

Abstract
Tyrosine kinase domain mutations are a common cause of acquired clinical resistance to tyrosine kinase inhibitors (TKIs) used to treat cancer, including the FLT3 inhibitor quizartinib. Mutation of kinase “gatekeeper” residues, which control access to an allosteric pocket adjacent to the ATP-binding site, have been frequently implicated in TKI resistance. The molecular underpinnings of gatekeeper mutation-mediated resistance are incompletely understood. We report the first co-crystal structure of FLT3 with the TKI quizartinib, which demonstrates that quizartinib binding relies on essential edge-to-face aromatic interactions with the gatekeeper F691 residue, and F830 within the highly conserved DFG motif in the activation loop. This reliance makes quizartinib critically vulnerable to gatekeeper and activation loop substitutions while minimizing the impact of mutations elsewhere. Moreover, we identify PLX3397, a novel FLT3 inhibitor that retains activity against the F691L mutant due to a binding mode that depends less vitally on specific interactions with the gatekeeper position.
Keywords: Gatekeeper Mutations, FLT3, Quizartinib, Resistance
Introduction
Secondary mutations in the tyrosine kinase domain (KD) remain the most commonly encountered cause of acquired clinical resistance to small molecule tyrosine kinase inhibitors (TKIs) in human cancer(1–4). Recent pharmaceutical efforts have focused on the development of “type II” kinase inhibitors, which bind to a relatively non-conserved inactive kinase conformation and exploit an allosteric site adjacent to the ATP-binding pocket as a potential means to increase kinase selectivity, although recent data suggest such efforts may be misguided(5). Commonly, KD mutations effect resistance to type II inhibitors via two mechanisms: (i) substitution of amino acid positions directly involved in binding inhibitor, or (ii) mutation of residues that stabilize the inactive kinase conformation required for binding(6). Type I inhibitors, which bind to the more conserved active kinase conformation, are typically vulnerable only to mutations at inhibitor contact residues and therefore may be generally impacted by a reduced spectrum of resistance-causing KD mutations(7, 8).
FMS-Like Tyrosine Kinase 3 (FLT3) is a class III receptor tyrosine kinase that is expressed in hematopoietic cells. Mutations in FLT3 represent the most common genetic alteration in patients with acute myeloid leukemia (AML)(9), and are comprised primarily of constitutively activating internal tandem duplication (ITD) mutations (of 1–100 amino acids) in the juxtamembrane domain, and less commonly, point mutations, typically within the kinase activation loop. We recently identified secondary KD mutations in FLT3-ITD that can cause preclinical and acquired clinical resistance to the highly potent type II FLT3 inhibitor quizartinib(10), which achieved a composite complete remission (CRc) rate of ~50% in relapsed or chemotherapy-refractory FLT3-ITD+ AML patients in monotherapy studies (11). An in vitro saturation mutagenesis screen of FLT3-ITD identified five quizartinib-resistant KD mutations at three residues: the “gatekeeper” F691 residue, and two amino acid positions within the kinase activation loop (D835 and Y842), a surprisingly limited spectrum of mutations for a type II inhibitor. Mutations at two of these residues (F691L and D835V/Y/F) were subsequently identified in each of eight samples analyzed at the time of acquired clinical resistance to quizartinib(10), a finding that definitively validated FLT3 as a therapeutic target in AML. The type II multikinase inhibitor sorafenib, which also has some clinical activity in FLT3-ITD+ AML, is ineffective against all identified quizartinib resistance-causing mutants, in addition to other mutant isoforms(10, 12). While we(8) and others(13, 14) recently identified the type I inhibitor crenolanib as an equipotent inhibitor of quizartinib-resistant D835 mutants, no FLT3 inhibitor has demonstrated equipotent inhibition of the F691L mutant, including the ABL/FLT3 inhibitor ponatinib, which was rationally designed to retain activity against the problematic gatekeeper T315I mutant in BCR-ABL(15).
Mutation of the conserved kinase gatekeeper residue in the ATP-binding pocket, which controls access to an allosteric site adjacent to this pocket, represents a well-documented cause of clinical resistance to inhibitors of a number of pathologically activated kinases in addition to FLT3-ITD and BCR-ABL, including FIPL1-PDGFRalpha(3), KIT(16), EGFR(2) and EML4-ALK(4). Co-crystal structure analyses of TKI-bound kinases have suggested that the molecular mechanism of TKI resistance due to gatekeeper mutations is largely attributable to steric hindrance resulting from substitution of a bulkier amino acid residue for a smaller (commonly threonine) gatekeeper residue(6, 17). In the case of the EGFR gatekeeper T790M mutation, increased affinity for ATP has been reported to also play a role(18). Notably, FLT3 is one of a limited number of kinases to harbor a bulky phenylalanine at the gatekeeper position. Although the non-ligand bound structure of intracellular FLT3 has been described(19), no co-crystal structure of FLT3 in complex with ATP or any other ligand has been reported, greatly limiting our structural understanding of this pathologically important kinase. To elucidate the structural mechanism of resistance mediated by F691L and other quizartinib-resistant mutants, we solved the co-crystal structure of quizartinib bound to the FLT3 kinase domain, with the ultimate aim of informing the rational development of novel FLT3 inhibitors with the potential to retain activity against these mutants.
Results
Co-Crystal Structure of FLT3 Bound to Quizartinib
In the co-structure with FLT3 (Supplemental Table 1, Figure 1a, Supplemental Figure 1), quizartinib adopts the canonical type II kinase inhibitor binding mode with the imidazobenzothiazole “head” occupying the adenine binding pocket and t-butyl-isoxazole “tail” residing in the allosteric back pocket of the kinase. The terminal t-butyl group is surrounded by hydrophobic side-chains including L802 from the αE helix that marks the distal end of the back pocket (Figure 1b). The urea linker between the phenyl and isoxazole rings are hydrogen-bonded to both E661 of the αC helix and the backbone NH of D829 of the DFG motif, a pattern shared by other urea- and amide-containing type II kinase inhibitors including imatinib(20), sorafenib(21) and ponatinib(22). The middle phenyl ring forms edge-to-face interactions with both gatekeeper F691 and F830 of the conserved DFG motif. Edge-to-face interaction is the preferred interface between non-sequential aromatic residues inside proteins(23). Perturbation of any component of this compact unit (either F691 or F830) is expected to cause substantial loss in binding affinity. This explains why mutations in FLT3 that confer clinical resistance to quizartinib have been thus far confined to two residues: the gatekeeper F691 and D835, a key residue of the activation loop that determines the conformational state of the DFG motif and thus the orientation of F830. This binding mode places the imidazobenzothiazole of quizartinib inside the narrow cleft between the N- and C-terminal lobes of the kinase abutting the hinge region (Figure 1a). Quizartinib is an unusual kinase inhibitor in that it lacks a readily identifiable adenine-like moiety that can simultaneously fit the interlobe cleft shape and the hinge hydrogen bonding requirements. Forcing quizartinib to conform to the adenine-like hinge interactions led to binding mode predictions that are not supported by crystallographic studies(10) (Supplemental Figure 2a,b). Unexpectedly, the co-crystal structure revealed a workaround – instead of engaging the hinge directly, the imidazobenzothiazole group recruits a water molecule to form bidentate hydrogen bonds with two polar backbone atoms of the hinge (Figure 1b, Supplemental Figure 3). This orientation of the imadazobenzothiazole confers site specificity to the attached morpholinoethoxy aqueous solubilizing group as observed previously(24).
Figure 1. Co-Crystal structure of quizartinib bound to FLT3 and inferred binding poses of other FLT3 Inhibitors.

A. Overview of FLT3 kinase domain in complex with quizartinib. B. A close-up view of quizartinib inside FLT3 (colors correspond to the structural features indicated in a) highlighting key active site residues, water-mediated hinge interactions, anchoring of the urea group, and the intramolecular hydrogen bond within the morpholinoethoxy solubilizing group. C. Inferred binding pose of sorafenib using sorafenib-KDR co-structure (PDB:4ASD) as a template. D. A view of quizartinib bound to native FLT3. E. F691L creates steric clashes with quizartinib (middle ring and urea linker); F. F691L is incompatible with the urea linker of sorafenib. G. Inferred binding pose of ponatinib using ponatinib-KIT co-structure as a template (PDB:4U0I). H. The steric effect of F691L is better tolerated by ponatinib.
The conjugation between the imidazobenzothiazole and the middle phenyl ring restricts the orientation of the phenyl ring and thus contributes to the edge-to-face interactions. Interestingly, sorafenib - another diarylurea - also exhibited ~1,000 fold loss in potency against the gatekeeper mutant F691L compared to native FLT3(10). The phenyl ring in sorafenib is linked to a conventional hinge-binding scaffold through a flexible ether linker. The optimal fit of sorafenib in FLT3 entails a tilted middle ring orientation relative to quizartinib (Figure 1c). The cause for F691L-induced resistance to diarylureas is not the orientation of the middle ring, but rather the urea NH proximal to the phenyl ring, which clashes with the Cδ atom of the mutated L691 (Figure 1d,e,f). Supporting the conjecture that the urea linker rather than the geometry of the phenyl ring is responsible for F691L resistance, ponatinib, which contains a shorter amide linker (i.e. lacking the corresponding NH of a urea linker, Figure 1g,h), is less affected (17-fold) by F691L despite having additional substitution on the middle ring(15).
PLX3397 is A Novel FLT3 Inhibitor with In Vitro Activity Against the Quizartinib-Resistant FLT3 F691L Mutation
To identify FLT3 inhibitors with activity against gatekeeper mutants, we thus turned to novel chemical structures that possess abridged connectors between the middle and tail rings. PLX3397, a triple kinase inhibitor of CSF1R (enzymatic IC50=13nM), KIT (enzymatic IC50=27nM) and FLT3-ITD (enzymatic IC50=11nM), represents a new chemotype containing a pyridine (rather than phenyl) middle ring and a methyl amine (rather than amide or urea as in other type II kinase inhibitors) linker(25) (Figure 2a). Based on in vitro phosphorylation assays, PLX3397 inhibits FLT3 signaling in cells harboring FLT3-ITD (e.g. biochemical IC50 =18nM in MV4;11 cells) but is significantly less effective in cells containing only native FLT3 (e.g. biochemical IC50 =1.8μM in RS4;11 cells), suggesting bias towards binding the ITD-mutant form of FLT3. This is corroborated by in vitro assays using recombinant proteins that show preferential binding of PLX3397 to FLT3-ITD (IC50 =10nM) compared to native, auto-inhibited FLT3 (0.4μM). The anti-FLT3-ITD activity was confirmed in vivo in a MV4;11 xenograft model (Figure 2b). Based on its chemical structure and inferred binding pose (Figure 2c,d; Supplemental Figure 4), we speculated that PLX3397 would retain activity against the F691L mutant. We performed proliferation studies of PLX3397 and quizartinib for Ba/F3 cells expressing FLT3-ITD and FLT3-ITD/F691L and confirmed that PLX3397, in contrast to quizartinib, has the ability to inhibit the proliferation of both isoforms (Supplemental Table 2). PLX3397 equally inhibited the proliferation of Molm14 F691L cells, which have an acquired F691L mutation in FLT3-ITD following selection in quizartinib, and parental Molm14 cells (Figure 2e and Supplemental Table 3). Importantly, PLX3397 achieved equipotent inhibition of FLT3 phosphorylation and downstream signaling in Molm14 F691L and parental Molm14 cells in the presence of human plasma, albeit at higher concentrations than those required to inhibit FLT3 signaling in media (Figure 2f; Supplemental Figure 5). This suggests that if sufficient plasma drug levels can be obtained, PLX3397 would be expected to be clinically active both in patients with native FLT3-ITD and patients who acquired the FLT3-ITD/F691L as a consequence of prior quizartinib or sorafenib therapy. In contrast, quizartinib demonstrated decreased activity against Molm14 F691L compared to parental cells in similar assays (Supplemental Figure 6a,b).
Figure 2. PLX3397 is an inhibitor with activity against the FLT3-ITD/F691L mutation.

A. The chemical structure of PLX3397. B. PLX3397 inhibition of FLT3-ITD-driven human AML MV4;11 xenografts in nude mice. Mice were injected with MV4;11 cell suspension in 100 μL PBS + 100 μL Matrigel (5×106 cells per mouse) in the lower left abdominal flank. Treatments (vehicle and PLX3397 at two dose levels, all given daily via oral gavage) started when the average tumor size reached 125 mm3. PLX3397 at 10mg/kg dose resulted in tumor stasis whereas PLX3397 at 30mg/kg dose caused tumor regression. Mean tumor sizes of each treatment group (+standard errors) are plotted over the course of 21-day dosing. C. Binding pose of PLX3397 in FLT3 inferred from its co-structure with CSF1R (PDB:4R7H). D. PLX3397 binding is minimally affected by F691L. E. Proliferation of Molm14, Molm14/F691L, MV4;11 and K562 cells after 48 hours in various concentrations of PLX3397 (error bars represent s.d. of triplicates from the same experiment). F. Western blot analysis for phosphotyrosine and total FLT3 performed after immunoprecipitation using anti-FLT3 antibody from lysates prepared from Molm14 and Molm14/F691L cells. Western Blot analysis using anti-phospho-S6, anti-phospho-STAT5, anti-S6 and anti-STAT5 antibody performed on whole cell lysates prepared from Molm14 and Molm14/F691L cells. Cells were exposed for 120 minutes to healthy control plasma with the indicated concentrations of PLX3397. Data shown represents a single experiment.
PLX3397 is Vulnerable to A Spectrum of FLT3 TKD Mutations at Non-Gatekeeper Residues
We next sought to assess the activity of PLX3397 against other quizartinib-resistant mutants. While PLX3397 potently inhibited growth and signaling of Ba/F3 cells expressing the FLT3-ITD/F691L mutant, cells expressing quizartinib-resistant activation loop mutants (at residues D835 and Y842) demonstrated substantial relative resistance to PLX3397 (Supplemental Figure 7a,b and Supplemental Table 4), suggesting that PLX3397 may be particularly vulnerable to activation loop substitutions.
To more broadly assess the vulnerability of PLX3397 to resistance-causing KD mutations within FLT3-ITD and to predict mutations that might confer clinical resistance to PLX3397, we utilized a well-validated in vitro saturation mutagenesis screen(10). Given the significant promise for clinical activity of PLX3397 in patients who develop resistance to quizartinib or sorafenib due to the F691L mutation, we subjected both FLT3-ITD and FLT3-ITD/F691L to mutagenesis. In contrast to the small number of resistance mutations identified in a similar screen for quizartinib resistance (5 substitutions at 3 residues)(10), we identified 18 distinct PLX3397 resistance-conferring substitutions at 10 different amino acid residues after selection in 2 μM PLX3397 (~20× IC50 for FLT3-ITD). Twelve mutations were identified on the background of FLT3-ITD (in 78 resistant clones analyzed); these substitutions, and an additional six mutations, were also identified on the background of FLT3-ITD/F691L (in 117 resistant clones analyzed) (Figure 3a,b). We re-created all identified mutations (in both the ITD and ITD/F691L backgrounds) and introduced them into Ba/F3 cells to confirm their ability to confer resistance to PLX3397. All mutations conferred relative resistance to PLX3397 in proliferation and biochemical studies when compared with FLT3-ITD alone (Figure 3c,d, Supplemental Table 4). The majority of identified mutations (15 of 18) occurred in the kinase activation loop (Figure 3a,b), suggesting that perturbing the conformation of the activation loop is a primary means to disrupt PLX3397 binding (Figure 4a,b). In addition, some mutations at these sites may favor or stabilize the active state of the kinase that is inaccessible to type II inhibitors. While the remaining three mutations were found in the N-terminal lobe and hinge, none makes direct contact with the inhibitor and their effects are mediated by long-range interactions (Figure 4b,c,d). In fact, M664I and N676S can both bias the conformation of the activation loop in favor of the active state either directly or indirectly (Figure 4b,c). Importantly, no resistance-conferring mutation at the F691 gatekeeper position was identified, whereas this residue was the most commonly mutated residue in our previous screen for quizartinib-resistant mutants. All mutations (with the exception of D835N) caused a higher degree of relative resistance in the ITD+F691L background compared to the ITD alone (at least ~2–5×), suggesting that in general, the aggregate resistance for any compound mutation is higher than for each component mutation alone.
Figure 3. Mutation Screen of FLT3-ITD and FLT3-ITD/F691L Reveals Kinase Domain Mutations that Cause Resistance to PLX3397.

A. Numbers of independently-derived PLX3397-resistant Ba/F3/FLT3-ITD subpopulations with amino acid substitution at the indicated residue obtained from a saturation mutagenesis assay for FLT3-ITD and B. FLT3-ITD/F691L. C. IC50s for proliferation of Ba/F3 cells expressing PLX3397-resistant mutants in FLT3-ITD and FLT3-ITD/F691L backgrounds. Bars with slanted lines indicate mutations observed only in clones with the ITD+F691L background. Error bars represent s.d. of 3 or more independent experiments. D. Western blot analysis using anti-phospho-FLT3, anti-phospho-STAT5, anti-FLT3 and anti-STAT5 antibody performed on lysates from IL-3-independent Ba/F3 populations expressing the FLT3-ITD mutant isoforms indicated. Cells were exposed to PLX3397 at the indicated concentrations for 90 minutes.
Figure 4. Structural mapping of the PLX3397-resistant mutations in FLT3.

A. The residues for which mutations have been identified from the in vitro saturation mutagenesis screen are represented as spheres (colors correspond to the structural features indicated in Figure 1a). Details of the mutations can be found in Figure 3 and Supplemental Table 4. No mutation makes a direct contact with the inhibitor. B. A majority of the mutations were found in the activation loop (yellow, depicted in the PLX3397-bound, inactive state). The red ribbon traces the extended conformation of activation loop during active state (modeled using the coordinates of active KIT structure, PDB:1PKG). The proximity of M664 suggests a role in biasing the activation loop conformation. C. Close-up view of the hydrogen bond network formed by N676, H671, E692 and K826 at the back of the ATP-binding pocket. N676S disengages this network, thus releasing a molecular brake that keeps the enzyme in an inactive state(30). D. Close-up view of D698 and its role in nucleating the structure between the end of hinge and the beginning of the αD helix. The long-range structural effect of D698N on state of the enzyme remains unknown.
PLX3397 is a Clinically Active FLT3 Inhibitor That Selects for Polyclonal Resistance Due to FLT3 KD Mutations in FLT3-ITD+ AML Patients
Based upon its encouraging preclinical activity, a phase I/II clinical trial of PLX3397 in relapsed or chemotherapy-refractory FLT3-ITD+ AML patients (NCT01349049) was initiated, testing doses ranging from 800mg to 5000mg daily. While the full results of this clinical trial will be reported elsewhere, we undertook translational studies of clinical samples to test our hypothesis that acquired resistance to PLX3397 would be due to mutations at residues other than the F691L gatekeeper position. Using a deep sequencing approach we previously successfully utilized to identify KD mutations occurring on the ITD-containing allele in patient samples(10), we analyzed pre-treatment and relapse blood or bone marrow samples from nine patients who achieved a partial or complete response on PLX3397 at any dose level (with or without recovery of peripheral blood counts) and who subsequently relapsed on drug (Supplemental Table 5). All assessed responders were treated with PLX3397 ≥ 1400 mg daily, the minimally effective dose. In six of nine patient samples, secondary FLT3 KD mutations were acquired at the time of disease relapse (Table 1), thereby demonstrating that PLX3397 is a clinically active FLT3 inhibitor that achieves clinical responses via suppression of FLT3 signaling. The majority of FLT3 KD mutations detected in relapse samples occurred at amino acid residues identified in our in vitro mutagenesis screen. Three patients (1.14, 5.03 and 7.08) also harbored low frequency mutations of uncertain significance at other residues in addition to those implicated in PLX3397 resistance (Table 1; Supplemental Table 6). However, the F691L substitution was not observed in any of the samples. Additionally, a striking degree of polyclonality was present in some samples, including one (5.03) that exhibited seven distinct mutations at three amino acid residues in FLT3-ITD at the time of relapse, illustrating the critical reliance of this leukemia on FLT3-ITD kinase activity, as well as confirming the vulnerability of PLX3397 to a larger spectrum of FLT3 KD mutations compared to quizartinib. One patient (1.14) was noted to have a triple compound mutation M837G/S838R/D839H acquired at the time of relapse. Recreation of the D839H mutation alone and the compound M837G/S838R/D839H mutations in Ba/F3 cells confirmed ability to cause PLX3397 resistance (Supplemental Table 4). None of the identified mutations in any relapse sample were detectable on ITD-containing alleles in pre-treatment samples (Table 1).
Table 1.
FLT3 Kinase Domain Mutations Acquired at the Time of Resistance in Patients Who Relapse After Response to PLX3397
| Patient (By Study Number) | Best Response | Mutation (FLT3-ITD-alleles) | Mutation Frequency (of FLT3-ITD-alleles) | Number of ITD-Reads | Mutation (FLT3-ITD+ alleles) | Mutation Frequency (of FLT3-ITD+ alleles) | Number of ITD+ Reads | Mutation Frequency in Pre-Treatment Sample |
|---|---|---|---|---|---|---|---|---|
| ¥1.14 | CRi | None | N/A | 6958 | £ M837G | 31–35% | 1188 | 0% |
| £ S838R | 31–35% | 0% | ||||||
| £ D839H | 31–35% | 0% | ||||||
| 3.08 | CRp | None | N/A | 5617 | None | N/A | 1048 | N/A |
| 5.02 | CRi | D835Y | 37.93% | 3417 | D835Y | 10.50% | 895 | 18.59% (ITD-alleles only) |
| 5.03 | PR | N609K | 1.19% | - | - | 0% | ||
| T628I | 1.19% | - | - | 0% | ||||
| N676K | 2.17% | N676K | 6.05% | 0% | ||||
| N676T | 1.58% | N676T | 1.76% | 0% | ||||
| D835H | 8.30% | 506 | D835H | 15.52% | 5341 | 0% | ||
| D835Y | 6.32% | D835Y | 8.43% | 0% | ||||
| D835V | 2.37% | D835V | 3.37% | 0% | ||||
| - | - | D835E | 2.04% | 0% | ||||
| - | - | N841Y | 1.52% | 0% | ||||
| 7.04 | PR | None | N/A | 102 | None | N/A | 87 | N/A |
| 7.08 | PR | - | - | S652G | 2.35% | 0% | ||
| N676K | 55% | 4885 | N676K | 23.53% | 89 | 0% | ||
| - | - | E708G | 2.35% | 0% | ||||
| - | - | T820A | 2.38% | 0% | ||||
| 7.11 | PR | None | N/A | 3958 | None | N/A | 2436 | N/A |
| 8.02 | CRi | D835Y | 37.10% | 6497 | - | - | 19 | 0% |
| 9.06 | CR | - | - | N676K | 1.00% | 0% | ||
| D835E (GAT->GAA) | 6.94 | 3693 | D835E | 26.26% | 1007 | 0% | ||
| D835E (GAT->GAG) | 5.68 | D835E | 16.41% | 0% | ||||
| D835H | 3.23 | D835H | 10.26% | 0% | ||||
| D835Y | 2.17 | D835Y | 7.59% | 0% |
All patients treated at dose of 1400mg or higher. All patients treated with PLX3397 who responded with PR or better and subsequently relapsed on study drug are included. PR = partial remission; CR = complete remission; CRi = CR with incomplete neutrophil recovery; CRp = CR with incomplete platelet recovery.
M837G/S838R/D839H mutations all co-occurred in compound on the same allele. Mutation frequency expressed as a range due to differences in local sequencing quality for each codon.
Multiple additional low frequency (~1–2%) mutations also found in this sample. For details, see Supplemental Table 6.
In four cases, PLX3397-resistant KD mutations were found in both ITD-containing (ITD+) and non-ITD-containing (ITD-) alleles, consistent with prior observations made by our group (unpublished data). In one sample obtained from a patient (8.02) treated with a relatively low dose of PLX3397 (1400mg), a D835Y mutation was identified only in ITD-alleles at relapse, which comprised the majority of the relapse sample (6497 ITD-reads versus 19 ITD+ reads). The pre-treatment sample from this patient was also comprised of a majority of ITD-alleles (3844 ITD- vs. 11 ITD+ reads), and no activating mutations in other PLX3397 targets (CSF1R or KIT) were observed (data not shown). These findings suggest that this patient may have responded primarily through inhibition of native FLT3, a phenomenon described with other FLT3 inhibitors (11). Interestingly, patient 5.02, who had a detectable D835Y mutation in ITD-alleles pre-treatment, achieved clearance of bone marrow blasts on 2000 mg of PLX3397, but subsequently relapsed with a highly resistant ITD+D835Y clone. These observations are consistent with our finding that D835V/Y mutations cause a lesser degree of resistance to PLX3397 in the absence of an ITD (Supplemental Figure 7a,b) and suggest that these mutations may retain clinical sensitivity to higher doses of PLX3397.
In three of the nine cases we analyzed, no secondary FLT3 KD mutations were identified at the time of PLX3397 resistance, suggesting that in these cases, non-FLT3-dependent resistance mechanisms may drive therapeutic resistance. However, in these patients, examination of the KD of other PLX3397 targets KIT or CSF1R revealed no mutations (data not shown). Efforts to define off-target, non-FLT3-dependent mechanisms of resistance in these samples are currently ongoing.
Clinically Achieved PLX3397 Concentrations Profoundly Inhibit the Kinase Activity of the FLT3-ITD F691L Mutant
The lack of secondary F691L mutations as a mechanism of acquired clinical resistance to PLX3397 in all nine patients available for analysis strongly suggests that this mutation can be effectively suppressed by PLX3397. In further support of this concept, steady-state plasma from two patients treated with PLX3397 at the phase 2 dose of 3000 mg contained sufficient drug to completely inhibit FLT3 phosphorylation in Molm14 F691L cells (Figure 5).
Figure 5. Plasma Inhibitory Assay Shows PLX3397 is Active Against FLT3-ITD/F691L at Clinically Achievable Plasma Concentrations.
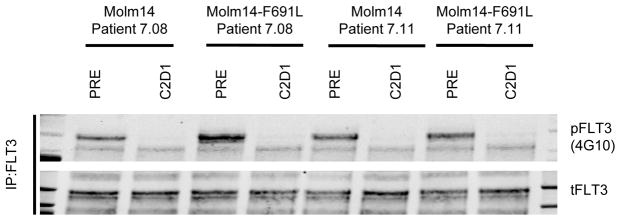
Western blot analysis for phosphotyrosine and total FLT3 performed after immunoprecipitation using anti-FLT3 antibody on lysates prepared from parental Molm14 cells and Molm14 cells expressing the FLT3-ITD/F691L mutation. Cells were exposed for 120 minutes to pre-treatment (PRE) and steady-state plasma from cycle 2 day 1 (C2D1) obtained from 2 patients (7.08 and 7.11) treated with PLX3397 at the phase 2 dose of 3000mg daily. Data shown represents a single experiment.
Discussion
The development of therapeutic resistance to clinically effective cancer therapies is a pervasive problem that has challenged cancer biologists since effective treatments were first identified. In the era of targeted therapy, mutations that prevent drug binding, particularly at kinase gatekeeper residues, have negatively impacted the durability of response achieved with numerous clinically active TKIs. The development of kinase inhibitors that retain activity against resistance-conferring KD mutations therefore represents a critical challenge.
Overriding clinical resistance through rational compound design necessitates a detailed structural understanding of the target kinase and its interaction with inhibitors. From a structural perspective, FLT3 is one of the least understood of all clinically relevant kinase targets, with only one reported crystal structure obtained in the inactive apo, non-ligand bound form. Here we report the co-crystal structure of FLT3 complexed with quizartinib, the first highly clinically active FLT3 TKI. This structure reveals features of quizartinib binding to FLT3 that differ from how the prototypic type II inhibitor imatinib interacts with BCR-ABL, including water-mediated interactions with the kinase hinge and remarkably optimized interactions with the ATP binding pocket and the allosteric pocket. Unexpectedly, the orientation of quizartinib bound to FLT3 is completely opposite to that predicted by previous molecular docking studies. Most surprisingly, the interaction of quizartinib with FLT3 appears to depend upon two edge-to-face interactions with the aromatic side-chains of the catalytically crucial F830 residue in the kinase activation loop and the F691 gatekeeper residue. Only strong disruptions of this critical interaction through substitutions at F691 and at the D835 residue, which is key to stabilizing the inactive conformation of the activation loop, are sufficient to induce clinical resistance to quizartinib while substitutions in other less critical regions of interaction appear insufficient to cause clinical resistance. The clinically active FLT3 inhibitor PLX3397, which relies upon a larger number of weak interactions for effective binding, nonetheless tolerates clinically problematic substitutions at the F691 residue. While this feature enables PLX3397 to retain activity against mutations at F691, its vulnerability to KD mutations at other residues is increased as a consequence.
Ultimately, the prevention of therapeutic resistance due to KD mutations may require combination therapy with clinically active inhibitors that exhibit non-overlapping resistance profiles. By virtue of its ability to retain activity against gatekeeper F691 substitutions, PLX3397 holds promise as a critical cornerstone therapy, that used in conjunction with an inhibitor that retains activity against FLT3 activation loop mutants may minimize KD mutation-mediated resistance and thereby improve response durability and therapeutic outcomes. To that end, our studies promise to further facilitate the rational development of the next generation of potent FLT3 inhibitors.
Materials and Methods
Inhibitors
Quizartinib/AC220, 1-(5-(tert-Butyl)isoxazol-3-yl)-3-(4-(7-(2-morpholinoethoxy)benzo[d]imidazo[2,1-b]thiazol-2-yl)phenyl)urea, was purchased from ChemShuttle (Wuxi, Jiangsu Province, China) and PLX3397, 5-[ (5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)methyl]-N-[[6-(trifluoromethyl)-3-pyridyl]methyl]pyridin-2-amine, was synthesized following the published procedures(25).
Quizartinib and FLT3 Co-Crystal Structure Determination
To enable crystallization, FLT3 kinase domain containing residues H564-V958 with internal deletion H711–H76l, an N-terminal 6×His-tag(19), and a TEV cleavage site was expressed in baculovirus-infected Sf9 insect cells. FMS proteins were purified through a combination of IMAC and further purified by ion-exchange and size-exclusion chromatography (SEC). The FLT3 SEC pool was auto-phosphorylated by the addition of 1 mM ATP and 5 mM MgCl2 (1 hour incubation at 4°C). Following kinase activation, the FLT3 protein was incubated with 100 μM quizartinib for 1 hour at room temperature. The protein was then concentrated to a final concentration of 10 mg/mL. Crystals were grown with vapor diffusion sitting drop method and incubated at 293K. Crystallization drops contained 200nl FLT3 native protein (10mg/ml) co-concentrated with quizartinib and mixed with 200nl of reservoir solution containing 100mM Na-Citrate, pH5.5 and 20% PEG3000. 0.10–0.15mm long needles were observed after 12 weeks. Crystals were cryo protected with 25% glycerol before freezing. Diffraction data was collected at the Advanced Light Source (Berkeley, CA) beamline 8.3.1. FLT3-quizartinib costructure was solved by molecular replacement with the program MOLREP and using the structure of apo-FLT3 (Protein Data Bank ID code 1RJB) as a starting model. The structure was refined with the program PHENIX. Cycles of manual rebuilding with COOT and structure refinement were continued until there was no further improvement, indicated by the convergence of the Rfree factor. The crystallographic data and refinement statistics are provided in Supplemental Table 1.
Model of PLX3397, sorafenib and ponatinib bound to FLT3
All three compounds have been previously co-crystallized with other class III receptor tyrosine kinases (PLX3397 in CSF1R, sorfenib in KDR, and ponatinib in KIT). Given the high degree of homology between these kinases and FLT3, we expect these inhibitors will exhibit the same overall binding modes. For each inhibitor, we performed structural alignment between the known co-structure containing the inhibitor and the co-structure of FLT3-quizartinib with an emphasis on optimal registration of ATP-binding apparatus. The pose of the inhibitor was then “copied” from its co-structure with another kinase and “pasted” into FLT3 to replace quizartinib. One round of energy minimization using the default parameter of MOE (Chemical Computing Group, Montreal, Quebec, Canada H3A 2R7) was used to refine the structural model.
DNA Constructs, Mutagenesis and Resistance Screen
Random mutagenesis was performed as previously described(10). Cells were selected in 2μM PLX3397 in soft agar. After 10–21 days, visible colonies were plucked and expanded in 2μM PLX3397.
Sequencing and Alignments
Sequencing was performed from amplified genomic DNA from colonies expanded from soft agar as previously described(10).
Generation of Mutants
Mutations isolated in the screen were engineered into pMSCVpuroFLT3-ITD by QuikChange mutagenesis (Agilent Technologies; Santa Clara, CA) according to manufacturers recommendations.
Cell Lines
Stable Ba/F3 lines were generated by retroviral spinfection with the appropriate mutated plasmid as previously described(10). Ba/F3 cell lines were originally obtained from the laboratory of Charles Sawyers in 2006 and have not been authenticated. MV4;11 and Molm14 cells for in vitro proliferation and Western Blot assays were obtained from the laboratory of Scott Kogan in 2008 and authenticated by Promega STR analysis in May 2014. All cell lines were mycoplasma-free.
MV4;11 Xenograft Study
MV4;11 cells were purchased from ATCC in April 2008 and have not been authenticated. Cells were expanded to a density of 0.5×106/ml. Cells were washed three times with PBS after which the cells were resuspended in to the final density of 50×106 cells/ml in PBS before inoculation. 8 male mice per group (24 mice total for vehicle and two dose groups), nu/nu outbred, 6 weeks old. With a sample size of 8 mice per group, it provides at least 80% power to detect a 2 standard deviation difference of mean tumor volume between two group with t-sided type I error = 1%. Feed is Laboratory Rodent Diet. Water is municipal tap water provided ad libitum. Housing is 18–26°C and 50±20% relative humidity, in rooms with at least ten room air changes per hour. Photoperiod is diurnal; 12 hours light, 12 hours dark. Mice were injected with MV4;11 cell suspension in 100 μL PBS + 100 μL Matrigel (5×106 cells per mouse) in the lower left abdominal flank. Treatments (vehicle and PLX3397 at two dose levels, all given daily via oral gavage) started when the average tumor size reached 125 mm3. Animals in the 3 groups of 8 were randomly assigned to match mean tumor volume measurements. Dosing duration is 21 days. Tumor measurements will be taken with electronic microcaliper 3 times weekly (Monday, Wed and Fri). Investigators were not blinded to animal group assignment. All animal studies were conducted in accordance with the Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and the US Department of Agriculture Animal Welfare Act and local animal ethics committees.
Cell-Viability Assay
Exponentially growing cells (5 × 103cells/well for Ba/F3, 1 × 104cells/well for others) were plated in each well of a 96-well plate with 0.1 mL of RPMI 1640 + 10% FCS containing the appropriate concentration of drug in triplicate. Cells were allowed to expand for 2 or 3 days (as indicated) and cellular proliferation was assessed using ATP content as a measure of cell viability using either CellTiter-Glo reagent (Promega; Madison, WI) or ATPlite 1step Luminescence Assay reagent (Perkin-Elmer) according to manufacturer’s recommendation on a SpectraMax M3 microplate reader using SpectraMax Pro Software (Molecular Devices; Sunnyvale, CA). The value at varying concentrations of drug was normalized to the median value in the no-drug sample for each mutant and a mean value was calculated. Numerical IC50 values were generated using non-linear best-fit regression analysis using Prism 5 software (GraphPad; San Diego, CA). IC50 values reported are the calculated mean result of 3 or more independent experiments. All cell viability data shown is representative of experiments replicated 3 or more times.
Immunoblotting
Exponentially growing Ba/F3 cells stably expressing FLT3 mutant isoforms were plated in RPMI medium 1640 + 10% FCS supplemented with kinase inhibitor at the indicated concentration. After a 90-minute incubation, the cells were washed in phosphate buffered saline (PBS) and lysed in buffer (50mM HEPES, pH 7.4, 10% glycerol, 150mM NaCl, 1% Triton X-100, 1mM EDTA, 1mM EGTA, 1.5mM MgCl2) supplemented with protease and phosphatase inhibitors. The lysate was clarified by centrifugation and quantitated by BCA assay (Thermo Scientific; Rockford, IL). Protein was subjected to sodium dodecyl sulfate polyacrylamide electrophoresis and transferred to nitrocellulose membranes. Immunoblotting was performed using anti-phospho-FLT3 (3464), anti-phospho-STAT5 (9351), anti-STAT5 (9363), anti-phospho-S6 (2211), anti-S6 (2317), anti-phospho-ERK (9101), anti-ERK (9102) (Cell Signaling; Beverly, MA), and anti-FLT3 S18 antibody (Santa Cruz Biotechnology; Santa Cruz, CA). Data shown in figures is representative of 3 technical replicates.
Plasma Inhibitory Assay
Plasma inhibitory assay was performed as previously described(26) with some modifications. Briefly, 7 × 106 parental Molm14 cells and Molm14 cells harboring the indicated mutations were re-suspended in 1 mL of plasma from either a healthy control containing the indicated concentration of drug or a patient treated with the indicated dose of inhibitor at steady state. All samples were collected under the UCSF institutional review board (IRB)–approved cell banking protocol (CC#112514). Informed consent was obtained in accordance with the Declaration of Helsinki. Cells were incubated at 37°C for 2 hours. FLT3 was immunoprecipitated from 400ug of total protein using anti-FLT3 S18 antibody and used for Western immunoblot with anti-FLT3 and anti-phospho-tyrosine antibody 4G10 (EMD Millipore; Billerica, MA). Remaining lysate was normalized and used for Western immunoblot as above.
Patients and sequencing analysis
Nine cases of acquired resistance to PLX3397 were analyzed, including all known patients with bone marrow responses who subsequently relapsed while still on drug treatment. Patients were enrolled on the multi-center Phase I/II clinical trial of PLX3397 in relapsed or refractory AML (NCT01349049). The clinical trial protocol, including analysis of resistance samples, was approved by the institutional review board of all participating institutions. Details of the clinical trials and results will be reported elsewhere. All patients were positive for FLT3-ITD mutation and negative for D835 mutation by standard clinical testing at the time of enrollment. Eligibility for the study required that patients be unsuitable for, relapsed or refractory to standard induction chemotherapy. Samples were collected pre-treatment and at the time of disease progression. Only patients who had achieved bone marrow partial (PR) or composite complete remission (CRc = CR + Cri + CRp) using modified International Working Group Criteria were analyzed.
Complete remission (CR) was defined as patients with bone marrow regenerating normal hematopoietic cells and a morphologic leukemia-free state with an absolute neutrophil count (ANC) >1 × 109/L and platelet count ≥ 100 × 109/L, and normal marrow differential with < 5% blasts, and red blood cell (RBC) and platelet transfusion independent (defined as 4 weeks without RBC transfusion and 1 week without platelet transfusion) with no evidence of extramedullary leukemia. CRp was defined as CR except for incomplete platelet recovery (<100 × 109/L). CRi was defined as CR except for incomplete hematological recovery with residual neutropenia (ANC ≤ 1 × 109/L) with or without residual thrombocytopenia (platelet count <100 × 109/L). PR was defined as bone marrow regenerating normal hematopoietic cells with evidence of peripheral recovery with no (or only a few regenerating) circulating blasts and with a decrease of at least 50% in the percentage of blasts in the bone marrow aspirate with the total marrow blasts between 5% and 25%. For CRp, CRi and PR, patients did not need to be RBC or platelet transfusion independent. All patients gave informed consent according to the Declaration of Helsinki to participate both in the clinical trials and for collection of samples.
For sequencing, frozen Ficoll-purified mononuclear cells obtained from blood or bone marrow were lysed in Trizol (Invitrogen) and RNA was isolated according to manufacturer protocol. cDNA was synthesized using Superscript II (Invitrogen) per manufacturer’s protocol. The FLT3 kinase domain and adjacent juxtamembrane domain were PCR amplified from cDNA as previously described(10). The CSF1R and KIT kinase domains were amplified from cDNA and sequenced by direct sequencing. Primer sequences are available upon request.
Sample Preparation and Sequencing
PCR product containing the FLT3 kinase domain was generated from patient cDNA as previously described above using high fidelity DNA polymerase(10). We prepared PCR products for Pacific Biosciences single-molecule real time sequencing(27) using standard commercial kits and reagents and sequenced on Pacific Biosciences RSII instrument as previously described, but using P4-C3 enzyme and chemistry alongside 3-hour collection times for CCS sequencing (10).
Computational Analysis of FLT3 Mutations
Data from SMRT sequencing was analyzed using an extension to the methods previously described in Smith et al.(10). CCS reads are separated by sample using barcoded primer sequences; and all reads with full length alignment to the targeted FLT3 interval are oriented and passed for downstream ITD analysis. We employ a variant of “dot plotting”(28), which is commonly used to visually compare DNA sequences; a match (dot) between two sequences is defined as a shared substring of size 10, but extensions to exact matches are permitted for 10 mers with up to 2 mismatches. This enables us to detect perfectly tandem duplications of a single codon and non-tandem duplications of 10 bp or more. In short, for a target sequence (t) and a query sequence (q), t is dot plotted against itself and q. The two resulting matrices are then summed column-wise, to generate a pair of distinct vectors, T and Q, of length |t|. T and Q represent the relative abundance of particular substrings in t and q, respectively, projected onto the target interval. Next, we normalize Q by element wise division of T, yielding QNorm. A two-state hidden markov model (HMM) is then applied on QNorm in order to identify putative intervals of duplication. Once all reads have been partitioned by sample into ITD+ and ITD-sets, variant analysis is performed using Quiver (29) on each read subset to identify candidate mutations.
Code Availability
Pacific Biosciences sequencing data was processed, mapped, and variant called using SMRT Analysis version 2.0.1. Custom scripts for detecting ITDs were written using Python version 2.7 and Cython version 0.21.1 and are available upon request.
Supplementary Material
Statement of Significance.
We report the first co-crystal structure of FLT3 with a kinase inhibitor, elucidating the structural mechanism of resistance due to the gatekeeper F691L mutation. PLX3397 is a novel FLT3 inhibitor with in vitro activity against this mutation but is vulnerable to kinase domain mutations in the FLT3 activation loop.
Acknowledgments
Financial support: This work was supported by grants from the National Cancer Institute (1RO1 CA166616-01) (N.P.S), the NIH T-32 Molecular Mechanisms of Cancer (E.A.L.), and the Leukemia and Lymphoma Society (C.C.S and N.P.S). C.C.S is an ASH Faculty Scholar and recipient of a Hellman Family Foundation Early Career Faculty Award.
N.P. Shah wishes to acknowledge the generous support of Arthur Kern, Mark Maymar, and the Edward S. Ageno family. This work is dedicated to Ilana Massi.
Grant Support. C.C.S is an American Society of Hematology Faculty Scholar and recipient of a Hellman Family Foundation Early Career Faculty Award. This work was supported by grants from the National Cancer Institute (1R01 CA166616-01) (N.P.S), the NIH T-32 Molecular Mechanisms of Cancer (E.A.L.), and the Leukemia and Lymphoma Society (C.C.S and N.P.S).
Abbreviations
- TKI
tyrosine kinase inhibitor
- KD
kinase domain
- FLT3
Fms-Like Tyrosine Kinase 3
- AML
acute myeloid leukemia
- ITD
internal tandem duplication
- CR
complete remission
- SEC
size exclusion chromatography
Footnotes
Author Contributions. C.C. Smith and C. Zhang designed experiments, performed research, analyzed data and wrote the manuscript. N.P. Shah, B.L. West, A. Kasarskis and G. Bollag designed experiments, analyzed data and wrote the manuscript. K. Lin, Y. Zhang, E.A. Lasater, E. Massi, L.E. Damon, R. Shellooe, G. Tsang, H. Carias, B. Powell, E.A. Burton, B. Matsuow, J. Zhang, W. Spevak, P.N. Ibrahim, G. Habets and R. Sebra performed research and reviewed the manuscript. A. Bashir and M. Pendleton performed bioinformatics analysis and wrote the manuscript. B.L. West, H.H. Hsu and M.H. Le analyzed and provided clinical data. A. Perl provided reagents, performed research and reviewed the manuscript.
Conflicts of interest: C.C.S and A.P. (research funding associated with conduct of a clinical trial, Plexxikon); N.P.S. (research funding associated with the conduct of clinical trials, ARIAD Pharmaceuticals, Ambit Biosciences; research funding, Plexxikon, Daiichi-Sankyo). C.Z., Y.Z., R.S., G.T., H.C., B.P., E.A.B., B.M., W.S., P.N.I, M.H.L, H.H., G.H., B.L.W. and G.B. are employees of Plexxikon, Inc.
Disclosure of Potential Conflicts of Interest. C.C. Smith and A. Perl have received research funding for the conduct of clinical trials from Plexxikon, Inc. N.P. Shah has received research funding for the conduct of clinical trials from ARIAD Pharmaceuticals and Ambit Biosciences. N.P. Shah has received research funding from Plexxikon, Inc. and Daiichi-Sankyo. C. Zhang, Y. Zhan, R. Shellooe, G. Tsang, H. Carias, B. Powell, E.A. Burton, B. Matusow, W. Spevak, P.N. Ibrahim, M.H. Le, H.H. Hsu, G. Habets, B.L. West and G. Bollag are employees of Plexxikon, Inc. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–25. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 2.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–14. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- 4.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, et al. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS chemical biology. 2014;9:1230–41. doi: 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–42. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 7.Burgess MR, Skaggs BJ, Shah NP, Lee FY, Sawyers CL. Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc Natl Acad Sci U S A. 2005;102:3395–400. doi: 10.1073/pnas.0409770102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith CC, Lasater EA, Lin KC, Wang Q, McCreery MQ, Stewart WK, et al. Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1320661111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(TCGA) TCGARN. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31:3681–7. doi: 10.1200/JCO.2013.48.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119:5133–43. doi: 10.1182/blood-2011-06-363960. [DOI] [PubMed] [Google Scholar]
- 13.Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123:94–100. doi: 10.1182/blood-2013-10-529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122:3607–15. doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dewey TM, Bowers B, Thourani VH, Babaliaros V, Smith CR, Leon MB, et al. Transapical aortic valve replacement for severe aortic stenosis: results from the nonrandomized continued access cohort of the PARTNER trial. Ann Thorac Surg. 2013;96:2083–9. doi: 10.1016/j.athoracsur.2013.05.093. [DOI] [PubMed] [Google Scholar]
- 16.Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127:294–9. doi: 10.1053/j.gastro.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 17.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–72. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 18.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–78. doi: 10.1016/s1097-2765(03)00505-7. [DOI] [PubMed] [Google Scholar]
- 20.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571) Cancer Res. 2002;62:4236–43. [PubMed] [Google Scholar]
- 21.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 22.Zhou T, Commodore L, Huang WS, Wang Y, Thomas M, Keats J, et al. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem Biol Drug Des. 2011;77:1–11. doi: 10.1111/j.1747-0285.2010.01054.x. [DOI] [PubMed] [Google Scholar]
- 23.Chelli R, Gervasio FL, Procacci P, Schettino V. Stacking and T-shape competition in aromatic-aromatic amino acid interactions. J Am Chem Soc. 2002;124:6133–43. doi: 10.1021/ja0121639. [DOI] [PubMed] [Google Scholar]
- 24.Chao Q, Sprankle KG, Grotzfeld RM, Lai AG, Carter TA, Velasco AM, et al. Identification of N-(5-tert-butyl-isoxazol-3-yl)-N′-{4-[7-(2-morpholin-4-yl-ethoxy)imidazo[2,1-b][1,3]benzothiazol-2-yl]phenyl}urea dihydrochloride (AC220), a uniquely potent, selective, and efficacious FMS-like tyrosine kinase-3 (FLT3) inhibitor. J Med Chem. 2009;52:7808–16. doi: 10.1021/jm9007533. [DOI] [PubMed] [Google Scholar]
- 25.Tap W, Wainberg Z, Anthony S, Ibrahim P, Zhang C, Healey J, et al. Structure-guided Blockade of CSF1R Kinase in Tenosynovial Giant Cell Tumor. New England Journal of Medicine. 2015 doi: 10.1056/NEJMoa1411366. In Press. [DOI] [PubMed] [Google Scholar]
- 26.Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108:3477–83. doi: 10.1182/blood-2006-04-015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 28.Gibbs AJ, McIntyre GA. The diagram, a method for comparing sequences. Its use with amino acid and nucleotide sequences. European journal of biochemistry/FEBS. 1970;16:1–11. doi: 10.1111/j.1432-1033.1970.tb01046.x. [DOI] [PubMed] [Google Scholar]
- 29.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature methods. 2013;10:563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–30. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.