

Abstract
Mutations in the leucine‐rich repeat kinase 2 (LRRK2)‐encoding gene are the most common cause of monogenic Parkinson's disease. The identification of LRRK2 polymorphisms associated with increased risk for sporadic Parkinson's disease, as well as the observation that LRRK2‐Parkinson's disease has a pathological phenotype that is almost indistinguishable from the sporadic form of disease, suggested LRRK2 as the culprit to provide understanding for both familial and sporadic Parkinson's disease cases. LRRK2 is a large protein with both GTPase and kinase functions. Mutations segregating with Parkinson's disease reside within the enzymatic core of LRRK2, suggesting that modification of its activity impacts greatly on disease onset and progression. Although progress has been made since its discovery in 2004, there is still much to be understood regarding LRRK2′s physiological and neurotoxic properties. Unsurprisingly, given the presence of multiple enzymatic domains, LRRK2 has been associated with a diverse set of cellular functions and signalling pathways including mitochondrial function, vesicle trafficking together with endocytosis, retromer complex modulation and autophagy. This review discusses the state of current knowledge on the role of LRRK2 in health and disease with discussion of potential substrates of phosphorylation and functional partners with particular emphasis on signalling mechanisms. In addition, the use of immune cells in LRRK2 research and the role of oxidative stress as a regulator of LRRK2 activity and cellular function are also discussed.
Keywords: autophagy, cytoskeleton, genetics, GTPase, kinase, LRRK2, LRRK2 kinase inhibiton, retromer complex, signalling mechanisms, vesicle trafficking
Abbreviations
- 4E‐BP
4E binding protein
- AMK
adenosine monophosphate‐activated protein kinase
- COR
C‐terminal of the Roc‐domain
- DLP1
dynamin‐like protein 1
- EndoA
endophilin A
- LRRK2
leucine‐rich repeat kinase 2
- LRR
leucine‐rich repeat
- MAPK
mitogen‐activated protein kinase
- PD
Parkinson's disease
- ROC
Ras of complex
- ROS
reactive oxygen species
- TOR
target of rapamycin
- Wnt pathway
Wingless signalling pathway
Introduction
Parkinson's disease (PD) is an insidious and progressive neurodegenerative disease, affecting around 1–2% of the population over the age of 65 1. The vast majority of PD is sporadic in origin, with only 5–10% being familial 2; because age is the most significant risk factor for the development of the disease, and with an ever‐increasing life span in the western world, disease prevalence is likely to increase. Current treatments ameliorate symptoms but are not capable of slowing disease progression. Moreover, it has been estimated that by the time that motor symptoms emerge, 50–70% of substantia nigra dopaminergic neurons have already degenerated 3; damage which is currently irreversible. It is clear that there is a direct need to increase understanding of PD aetiology and the molecular mechanisms of pathogenesis in order to achieve early diagnosis and identify potential neuroprotective therapies.
Mutations in the gene encoding for leucine‐rich repeat kinase 2 (LRRK2) are the most frequent cause of familial PD 4. LRRK2‐PD has an almost indistinguishable clinico‐pathological phenotype from sporadic PD regarding age of onset, the presence of Lewy bodies (although a minority of cases have been reported without Lewy bodies) 5 and responsiveness to dopamine replacement therapy. These observations, paired with the identification of LRRK2 polymorphisms associated with increased lifetime risk for developing sporadic PD 6, suggested that LRRK2 may provide a deep understanding of the molecular mechanisms of PD. The exact physiological role of LRRK2 is still unknown, although it has been implicated in many cellular functions. In the effort to tackle PD, there is a compelling need for a profound understanding of LRRK2 functions, as well as a description of the signalling pathways in which LRRK2 may be involved in diverse cell types, the identification of regulators of LRRK2 activity, binding partners and phosphorylation targets.
LRRK2 genetics, protein domain structure; kinase and GTPase activities
In 2004, LRRK2 was identified as the gene responsible for PD inheritance associated with the PARK8 locus 7, 8 and was found to be comprised of 51 exons, giving rise to a large (268 kDa) protein. Subsequently, many variants in LRRK2 primary structure have been identified, including dominant mutations segregating with familial PD that also occur in sporadic PD and in cancer 9, together with polymorphisms at the LRRK2 locus that increase the lifetime risk for the development of sporadic PD, but also inflammatory bowel disorder and leprosy 4, 10, 11.
LRRK2 is a multidomain protein encompassing two enzymatic functions at its core. The GTPase domain, comprising of Ras of complex protein (ROC) terminating with a spacer domain called the C‐terminal of the Roc‐domain (COR), is immediately followed by the kinase domain, belonging to the serine/threonine kinases. This enzymatic core is surrounded by protein–protein interaction domains comprising the armadillo, ankyrin and leucine‐rich repeat (LRR) domains at the LRRK2 N terminus 12. The LRRK2 C terminus harbours the WD40 domain, which is deemed essential for protein folding, thus controlling LRRK2 function and kinase activity 13 (Fig. 1). Interestingly, the dominant, pathogenic mutations described up to date, occur within the enzymatic core of LRRK2 (Fig. 1), suggesting that modification of LRRK2 activity greatly impacts PD onset and progression. The similarity in PD phenotype and age of onset between homozygous and heterozygous mutation carriers suggests that pathogenic mutations might act by conferring a toxic function on LRRK2 14, 15.
Figure 1.
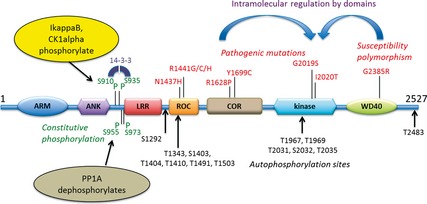
LRRK2 domian structure, pathogenic mutations, constitutive and autophosphorylation sites important for understanding LRRK2 function and dysfunction. Pathogenic mutations and susceptibility polymorphisms are shown in red, constitutive phosphorylation in green and a selection of autophosphorylation sites shown in black. The blue curved arrows depicts intramolecular regulation of kinase activity by other LRRK2 domians; IkappaB and casein kinase 1 alpha (shown in yellow) phosphorylate and PP1 alpha dephosphorylates (shown in fawn) LRRK2 at S910/S935 sites.
In the overall LRRK2‐PD population, the G2019S mutation is the most frequent pathogenic mutation 4. Its occurrence differs among groups; the G2019S mutation is rare in Asia, although it is relatively frequent in Southern Europe, reaching a maximum frequency in Ashkenazi Jewish (10–30% of PD patients are G2019S carriers) 16 and North African Berber populations (35–40% of PD patients are G2019S carriers) 17. The penetrance of the G2019S‐LRRK2 mutation appears to have a clear age‐dependent effect and varies from around 50% at age 50, to ~ 74% at age 79 16; although some patients do not manifest any clinical features even in their 80s 18. The G2019S mutation occurs in the kinase domain of LRRK2, leading to an increase in kinase activity 19. Cellular toxicity, in both the absence and presence of oxidative stress, and the formation of inclusion bodies were observed when overexpressing G2019S‐LRRK2 in cell lines and primary neuronal cultures 20, 21. These results, and the fact that genetic inactivation of LRRK2 kinase activity showed a protective effect against such a toxic phenotype, suggest that an alteration in LRRK2 kinase activity is potentially involved in the neurotoxic and pathogenic mechanisms of LRRK2‐PD. A second mutation in the kinase domain (I2020T) was isolated in the Japanese family in which the PARK8 locus was first described as being associated with PD 22. The effect of this mutation on LRRK2 kinase activity remains controversial 23, 24, 25 and, even with a confirmed increase in kinase activity, it would not be as striking as that reported for G2019S‐LRRK2 under the same experimental conditions 26. Nevertheless, I2020T‐LRRK2 has been shown to induce toxicity in overexpressing models 19, 27. Combined analysis of LRRK2 toxicity when carrying the G2019S or I2020T mutation suggests that an increase in LRRK2 kinase activity is sufficient per se, but not essential to trigger neurotoxicity.
The kinase function of LRRK2 is of particular interest, especially to pharmacologists, because kinases are typical targets of pharmaceuticals. The LRRK2 kinase domain is thought to assume a typical kinase fold, in which an N‐terminal and a C‐terminal sub‐domain can be identified, with the active site sitting in the cleft between the two. The activation loop, thought to be situated in the C terminus of the kinase domain 28, possesses a DYG motif. In some proteins, this motif undergoes conformational changes that may be related to kinase activity regulation 29. Interestingly, the G2019S mutation is situated within this segment of the activation loop and it has been speculated that the glycine residue here imparts conformational flexibility 30; therefore, replacement of the glycine with a serine residue may alter LRRK2 dynamics.
The R1441C, R1441G and R1441H mutations are located in the GTPase domain of LRRK2; the R1441G mutation is especially frequent in the Basque population where it accounts for > 40% of familial PD cases 31. Finally, the Y1699C mutation lies within the spacer domain, between the GTPase and kinase domains, and is responsible for one of the largest PD pedigrees in the UK with 25 affected subjects over four generations 32. The R1441C/G/H mutations showed decreased GTP hydrolysis 33, 34, as did the Y1699C mutation 35. Their impact on the kinase function of LRRK2 is still debatable 26; even if an increase in kinase activity is present, it is recorded as a moderate effect in comparison with G2019S‐LRRK2. R1441C, R1441G and Y1699C LRRK2 have been associated with cellular toxicity 27, 36 thus reinforcing the idea that kinase activity is not the only culprit for LRRK2‐induced neurotoxicity. Collectively, this suggests that the pathobiology of LRRK2 is likely to involve the entire enzymatic core, its activity, its folding and potentially its interactions with functional partners.
The presence of a double enzymatic core within the LRRK2 protein suggests that these two functions might influence each other's activities. Autophosphorylation of specific residues (Fig. 1) within the ROC domain has been found to modulate GTP binding 37 and it has been suggested that LRRK2 GTPase activity is regulated by its kinase activity. However, this should be viewed with some caution; autophosphorylation has been observed predominantly in in vitro assays as opposed to cellular systems. Moreover, if GTPase activity were regulated solely by autophosphorylation, it would be logical to assume that mutations within the kinase domain would subsequently alter GTP binding/hydrolysis. The hydrolysis of GTP to GDP has been shown to be altered in cell cultures expressing mutations in the GTPase domain 33, 35, 38. However, mutations such as G2019S, which occur in the kinase domain of the protein, do not disrupt this 39. Recently, LRRK2 kinase activity was shown to be dependent on GTP binding to the ROC domain 40. In addition, ARHGEF7, the rho guanine nucleotide exchange factor, was identified as an interactor of LRRK2 that could influence GTP hydrolysis activity 41, whereas the guanine exchange nuclear factor GAP (ArfGAP1) markedly reduced GTP hydrolysis and promoted the kinase activity of LRRK2 in vitro 42. Furthermore, using a systems biology approach, Dusonchet et al. 43 identified regulator of G‐protein signalling 2 (RGS2) as an interactor able to regulate LRRK2 kinase and GTPase activities in vitro in a synergistic manner. Clearly, the enzyme activities of LRRK2 undergo intramolecular regulations that can also be influenced by other LRRK2 interactors.
In addition to the known pathogenic mutations, there are a number of coding variants within the LRRK2 gene which are very rare, and some of these are also present in controls (see Paisan‐Ruiz et al. 18 for a detailed review on LRRK2 genetics). Among these, the coding variants G2385R in the WD40 domain and R1628P in the COR domain act as common PD risk factors among Asian populations 44, 45, 46. The G2385R variant essentially doubles the lifetime risk of getting PD 47. It has been shown that the C‐terminally truncated constructs that include the WD40 domain impacts on LRRK2 kinase activity, and a G2385R substitution resulted in ~ 50% loss of kinase activity 13. Interestingly, a combined G2019S/G2385R construct harboured kinase activity similar to WT‐LRRK2 protein, suggesting that these mutations were in fact opposing each other's functions 13. This is further suggestive of a complex interplay of intramolecular interactions within the LRRK2 molecule that is likely to reflect on cellular functions of LRRK2. The rare N1437H polymorphism has been found in Scandinavian families 48, 49, but the lack of detailed genetic data makes the pathogenic prediction uncertain. However, overexpression of the construct in HEK293 cells led to increased phosphorylation at Ser1292 50. The genome‐wide association study by Ross et al. 47 showed the LRRK2 locus to be an independent risk factor for sporadic PD. This important finding, together with a similar phenotypic spectrum of LRRK2 patients compared with sporadic cases 51, has fuelled the hypothesis that LRRK2 might also play a role in the pathogenesis of sporadic PD.
One final remark should be on LRRK2′s closest paralogue LRRK1. Despite rare variants in LRRK1 having been proposed to segregate with PD, there is no genetic support for the causal involvement of LRRK1 in disease 52. LRRK1 displays a similar domain organization as LRRK2 53, however, it has been observed that they have specific and independent interactors and are implicated in unique cellular pathways 54. Unfortunately, because of its nonpathogenic relevance, LRRK1 is not a subject of intense study like LRRK2. There are also limited tools developed to study LRRK1; antibodies are not as reliable as those validated for LRRK2, LRRK1 knockout mice 55 and double LRRK2/LRRK1 knockout mice are available (Jackson laboratories) but they have not been extensively studied and there are currently no LRRK1 kinase inhibitors. However, given the close similarity between LRRK1 and LRRK2 and the apparent absence of involvement of LRRK1 in PD, it would be extremely interesting to analyse functional differences between the two enzymes.
Importance of LRRK2 autophosphorylation and constitutive phosphorylation
Research efforts focussed towards finding molecular substrates of LRRK2 phosphorylation have proven difficult and at the moment, the only recognized target for LRRK2 kinase activity is LRRK2 itself. LRRK2 has been found to autophosphorylate > 20 serine and threonine residues in vitro 37, 50, 56, 57, 58, 59, 60, 61. The majority of the autophosphorylation sites reside in the ROC domain, with only a few in the COR and kinase domains (Fig. 1). The physiological relevance of these phosphorylation sites is still not clear; some of them have failed to be detected in vivo. In the cellular context, autophosphorylation was observed at T1410 61 and at Ser1292 and this was proposed as a potential measure of LRRK2 kinase activity 50.
Immediately prior to the LRRK2 ROC domain, there is a cluster of serine residues – Ser910, Ser935, Ser955 and Ser973 60, 62, 63, 64 (Fig. 1), which are constitutively phosphorylated; however, they are not autophosphorylation sites. Phosphorylation of these residues is affected by LRRK2 mutations in the ROC/COR/kinase domains, by LRRK2 kinase inhibition and also by extrinsic stressors 62, 65, 66; they are, therefore, used as indirect measures of LRRK2 kinase activity. Dephosphorylation of Ser910/Ser935 affects 14‐3‐3 binding and impacts on downstream signalling 65, 66. The Ikappa B family of kinases has been shown to phosphorylate LRRK2 at Ser910/Ser935 67 and more recently, casein kinase 1 alpha was proposed as the kinase that phosphorylates LRRK2 at these sites 68. However, Protein phosphatase 1A (PPIA) dephosphorylated LRRK2 at Ser910/Ser935 which was reversed using calyculin A 69, a PP1A inhibitor. Further confirmation of PP1A as the phosphatase for LRRK2 constitutive phosphorylation was shown when calyculin A prevented dephosphorylation of LRRK2 at Ser910/Ser935 as a consequence of arsenite‐induced oxidative stress 66. Finding kinases and phosphatases modulating LRRK2 function in a variety of brain cell types and under different cellular contexts will be an important avenue of LRRK2 research that should aid in our understanding of the pathobiology of LRRK2.
Signalling pathways through LRRK2
The presence of active kinase and GTPase domains surrounded by protein–protein interaction motifs, suggested analysing LRRK2 in the context of signalling pathways. Indeed, over the past decade, many different signal transduction cascades have been associated with LRRK2 (Fig. 2).
Figure 2.
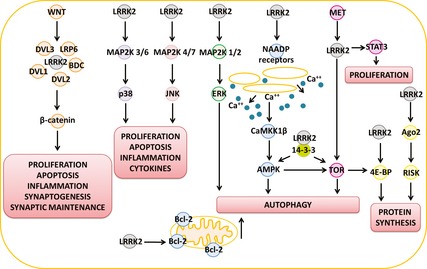
Implication of LRRK2 in signalling pathways. The cartoon depicts multiple signalling pathways that have been associated with LRRK2 function in physiology and/or disease. Signalling pathways are sometimes interconnected and they coordinate the control over multiple cellular activities as reported in the red boxes.
The mitogen‐activated protein kinase pathways (MAPK) were among the first to be investigated as potentially related to LRRK2. MAPK pathways are composed of three layers of proteins able to activate each other in a cascade. At the top layer is a kinase identified as MAP3K that is able to phosphorylate and activate a MAP2K in the second layer, which consequently activates the final MAPK, thus inducing a change in transcription. LRRK2 was able to bind to and phosphorylate MAP2K, ‐3, ‐4, ‐6 and potentially ‐7 in vitro 70, 71. Furthermore, a G2019S‐LRRK2 transgenic mouse model showed degeneration of dopaminergic neurons in the substantia nigra concomitant with hyperphosphorylation of MAP2K4 72. Activation of MAP2K3 and MAP2K6 is stimulated by stress, cytokines and growth factors, leading to the activation of the downstream effector p38. Activation of MAP2K4 and MAP2K7 is equally sensitive to stress, cytokines and growth factors, leading to the activation of the downstream effector JNK. JNK, and p38, are known to control cell proliferation and differentiation, apoptosis, inflammation, immune responses and the production of cytokines 73.
Another MAPK pathway was recently associated with LRRK2 after MAP2K1 and MAP2K2 (also known as MEK1 and MEK2) were found to be activated by G2019S‐LRRK2, leading to hyperphosphorylation of their effectors; ERK1 and ERK2. The alteration of this MAPK pathway was considered responsible for the G2019S‐LRRK2 mediated increase in basal autophagy 74.
The Wingless signalling pathway (Wnt) is responsible for the activation of the transcription factor β‐catenin, and is able to regulate nearly 400 genes 73 involved in cell growth, apoptosis, immune functions and inflammation, synaptogenesis during embryonic development and synaptic maintenance in adulthood; alterations in the Wnt pathway have been linked to a loss of synapses during Alzheimer's disease 75. First, the ROC–COR domain of LRRK2 was shown to interact with Dishevelled proteins 1, ‐2 and ‐3 (DVL1–3), key components of the Wnt pathway, whereas LRRK2 pathogenic mutations were sufficient to alter this binding 76. This association has been further characterized and LRRK2 is now hypothesized to have a role interacting with DVL1–3 and other proteins in the canonical Wnt cascade (LRP6 and BDC), thus enhancing the transduction of the Wnt signal 77.
It is interesting to note that LRRK2 is also involved in cancer signalling pathways. Thus, LRRK2 has been associated with MET signalling pathways in papillary renal and thyroid carcinomas; in particular, LRRK2 knockdown was shown to reduce tumour proliferation with concomitant increase in cell death, and reduction of MET signalling through the effectors target of rapamycin (TOR) and signal transducer and activator of transcription 3 (STAT3) 78. In this respect, MET is an oncogene tyrosine kinase receptor that controls multiple pathways involved in cell proliferation and actin organization. MET works by stimulating a variety of downstream effectors, among which are β‐catenin and ERK, thus linking MET with the Wnt and MAPK pathways 79.
LRRK2 has been suggested to modulate nicotinic adenine acid dinucleotide phosphate receptors, with consequent activation of a calcium signalling cascade (mediated by CaMKKβ) that would eventually activate adenosine monophosphate‐activated protein kinase (AMPK) to coordinate different cellular functions related to nutrient homeostasis and energetic balance 80. The CaMKKβ/AMPK pathway for induction of autophagy is sensitive to inhibition by endoplasmic reticulum‐located Bcl‐2 81; intriguingly, it has recently been proposed that G2019S‐LRRK2 may be able to bind to and phosphorylate Bcl‐2, with a consequent reduction of the mitochondrial membrane potential, thus stimulating mitophagy 82.
Human LRRK2 and its Drosophila orthologue were shown to be able to phosphorylate eukaryotic initiation transcription factor 4E binding protein (4E‐BP) 83. 4E‐BP is a downstream effector in the TOR pathway and its dephosphorylation during nutrient deprivation reduces protein synthesis. However, these data was not reproduced in mammalian cells 84. LRRK2 control over phosphorylation of 4E‐BP was not supported by analysis of sporadic or G2019S‐LRRK2 PD brains, or in LRRK2 knockdown or G2019S overexpressing mice 85. It is, therefore, possible to argue that results from in vitro assays and low‐complex model organisms may not be directly transferable to mammalian systems. However, there are other plausible hypotheses to justify this discrepancy; there may be specific signals needed to activate the function of LRRK2 over 4E‐BP in mammalian systems, or the involvement of LRRK2 within this branch of the TOR pathway may be cell specific, meaning that a cautious selection of cell type is needed to be able to detect it.
Knockin transgenic mice expressing G2019S‐LRRK2, as well as LRRK2 knockout mice, exhibited increased expression of mTOR in the kidneys, whereas knockin transgenic mice expressing a kinase inactive form of LRRK2 showed the opposite 86. In the same publication, changes were also described, in the kidneys, for 4E‐BP1 and for protein kinase B (Akt), another hub protein that shares functions with mTOR in the control of the cell metabolism. However, the tissue specificity of these alterations and the complexity of results coming from different genotypes, make the physiological relevance of these results difficult to contextualize without further investigations.
Another study in Drosophila proposed that LRRK2 associates with Drosophila Argonaute‐1 (dAgo1) and human Argonaute‐2 (hAgo2), thus modulating the RNA‐induced silencing complex 87. A recent finding suggested a component of the protein synthesis pathway, the ribosomal small subunit s15, to be phosphorylated by LRRK2 and sustain cell toxicity in both Drosophila model and human neurons 88. Interestingly, the same study reported that s15 was hyperphosphorylated in ribosomal fractions from a small number of G2019S brains compared with controls 88, although this would need further verification using a larger cohort. Another study performed by microarray‐based protein interaction technology and affinity purification coupled by tandem mass spectrometry isolated, among many other positive hits, few ribosomal proteins as possible LRRK2 interaction partners 54, even though they were not selected to be carried on to validation. Similarly, previous immunoprecipitation tandem mass spectrometry isolated, but did not validate, translation initiation factor 2C1 and 2C2 as possible LRRK2 interactors 89. Overall, these reports suggest a possible control of LRRK2 over protein synthesis; but nonetheless, these will need further functional confirmation in mammalian systems for a true physiological function.
A recent study in LRRK2 knockout mice showed that LRRK2 interacts with the PKARIIβ subunit of the protein kinase A holoenzyme regulating its localization. Aberrant localization of protein kinase A in knockout mice increased cofilin and glutamate receptor 1 (GluR1) phosphorylation, thus interfering with synaptogenesis and dopamine signalling through the dopamine receptor Drd1 90.
A final remark should be for the 14‐3‐3 proteins that have been demonstrated to be LRRK2 interaction partners involved in the regulation of LRRK2 cellular localization 62. 14‐3‐3 proteins are able to interact with a plethora of target proteins, thus supporting the function of many different signalling cascades; amongst which are the AMPK 91 and the TOR 92 pathways.
An interesting variety of signalling pathways have been associated with LRRK2 thus far from various reports (see Fig. 2), however, these data have to be considered cautiously because there is still incomplete agreement regarding their relevance. Reproducibility problems arise by the use of different cellular models, different species and sometimes in vitro kinase assays that have been difficult to replicate under expression of physiological levels of LRRK2. However, the intriguing idea behind this plethora of possible LRRK2‐modulated signalling pathways is that LRRK2 might play more than one role depending on the cell type in which it is expressed. Moreover, the presence of mutations in the LRRK2 sequence may cause a gain of novel functions, thus implicating LRRK2 in pathology pathways that may not be relevant under physiological conditions. This may support the existence of different functions for LRRK2 in PD, cancer and immune disorders, and it suggests caution in extrapolating general information from different model systems and from experiments involving the use of mutant LRRK2.
LRRK2 and cytoskeleton
Abnormalities in neurite outgrowth and branching were among the earliest observed LRRK2 cellular phenotypes 93, 94, 95. It was initially proposed that the source of such morphological changes could be a consequence of apoptotic processes 94; however, further studies provided evidence for an association of LRRK2 with tubulin/actin, thus suggesting that such morphological changes may be consequences of LRRK2‐modulation of cytoskeletal dynamics. The GTPase domain of LRRK2 was shown to pull‐down α/β tubulin from cell lysates 96; LRRK2 was coprecipitated with β tubulin from wild‐type mouse brain and recombinant LRRK2 has been proposed to phosphorylate β tubulin in vitro 97; a high‐throughput screening to decipher LRRK2 interactome revealed proteins of the actin family and from the actin‐regulatory network to be LRRK2 interactors with LRRK2 able to affect actin polymerization in vitro 98, and finally, LRRK2 carrying pathogenic mutations was found to decorate microtubules in cell models 99.
Further work has demonstrated that the interaction between LRRK2 and the cytoskeleton components is not just for the purpose of localization; LRRK2 was found to be able to modulate cytoskeletal dynamics. Disassembly of actin filaments in a process mediated by the GTPase Rac1, was observed in cell lines after LRRK2 knockdown or expression of mutant LRRK2 100. In neuronal cells from R1441G‐LRRK2 transgenic mice, as well as in G2019S‐LRRK2 fibroblasts, LRRK2 sensitized the actin cytoskeleton to depolymerizing agents 101. LRRK2 was shown to be able to phosphorylate moesin 25, a member of the ezrin/radixin/moesin (ERM) protein family involved in regulation of actin and microtubule structure; G2019S‐LRRK2 transgenic and LRRK2 knockout mice were shown to have alterations in the pool of filamentous actin in the filopodia as a consequence of alterations of ERM proteins phosphorylation 95.
R1441C and Y1699C‐LRRK2, but not G2019S or wild‐type LRRK2, were found to decorate nonacetylated microtubules in cell lines and to alter axonal transport in rat neuronal cultures and in Drosophila with a mechanism dependent on microtubules acetylation 102. LRRK2 binding to tubulin was associated with modulation of microtubule stability and acetylation 103. The stabilization of microtubules by LRRK2 may be mediated by LRRK2′s interaction with microtubule‐associated protein tau, because it has been demonstrated that LRRK2 is capable of phosphorylating tau in the presence of tubulin, thus altering microtubule–tau binding dynamics 104. Furthermore, introduction of human LRRK2 into a mouse model of tauopathy increased tau phosphorylation at various epitopes and changed its aggregation properties 105.
It is difficult to find a reoccurring theme with LRRK2 findings, and make unique sense of the results obtained in experiments performed with wild‐type LRRK2, mutated forms of LRRK2 and LRRK2 knockout. It is, therefore, difficult to determine whether the regulation of cytoskeleton dynamics is a normal LRRK2 physiological feature, if it is altered during disease, thus contributing to pathogenesis, or if it is a function that gains relevance during disease only. Moreover, it is still not clear whether mutations in the GTPase or kinase domain of LRRK2 affect the regulation of cytoskeleton dynamics to the same extent.
However, a putative function of LRRK2 in cytoskeletal dynamics is intriguing, not only because it could elegantly recapitulate morphological alterations observed in cellular models of LRRK2, but also because it lends to the possibility that LRRK2 may be involved in development and even govern different functions in development and adult life.
LRRK2 and autophagy
Autophagy was initially associated with LRRK2 when blocking macroautophagy through the knockdown of essential autophagy proteins (Atg7 and Atg8) was sufficient to attenuate the toxicity of overexpressed G2019S‐LRRK2 in SHSY5Y cells 106. A following report localized LRRK2 to autophagic vesicles and multivesicular bodies, whereas the knockdown of endogenous LRRK2 was found to be sufficient to induce macroautophagy in HEK293 cells 107. Numerous studies have followed this route of investigation, analysing the role of LRRK2 in autophagy. However, different approaches and model systems have been used to study LRRK2 and it is still not known whether the role of LRRK2 may be different throughout cell lines. The effect of LRRK2 overexpression may be different with respect to studies at endogenous levels, and it is not known whether the two enzymatic domains within LRRK2 orchestrate facilitating or opposing functions. Thus, although there is evidence implicating LRRK2 in autophagy, the extant literature is not sufficient, and at times controversial, in describing the molecular mechanisms through which this association happens. First, LRRK2 has been found to be a degradation substrate of chaperone‐mediated autophagy. Overexpression of G2019S or WT‐LRRK2 was able to reduce the payload of chaperone‐mediated autophagy, indicating that an accumulation of α‐synuclein, and misfolded proteins in general, as seen in PD, may be a partial consequence of a LRRK2‐mediated alteration of cellular proteolytic pathways 108. LRRK2 kinase inhibitors and knockdown 109, 110, 111, as well as LRRK2 overexpression in cell models 80, were able to modify the macroautophagic flux in vitro; however, it is still debatable whether LRRK2 possesses a positive or negative regulatory role in the control of macroautophagy and if the role of LRRK2 resides within the initiation or the clearance steps. This open debate has been further emphasized by the study of LRRK2 knockout animal models. Even though the brain of LRRK2 knockout mice did not recapitulate the pathological hallmarks of PD, a biphasic alteration in macroautophagy has been observed in the kidneys, with enhanced autophagy at young ages and reduced autophagy at old ages 112. The use of human fibroblasts carrying LRRK2 pathogenic mutations has confirmed an alteration in autophagy with reports suggesting an increase in basal macroautophagy in G2019S carriers 113, or an impaired response to starvation‐induced macroautophagy across mutations in the LRRK2 catalytic core (G2019S, Y1699C and R1441G) 114. The use of induced Pluripotent Stem cell (iPSC)‐derived, human dopaminergic neurons carrying G2019S‐LRRK2 has confirmed a reduction in macroautophagy in comparison with healthy controls 115; but again, details of the molecular mechanism underlying this are still ambiguous. The recent identification of potential interactors of LRRK2 such as Rab7L1, GAK, BAG5, Rab32 and endophilin A (EndoA) 116, 117, 118, and the description of an autophagy/lysosomal phenotype that can be corrected by Rab9 in mutant Drosophila 119 suggest that the study of LRRK2 in autophagy should probably be considered with a much wider prospective, taking into account a possible involvement of LRRK2 in vesicles dynamics in general.
LRRK2 function in vesicle dynamics and retromer function
Recent accruing evidence suggests a role for LRRK2 in vesicle dynamics and retromer function. LRRK2 was found to be associated with membranous structures and vesicles in the mammalian brain 120 and enriched in the Golgi complex 42, 94 at the extent that mice with wild‐type and G2019S‐LRRK2 overexpression presented fragmentation of the Golgi complex 121.
LRRK2 has been described as regulating synaptic endocytosis via association with Rab5b; siRNA knockdown of LRRK2 markedly reduced synaptic vesicle endocytosis 122, which was reversed by the introduction of Rab5B. In mammalian cells, interaction was seen between LRRK2 and the dynamin GTPase superfamily 123 involved in membrane scission during clathrin‐associated endocytosis. In Drosophila, LRRK2 was shown to phosphorylate EndoA, decreasing EndoA affinity for membranes and affecting EndoA‐dependent membrane tubulation. The G2019S mutation impeded synaptic endocytosis 116 that was restored by pharmacological inhibition of LRRK2 kinase activity in G2019S overexpressing flies. Knockout of EndoA led to neurodegeneration 73, thus linking LRRK2‐associated defects to PD. These results were recently further validated in mammalian cells in which LRRK2 was described as being able to phosphorylate neuronal‐specific EndoA1 124.
LRRK2 has been proposed to participate in the control of synaptic vesicle exocytosis by phosphorylating Snapin, and thus regulating soluble NSF attachment protein receptor (SNARE) complex functionality and late endosomal transport 125. Alterations in the amount of ready releasable vesicles have been described in cell models overexpressing G2019S‐LRRK2 126. LRRK2 silencing in primary cortical neurons showed altered vesicle‐recycling dynamics and increased vesicle kinetics, suggesting a role for LRRK2 in the control of vesicle pools within the presynaptic bouton 127. Furthermore, inhibition of LRRK2 kinase activity was proven to reduce neurotransmitter release, thus impacting on presynaptic functionality (103). Studies in LRRK2 knockout rats proposed LRRK2 to take part in the control of vesicles exocytosis in lung cells 128.
Recently, LRRK2 has been described as colocalizing with Sec16A, a protein involved in the formation of the endoplasmic reticulum exit site. Loss of LRRK2 led to a reduction in protein transport to the dendritic spine with a consequent reduction in glutamate receptors onto the synapse surface 129.
Two interesting studies by MacLeod et al. 130 and Beilina et al. 118 showed a genetic interaction between LRRK2 and Rab7L1; a genetic risk factor for sporadic PD. Expression of G2019S‐LRRK2 in primary neurons induced lysosomal swelling and accumulation of a component of the retromer complex; the mannose phosphate receptor 130. The mannose phosphate receptor is normally recycled between endolysosomes and the Golgi apparatus 131. The sorting defect was rescued by the overexpression of the retromer component VPS35, as well as the overexpression of Rab7L1 130. Subsequently, Rab7 was found in complex with LRRK2 to cooperatively promote clearance of Golgi‐derived vesicles through the autophagy–lysosomal system. The pathogenic mutations, G2019S, R1441C and Y1699C enhanced Golgi clearance, but the hypothesis‐testing mutations that decrease GTP binding, the T1348N, or the kinase‐inactive K1906M, did not sustain Golgi clearance, suggesting that both kinase and GTPase activities are required for maintaining this cellular process 118. Overexpression of VPS35 exhibited protective effects in mutant LRRK2 Drosophila 132; it is of interest to note that mutations in the VPS35 encoding gene have been identified in PD families 133, 134, further implicating the disruption of retromer mediated protein sorting as potentially leading to PD.
Many LRRK2 transgenic models have been created in an attempt to model PD. Although none have alterations resembling PD and the vast majority show no signs of neurodegeneration either, some exhibit a variety of synaptic alterations 135 such as altered striatal dopamine release and/or uptake 136, impairment of dopamine signalling through D2 receptors 137, impaired dopamine reuptake 138, alteration of glutamatergic transmission 139, impaired synaptic vesicles endocytosis with ultrastructural abnormalities in striatal neurons 124 and decreased extracellular dopamine levels in the presence of unaltered synthesis, storage and uptake 140. These findings may be seen as a confirmation of a putative LRRK2 function at the synapses; however, it is very difficult to harmonize different results coming from different models and combine them to eventually draw an exhaustive picture of the molecular mechanism supported by LRRK2. At the moment, we can only confidently state that the bulk of all these studies suggest that LRRK2 may be associated with a complex array of cellular functions involving vesicle dynamics, trans‐Golgi networks and autophagy/lysosomal homeostasis. The intriguing hypothesis is that the synergism of all these membrane dynamics may be controlled by LRRK2 and may be at the molecular base of PD neurodegeneration.
LRRK2, reactive oxygen species and mitochondria
LRRK2′s putative association with mitochondria suggests that it might play a role in mitochondrial dysfunction driving PD pathogenesis. Indeed, fibroblasts from PD patients carrying the G2019S mutation showed abnormal mitochondrial morphology 141. Similarly, wild‐type LRRK2 overexpression in SH‐SY5Y cells caused mitochondrial fragmentation, which was further exaggerated by the R1441C and G2019S mutations 142. In G2019S transgenic mice, ultrastructure examination showed an accumulation of damaged mitochondria, consistent with altered mitophagy in aged mice 143. In a double‐transgenic mouse expressing G2019S‐LRRK2 and A53T α‐synuclein, structural and functional abnormalities within the brain mitochondria suggested LRRK2 to induce a mitochondrial phenotype 121. Overexpression of G2019S‐LRRK2 in SH‐SY5Y cells caused mitochondrial uncoupling, leading to reduced membrane potential and increased oxygen consumption 144. Primary mouse cortical neurons expressing either G2019S or R1441C‐LRRK2 demonstrated increased mitophagy associated with altered calcium levels 145. Although human iPSC‐derived neurons carrying G2019S or R1441C‐LRRK2 showed normal mitochondrial electron transport chain, they showed increased vulnerability to chemical stressors and disrupted mitochondrial movement 146. LRRK2 overexpression caused the recruitment of dynamin‐like protein 1 (DLP1) protein to the mitochondria 142. Similarly, coexpression of DLP1 and LRRK2 induced increased oxidative stress, DLP1 relocation to the mitochondria and promoted mitochondria clearance 147. Finally, inhibition of DRP1 was able to rescue mitochondrial fragmentation in both G2019S expressing HEK cells and G2019S‐LRRK2 fibroblasts 148. These observations suggest that LRRK2 might be responsible for mitochondria homeostasis, possibly via DLP1‐dependent, mitochondrial quality control. However, it remains to be determined whether this truly acts as a primary pathogenic event in LRRK2‐PD, or if mitochondrial damage happens just as a secondary consequence of LRRK2‐induced toxicity.
The same causative hierarchy is yet to be determined for the association of LRRK2 with reactive oxygen species (ROS). Elevated ROS has been implicated as a pathological feature of PD; WT‐LRRK2 may be neuroprotective, attenuating H2O2‐induced cell‐death in HEK293 and SH‐SY5Y cells 149, whereas iPS cells carrying the G2019S mutation were found to be more sensitive to H2O2 exposure with increased caspase 3 activation and cell death 150. Similarly, mitochondrial dysfunction has been linked to increased ROS production in LRRK2 mutant cells 147.
Several mechanisms have been proposed to control the link between increased vulnerability to ROS and LRRK2 neurotoxicity. For example, increased kinase‐dependent interactions were shown between LRRK2 and two of its hypothetical substrates, DLP1 147 and peroxiredoxin 3 (PRDX3) 151. However, increased vulnerability to ROS was also described after overexpression of mutations outside the kinase domain as well as with kinase dead mutations in Caenorhabditis elegans 152, again suggesting that the molecular function of LRRK2 is likely to be governed by a delicate balance between kinase and GTPase activities. More recently, studies from our laboratory have shown that oxidative stress caused by arsenite led to altered biochemical properties of LRRK2 protein. Arsenite‐induced stress caused LRRK2 self‐association, inhibited its kinase activity, abrogated the GTP binding and translocated LRRK2 into centrosomes 66. In the context of exogenous stress and LRRK2 properties, it will be important to study other relevant PD stressors in different cellular phenotypes.
LRRK2 and the immune system
Despite LRRK2 having ubiquitous expression, substantial levels of LRRK2 protein and mRNA are present in peripheral blood mononuclear cells, lymph nodes, spleen 153 and primary microglia 154. There is no definitive description of LRRK2 function in immune cell lineages and how this may contribute to disease pathogenesis. It has been proposed that within the immune system, LRRK2 may be involved in the activation and maturation of immune cells 155, in controlling the radical burst against pathogens in macrophages 156, and in modulating neuroinflammation through cytokine signalling 157, 158. A more detailed link between LRRK2 and neuroinflammation in PD has been discussed recently by Greggio and colleagues 159.
The manifestation of these very specific functions of LRRK2 within the immune system opens up the intriguing hypothesis that LRRK2 may play different roles within different cell types and tissues. This scenario could be explained only by the presence of molecular mechanisms allowing the same LRRK2 protein to behave differently in different tissues; those mechanisms may be based on cell‐type‐specific LRRK2 differential splicing, or on tissue‐specific expression of LRRK2 activators, substrates and partners. Little is known about LRRK2 splicing and it may be reasonable to suppose that different cell types express different LRRK2 isoforms potentially involved in different cellular functions. The first study performed in mice showed indeed a differential expression of two splicing variants of LRRK2 in primary neurons, astrocytes and microglia 160, but more investigations are required.
Different cell lineages may express different LRRK2 partners able to specifically regulate LRRK2 levels and phosphorylation; features that are supposed to be linked with LRRK2 activation/repression. This assumption was demonstrated by the observation that LRRK2 expression and activity can be modulated via immune‐cell‐specific signalling pathways (Fig. 3). For example, LRRK2 expression can be induced in peripheral blood mononuclear cells by interferon‐γ 156, 161; or in monocytes after triggering their maturation to dendritic cells and macrophages 155. It was also demonstrated that in a macrophagic cell line (RAW264.7) and a microglial cell line (BV2), stimulation of toll‐like receptors 2 and 4 (TLR2, TLR4) was able to increase phosphorylation of LRRK2 and induce its recruitment to membranes 67, 109. Finally, toll‐like receptor 4 stimulation was able to increase LRRK2 expression and phosphorylation in primary rat microglia 158. LRRK2 was described as a negative regulator of nuclear factor of activated T cells (NFAT), a protein involved in transcriptional regulation in T cells, macrophages, dendritic cells and neutrophils 162; reinforcing the hypothesis that LRRK2 may have a precise role in the immune system because of the peculiar interaction partners through which it can exert tissue‐specific functions. The pathological implications of these observations are intriguing in two ways. First, this enforces the relevance of astrocytes and microglia in PD alongside with neurons. Indeed, activated microglia and monocytes, as well as increased cytokine levels, were reported in PD brains 26. Second, a putative role of LRRK2 in the regulation of the immune response may justify the genetic association of LRRK2 with the susceptibility to inflammatory bowel disorder 10 and leprosy 11 other than with PD.
Figure 3.
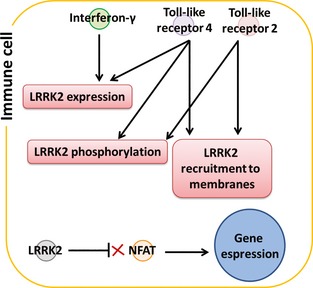
Implication of LRRK2 in immune‐specific functions. The cartoon summarizes the LRRK2‐specific events that have been described to occur in cells from the immune system.
LRRK2 kinase inhibition: therapeutic potential
The fact that LRRK2‐PD has an almost indistinguishable pathological phenotype from sporadic PD and the presence of a druggable kinase activity within LRRK2 were sufficient reasons for researchers to look at this protein as a potential target for a neuroprotective treatment in PD. Most notably, the development of selective pharmacological inhibitors of LRRK2 kinase activity is an active area of research with at least six potential LRRK2 inhibitors described in the literature and many leading pharmaceutical companies working on LRRK2 research programmes (see Ref. 163 for a review). Although some of the earlier inhibitors had rather limited use because of their nonselective effects in cells and their inability to cross the blood–brain barrier 164, 165, the development of LRRK2‐IN‐1 166 provided the first step towards a pharmacological tool to define the biological role of LRRK2, at least in cell models. On the flip side, LRRK2‐IN‐1 is not brain penetrant and also has inhibitory effects on ERK5 161, a critical enzyme and therefore ruling out its usefulness in clinical settings. Furthermore, it has recently been demonstrated that LRRK2‐IN‐1 exhibits significant off‐target effects, independently of LRRK2, including the inhibition of tumour necrosis factor alpha in astrocytes and increased neurite branching and length in neurons 167; perceived pathways of LRRK2 pathology. Not only does this highlight the problematic use of LRRK2‐IN‐1 when investigating LRRK2 function, but also negative off‐target effects in a therapeutic context.
Following the initial screening for LRRK2 inhibitor compounds, more brain‐penetrant LRRK2‐specific inhibitors were developed soon thereafter. The compound HG‐10‐102‐01(4) showed selective inhibition of WT and G2019S‐LRRK2 at micromolar concentrations in mouse brain and also inhibited Ser910/Ser935 phosphorylation 168. The compound GSK2578215A 169 was highly selective for LRRK2 kinase inhibition when compared with more than 450 other kinases tested. However, both these compounds have limited pharmacokinetic properties that exclude these from testing in clinical trials in humans.
Based on the structure of the HG‐10‐102‐01(4) compound, two further compounds were developed recently; the GNE‐0877 and GNE‐9605 170. Both these compounds demonstrated high selectivity, potency, brain penetrance and good metabolic clearance and stability when tested in vivo in rat models expressing human LRRK2 (see Ref. 171 for a review). The improved pharmacokinetic profile supported the hypothesis that these compounds can be safely tested in higher order animals and could potentially be entered into the preliminary stages of drug screening.
Clearly, the developments in identifying LRRK2 kinase inhibitors and a potential to treat PD have now entered an exciting phase but should be viewed with cautious optimism. A recent report demonstrated abnormal lung, kidney and liver pathology in LRRK2 knockout rats compared with rats expressing physiological levels of LRRK2 172. Crohn's T2397M‐mutant LRRK2 patients have reportedly lower levels of LRRK2 protein activity within their immune cells 162, suggesting that one harmful side effect of a complete inhibition of LRRK2 as a treatment for PD could be the development of intestinal‐immune diseases. What would be necessary now is the investigation of LRRK2 inhibitors in higher order mammalian models including their long‐term effects on the immune cells and peripheral organs in order to assess safety 173. The efficacy of such a therapy will also need to be demonstrated, and confirmation of the physical substrates of LRRK2 will aid in designing alternative activity assays for such inhibitors. As well, a general consensus of a model for LRRK2‐mediated pathology will need to be agreed upon in order for a validation criteria to be established 173. Importantly, for clinical use, the long‐term benefits of LRRK2 inhibitor treatment should outweigh the advantages of the already existing symptomatic treatments for PD 174. In this endeavour, it will be important to broaden the search for LRRK2 inhibition to include domains outside of the kinase domain 175, 176.
Concluding remarks
It is evident that there is still much to be understood about the LRRK2 protein regarding both its physiological and neurotoxic properties. A large variety of functions have been associated with LRRK2, both in terms of its physiological and cellular roles, and its pathological role during neurodegeneration (Fig. 4). These constitute an intricate network of LRRK2 putative functions making it central to two recent bioinformatics articles that have analysed LRRK2 interactome by means of protein–protein interaction databases and repositories of cellular pathways 177, 178. Instead of a clarifying mechanism of LRRK2‐PD, this wide range of implicated functions makes interpreting LRRK2′s role in disease even more challenging. Many questions are still unanswered. This is, in part, due to the complex protein structural domains of LRRK2, as well as the variety of models used in LRRK2 research. What needs to be resolved now is the ability to distinguish between LRRK2 physiological functions and those that are acquired during pathology, and to discriminate between primary and secondary pathophysiological events.
Figure 4.
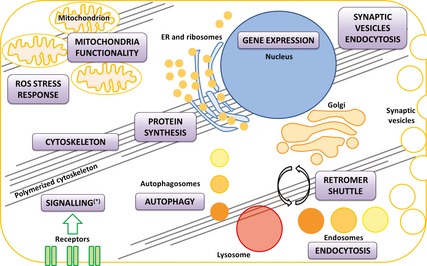
Implication of LRRK2 in cellular functions. The cartoon represents the cellular processes (red boxes) that have been associated with LRRK2 function in physiology and/or disease. The data are from a multitude of experiments run by laboratories across the world over the past decade and are based on different model systems and experimental approaches. It is not possible to describe a hierarchy among these processes, or score them based on reliability. More investigations are needed to determine which of these processes are directly controlled by LRRK2 and which appear to be LRRK2 regulated or whether they are just consequences of other LRRK2 primary functions. It remains to be determined if all of these functions co‐occur in a single cell type or whether LRRK2 orchestrate different, specific functions in different cell types.
It is still unknown whether LRRK2 may interpret different roles in different cell types, thus giving origin to cell‐specific phenotypes; in light of this, LRRK2 functions may be tissue specific, thus giving an explanation for the plethora of activities described up to now in diverse model systems (Fig. 4). Analysis of LRRK2 in different cell types is of extreme interest, as is the investigation of different LRRK2 isoforms in different brain regions, as recently shown by Trabzuni et al. 179 in control brains. This, together with further studies of LRRK2‐related biology associated with ROS and PD related toxins, phosphatases and kinases that modulate LRRK2 biology, should lead to increased understanding on LRRK2 function and dysfunction.
It will be crucial for future research on LRRK2 to consider the early events in neurodegeneration, as LRRK2 genetic penetrance varies between 30% and 80% depending on age. Although the loss of dopaminergic neurons is a key pathological characteristic of PD, it is preceded by many other dysregulations, which occur prior to the development of motor symptoms. It is the mechanism of LRRK2 associated with these events that needs to be uncovered. For example, LRRK2 has been associated with Wnt signalling pathways 76, which are essential for the acute regulation of synaptic function; a function that is dysregulated in the premotor symptom stages of rodent models of PD 180. From this prospective, the involvement of LRRK2 in Wnt signalling is an exciting possibility that may give a reason for the alteration in gene expression, as well as disruption of vesicle trafficking 181, which are implicated in PD. Another interesting connection is between LRRK2 and the autophagy/lysosomal/Golgi network, because alteration of the proteolytic balance within the cell may be the reason for the build‐up of toxic aggregates of amyloidogenic α‐synuclein, as implicated in PD. Indeed, the majority of LRRK2 mutation cases show an abnormal accumulation of α‐synuclein‐positive Lewy bodies 182, although it is unclear which α‐synuclein species is more relevant in G2019S pathology 183. Identifying signalling molecules that regulate the normal and pathophysiological functions of LRRK2 is a critical unmet need for developing novel therapies and the choice of the most relevant disease models will be critical in this endeavour.
Finally, it is important that all progress in LRRK2 research is interpreted carefully. The development of LRRK2 kinase inhibitors gives us cause for optimism for potential treatment for PD, but clearly the effects of inhibiting kinase activity should be gauged in the context of the entire LRRK2 protein.
Author contributions
Rebecca Wallings and Claudia Manzoni have contributed equally to the review. Rina Bandopadhyay has thoroughly revised and proof read the manuscript. All three authors have contributed to parts of the manuscript.
Acknowledgements
The authors acknowledge critical reading of the manuscript by Professor John Hardy (UCL, ION), Dr Mark Cookson (NIA/NIH, Bethesda) and Dr Patrick A. Lewis (University of Reading). The authors would like to acknowledge generous research support from the Michael J. Fox Foundation, Parkinson's UK and the Rosetrees Trust. This work was supported in part by the Wellcome Trust/MRC Joint Call in Neurodegeneration Award (WT089698) to the UK Parkinson's Disease Consortium (UKPDC) whose members are from the UCL Institute of Neurology, the University of Sheffield and the MRC Protein Phosphorylation Unit at the University of Dundee. RW is currently funded by the Pitts‐Tucker Studentship in association with the Wade‐Martins Group; Laboratory of Molecular Neurodegeneration and Gene Therapy, Oxford. CM is funded by an MRC Grant (MR/L010933/1) awarded to Dr Patrick A. Lewis. RB is funded by the Reta Lila Weston Trust.
R. Wallings and C. Manzoni contributed equally to this work.
References
- 1. von Campenhausen S, Bornschein B, Wick R, Botzel K, Sampaio C, Poewe W, Oertel W, Siebert U, Berger K & Dodel R (2005) Prevalence and incidence of Parkinson's disease in Europe. Eur Neuropsychopharmacol 15, 473–490. [DOI] [PubMed] [Google Scholar]
- 2. Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA & Nelson LM (2003) Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am J Epidemiol 157, 1015–1022. [DOI] [PubMed] [Google Scholar]
- 3. Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM & Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain J Neurol 136, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Singleton AB, Farrer MJ & Bonifati V (2013) The genetics of Parkinson's disease: progress and therapeutic implications. Mov Disord 28, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, Ross OA, Van Deerlin VM, Trojanowski JQ, Hurtig HI et al (2014) Clinical correlations with lewy body pathology in LRRK2‐related parkinson disease. JAMA Neurol 72, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M et al (2014) Large‐scale meta‐analysis of genome‐wide association data identifies six new risk loci for Parkinson's disease. Nat Genet 46, 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paisan‐Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N et al (2004) Cloning of the gene containing mutations that cause PARK8‐linked Parkinson's disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- 8. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB et al (2004) Mutations in LRRK2 cause autosomal‐dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
- 9. Agalliu I, San Luciano M, Mirelman A, Giladi N, Waro B, Aasly J, Inzelberg R, Hassin‐Baer S, Friedman E, Ruiz‐Martinez J et al (2015) Higher frequency of certain cancers in LRRK2 G2019S mutation carriers with Parkinson disease: a pooled analysis. JAMA Neurol 72, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA et al (2012) Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marcinek P, Jha AN, Shinde V, Sundaramoorthy A, Rajkumar R, Suryadevara NC, Neela SK, van Tong H, Balachander V, Valluri VL et al (2013) LRRK2 and RIPK2 variants in the NOD 2‐mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PLoS One 8, e73103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marin I (2006) The Parkinson disease gene LRRK2: evolutionary and structural insights. Mol Biol Evol 23, 2423–2433. [DOI] [PubMed] [Google Scholar]
- 13. Rudenko IN, Kaganovich A, Hauser DN, Beylina A, Chia R, Ding J, Maric D, Jaffe H & Cookson MR (2012) The G2385R variant of leucine‐rich repeat kinase 2 associated with Parkinson's disease is a partial loss‐of‐function mutation. Biochem J 446, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsika E & Moore DJ (2012) Mechanisms of LRRK2‐mediated neurodegeneration. Curr Neurol Neurosci Rep 12, 251–260. [DOI] [PubMed] [Google Scholar]
- 15. Tsika E & Moore DJ (2013) Contribution of GTPase activity to LRRK2‐associated Parkinson disease. Small GTPases 4, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2‐associated Parkinson's disease: a case‐control study. Lancet Neurol 7, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benamer HT & de Silva R (2010) LRRK2 G2019S in the North African population: a review. Eur Neurol 63, 321–325. [DOI] [PubMed] [Google Scholar]
- 18. Paisan‐Ruiz C, Lewis PA & Singleton AB (2013) LRRK2: cause, risk, and mechanism. J Parkinsons Dis 3, 85–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL et al (2007) Parkinson's disease‐associated mutations in LRRK2 link enhanced GTP‐binding and kinase activities to neuronal toxicity. Hum Mol Genet 16, 223–232. [DOI] [PubMed] [Google Scholar]
- 20. Heo HY, Park JM, Kim CH, Han BS, Kim KS & Seol W (2010) LRRK2 enhances oxidative stress‐induced neurotoxicity via its kinase activity. Exp Cell Res 316, 649–656. [DOI] [PubMed] [Google Scholar]
- 21. Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ et al (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 23, 329–341. [DOI] [PubMed] [Google Scholar]
- 22. Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, Tsuji S & Obata F (2005) An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 57, 918–921. [DOI] [PubMed] [Google Scholar]
- 23. Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H & Ueffing M (2006) The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet 15, 223–232. [DOI] [PubMed] [Google Scholar]
- 24. Ray S, Bender S, Kang S, Lin R, Glicksman MA & Liu M (2014) The Parkinson disease‐linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J Biol Chem 289, 13042–13053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A & Alessi DR (2007) LRRK2 phosphorylates moesin at threonine‐558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J 405, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greggio E & Cookson MR (2009) Leucine‐rich repeat kinase 2 mutations and Parkinson's disease: three questions. ASN Neuro 1, pii: e00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chan SL, Chua LL, Angeles DC & Tan EK (2014) MAP1B rescues LRRK2 mutant‐mediated cytotoxicity. Mol Brain 7, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hudkins RL, Diebold JL, Tao M, Josef KA, Park CH, Angeles TS, Aimone LD, Husten J, Ator MA, Meyer SL et al (2008) Mixed‐lineage kinase 1 and mixed‐lineage kinase 3 subtype‐selective dihydronaphthyl[3,4‐a]pyrrolo[3,4‐c]carbazole‐5‐ones: optimization, mixed‐lineage kinase 1 crystallography, and oral in vivo activity in 1‐methyl‐4‐phenyltetrahydropyridine models. J Med Chem 51, 5680–5689. [DOI] [PubMed] [Google Scholar]
- 29. Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T & Kuriyan J (2011) Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell 42, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shan Y, Seeliger MA, Eastwood MP, Frank F, Xu H, Jensen MO, Dror RO, Kuriyan J & Shaw DE (2009) A conserved protonation‐dependent switch controls drug binding in the Abl kinase. Proc Natl Acad Sci USA 106, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gorostidi A, Ruiz‐Martinez J, Lopez de Munain A, Alzualde A & Marti Masso JF (2009) LRRK2 G2019S and R1441G mutations associated with Parkinson's disease are common in the Basque Country, but relative prevalence is determined by ethnicity. Neurogenetics 10, 157–159. [DOI] [PubMed] [Google Scholar]
- 32. Khan NL, Jain S, Lynch JM, Pavese N, Abou‐Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M et al (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128, 2786–2796. [DOI] [PubMed] [Google Scholar]
- 33. Lewis PA, Greggio E, Beilina A, Jain S, Baker A & Cookson MR (2007) The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun 357, 668–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liao J, Wu CX, Burlak C, Zhang S, Sahm H, Wang M, Zhang ZY, Vogel KW, Federici M, Riddle SM et al (2014) Parkinson disease‐associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc Natl Acad Sci USA 111, 4055–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daniels V, Vancraenenbroeck R, Law BM, Greggio E, Lobbestael E, Gao F, De Maeyer M, Cookson MR, Harvey K, Baekelandt V et al (2011) Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. J Neurochem 116, 304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL & Dawson TM (2005) Parkinson's disease‐associated mutations in leucine‐rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Webber PJ, Smith AD, Sen S, Renfrow MB, Mobley JA & West AB (2011) Autophosphorylation in the leucine‐rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP‐binding activities. J Mol Biol 412, 94–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Inzelberg R, Cohen OS, Aharon‐Peretz J, Schlesinger I, Gershoni‐Baruch R, Djaldetti R, Nitsan Z, Ephraty L, Tunkel O, Kozlova E et al (2012) The LRRK2 G2019S mutation is associated with Parkinson disease and concomitant non‐skin cancers. Neurology 78, 781–786. [DOI] [PubMed] [Google Scholar]
- 39. Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM & Moore DJ (2010) GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet 6, e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Taymans JM, Vancraenenbroeck R, Ollikainen P, Beilina A, Lobbestael E, De Maeyer M, Baekelandt V & Cookson MR (2011) LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS One 6, e23207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haebig K, Gloeckner CJ, Miralles MG, Gillardon F, Schulte C, Riess O, Ueffing M, Biskup S & Bonin M (2010) ARHGEF7 (Beta‐PIX) acts as guanine nucleotide exchange factor for leucine‐rich repeat kinase 2. PLoS One 5, e13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stafa K, Trancikova A, Webber PJ, Glauser L, West AB & Moore DJ (2012) GTPase activity and neuronal toxicity of Parkinson's disease‐associated LRRK2 is regulated by ArfGAP1. PLoS Genet 8, e1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dusonchet J, Li H, Guillily M, Liu M, Stafa K, Derada Troletti C, Boon JY, Saha S, Glauser L, Mamais A et al (2014) A Parkinson's disease gene regulatory network identifies the signaling protein RGS2 as a modulator of LRRK2 activity and neuronal toxicity. Hum Mol Genet 23, 4887–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fu X, Zheng Y, Hong H, He Y, Zhou S, Guo C, Liu Y, Xian W, Zeng J, Li J et al (2013) LRRK2 G2385R and LRRK2 R1628P increase risk of Parkinson's disease in a Han Chinese population from Southern Mainland China. Parkinsonism Relat Disord 19, 397–398. [DOI] [PubMed] [Google Scholar]
- 45. Ross OA, Wu YR, Lee MC, Funayama M, Chen ML, Soto AI, Mata IF, Lee‐Chen GJ, Chen CM, Tang M et al (2008) Analysis of Lrrk2 R1628P as a risk factor for Parkinson's disease. Ann Neurol 64, 88–92. [DOI] [PubMed] [Google Scholar]
- 46. Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, Murata M, Toda T, Mizuno Y & Hattori N (2007) Leucine‐rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. NeuroReport 18, 273–275. [DOI] [PubMed] [Google Scholar]
- 47. Ross OA, Soto‐Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, Bozi M, Brice A et al (2011) Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case‐control study. Lancet Neurol 10, 898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aasly JO, Vilarino‐Guell C, Dachsel JC, Webber PJ, West AB, Haugarvoll K, Johansen KK, Toft M, Nutt JG, Payami H et al (2010) Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson's disease. Mov Disord 25, 2156–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Puschmann A, Englund E, Ross OA, Vilarino‐Guell C, Lincoln SJ, Kachergus JM, Cobb SA, Tornqvist AL, Rehncrona S, Widner H et al (2012) First neuropathological description of a patient with Parkinson's disease and LRRK2 p. N1437H mutation. Parkinsonism Relat Disord 18, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, Solanoy HO, Drummond J, Zhang X, Ding X et al (2012) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med 4, 164ra161. [DOI] [PubMed] [Google Scholar]
- 51. San Luciano M, Lipton RB, Wang C, Katz M, Zimmerman ME, Sanders AE, Ozelius LJ, Bressman SB & Saunders‐Pullman R (2010) Clinical expression of LRRK2 G2019S mutations in the elderly. Mov Disord 25, 2571–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schulte EC, Ellwanger DC, Dihanich S, Manzoni C, Stangl K, Schormair B, Graf E, Eck S, Mollenhauer B, Haubenberger D et al (2014) Rare variants in LRRK1 and Parkinson's disease. Neurogenetics 15, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Civiero L, Vancraenenbroeck R, Belluzzi E, Beilina A, Lobbestael E, Reyniers L, Gao F, Micetic I, De Maeyer M, Bubacco L et al (2012) Biochemical characterization of highly purified leucine‐rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS One 7, e43472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reyniers L, Del Giudice MG, Civiero L, Belluzzi E, Lobbestael E, Beilina A, Arrigoni G, Derua R, Waelkens E, Li Y et al (2014) Differential protein‐protein interactions of LRRK1 and LRRK2 indicate roles in distinct cellular signaling pathways. J Neurochem 131, 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xing W, Liu J, Cheng S, Vogel P, Mohan S & Brommage R (2013) Targeted disruption of leucine‐rich repeat kinase 1 but not leucine‐rich repeat kinase 2 in mice causes severe osteopetrosis. J Bone Miner Res 28, 1962–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniels V, Lewis P, Jain S, Ding J, Syed A et al (2008) The Parkinson disease‐associated leucine‐rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem 283, 16906–16914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Greggio E, Taymans JM, Zhen EY, Ryder J, Vancraenenbroeck R, Beilina A, Sun P, Deng J, Jaffe H, Baekelandt V et al (2009) The Parkinson's disease kinase LRRK2 autophosphorylates its GTPase domain at multiple sites. Biochem Biophys Res Commun 389, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kamikawaji S, Ito G & Iwatsubo T (2009) Identification of the autophosphorylation sites of LRRK2. Biochemistry 48, 10963–10975. [DOI] [PubMed] [Google Scholar]
- 59. Kamikawaji S, Ito G, Sano T & Iwatsubo T (2013) Differential effects of familial parkinson mutations in LRRK2 revealed by a systematic analysis of autophosphorylation. Biochemistry 52, 6052–6062. [DOI] [PubMed] [Google Scholar]
- 60. Gloeckner CJ, Boldt K, von Zweydorf F, Helm S, Wiesent L, Sarioglu H & Ueffing M (2010) Phosphopeptide analysis reveals two discrete clusters of phosphorylation in the N terminus and the Roc domain of the Parkinson‐disease associated protein kinase LRRK2. J Proteome Res 9, 1738–1745. [DOI] [PubMed] [Google Scholar]
- 61. Pungaliya PP, Bai Y, Lipinski K, Anand VS, Sen S, Brown EL, Bates B, Reinhart PH, West AB, Hirst WD et al (2010) Identification and characterization of a leucine‐rich repeat kinase 2 (LRRK2) consensus phosphorylation motif. PLoS One 5, e13672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nichols RJ, Dzamko N, Morrice NA, Campbell DG, Deak M, Ordureau A, Macartney T, Tong Y, Shen J, Prescott AR et al (2010) 14‐3‐3 binding to LRRK2 is disrupted by multiple Parkinson's disease‐associated mutations and regulates cytoplasmic localization. Biochem J 430, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Doggett EA, Zhao J, Mork CN, Hu D & Nichols RJ (2012) Phosphorylation of LRRK2 serines 955 and 973 is disrupted by Parkinson's disease mutations and LRRK2 pharmacological inhibition. J Neurochem 120, 37–45. [DOI] [PubMed] [Google Scholar]
- 64. Li X, Wang QJ, Pan N, Lee S, Zhao Y, Chait BT & Yue Z (2011) Phosphorylation‐dependent 14‐3‐3 binding to LRRK2 is impaired by common mutations of familial Parkinson's disease. PLoS One 6, e17153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR & Nichols RJ (2010) Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14‐3‐3 binding and altered cytoplasmic localization. Biochem J 430, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mamais A, Chia R, Beilina A, Hauser DN, Hall C, Lewis PA, Cookson MR & Bandopadhyay R (2014) Arsenite stress down‐regulates phosphorylation and 14‐3‐3 binding of leucine‐rich repeat kinase 2 (LRRK2), promoting self‐association and cellular redistribution. J Biol Chem 289, 21386–21400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dzamko N, Inesta‐Vaquera F, Zhang J, Xie C, Cai H, Arthur S, Tan L, Choi H, Gray N, Cohen P et al (2012) The IkappaB kinase family phosphorylates the Parkinson's disease kinase LRRK2 at Ser935 and Ser910 during Toll‐like receptor signaling. PLoS One 7, e39132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chia R, Haddock S, Beilina A, Rudenko IN, Mamais A, Kaganovich A, Li Y, Kumaran R, Nalls MA & Cookson MR (2014) Phosphorylation of LRRK2 by casein kinase 1alpha regulates trans‐Golgi clustering via differential interaction with ARHGEF7. Nat Commun 5, 5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lobbestael E, Zhao J, Rudenko IN, Beylina A, Gao F, Wetter J, Beullens M, Bollen M, Cookson MR, Baekelandt V et al (2013) Identification of protein phosphatase 1 as a regulator of the LRRK2 phosphorylation cycle. Biochem J 456, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hsu CH, Chan D, Greggio E, Saha S, Guillily MD, Ferree A, Raghavan K, Shen GC, Segal L, Ryu H et al (2010) MKK6 binds and regulates expression of Parkinson's disease‐related protein LRRK2. J Neurochem 112, 1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gloeckner CJ, Schumacher A, Boldt K & Ueffing M (2009) The Parkinson disease‐associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J Neurochem 109, 959–968. [DOI] [PubMed] [Google Scholar]
- 72. Chen CY, Weng YH, Chien KY, Lin KJ, Yeh TH, Cheng YP, Lu CS & Wang HL (2012) (G2019S) LRRK2 activates MKK4‐JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ 19, 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Milosevic I, Giovedi S, Lou X, Raimondi A, Collesi C, Shen H, Paradise S, O'Toole E, Ferguson S, Cremona O et al (2011) Recruitment of endophilin to clathrin‐coated pit necks is required for efficient vesicle uncoating after fission. Neuron 72, 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bravo‐San Pedro JM, Niso‐Santano M, Gomez‐Sanchez R, Pizarro‐Estrella E, Aiastui‐Pujana A, Gorostidi A, Climent V, Lopez de Maturana R, Sanchez‐Pernaute R, Lopez de Munain A et al (2012) The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci 70, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Purro SA, Galli S & Salinas PC (2014) Dysfunction of Wnt signaling and synaptic disassembly in neurodegenerative diseases. J Mol Cell Biol 6, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sancho RM, Law BM & Harvey K (2009) Mutations in the LRRK2 Roc‐COR tandem domain link Parkinson's disease to Wnt signalling pathways. Hum Mol Genet 18, 3955–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Berwick DC & Harvey K (2012) LRRK2 functions as a Wnt signaling scaffold, bridging cytosolic proteins and membrane‐localized LRP6. Hum Mol Genet 21, 4966–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Looyenga BD, Furge KA, Dykema KJ, Koeman J, Swiatek PJ, Giordano TJ, West AB, Resau JH, Teh BT & MacKeigan JP (2011) Chromosomal amplification of leucine‐rich repeat kinase‐2 (LRRK2) is required for oncogenic MET signaling in papillary renal and thyroid carcinomas. Proc Natl Acad Sci USA 108, 1439–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Graveel CR, Tolbert D & Vande Woude GF (2013) MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol 5, pii:a009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gomez‐Suaga P, Luzon‐Toro B, Churamani D, Zhang L, Bloor‐Young D, Patel S, Woodman PG, Churchill GC & Hilfiker S (2012) Leucine‐rich repeat kinase 2 regulates autophagy through a calcium‐dependent pathway involving NAADP. Hum Mol Genet 21, 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hoyer‐Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R et al (2007) Control of macroautophagy by calcium, calmodulin‐dependent kinase kinase‐beta, and Bcl‐2. Mol Cell 25, 193–205. [DOI] [PubMed] [Google Scholar]
- 82. Su YC, Guo X & Qi X (2015) Threonine 56 phosphorylation of Bcl‐2 is required for LRRK2 G2019S‐induced mitochondrial depolarization and autophagy. Biochim Biophys Acta 1852, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E & Lu B (2008) Phosphorylation of 4E‐BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J 27, 2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kumar A, Greggio E, Beilina A, Kaganovich A, Chan D, Taymans JM, Wolozin B & Cookson MR (2010) The Parkinson's disease associated LRRK2 exhibits weaker in vitro phosphorylation of 4E‐BP compared to autophosphorylation. PLoS One 5, e8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Trancikova A, Mamais A, Webber PJ, Stafa K, Tsika E, Glauser L, West AB, Bandopadhyay R & Moore DJ (2012) Phosphorylation of 4E‐BP1 in the mammalian brain is not altered by LRRK2 expression or pathogenic mutations. PLoS One 7, e47784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S et al (2011) LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet 20, 4209–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gehrke S, Imai Y, Sokol N & Lu B (2010) Pathogenic LRRK2 negatively regulates microRNA‐mediated translational repression. Nature 466, 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M et al (2014) Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson's disease. Cell 157, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dachsel JC, Taylor JP, Mok SS, Ross OA, Hinkle KM, Bailey RM, Hines JH, Szutu J, Madden B, Petrucelli L et al (2007) Identification of potential protein interactors of Lrrk2. Parkinsonism Relat Disord 13, 382–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM et al (2014) LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci 17, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bright NJ, Thornton C & Carling D (2009) The regulation and function of mammalian AMPK‐related kinases. Acta Physiol (Oxf) 196, 15–26. [DOI] [PubMed] [Google Scholar]
- 92. Kleppe R, Martinez A, Doskeland SO & Haavik J (2011) The 14‐3‐3 proteins in regulation of cellular metabolism. Semin Cell Dev Biol 22, 713–719. [DOI] [PubMed] [Google Scholar]
- 93. Tsika E, Kannan M, Foo CS, Dikeman D, Glauser L, Gellhaar S, Galter D, Knott GW, Dawson TM, Dawson VL et al (2014) Conditional expression of Parkinson's disease‐related R1441C LRRK2 in midbrain dopaminergic neurons of mice causes nuclear abnormalities without neurodegeneration. Neurobiol Dis 71, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. MacLeod D, Dowman J, Hammond R, Leete T, Inoue K & Abeliovich A (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593. [DOI] [PubMed] [Google Scholar]
- 95. Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L et al (2009) Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci 29, 13971–13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gandhi PN, Chen SG & Wilson‐Delfosse AL (2009) Leucine‐rich repeat kinase 2 (LRRK2): a key player in the pathogenesis of Parkinson's disease. J Neurosci Res 87, 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gillardon F (2009) Interaction of elongation factor 1‐alpha with leucine‐rich repeat kinase 2 impairs kinase activity and microtubule bundling in vitro. Neuroscience 163, 533–539. [DOI] [PubMed] [Google Scholar]
- 98. Meixner A, Boldt K, Van Troys M, Askenazi M, Gloeckner CJ, Bauer M, Marto JA, Ampe C, Kinkl N & Ueffing M (2011) A QUICK screen for Lrrk2 interaction partners–leucine‐rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Mol Cell Proteomics 10 (M110), 001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kett LR, Boassa D, Ho CC, Rideout HJ, Hu J, Terada M, Ellisman M & Dauer WT (2012) LRRK2 Parkinson disease mutations enhance its microtubule association. Hum Mol Genet 21, 890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chan D, Citro A, Cordy JM, Shen GC & Wolozin B (2011) Rac1 protein rescues neurite retraction caused by G2019S leucine‐rich repeat kinase 2 (LRRK2). J Biol Chem 286, 16140–16149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Caesar M, Felk S, Aasly JO & Gillardon F (2015) Changes in actin dynamics and F‐actin structure both in synaptoneurosomes of LRRK2(R1441G) mutant mice and in primary human fibroblasts of LRRK2(G2019S) mutation carriers. Neuroscience 284, 311–324. [DOI] [PubMed] [Google Scholar]
- 102. Godena VK, Brookes‐Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ & De Vos KJ (2014) Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc‐COR domain mutations. Nat Commun 5, 5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Law BM, Spain VA, Leinster VH, Chia R, Beilina A, Cho HJ, Taymans JM, Urban MK, Sancho RM, Blanca Ramirez M et al (2014) A direct interaction between leucine‐rich repeat kinase 2 and specific beta‐tubulin isoforms regulates tubulin acetylation. J Biol Chem 289, 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kawakami F, Yabata T, Ohta E, Maekawa T, Shimada N, Suzuki M, Maruyama H, Ichikawa T & Obata F (2012) LRRK2 phosphorylates tubulin‐associated tau but not the free molecule: LRRK2‐mediated regulation of the tau‐tubulin association and neurite outgrowth. PLoS One 7, e30834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bailey RM, Covy JP, Melrose HL, Rousseau L, Watkinson R, Knight J, Miles S, Farrer MJ, Dickson DW, Giasson BI et al (2013) LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol 126, 809–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Plowey ED, Cherra SJ 3rd, Liu YJ & Chu CT (2008) Role of autophagy in G2019S‐LRRK2‐associated neurite shortening in differentiated SH‐SY5Y cells. J Neurochem 105, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Alegre‐Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O & Wade‐Martins R (2009) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet 18, 4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez‐Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A et al (2013) Interplay of LRRK2 with chaperone‐mediated autophagy. Nat Neurosci 16, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Schapansky J, Nardozzi JD, Felizia F & LaVoie MJ (2014) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet 23, 4201–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Manzoni C, Mamais A, Dihanich S, Abeti R, Soutar MP, Plun‐Favreau H, Giunti P, Tooze SA, Bandopadhyay R & Lewis PA (2013) Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta 1833, 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Saez‐Atienzar S, Bonet‐Ponce L, Blesa JR, Romero FJ, Murphy MP, Jordan J & Galindo MF (2014) The LRRK2 inhibitor GSK2578215A induces protective autophagy in SH‐SY5Y cells: involvement of Drp‐1‐mediated mitochondrial fission and mitochondrial‐derived ROS signaling. Cell Death Dis 5, e1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ 3rd & Shen J (2010) Loss of leucine‐rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha‐synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci USA 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bravo‐San Pedro JM, Niso‐Santano M, Gomez‐Sanchez R, Pizarro‐Estrella E, Aiastui‐Pujana A, Gorostidi A, Climent V, Lopez de Maturana R, Sanchez‐Pernaute R, Lopez de Munain A et al (2013) The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci 70, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Manzoni C, Mamais A, Dihanich S, McGoldrick P, Devine MJ, Zerle J, Kara E, Taanman JW, Healy DG, Marti‐Masso JF et al (2013) Pathogenic Parkinson's disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem Biophys Res Commun 441, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sanchez‐Danes A, Richaud‐Patin Y, Carballo‐Carbajal I, Jimenez‐Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A et al (2012) Disease‐specific phenotypes in dopamine neurons from human iPS‐based models of genetic and sporadic Parkinson's disease. EMBO Mol Med 4, 380–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D et al (2012) LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75, 1008–1021. [DOI] [PubMed] [Google Scholar]
- 117. Waschbusch D, Michels H, Strassheim S, Ossendorf E, Kessler D, Gloeckner CJ & Barnekow A (2014) LRRK2 transport is regulated by its novel interacting partner Rab32. PLoS One 9, e111632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K et al (2014) Unbiased screen for interactors of leucine‐rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci USA 111, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Dodson MW, Leung LK, Lone M, Lizzio MA & Guo M (2014) Novel alleles of the Drosophila LRRK2 homolog reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis Model Mech 7, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ et al (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol 60, 557–569. [DOI] [PubMed] [Google Scholar]
- 121. Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J et al (2009) Leucine‐rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's‐disease‐related mutant alpha‐synuclein. Neuron 64, 807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J et al (2008) LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res 314, 2055–2065. [DOI] [PubMed] [Google Scholar]
- 123. Stafa K, Tsika E, Moser R, Musso A, Glauser L, Jones A, Biskup S, Xiong Y, Bandopadhyay R, Dawson VL et al (2014) Functional interaction of Parkinson's disease‐associated LRRK2 with members of the dynamin GTPase superfamily. Hum Mol Genet 23, 2055–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Arranz AM, Delbroek L, Van Kolen K, Guimaraes MR, Mandemakers W, Daneels G, Matta S, Calafate S, Shaban H, Baatsen P et al (2014) LRRK2 functions in synaptic vesicle endocytosis through a kinase‐dependent mechanism. J Cell Sci 128, 541–552. [DOI] [PubMed] [Google Scholar]
- 125. Yun HJ, Park J, Ho DH, Kim H, Kim CH, Oh H, Ga I, Seo H, Chang S, Son I et al (2013) LRRK2 phosphorylates Snapin and inhibits interaction of Snapin with SNAP‐25. Exp Mol Med 45, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Migheli R, Del Giudice MG, Spissu Y, Sanna G, Xiong Y, Dawson TM, Dawson VL, Galioto M, Rocchitta G, Biosa A et al (2013) LRRK2 affects vesicle trafficking, neurotransmitter extracellular level and membrane receptor localization. PLoS One 8, e77198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Piccoli G, Condliffe SB, Bauer M, Giesert F, Boldt K, De Astis S, Meixner A, Sarioglu H, Vogt‐Weisenhorn DM, Wurst W et al (2011) LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci 31, 2225–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Miklavc P, Ehinger K, Thompson KE, Hobi N, Shimshek DR & Frick M (2014) Surfactant secretion in LRRK2 knock‐out rats: changes in lamellar body morphology and rate of exocytosis. PLoS One 9, e84926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Cho HJ, Yu J, Xie C, Rudrabhatla P, Chen X, Wu J, Parisiadou L, Liu G, Sun L, Ma B et al (2014) Leucine‐rich repeat kinase 2 regulates Sec16A at ER exit sites to allow ER‐Golgi export. EMBO J 33, 2314–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA et al (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron 77, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Seaman MN, Harbour ME, Tattersall D, Read E & Bright N (2009) Membrane recruitment of the cargo‐selective retromer subcomplex is catalysed by the small GTPase Rab7 and inhibited by the Rab‐GAP TBC1D5. J Cell Sci 122, 2371–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Linhart R, Wong SA, Cao J, Tran M, Huynh A, Ardrey C, Park JM, Hsu C, Taha S, Peterson R et al (2014) Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson's disease mutant of Leucine‐Rich Repeat Kinase 2 (LRRK2). Mol Neurodegener 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zimprich A, Benet‐Pages A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P et al (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late‐onset Parkinson disease. Am J Hum Genet 89, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Vilarino‐Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto‐Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA et al (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Beccano‐Kelly DA, Kuhlmann N, Tatarnikov I, Volta M, Munsie LN, Chou P, Cao LP, Han H, Tapia L, Farrer MJ et al (2014) Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock‐in mice. Front Cell Neurosci 8, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME & Yue Z (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci 30, 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN & Shen J (2009) R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci USA 106, 14622–14627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Zhou H, Huang C, Tong J, Hong WC, Liu YJ & Xia XG (2011) Temporal expression of mutant LRRK2 in adult rats impairs dopamine reuptake. Int J Biol Sci 7, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Plowey ED, Johnson JW, Steer E, Zhu W, Eisenberg DA, Valentino NM, Liu YJ & Chu CT (2014) Mutant LRRK2 enhances glutamatergic synapse activity and evokes excitotoxic dendrite degeneration. Biochim Biophys Acta 1842, 1596–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Melrose HL, Dachsel JC, Behrouz B, Lincoln SJ, Yue M, Hinkle KM, Kent CB, Korvatska E, Taylor JP, Witten L et al (2010) Impaired dopaminergic neurotransmission and microtubule‐associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis 40, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Mortiboys H, Johansen KK, Aasly JO & Bandmann O (2010) Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75, 2017–2020. [DOI] [PubMed] [Google Scholar]
- 142. Wang X, Yan MH, Fujioka H, Liu J, Wilson‐Delfosse A, Chen SG, Perry G, Casadesus G & Zhu X (2012) LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 21, 1931–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L et al (2011) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 6, e18568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Papkovskaia TD, Chau KY, Inesta‐Vaquera F, Papkovsky DB, Healy DG, Nishio K, Staddon J, Duchen MR, Hardy J, Schapira AH et al (2012) G2019S leucine‐rich repeat kinase 2 causes uncoupling protein‐mediated mitochondrial depolarization. Hum Mol Genet 21, 4201–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cherra SJ 3rd, Steer E, Gusdon AM, Kiselyov K & Chu CT (2013) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Cooper O, Seo H, Andrabi S, Guardia‐Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo‐Reid L, Xie Z, Osborn T et al (2012) Pharmacological rescue of mitochondrial deficits in iPSC‐derived neural cells from patients with familial Parkinson's disease. Sci Transl Med 4, 141ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Niu J, Yu M, Wang C & Xu Z (2012) Leucine‐rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin‐like protein. J Neurochem 122, 650–658. [DOI] [PubMed] [Google Scholar]
- 148. Su YC & Qi X (2013) Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet 22, 4545–4561. [DOI] [PubMed] [Google Scholar]
- 149. Liou AK, Leak RK, Li L & Zigmond MJ (2008) Wild‐type LRRK2 but not its mutant attenuates stress‐induced cell death via ERK pathway. Neurobiol Dis 32, 116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W et al (2011) LRRK2 mutant iPSC‐derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Angeles DC, Gan BH, Onstead L, Zhao Y, Lim KL, Dachsel J, Melrose H, Farrer M, Wszolek ZK, Dickson DW et al (2011) Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress‐induced neuronal death. Hum Mutat 32, 1390–1397. [DOI] [PubMed] [Google Scholar]
- 152. Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K et al (2009) LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci 29, 9210–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR et al (2011) Parkinson's disease‐linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm 118, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Gillardon F, Schmid R & Draheim H (2012) Parkinson's disease‐linked leucine‐rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 208, 41–48. [DOI] [PubMed] [Google Scholar]
- 155. Thevenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C & Sagot YJ (2011) Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One 6, e21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ et al (2010) LRRK2 is involved in the IFN‐gamma response and host response to pathogens. J Immunol 185, 5577–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gillardon F, Kremmer E, Froehlich T, Ueffing M, Hengerer B & Gloeckner CJ (2013) ATP‐competitive LRRK2 inhibitors interfere with monoclonal antibody binding to the kinase domain of LRRK2 under native conditions. A method to directly monitor the active conformation of LRRK2? J Neurosci Methods 214, 62–68. [DOI] [PubMed] [Google Scholar]
- 158. Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM & West AB (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32, 1602–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Russo I, Bubacco L & Greggio E (2014) LRRK2 and neuroinflammation: partners in crime in Parkinson's disease? J Neuroinflammation 11, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Giesert F, Hofmann A, Burger A, Zerle J, Kloos K, Hafen U, Ernst L, Zhang J, Vogt‐Weisenhorn DM & Wurst W (2013) Expression analysis of Lrrk1, Lrrk2 and Lrrk2 splice variants in mice. PLoS One 8, e63778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Kuss M, Adamopoulou E & Kahle PJ (2014) Interferon‐gamma induces leucine‐rich repeat kinase LRRK2 via extracellular signal‐regulated kinase ERK5 in macrophages. J Neurochem 129, 980–987. [DOI] [PubMed] [Google Scholar]
- 162. Liu Z, Lee J, Krummey S, Lu W, Cai H & Lenardo MJ (2011) The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol 12, 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. West AB (2014) Ten years and counting: moving leucine‐rich repeat kinase 2 inhibitors to the clinic. Mov Disord 30, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Zhang J, Yang PL & Gray NS (2009) Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 9, 28–39. [DOI] [PubMed] [Google Scholar]
- 165. Ramsden N, Perrin J, Ren Z, Lee BD, Zinn N, Dawson VL, Tam D, Bova M, Lang M, Drewes G et al (2011) Chemoproteomics‐based design of potent LRRK2‐selective lead compounds that attenuate Parkinson's disease‐related toxicity in human neurons. ACS Chem Biol 6, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR et al (2011) Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Luerman GC, Nguyen C, Samaroo H, Loos P, Xi H, Hurtado‐Lorenzo A, Needle E, Stephen Noell G, Galatsis P, Dunlop J et al (2014) Phosphoproteomic evaluation of pharmacological inhibition of leucine‐rich repeat kinase 2 reveals significant off‐target effects of LRRK‐2‐IN‐1. J Neurochem 128, 561–576. [DOI] [PubMed] [Google Scholar]
- 168. Choi HG, Zhang J, Deng X, Hatcher JM, Patricelli MP, Zhao Z, Alessi DR & Gray NS (2012) Brain Penetrant LRRK2 Inhibitor. ACS Med Chem Lett 3, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Reith AD, Bamborough P, Jandu K, Andreotti D, Mensah L, Dossang P, Choi HG, Deng X, Zhang J, Alessi DR et al (2012) GSK2578215A; a potent and highly selective 2‐arylmethyloxy‐5‐substitutent‐N‐arylbenzamide LRRK2 kinase inhibitor. Bioorg Med Chem Lett 22, 5625–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Estrada AA, Chan BK, Baker‐Glenn C, Beresford A, Burdick DJ, Chambers M, Chen H, Dominguez SL, Dotson J, Drummond J et al (2014) Discovery of highly potent, selective, and brain‐penetrant aminopyrazole leucine‐rich repeat kinase 2 (LRRK2) small molecule inhibitors. J Med Chem 57, 921–936. [DOI] [PubMed] [Google Scholar]
- 171. Kethiri RR & Bakthavatchalam R (2014) Leucine‐rich repeat kinase 2 inhibitors: a review of recent patents (2011–2013). Expert Opin Ther Pat 24, 745–757. [DOI] [PubMed] [Google Scholar]
- 172. Baptista MA, Dave KD, Frasier MA, Sherer TB, Greeley M, Beck MJ, Varsho JS, Parker GA, Moore C, Churchill MJ et al (2013) Loss of leucine‐rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS One 8, e80705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Taymans JM (2014) Can the increasing number of newly developed leucine‐rich repeat kinase 2 inhibitors validate or invalidate a potential disease‐modifying therapeutic approach for Parkinson's disease? Expert Opin Ther Pat 24, 727–730. [DOI] [PubMed] [Google Scholar]
- 174. Rudenko IN, Chia R & Cookson MR (2012) Is inhibition of kinase activity the only therapeutic strategy for LRRK2‐associated Parkinson's disease? BMC Med 10, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ray S & Liu M (2012) Current understanding of LRRK2 in Parkinson's disease: biochemical and structural features and inhibitor design. Future Med Chem 4, 1701–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Lee BD, Dawson VL & Dawson TM (2012) Leucine‐rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson's disease. Trends Pharmacol Sci 33, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Manzoni C, Denny P, Lovering RC & Lewis PA (2015) Computational analysis of the LRRK2 interactome. PeerJ 3, e778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Porras P, Duesbury M, Fabregat A, Ueffing M, Orchard S, Gloeckner CJ & Hermjakob H (2015) A visual review of the interactome of LRRK2: Using deep‐curated molecular interactions data to represent biology. Proteomics 15, 1390–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Trabzuni D, Ryten M, Emmett W, Ramasamy A, Lackner KJ, Zeller T, Walker R, Smith C, Lewis PA, Mamais A et al (2013) Fine‐mapping, gene expression and splicing analysis of the disease associated LRRK2 locus. PLoS One 8, e70724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B et al (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci USA 110, E4016–E4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Inestrosa NC & Arenas E (2010) Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci 11, 77–86. [DOI] [PubMed] [Google Scholar]
- 182. Poulopoulos M, Levy OA & Alcalay RN (2012) The neuropathology of genetic Parkinson's disease. Mov Disord 27, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Mamais A, Raja M, Manzoni C, Dihanich S, Lees A, Moore D, Lewis PA & Bandopadhyay R (2013) Divergent alpha‐synuclein solubility and aggregation properties in G2019S LRRK2 Parkinson's disease brains with Lewy Body pathology compared to idiopathic cases. Neurobiol Dis 58, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]