in an in vitro model system can be seen even in the absence of MMR proteins [80, 81]. Furthermore, in a HeLa nuclear extract, excess MutSβ does not inhibit or promote CTG or CAG repair of preformed hairpin substrates [78, 82]. This would be consistent with the idea that MutSβ promotes a process such as the formation of secondary structures rather than determining repair outcome.
5.4. Base Excision Repair (BER) proteins
Loss of the DNA glycosylases OGG1 and nei endonuclease VIII-like 1 (NEIL1) reduces somatic expansions in a mouse model of HD [83, 84]. OGG1 and NEIL1 are involved in BER, the major pathway by which oxidative damage is repaired in mammals. Both enzymes are involved in the removal of oxidized bases from DNA, one of the first steps in the BER pathway. A more general role of oxidative damage repair in generating expansions is suggested by the observation that potassium bromate (KBrO3), a potent DNA oxidizing agent, increases germ line expansions in a mouse model of the FXDs [21]. The loss of NEIL1 in the HD mouse model also reduces the average size of expansions seen on intergenerational transmission although it does not affect the absolute germline expansion frequency [84]. The fact that neither OGG1 nor NEIL1 reduce germ line expansion frequencies may reflect the contribution of other DNA glycosylases to the expansion process. However, mutations in alkyladenine glycosylase (AAG), a DNA glycosylase that excises a variety of alkylated bases, or homologue of Escherichia coli endonuclease III (NTH1), which prefers to excise thymine glycol, did not reduce expansions in the HD mouse model [83]. The action of DNA glycosylases results in the generation of an a basic site that is the substrate for the apurinic/apyrimidinic endonuclease 1 (APE1). The nicks generated by APE1 are then channeled into either the single nucleotide (SN) or short patch (SP) BER pathway or the long patch (LP) BER pathway. It is possible that BER-mediated expansion could be initiated in response to other sources of a basic sites or nicks. One potential source of such a basic sites is depurination which is thought to be very common in GC-rich regions [85]. Such sites can be processed by APE1 to generate a nick upon which the BER process can act. However, at this time there is no data implicating proteins that act downstream of the generation of a basic sites in the repeat expansion process in any model or tissue culture model.
5.5. Nucleotide Excision Repair (NER) proteins
Proteins involved in NER also affect repeat expansion. Two overlapping NER pathways operate in mammalian cells, Global Genome Repair (GGR) and Transcription Coupled Repair (TCR) (reviewed in [86]). The TCR pathway is confined to the repair of transcription blocking lesions on the transcribed strand of active genes, while GGR occurs genome wide. The GGR and TCR pathways converge downstream of the DNA damage recognition step.
The only NER protein specific for GGR that has been examined in mice is xeroderma pigmentosum, complementation group C (XPC), the earliest DNA damage detector in the initiation of GGR. Loss of XPC did not affect striatal expansions or germ line expansions in a mouse model of HD [64]. However, loss of the xeroderma pigmentosum, complementation group A (XPA) protein that acts in both GGR and TCR does affect repeat expansion in a mouse model of SCA1 [88]. Loss of XPA did not affect the intergenerational expansion frequency or the extent of somatic expansion in liver and kidney, but it did dramatically reduce the expansions seen in striatum, hippocampus and cerebral cortex [88]. Although expansions were dramatically reduced, they were not eliminated completely even in neural tissue suggesting that XPA may be playing an auxiliary role in expansion outside of NER. For example, XPA is known to bind with higher affinity to DNA junctions than to DNA damage [89]. XPA may thus help stabilize the secondary structures thought to be the substrates for expansion. While the different effects of the loss of XPA in different tissues could suggest the existence of tissue-specific expansion mechanisms, it is also possible that in some tissues the core factors necessary for expansions are present in sufficient quantities such that auxiliary proteins are not needed.
Cockayne Syndrome B (CSB), a transcription elongation factor that is essential for early steps in TCR, promotes both germ line and somatic expansions in the FXD mouse [90]. However, CSB is not essential for expansions either since its loss does not eliminate them all. Thus CSB is also likely acting as an accessory factor to promote expansions in a pathway other than TCR. It might facilitate expansion via a BER-based mechanism since CSB is known to up-regulate OGG1 expression [91] and to promote the incision activities of OGG1, NEIL1 [92, 93] and APE1 [94]. It could also act via its ability to modify chromatin and/or increase transcription elongation [95, 96]. While very few genetic modifiers of expansion risk in humans have thus far been identified, it is interesting to note that single nucleotide polymorphisms (SNPs) in three TCR-related genes are associated with an increased expansion risk in Machado-Joseph Disease (MJD/SCA3) alleles, including one in the ERCC6/CSB gene [97].
6. An integrated model for repeat expansion
Thus, while DSBR does not seem to be involved in repeat expansion in mammalian systems, proteins from a variety of other DNA repair pathways have been shown to contribute significantly to expansion. Given the different contributions of some of these proteins in different disease models, in males and females and in different cell types, it is possible that more than one expansion mechanism is responsible. However, since expansions have a number of unique features and share a common dependence on MMR factors, it could be argued that a single mechanism is at work. Most of the reported differences between disease models, tissue and genders could be related to the effects of different genetic backgrounds, the differential expression of the proteins involved and/or the variable contribution of proteins that promote but are not essential for expansion. Furthermore, although the proteins implicated in repeat expansion act in a wide variety of different DNA repair pathways, it is becoming increasingly apparent that there is a lot of cross talk between these pathways (e.g., [98, 99]).
Thus, it may be possible to reconcile all of the data described above into a single model. Such a model would have to accommodate the fact that expansions can occur independent of genomic replication via a process in which transcription or an open chromatin configuration is important. It should also account for the strong expansion bias that is seen in the Repeat Expansion Diseases. It would also have to accommodate the contribution of MMR proteins and the involvement of proteins more typically associated with BER and TCR. The requirement for transcriptionally competent chromatin may simply reflect the fact that transcription through the repeat creates the opportunity for the secondary structures that are thought to be the expansion substrates to form. However, it is also possible that it reflects a DNA repair process other than TCR that is also confined to actively transcribed regions of the genome. Models for repeat expansion that involve LP BER are thus appealing since recent work suggests this process occurs preferentially in actively transcribed regions of the genome [100, 101].
The role of DNA glycosylases normally involved in the initiation of BER in response to oxidative stress suggests that an early step in the expansion process may be the recognition and removal of damaged bases in DNA and the generation of nicks as illustrated in Fig. 1. It has been suggested that LP BER is the major BER pathway for the repair of 8-oxoG lesions and AP sites [102]. LP BER occurs via one of two sub-pathways. Both sub-pathways involve Polβ, the polymerase responsible for SP BER. One sub-pathway also involves Polδ/Polε, two processive polymerases with stronger strand displacement activities than Polβ. This generally results in the synthesis of 2-13 nucleotides [103]. The second sub-pathway involves Polβ acting without Polδ or Polε to carry out a more limited gap-filling reaction that involves the synthesis of fewer nucleotides [104]. There is in vitro data to support the idea that expansions could arise in the Polβ/Polδ/Polε-dependent pathway if strand-slippage occurred on the nascent strand, an event that would be facilitated by secondary structure formation, and if priming by Polβ then occurred from the slipped position [105]. Expansions may also arise by structure formation on the displaced strand that prevents proper flap processing. Expansions could arise even via the Polδ/Polε-independent sub-pathway if some Polβ-mediated strand displacement occurs that results in the formation of a secondary structure that is not properly processed by FEN1 as illustrated in the bottom right hand side of Fig. 1. A form of alternate FEN1 cleavage has been suggested to facilitate this process [106]. A role for both sub-pathways may help explain the different “jump sizes” are seen in different organs of mouse models (e.g., [36, 83, 107]). For example, expansion in organs showing large “jumps” could occur via the use of the first pathway while small “jumps” occur via the use of the second. The choice of which pathway is used may depend on the relative levels of Polβ, Polδ and/or Polε. Models in which expansions result from the failure to properly process 5’ flaps generated by strand-displacement synthesis are appealing since they could account for the strong expansion bias observed in many Repeat Expansion Diseases.
Fig. 1. An integrated model for the repeat expansion mechanism in mammals.
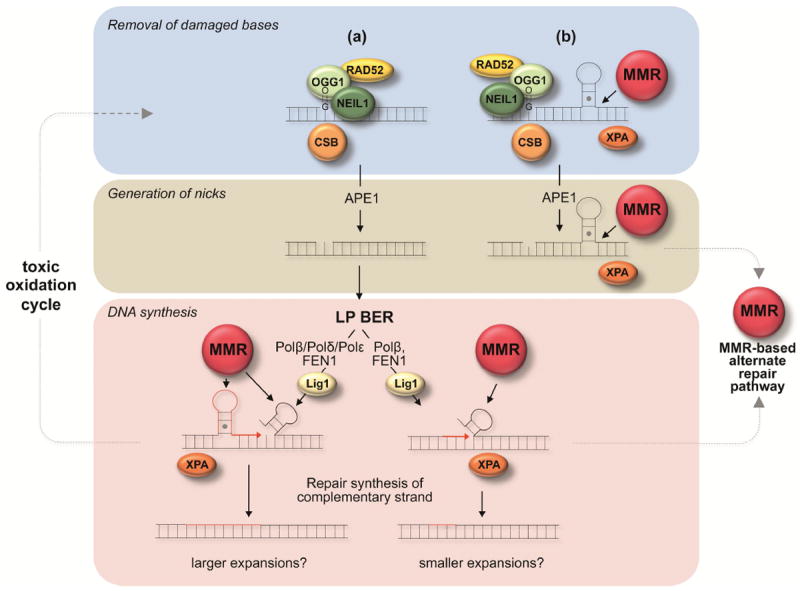
Proteins directly implicated in generating expansions in mouse models or human cells are shown as colored spheres. MMR refers to a complex consisting of the MMR proteins MutSα or MutSβ together with either MutLα or MutLγ. Red strands represent newly synthesized DNA. The repair of DNA damage within the repeat is initiated by DNA glycosylases in response to oxidized bases. This is followed by the removal of the abasic site by APE1 to generate a nick. (a) Repair of the nick may proceed via an LP BER pathway that involves Polβ, Polδ and perhaps Polε [111]. Strand-slippage/hairpin formation at the 3’ terminus of the nascent strand arising during strand displacement synthesis by Polδ/Polε could result in expansion if the hairpin is not removed because Polβ synthesis prevents proof-reading by Polδ/Polε [105]. Formation of a secondary structure on a displaced flap could also result in expansion if proper processing were blocked. A second LP BER pathway that involves Polβ but not Polδ/Polε may generate small expansions by stepwise and distributive gap-filling by Polβ and single-nucleotide gap formation by FEN1 [106]. Improper coordination between Polβ and FEN1 could lead to a small amount of strand displacement with the formation of a small hairpin in the displaced flap. FEN1 “alternate cleavage” of a short 5’ flap at the base of the hairpin could produce a ligatable nick that after ligation results in incorporation of hairpin bases into the “repaired” strand.(b) Alternatively, a nick close to a repeat loop-out formed during transcription or replication may allow loading of MutSβ/MutSα complexes and the diversion of the normal BER process to produce an MMR-dependent expansion. MMR proteins may also act in the LP BER-based expansion processes shown in (a) to stabilize secondary structures formed by the repeats and perhaps to prevent their removal by enzymes like FEN1. XPA, CSB and RAD52 may act in an auxiliary capacity in either of these pathways via the ability to stabilize secondary structures in the case of XPA, to facilitate incision by OGG1 in the case of RAD52 [61] and to increase incision or to facilitate the formation of an optimal chromatin or transcriptional state in the case of CSB (reviewed in [112]). Loops generated at any stage of the expansion process are susceptible to oxidative damage that could produce result in repeated “toxic oxidation cycles” that could result in multiple rounds of BER-mediated expansions [108].
CSB may contribute to the generation of nicks via its ability to enhance the activity of DNA glycosylases and APE1, while RAD52 may also act at this step to enhance OGG1 incision. The effect of CSB and RAD52 may only be apparent in cells in which the incision process is somehow limiting. MMR proteins may increase the likelihood that oxidative damage will occur by stabilizing secondary structures that are sensitive to such damage [108]. XPA may act in an auxiliary capacity to facilitate expansion by contributing to this stabilization since MutSβ generates a strong bend when it binds to an INDEL [109] and XPA binds preferentially to bent DNA [87]. The effect of XPA may only be apparent in neural cells if MutSβ is limiting there but not in other cells. Alternatively another protein may substitute for XPA outside the central nervous system (CNS). MMR proteins may also act later in the LP BER pathway by stabilizing the hairpins generated by strand-slippage thus increasing the likelihood that priming will occur from the slipped position during repair synthesis. They may also reduce the likelihood that FEN1 would be able to properly process any flaps generated by strand-displacement during LP BER.
However, the fact that MutL complexes that act downstream of MutSβ/MutSα in the MMR pathway are important for expansions suggests that the role of MMR proteins may extend beyond simply stabilizing the substrates for expansion. Thus it is possible that MMR can use the nicks generated during early steps of BER to load EXO1 and other proteins required for MMR as illustrated in Fig. 1(b).
Perhaps the most intriguing finding in the field in recent months is that there is a specific requirement for MutLγ rather than MutLα for expansion in some mouse models. This is of interest since MutLγ is present only in very low levels in mammalian cells and while it colocalizes with sites of DNA damage, it has been suggested that it does not contribute significantly to normal MMR in mammals [110]. The role of MutLγ is thus enigmatic and a better understanding of the pathways in which it acts is essential to our understanding of the expansion mechanism.
Note added in Proof
We have recently demonstrated that heterozygosity for a hypomorphic PolB mutation reduces the expansion frequency in a FXD mouse model (Lokanga, Senejani, Sweasy and Usdin. Heterozygosity for a hypomorphic PolB mutation reduces the expansion frequency in a mouse model of the Fragile X-related disorders. PLoS Genetics. 11, 2015, e1005181). It also results in a preferential loss of smaller expansions. These data lend support to the model shown in Fig. 1.
Acknowledgments
In a review of a subject as broad as this one, it is difficult to cite all of the relevant literature. We have thus tried to provide illustrative examples rather than generating an exhaustive citation list. We apologize to those whose contributions to the field, for reasons of space, were not acknowledged specifically.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fry M, Usdin K. Human Nucleotide Expansion Disorders. Springer; Heidelberg: 2006. [Google Scholar]
- 2.Plotz G, Zeuzem S, Raedle J. DNA mismatch repair and Lynch syndrome. J Mol Histol. 2006;37:271–283. doi: 10.1007/s10735-006-9038-5. [DOI] [PubMed] [Google Scholar]
- 3.McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–799. doi: 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mirkin SM. DNA structures, repeat expansions and human hereditary disorders. Curr Opin Struct Biol. 2006;16:351–358. doi: 10.1016/j.sbi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 5.LópezCastel A, Clear JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. 2010;11:165–170. doi: 10.1038/nrm2854. [DOI] [PubMed] [Google Scholar]
- 6.Kim JC, Mirkin SM. The balancing act of DNA repeat expansions. Curr Opin Genet Dev. 2013;23:280–288. doi: 10.1016/j.gde.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Usdin K, House NC, Freudenreich CH. Repeat instability during DNA repair: Insights from model systems. Crit Rev Biochem Mol Biol. 2015:1–26. doi: 10.3109/10409238.2014.999192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loomis EW, Sanz LA, Chedin F, Hagerman PJ. Transcription-Associated R-Loop Formation across the Human FMR1 CGG-Repeat Region. PLoS Genet. 2014;10:e1004294. doi: 10.1371/journal.pgen.1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu G, Chen X, Bissler JJ, Sinden RR, Leffak M. Replication-dependent instability at (CTG) × (CAG) repeat hairpins in human cells. Nat Chem Biol. 2010;6:652–659. doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pearson CE, Eichler EE, Lorenzetti D, Kramer SF, Zoghbi HY, Nelson DL, Sinden RR. Interruptions in the triplet repeats of SCA1 and FRAXA reduce the propensity and complexity of slipped strand DNA (S-DNA) formation. Biochem. 1998;37:2701–2708. doi: 10.1021/bi972546c. [DOI] [PubMed] [Google Scholar]
- 11.Jarem DA, Huckaby LV, Delaney S. AGG interruptions in (CGG)(n) DNA repeat tracts modulate the structure and thermodynamics of non-B conformations in vitro. Biochem Biophys Res Comm. 2010;49:6826–6837. doi: 10.1021/bi1007782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Latham GJ, Coppinger J, Hadd AG, Nolin SL. The role of AGG interruptions in fragile X repeat expansions: a twenty-year perspective. Front Genet. 2014;5:244. doi: 10.3389/fgene.2014.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yrigollen CM, Martorell L, Durbin-Johnson B, Naudo M, Genoves J, Murgia A, Polli R, Zhou L, Barbouth D, Rupchock A, Finucane B, Latham GJ, Hadd A, Berry-Kravis E, Tassone F. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J Neurodev Disord. 2014;6:24. doi: 10.1186/1866-1955-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choudhry S, Mukerji M, Srivastava AK, Jain S, Brahmachari SK. CAG repeat instability at SCA2 locus: anchoring CAA interruptions and linked single nucleotide polymorphisms. Hum Mol Genet. 2001;10:2437–2446. doi: 10.1093/hmg/10.21.2437. [DOI] [PubMed] [Google Scholar]
- 15.Chung MY, Ranum LP, Duvick LA, Servadio A, Zoghbi HY, Orr HT. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat Genet. 1993;5:254–258. doi: 10.1038/ng1193-254. [DOI] [PubMed] [Google Scholar]
- 16.Groh M, Lufino MM, Wade-Martins R, Gromak N. R-loops Associated with Triplet Repeat Expansions Promote Gene Silencing in Friedreich Ataxia and Fragile X Syndrome. PLoS Genet. 2014;10:e1004318. doi: 10.1371/journal.pgen.1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grabczy E, Mancus M, Sammarc MC. A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 2007;35:5351–5359. doi: 10.1093/nar/gkm589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grabczyk E, Usdin K. The GAA*TTC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000;28:2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy K, Schmidt MH, Geist JM, Thakkar NP, Panigrahi GB, Wang YH, Pearson CE. Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC). (GGCCCC) repeats causes instability. Nucleic Acids Res. 2014;42:10473–10487. doi: 10.1093/nar/gku658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Entezam A, Lokanga AR, Le W, Hoffman G, Usdin K. Potassium bromate, a potent DNA oxidizing agent, exacerbates germline repeat expansion in a fragile X premutation mouse model. Hum Mutat. 2010;31:611–616. doi: 10.1002/humu.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lokanga RA, Zhao X-N, Usdin K. The mismatch repair protein, MSH2, is rate-limiting for repeat expansion in a Fragile X premutation mouse model. Hum Mutat. 2014;35:129–136. doi: 10.1002/humu.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kovtun IV, Thornhill AR, McMurray CT. Somatic deletion events occur during early embryonic development and modify the extent of CAG expansion in subsequent generations. Hum Mol Genet. 2004;13:3057–3068. doi: 10.1093/hmg/ddh325. [DOI] [PubMed] [Google Scholar]
- 24.Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, Adam S, Greenberg C, Ives EJ, Clarke LA, et al. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. 1994;6:409–414. doi: 10.1038/ng0494-409. [DOI] [PubMed] [Google Scholar]
- 25.Thornton CA, Johnson K, Moxley RT., 3rd Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes. Ann Neurol. 1994;35:104–107. doi: 10.1002/ana.410350116. [DOI] [PubMed] [Google Scholar]
- 26.Trang H, Stanley SY, Thorner P, Faghfoury H, Schulze A, Hawkins C, Pearson CE, Yoon G. Massive CAG Repeat Expansion and Somatic Instability in Maternally Transmitted Infantile Spinocerebellar Ataxia Type 7. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.1902. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka F, Reeves MF, Ito Y, Matsumoto M, Li M, Miwa S, Inukai A, Yamamoto M, Doyu M, Yoshida M, Hashizume Y, Terao S, Mitsuma T, Sobue G. Tissue-specific somatic mosaicism in spinal and bulbar muscular atrophy is dependent on CAG-repeat length and androgen receptor--gene expression level. Am J Hum Genet. 1999;65:966–973. doi: 10.1086/302578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shelbourne PF, Keller-McGandy C, Bi WL, Yoon SR, Dubeau L, Veitch NJ, Vonsattel JP, Wexler NS, Arnheim N, Augood SJ. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. 2007;16:1133–1142. doi: 10.1093/hmg/ddm054. [DOI] [PubMed] [Google Scholar]
- 29.Gonitel R, Moffitt H, Sathasivam K, Woodman B, Detloff PJ, Faull RL, Bates GP. DNA instability in postmitotic neurons. Proc Natl Acad Sci U S A. 2008;105:3467–3472. doi: 10.1073/pnas.0800048105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hashida H, Goto J, Suzuki T, Jeong S, Masuda N, Ooie T, Tachiiri Y, Tsuchiya H, Kanazawa I. Single cell analysis of CAG repeat in brains of dentatorubral-pallidoluysian atrophy (DRPLA) J Neurol Sci. 2001;190:87–93. doi: 10.1016/s0022-510x(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 31.De Biase I, Rasmussen A, Endres D, Al-Mahdawi S, Monticelli A, Cocozza S, Pook M, Bidichandani SI. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. 2007;61:55–60. doi: 10.1002/ana.21052. [DOI] [PubMed] [Google Scholar]
- 32.De Temmerman N, Sermon K, Seneca S, De Rycke M, Hilven P, Lissens W, Van Steirteghem A, Liebaers I. Intergenerational instability of the expanded CTG repeat in the DMPK gene: studies in human gametes and preimplantation embryos. Am J Hum Genet. 2004;75:325–329. doi: 10.1086/422762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dean NL, Tan SL, Ao A. Instability in the transmission of the myotonic dystrophy CTG repeat in human oocytes and preimplantation embryos. Fertil Steril. 2006;86:98–105. doi: 10.1016/j.fertnstert.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 34.Morales F, Vasquez M, Cuenca P, Campos D, Santamaria C, Del Valle G, Brian R, Sittenfeld M, Monckton DG. Parental age effects, but no evidence for an intrauterine effect in the transmission of myotonic dystrophy type 1. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Michele G, Cavalcanti F, Criscuolo C, Pianese L, Monticelli A, Filla A, Cocozza S. Parental gender, age at birth and expansion length influence GAA repeat intergenerational instability in the X25 gene: pedigree studies and analysis of sperm from patients with Friedreich’s ataxia. Hum Mol Genet. 1998;7:1901–1906. doi: 10.1093/hmg/7.12.1901. [DOI] [PubMed] [Google Scholar]
- 36.Lokanga RA, Entezam A, Kumari D, Yudkin D, Qin M, Smith CB, Usdin K. Somatic expansion in mouse and human carriers of Fragile X premutation alleles. Hum Mutat. 2013;34:157–166. doi: 10.1002/humu.22177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lia AS, Seznec H, Hofmann-Radvanyi H, Radvanyi F, Duros C, Saquet C, Blanche M, Junien C, Gourdon G. Somatic instability of the CTG repeat in mice transgenic for the myotonic dystrophy region is age dependent but not correlated to the relative intertissue transcription levels and proliferative capacities. Hum Mol Genet. 1998;7:1285–1291. doi: 10.1093/hmg/7.8.1285. [DOI] [PubMed] [Google Scholar]
- 38.van den Broek WJ, Wansink DG, Wieringa B. Somatic CTG*CAG repeat instability in a mouse model for myotonic dystrophy type 1 is associated with changes in cell nuclearity and DNA ploidy. BMC Mol Biol. 2007;8:61. doi: 10.1186/1471-2199-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovalenko M, Dragileva E, St Claire J, Gillis T, Guide JR, New J, Dong H, Kucherlapati R, Kucherlapati MH, Ehrlich ME, Lee JM, Wheeler VC. Msh2 acts in medium-spiny striatal neurons as an enhancer of CAG instability and mutant huntingtin phenotypes in Huntington’s disease knock-in mice. PLoS One. 2012;7:e44273. doi: 10.1371/journal.pone.0044273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kovtun IV, McMurray CT. Trinucleotide expansion in haploid germ cells by gap repair. Nat Genet. 2001;27:407–411. doi: 10.1038/86906. [DOI] [PubMed] [Google Scholar]
- 41.Gomes-Pereira M, Hilley JD, Morales F, Adam B, James HE, Monckton DG. Disease-associated CAG.CTG triplet repeats expand rapidly in non-dividing mouse cells, but cell cycle arrest is insufficient to drive expansion. Nucleic Acids Res. 2014;42:7047–7056. doi: 10.1093/nar/gku285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dion V, Lin Y, Hubert L, Jr, Waterland RA, Wilson JH. Dnmt1 deficiency promotes CAG repeat expansion in the mouse germline. Hum Mol Genet. 2008;17:1306–1317. doi: 10.1093/hmg/ddn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorbunova V, Seluanov A, Mittelman D, Wilson JH. Genome-wide demethylation destabilizes CTG.CAG trinucleotide repeats in mammalian cells. Hum Mol Genet. 2004;13:2979–2989. doi: 10.1093/hmg/ddh317. [DOI] [PubMed] [Google Scholar]
- 44.Gomes-Pereira M, Monckton DG. Chemically induced increases and decreases in the rate of expansion of a CAG*CTG triplet repeat. Nucleic Acids Res. 2004;32:2865–2872. doi: 10.1093/nar/gkh612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gannon AM, Frizzell A, Healy E, Lahue RS. MutSbeta and histone deacetylase complexes promote expansions of trinucleotide repeats in human cells. Nucleic Acids Res. 2012;40:10324–10333. doi: 10.1093/nar/gks810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas M, Mandel J. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 47.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 48.Glaser D, Wohrle D, Salat U, Vogel W, Steinbach P. Mitotic behavior of expanded CGG repeats studied on cultured cells: further evidence for methylation-mediated triplet repeat stability in fragile X syndrome. Am J Med Genet. 1999;84:226–228. [PubMed] [Google Scholar]
- 49.Wohrle D, Salat U, Hameister H, Vogel W, Steinbach P. Demethylation, reactivation, and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am J Hum Genet. 2001;69:504–515. doi: 10.1086/322739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lokanga AR, Zhao X-N, Entezam A, Usdin K. X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the Fragile X-related Disorders: implications for the mechanism of repeat expansion. Hum Mol Genet. 2014;23:4985–4994. doi: 10.1093/hmg/ddu213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grasso M, Boon EM, Filipovic-Sadic S, van Bunderen PA, Gennaro E, Cao R, Latham GJ, Hadd AG, Coviello DA. A novel methylation PCR that offers standardized determination of FMR1 methylation and CGG repeat length without southern blot analysis. J Mol Diagn. 2014;16:23–31. doi: 10.1016/j.jmoldx.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dure LSt, Landwehrmeyer GB, Golden J, McNeil SM, Ge P, Aizawa H, Huang Q, Ambrose CM, Duyao MP, Bird ED, et al. IT15 gene expression in fetal human brain. Brain Res. 1994;659:33–41. doi: 10.1016/0006-8993(94)90860-5. [DOI] [PubMed] [Google Scholar]
- 53.Goula AV, Festenstein R, Merienne K. Tissue-dependent regulation of RNAP II dynamics: the missing link between transcription and trinucleotide repeat instability in diseases? Transcription. 2013;4:172–176. doi: 10.4161/trns.25971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mason AG, Tomé S, Simard JP, Libby RT, Bammler TK, Beyer RP, Morton AJ, Pearson CE, La Spada AR. Expression levels of DNA replication and repair genes predict regional somatic repeat instability in the brain but are not altered by polyglutamine disease protein expression or age. Hum Mol Genet. 2014;23:1606–1618. doi: 10.1093/hmg/ddt551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goula AV, Berquist BR, Wilson DM, 3rd, Wheeler VC, Trottier Y, Merienne K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. 2009;5:e1000749. doi: 10.1371/journal.pgen.1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomé S, Panigrahi GB, Castel AL, Foiry L, Melton DW, Gourdon G, Pearson CE. Maternal germline-specific effect of DNA ligase I on CTG/CAG instability. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr099. [DOI] [PubMed] [Google Scholar]
- 57.Spiro C, Pelletier R, Rolfsmeier ML, Dixon MJ, Lahue RS, Gupta G, Park MS, Chen X, Mariappan SV, McMurray CT. Inhibition of FEN-1 processing by DNA secondary structure at trinucleotide repeats. Mol Cell. 1999;4:1079–1085. doi: 10.1016/s1097-2765(00)80236-1. [DOI] [PubMed] [Google Scholar]
- 58.van den Broek WJ, Nelen MR, van der Heijden GW, Wansink DG, Wieringa B. Fen1 does not control somatic hypermutability of the (CTG)(n)*(CAG)(n) repeat in a knock-in mouse model for DM1. FEBS Lett. 2006;580:5208–5214. doi: 10.1016/j.febslet.2006.08.059. [DOI] [PubMed] [Google Scholar]
- 59.Spiro C, McMurray CT. Nuclease-deficient FEN-1 blocks Rad51/BRCA1-mediated repair and causes trinucleotide repeat instability. Mol Cell Biol. 2003;23:6063–6074. doi: 10.1128/MCB.23.17.6063-6074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Savouret C, Brisson E, Essers J, Kanaar R, Pastink A, te Riele H, Junien C, Gourdon G. CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. 2003;22:2264–2273. doi: 10.1093/emboj/cdg202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Souza-Pinto NC, Maynard S, Hashiguchi K, Hu J, Muftuoglu M, Bohr VA. The recombination protein RAD52 cooperates with the excision repair protein OGG1 for the repair of oxidative lesions in mammalian cells. Mol Cell Biol. 2009;29:4441–4454. doi: 10.1128/MCB.00265-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fishel R, Wilson T. MutS homologs in mammalian cells. Curr Opin Genet Dev. 1997;7:105–113. doi: 10.1016/s0959-437x(97)80117-7. [DOI] [PubMed] [Google Scholar]
- 63.Ezzatizadeh V, Pinto RM, Sandi C, Sandi M, Al-Mahdawi S, Te Riele H, Pook MA. The mismatch repair system protects against intergenerational GAA repeat instability in a Friedreich ataxia mouse model. Neurobiol Dis. 2012;46:165–171. doi: 10.1016/j.nbd.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dragileva E, Hendricks A, Teed A, Gillis T, Lopez ET, Friedberg EC, Kucherlapati R, Edelmann W, Lunetta KL, MacDonald ME, Wheeler VC. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiol Dis. 2009;33:37–47. doi: 10.1016/j.nbd.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Foiry L, Dong L, Savouret C, Hubert L, te Riele H, Junien C, Gourdon G. Msh3 is a limiting factor in the formation of intergenerational CTG expansions in DM1 transgenic mice. Hum Genet. 2006;119:520–526. doi: 10.1007/s00439-006-0164-7. [DOI] [PubMed] [Google Scholar]
- 66.Tomé S, Holt I, Edelmann W, Morris GE, Munnich A, Pearson CE, Gourdon G. MSH2 ATPase domain mutation affects CTG*CAG repeat instability in transgenic mice. PLoS Genet. 2009;5:e1000482. doi: 10.1371/journal.pgen.1000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tomé S, Manley K, Simard JP, Clark GW, Slean MM, Swami M, Shelbourne PF, Tillier ER, Monckton DG, Messer A, Pearson CE. MSH3 polymorphisms and protein levels affect CAG repeat instability in Huntington’s disease mice. PLoS Genet. 2013;9:e1003280. doi: 10.1371/journal.pgen.1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van den Broek WJ, Nelen MR, Wansink DG, Coerwinkel MM, te Riele H, Groenen PJ, Wieringa B. Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum Mol Genet. 2002;11:191–198. doi: 10.1093/hmg/11.2.191. [DOI] [PubMed] [Google Scholar]
- 69.Bourn RL, De Biase I, Pinto RM, Sandi C, Al-Mahdawi S, Pook MA, Bidichandani SI. Pms2 suppresses large expansions of the (GAA.TTC)n sequence in neuronal tissues. PLoS One. 2012;7:e47085. doi: 10.1371/journal.pone.0047085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Du J, Campau E, Soragni E, Ku S, Puckett JW, Dervan PB, Gottesfeld JM. Role of mismatch repair enzymes in GAA.TTC triplet-repeat expansion in Friedreich ataxia induced pluripotent stem cells. J Biol Chem. 2012;287:29861–29872. doi: 10.1074/jbc.M112.391961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Halabi A, Ditch S, Wang J, Grabczyk E. DNA mismatch repair complex MutSbeta promotes GAA.TTC repeat expansion in human cells. J Biol Chem. 2012;287:29958–29967. doi: 10.1074/jbc.M112.356758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gomes-Pereira M, Fortune MT, Ingram L, McAbney JP, Monckton DG. Pms2 is a genetic enhancer of trinucleotide CAG.CTG repeat somatic mosaicism: implications for the mechanism of triplet repeat expansion. Hum Mol Genet. 2004;13:1815–1825. doi: 10.1093/hmg/ddh186. [DOI] [PubMed] [Google Scholar]
- 73.Pinto RM, Dragileva E, Kirby A, Lloret A, Lopez E, St Claire J, Panigrahi GB, Hou C, Holloway K, Gillis T, Guide JR, Cohen PE, Li GM, Pearson CE, Daly MJ, Wheeler VC. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: genome-wide and candidate approaches. PLoS Genet. 2013;9:e1003930. doi: 10.1371/journal.pgen.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ezzatizadeh V, Sandi C, Sandi M, Anjomani-Virmouni S, Al-Mahdawi S, Pook MA. MutLalpha heterodimers modify the molecular phenotype of Friedreich ataxia. PLoS One. 2014;9:e100523. doi: 10.1371/journal.pone.0100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charbonneau N, Amunugama R, Schmutte C, Yoder K, Fishel R. Evidence that hMLH3 functions primarily in meiosis and in hMSH2-hMSH3 mismatch repair. Cancer Biol Ther. 2009;8:1411–1420. doi: 10.4161/cbt.8.14.8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pluciennik A, Burdett V, Baitinger C, Iyer RR, Shi K, Modrich P. Extrahelical (CAG)/(CTG) triplet repeat elements support proliferating cell nuclear antigen loading and MutLalpha endonuclease activation. Proc Natl Acad Sci U S A. 2013;110:12277–12282. doi: 10.1073/pnas.1311325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Owen BA, Yang Z, Lai M, Gajec M, Badger JD, 2nd, Hayes JJ, Edelmann W, Kucherlapati R, Wilson TM, McMurray CT. (CAG)(n)-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. 2005;12:663–670. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- 78.Tian L, Hou C, Tian K, Holcomb NC, Gu L, Li GM. Mismatch recognition protein MutSbeta does not hijack (CAG)n hairpin repair in vitro. J Biol Chem. 2009;284:20452–20456. doi: 10.1074/jbc.C109.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lang WH, Coats JE, Majka J, Hura GL, Lin Y, Rasnik I, McMurray CT. Conformational trapping of mismatch recognition complex MSH2/MSH3 on repair-resistant DNA loops. Proc Natl Acad Sci U S A. 2011;108:E837–844. doi: 10.1073/pnas.1105461108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Panigrahi GB, Lau R, Montgomery SE, Leonard MR, Pearson CE. Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat Struct Mol Biol. 2005;12:654–662. doi: 10.1038/nsmb959. [DOI] [PubMed] [Google Scholar]
- 81.Panigrahi GB, Slean MM, Simard JP, Gileadi O, Pearson CE. Isolated short CTG/CAG DNA slip-outs are repaired efficiently by hMutSbeta, but clustered slip-outs are poorly repaired. Proc Natl Acad Sci USA. 2010;107:12593–12598. doi: 10.1073/pnas.0909087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang T, Huang J, Gu L, Li GM. In vitro repair of DNA hairpins containing various numbers of CAG/CTG trinucleotide repeats. DNA Repair (Amst) 2012;11:201–209. doi: 10.1016/j.dnarep.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mollersen L, Rowe AD, Illuzzi JL, Hildrestrand GA, Gerhold KJ, Tveteras L, Bjolgerud A, Wilson DM, 3rd, Bjoras M, Klungland A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum Mol Genet. 2012;21:4939–4947. doi: 10.1093/hmg/dds337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loeb LA, Preston BD. Mutagenesis by apurinic/apyrimidinic sites. Annu Rev Genet. 1986;20:201–230. doi: 10.1146/annurev.ge.20.120186.001221. [DOI] [PubMed] [Google Scholar]
- 86.Hanawalt PC, Crowley DJ, Ford JM, Ganesan AK, Lloyd DR, Nouspikel T, Smith CA, Spivak G, Tornaletti S. Regulation of nucleotide excision repair in bacteria and mammalian cells. Cold Spring Harb Symp Quant Biol. 2000;65:183–191. doi: 10.1101/sqb.2000.65.183. [DOI] [PubMed] [Google Scholar]
- 87.Camenisch U, Nageli H. XPA gene, its product and biological roles. Adv Exp Med Biol. 2008;637:28–38. doi: 10.1007/978-0-387-09599-8_4. [DOI] [PubMed] [Google Scholar]
- 88.Hubert L, Jr, Lin Y, Dion V, Wilson JH. Xpa deficiency reduces CAG trinucleotide repeat instability in neuronal tissues in a mouse model of SCA1. Hum Mol Genet. 2011;20:4822–4830. doi: 10.1093/hmg/ddr421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang Z, Roginskaya M, Colis LC, Basu AK, Shell SM, Liu Y, Musich PR, Harris CM, Harris TM, Zou Y. Specific and efficient binding of xeroderma pigmentosum complementation group A to double-strand/single-strand DNA junctions with 3’- and/or 5’-ssDNA branches. Biochemistry. 2006;45:15921–15930. doi: 10.1021/bi061626q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao X-N, Usdin K. Gender and cell-type specific effects of the transcription coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the Fragile X-related disorders. Hum Mutat. 2014;35:341–349. doi: 10.1002/humu.22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Javeri A, Lyons JG, Huang XX, Halliday GM. Downregulation of Cockayne syndrome B protein reduces human 8-oxoguanine DNA glycosylase-1 expression and repair of UV radiation-induced 8-oxo-7,8-dihydro-2’-deoxyguanine. Cancer Sci. 2011;102:1651–1658. doi: 10.1111/j.1349-7006.2011.02005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Muftuoglu M, de Souza-Pinto NC, Dogan A, Aamann M, Stevnsner T, Rybanska I, Kirkali G, Dizdaroglu M, Bohr VA. Cockayne syndrome group B protein stimulates repair of formamidopyrimidines by NEIL1 DNA glycosylase. J Biol Chem. 2009;284:9270–9279. doi: 10.1074/jbc.M807006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tuo J, Chen C, Zeng X, Christiansen M, Bohr VA. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst) 2002;1:913–927. doi: 10.1016/s1568-7864(02)00116-7. [DOI] [PubMed] [Google Scholar]
- 94.Wong HK, Muftuoglu M, Beck G, Imam SZ, Bohr VA, Wilson DM., 3rd Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35:4103–4113. doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103:9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Selby CP, Sancar A. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc Natl Acad Sci U S A. 1997;94:11205–11209. doi: 10.1073/pnas.94.21.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martins S, Pearson CE, Coutinho P, Provost S, Amorim A, Dube MP, Sequeiros J, Rouleau GA. Modifiers of (CAG)(n) instability in Machado-Joseph disease (MJD/SCA3) transmissions: an association study with DNA replication, repair and recombination genes. Hum Genet. 2014;133:1311–1318. doi: 10.1007/s00439-014-1467-8. [DOI] [PubMed] [Google Scholar]
- 98.Guo J, Hanawalt PC, Spivak G. Comet-FISH with strand-specific probes reveals transcription-coupled repair of 8-oxoGuanine in human cells. Nucleic Acids Res. 2013;41:7700–7712. doi: 10.1093/nar/gkt524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Parlanti E, D’Errico M, Degan P, Calcagnile A, Zijno A, van der Pluijm I, van der Horst GT, Biard DS, Dogliotti E. The cross talk between pathways in the repair of 8-oxo-7,8-dihydroguanine in mouse and human cells. Free Rad Biol Med. 2012;53:2171–2177. doi: 10.1016/j.freeradbiomed.2012.08.593. [DOI] [PubMed] [Google Scholar]
- 100.Lan L, Nakajima S, Wei L, Sun L, Hsieh CL, Sobol RW, Bruchez M, Van Houten B, Yasui A, Levine AS. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Res. 2014;42:2330–2345. doi: 10.1093/nar/gkt1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Amouroux R, Campalans A, Epe B, Radicella JP. Oxidative stress triggers the preferential assembly of base excision repair complexes on open chromatin regions. Nucleic Acids Res. 2010;38:2878–2890. doi: 10.1093/nar/gkp1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sattler U, Frit P, Salles B, Calsou P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO Rep. 2003;4:363–367. doi: 10.1038/sj.embor.embor796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gary R, Kim K, Cornelius HL, Park MS, Matsumoto Y. Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. J Biol Chem. 1999;274:4354–4363. doi: 10.1074/jbc.274.7.4354. [DOI] [PubMed] [Google Scholar]
- 104.Fan J, Wilson DM., 3rd Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radic Biol Med. 2005;38:1121–1138. doi: 10.1016/j.freeradbiomed.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 105.Chan NL, Guo J, Zhang T, Mao G, Hou C, Yuan F, Huang J, Zhang Y, Wu J, Gu L, Li GM. Coordinated processing of 3’ slipped (CAG)n/(CTG)n hairpins by DNA polymerases beta and delta preferentially induces repeat expansions. J Biol Chem. 2013;288:15015–15022. doi: 10.1074/jbc.M113.464370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Y, Wilson SH. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci. 2012;37:162–172. doi: 10.1016/j.tibs.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee JM, Pinto RM, Gillis T, St Claire JC, Wheeler VC. Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One. 2011;6:e23647. doi: 10.1371/journal.pone.0023647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jarem DA, Wilson NR, Delaney S. Structure-dependent DNA damage and repair in a trinucleotide repeat sequence. Biochemistry. 2009;48:6655–6663. doi: 10.1021/bi9007403. [DOI] [PubMed] [Google Scholar]
- 109.Gupta S, Gellert M, Yang W. Mechanism of mismatch recognition revealed by human MutSbeta bound to unpaired DNA loops. Nat Struct Mol Biol. 2012;19:72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roesner LM, Mielke C, Fahnrich S, Merkhoffer Y, Dittmar KE, Drexler HG, Dirks WG. Stable expression of MutLgamma in human cells reveals no specific response to mismatched DNA, but distinct recruitment to damage sites. J Cell Biochem. 2013;114:2405–2414. doi: 10.1002/jcb.24591. [DOI] [PubMed] [Google Scholar]
- 111.Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hubscher U, Dogliotti E. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene. 1998;17:835–843. doi: 10.1038/sj.onc.1202001. [DOI] [PubMed] [Google Scholar]
- 112.Lake RJ, Fan HY. Structure, function and regulation of CSB: a multi-talented gymnast. Mech Ageing Dev. 2013;134:202–211. doi: 10.1016/j.mad.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T, et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA) Nat Genet. 1994;6:9–13. doi: 10.1038/ng0194-9. [DOI] [PubMed] [Google Scholar]
- 114.Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M, Takeda T, Tadokoro K, Kondo I, Murayama N, et al. Dentatorubral and pallidoluysian atrophy expansion of an unstable CAG trinucleotide on chromosome 12p. Nat Genet. 1994;6:14–18. doi: 10.1038/ng0194-14. [DOI] [PubMed] [Google Scholar]
- 115.The Huntington’s Disease Collaborative Research Group, A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 116.Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, Squitieri F, Lin B, Bassett A, Almqvist E, et al. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. New Eng J Med. 1994;330:1401–1406. doi: 10.1056/NEJM199405193302001. [DOI] [PubMed] [Google Scholar]
- 117.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 118.Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, Hughes MR, Zoghbi HY. Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1995;10:344–350. doi: 10.1038/ng0795-344. [DOI] [PubMed] [Google Scholar]
- 119.Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, Garnier JM, Weber C, Mandel JL, Cancel G, Abbas N, Durr A, Didierjean O, Stevanin G, Agid Y, Brice A. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet. 1996;14:285–291. doi: 10.1038/ng1196-285. [DOI] [PubMed] [Google Scholar]
- 120.Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A, Tashiro K, Ishida Y, Ikeuchi T, Koide R, Saito M, Sato A, Tanaka T, Hanyu S, Takiyama Y, Nishizawa M, Shimizu N, Nomura Y, Segawa M, Iwabuchi K, Eguchi I, Tanaka H, Takahashi H, Tsuji S. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique. DIRECT, Nat Genet. 1996;14:277–284. doi: 10.1038/ng1196-277. [DOI] [PubMed] [Google Scholar]
- 121.Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, Chneiweiss H, Benomar A, Lyon-Caen O, Julien J, Serdaru M, Penet C, Agid Y, Brice A. Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol. 1996;39:490–499. doi: 10.1002/ana.410390411. [DOI] [PubMed] [Google Scholar]
- 122.Takano H, Cancel G, Ikeuchi T, Lorenzetti D, Mawad R, Stevanin G, Didierjean O, Durr A, Oyake M, Shimohata T, Sasaki R, Koide R, Igarashi S, Hayashi S, Takiyama Y, Nishizawa M, Tanaka H, Zoghbi H, Brice A, Tsuji S. Close associations between prevalences of dominantly inherited spinocerebellar ataxias with CAG-repeat expansions and frequencies of large normal CAG alleles in Japanese and Caucasian populations. Am J Hum Genet. 1998;63:1060–1066. doi: 10.1086/302067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jodice C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L, Francia A, Spadaro M, Pierelli F, Salvi F, Ophoff RA, Frants RR, Frontali M. Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet. 1997;6:1973–1978. doi: 10.1093/hmg/6.11.1973. [DOI] [PubMed] [Google Scholar]
- 124.Riess O, Schols L, Bottger H, Nolte D, Vieira-Saecker AM, Schimming C, Kreuz F, Macek M, Jr, Krebsova A, Macek MS, Klockgether T, Zuhlke C, Laccone FA. SCA6 is caused by moderate CAG expansion in the alpha1A-voltage-dependent calcium channel gene. Hum Mol Genet. 1997;6:1289–1293. doi: 10.1093/hmg/6.8.1289. [DOI] [PubMed] [Google Scholar]
- 125.David G, Durr A, Stevanin G, Cancel G, Abbas N, Benomar A, Belal S, Lebre AS, Abada-Bendib M, Grid D, Holmberg M, Yahyaoui M, Hentati F, Chkili T, Agid Y, Brice A. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum Mol Genet. 1998;7:165–170. doi: 10.1093/hmg/7.2.165. [DOI] [PubMed] [Google Scholar]
- 126.Fujigasaki H, Verma IC, Camuzat A, Margolis RL, Zander C, Lebre AS, Jamot L, Saxena R, Anand I, Holmes SE, Ross CA, Durr A, Brice A. SCA12 is a rare locus for autosomal dominant cerebellar ataxia: a study of an Indian family. Ann Neurol. 2001;49:117–121. [PubMed] [Google Scholar]
- 127.Holmes SE, O’Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, Kwak NG, Ingersoll-Ashworth RG, Sherr M, Sumner AJ, Sharp AH, Ananth U, Seltzer WK, Boss MA, Vieria-Saecker AM, Epplen JT, Riess O, Ross CA, Margolis RL. Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat Genet. 1999;23:391–392. doi: 10.1038/70493. [DOI] [PubMed] [Google Scholar]
- 128.Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–1448. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 129.Maltecca F, Filla A, Castaldo I, Coppola G, Fragassi NA, Carella M, Bruni A, Cocozza S, Casari G, Servadio A, De Michele G. Intergenerational instability and marked anticipation in SCA-17. Neurology. 2003;61:1441–1443. doi: 10.1212/01.wnl.0000094123.09098.a0. [DOI] [PubMed] [Google Scholar]
- 130.Stevanin G, Brice A. Spinocerebellar ataxia 17 (SCA17) and Huntington’s disease-like 4 (HDL4) Cerebellum. 2008;7:170–178. doi: 10.1007/s12311-008-0016-1. [DOI] [PubMed] [Google Scholar]
- 131.Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, Baratz KH. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012;7:e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- 133.Margolis RL, Holmes SE, Rosenblatt A, Gourley L, O’Hearn E, Ross CA, Seltzer WK, Walker RH, Ashizawa T, Rasmussen A, Hayden M, Almqvist EW, Harris J, Fahn S, MacDonald ME, Mysore J, Shimohata T, Tsuji S, Potter N, Nakaso K, Adachi Y, Nakashima K, Bird T, Krause A, Greenstein P. Huntington’s Disease-like 2 (HDL2) in North America and Japan. Ann Neurol. 2004;56:670–674. doi: 10.1002/ana.20248. [DOI] [PubMed] [Google Scholar]
- 134.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 135.Fu YH, Pizzuti A, Fenwick RG, Jr, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 136.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 137.Tsilfidis C, MacKenzie AE, Mettler G, Barcelo J, Korneluk RG. Correlation between CTG trinucleotide repeat length and frequency of severe congenital myotonic dystrophy. Nat Genet. 1992;1:192–195. doi: 10.1038/ng0692-192. [DOI] [PubMed] [Google Scholar]
- 138.Worth PF, Houlden H, Giunti P, Davis MB, Wood NW. Large, expanded repeats in SCA8 are not confined to patients with cerebellar ataxia. Nat Genet. 2000;24:214–215. doi: 10.1038/73411. [DOI] [PubMed] [Google Scholar]
- 139.Ikeda Y, Dalton JC, Moseley ML, Gardner KL, Bird TD, Ashizawa T, Seltzer WK, Pandolfo M, Milunsky A, Potter NT, Shoji M, Vincent JB, Day JW, Ranum LP. Spinocerebellar ataxia type 8: molecular genetic comparisons and haplotype analysis of 37 families with ataxia. Am J Hum Genet. 2004;75:3–16. doi: 10.1086/422014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mutsuddi M, Rebay I. Molecular genetics of spinocerebellar ataxia type 8 (SCA8) RNA Biol. 2005;2:49–52. doi: 10.4161/rna.2.2.1682. [DOI] [PubMed] [Google Scholar]
- 141.Metsu S, Rainger JK, Debacker K, Bernhard B, Rooms L, Grafodatskaya D, Weksberg R, Fombonne E, Taylor MS, Scherer SW, Kooy RF, FitzPatrick DR. A CGG-repeat expansion mutation in ZNF713 causes FRA7A: association with autistic spectrum disorder in two families. Hum Mutat. 2014;35:1295–1300. doi: 10.1002/humu.22683. [DOI] [PubMed] [Google Scholar]
- 142.Murray A, Webb J, Grimley S, Conway G, Jacobs P. Studies of FRAXA and FRAXE in women with premature ovarian failure. J Med Genet. 1998;35:637–640. doi: 10.1136/jmg.35.8.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97:189–194. doi: 10.1002/1096-8628(200023)97:3<189::AID-AJMG1036>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 144.Hagerman PJ, Hagerman RJ. Fragile X-associated tremor/ataxia syndrome (FXTAS) Ment Retard Dev Disabil Res Rev. 2004;10:25–30. doi: 10.1002/mrdd.20005. [DOI] [PubMed] [Google Scholar]
- 145.Yu S, Pritchard M, Kremer E, Lynch M, Nancarrow J, Baker E, Holman K, Mulley JC, Warren ST, Schlessinger D, et al. Fragile X genotype characterized by an unstable region of DNA. Science. 1991;252:1179–1181. doi: 10.1126/science.252.5009.1179. [DOI] [PubMed] [Google Scholar]
- 146.Knight SJ, Flannery AV, Hirst MC, Campbell L, Christodoulou Z, Phelps SR, Pointon J, Middleton-Price HR, Barnicoat A, Pembrey ME, et al. Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell. 1993;74:127–134. doi: 10.1016/0092-8674(93)90300-f. [DOI] [PubMed] [Google Scholar]
- 147.Gecz J, Gedeon AK, Sutherland GR, Mulley JC. Identification of the gene FMR2, associated with FRAXE mental retardation. Nat Genet. 1996;13:105–108. doi: 10.1038/ng0596-105. [DOI] [PubMed] [Google Scholar]
- 148.Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 149.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 150.Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, Schneider C, Koch MC, Beilman GJ, Harrison AR, Dalton JC, Ranum LP. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657–664. doi: 10.1212/01.wnl.0000054481.84978.f9. [DOI] [PubMed] [Google Scholar]
- 151.Liquori CL, Ikeda Y, Weatherspoon M, Ricker K, Schoser BG, Dalton JC, Day JW, Ranum LP. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am J Hum Genet. 2003;73:849–862. doi: 10.1086/378720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 153.Matsuura T, Fang P, Pearson CE, Jayakar P, Ashizawa T, Roa BB, Nelson DL. Interruptions in the expanded ATTCT repeat of spinocerebellar ataxia type 10: repeat purity as a disease modifier? Am J Hum Genet. 2006;78:125–129. doi: 10.1086/498654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Obayashi M, Stevanin G, Synofzik M, Monin ML, Duyckaerts C, Sato N, Streichenberger N, Vighetto A, Desestret V, Tesson C, Wichmann HE, Illig T, Huttenlocher J, Kita Y, Izumi Y, Mizusawa H, Schols L, Klopstock T, Brice A, Ishikawa K, Durr A. Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry. 2014 doi: 10.1136/jnnp-2014-309153. [DOI] [PubMed] [Google Scholar]
- 156.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lalioti MD, Mirotsou M, Buresi C, Peitsch MC, Rossier C, Ouazzani R, Baldy-Moulinier M, Bottani A, Malafosse A, Antonarakis SE. Identification of mutations in cystatin B, the gene responsible for the Unverricht-Lundborg type of progressive myoclonus epilepsy (EPM1) Am J Hum Genet. 1997;60:342–351. [PMC free article] [PubMed] [Google Scholar]
- 159.Lalioti MD, Scott HS, Genton P, Grid D, Ouazzani R, M’Rabet A, Ibrahim S, Gouider R, Dravet C, Chkili T, Bottani A, Buresi C, Malafosse A, Antonarakis SE. A PCR amplification method reveals instability of the dodecamer repeat in progressive myoclonus epilepsy (EPM1) and no correlation between the size of the repeat and age at onset. Am J Hum Genet. 1998;62:842–847. doi: 10.1086/301798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, Ishino F, Saito Y, Murayama S, Yoshida M, Hashizume Y, Takahashi Y, Tsuji S, Shimizu N, Toda T, Ishikawa K, Mizusawa H. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Messaed C, Rouleau GA. Molecular mechanisms underlying polyalanine diseases. Neurobiol Dis. 2009;34:397–405. doi: 10.1016/j.nbd.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 162.Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. 1999;23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- 163.Savouret C, Garcia-Cordier C, Megret J, te Riele H, Junien C, Gourdon G. MSH2-dependent germinal CTG repeat expansions are produced continuously in spermatogonia from DM1 transgenic mice. Mol Cell Biol. 2004;24:629–637. doi: 10.1128/MCB.24.2.629-637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kovtun IV, Johnson KO, McMurray CT. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging (Albany NY) 2011;3:509–514. doi: 10.18632/aging.100324. [DOI] [PMC free article] [PubMed] [Google Scholar]