

Abstract
The fourth conserved region (C4) in the HIV-1 envelope glycoprotein (Env) gp120 is a structural element that is important for its function, as it binds to both the receptor CD4 and the co-receptor CCR5/CXCR4. It has long been known that this region is highly immunogenic and that it harbors B-cell as well as T-cell epitopes. It is the target of a number of antibodies in animal studies, which are called CD4-blockers. However, the mechanism by which the virus shields itself from such antibody responses is not known. Here, we determined the crystal structure of R53 in complex with its epitope peptide using a novel anti-C4 rabbit monoclonal antibody R53. Our data show that although the epitope of R53 covers a highly conserved sequence 433AMYAPPI439, it is not available in the gp120 trimer and in the CD4-bound conformation. Our results suggest a masking mechanism to explain how HIV-1 protects this critical region from the human immune system.
Keywords: C4, CD4, Env, HIV-1, monoclonal antibody
Introduction
The HIV-1 envelope glycoprotein (Env) gp120 initiates viral entry into host cells by binding to its receptor CD4 and to its co-receptor CCR5/CXCR4, and it is the major target for acquired immune deficiency syndrome vaccine development. However, gp120 uses many decoys to evade immune surveillance in humans, rendering the development of a protective vaccine very challenging. Conformational masking, by either covering immunogenic epitope regions with other domains, or by having them adopt different conformations, is one of the decoys gp120 uses to evade the immune responses.1,2 For example, variable loops can often adopt different conformations, and antibodies that recognize one conformation will not be able to effectively target another conformation.3,4,5 Conformational masking can also protect functionally conserved sites within gp120. The CD4 receptor-binding site is protected by entropy masking,1 and the co-receptor-binding site in the pre-fusion complex is completely buried under variable loops.6,7,8 CD4 receptor binding will expose the co-receptor binding site, which is comprised of various conserved regions including the fourth conserved region (C4). The C4 region of gp120, which consists of residues 416-4599 (HxB2 numbering10), has many important functional roles. For example, it is directly involved in receptor binding, co-receptor binding and co-receptor selection (tropism).11,12 Crystal structures of gp120 complexes have revealed that residues 425 (Asn), 426 (Met), and 427 (Trp) in the C4 region have direct contact with CD4.13 The C4 region, together with the third variable loop (V3), is also involved in co-receptor binding. Early mutagenesis studies indicated that residues 438 (Pro) and 441 (Gly) in the C4 region are important for CCR5 binding.14 Structural studies of gp120 in complex with CD4 and monoclonal antibody (mAb) 412d showed that residues 439 (Ile), 440 (Arg), and 441 (Gly) in the C4 region are involved in binding with the N-terminus of CCR5.6 A slight conformational change in the C4 region can influence the structure of V3, and even a single amino acid mutation in the C4 region can increase the neutralization sensitivities of anti-V3 antibodies.15,16 The C4 region is also involved in co-receptor selection, and mutations of residue 440 in the C4 region can alter co-receptor specificity.17
The C4 region is highly immunogenic. It can induce cell-mediated immunities in HIV-1 infected patients and in immunized animals.18,19 For example, monomeric gp120 can elicit mouse helper T-cell immune responses reactive with a C4 peptide, named T1 (a 16-mer containing the region of residues 428-443).18 The C4 region can also induce humoral immune responses.20,21 In fact, the CD4 binding region of gp120 was first identified by an anti-C4 mAb, 5C2E5, which was raised by immunizing mice with a recombinant gp120, and its epitope region was identified by competition with CD4 binding.11 Since then, several antibodies targeting the C4 region have been generated in animals, including rabbit polyclonal antibodies R10-12 and R19-21 that were raised with a poliovirus chimaera expressing a region of 17 amino acids of C4,22 mouse mAbs G3-42, G3-299, G3-508, and G3-536 that were raised with a recombinant BH10 gp120,23,24 and rat mAbs ICR 38.8f and ICR38.1a that were raised with the recombinant BH10 gp120.25 One of the characteristics of these antibodies is that they can block CD4 binding of gp120, and thus, they were collectively named CD4-blocking antibodies.26
The C4 region was initially suggested to form amphipathic helices;19 however, crystal structures of CD4-bound gp120 molecules have shown that it actually forms two beta strands, numbered 20 and 21, of the bridging sheet and loop F.13 Strand 20 is involved in CD4 binding, while strand 21 is involved in co-receptor binding. The structures of gp120 in complex with various mAbs showed that the C4 region could display distinct conformations among the unliganded, CD4-bound, and different antibody-bound states.7,13,27,28,29
Here, we present functional and structural characterizations of a recently generated rabbit anti-C4 mAb, R53, which was elicited by JR-FL gp120 using a DNA prime-protein boost regimen.30 It can neutralize sensitive viruses quite potently but cannot neutralize tier 2 viruses. We show here that R53 has broad cross-clade binding activities against gp120 proteins from clade A, B, C, D, and AE. We have determined crystal structures of R53 Fab/epitope complex and R53 Fab alone, and have structurally defined the R53 epitope. We found that this epitope harbors a conserved motif, AMYAPPI (residues 433-439), located at the C-terminus of the bridging sheet and loop F of gp120. Our data provide a structural understanding of this immunogenic and functionally critical region and the mechanism by which it is masked.
Materials and methods
Enzyme-linked immunosorbent assay (ELISA)
Gp120 proteins produced from 293F cells were coated onto 96-well microtiter plates (Sigma-Aldrich, St. Louis, MO, USA) at 1 µg/mL in 100 µL of phosphate-buffered saline (PBS) as previously described.30 Plates were washed five times with PBS containing 0.1% Triton-X (EWB) and blocked overnight at 4 °C in PBS containing 4% whey and 5% powdered milk. R53, biotinylated anti-rabbit secondary antibody (Vector Labs BA-1000) at 1.5 µg/mL, and a streptavidin horseradish peroxidase construct (Vector Labs SA-5004, Burlingame, CA, USA) at 500 ng/mL were added sequentially to the wells in a volume of 100 µL. Plates were incubated for 1 h at room temperature and washed five times after each step with EWB and then developed for 3 min in 100 µL of a 3,3′5,5′-tetramethylbenzidine substrate solution (Sigma-Aldrich T3405, St. Louis, MO, USA). The reactions were stopped with 25 µL of 2N H2SO4.
Western blot
The gp120 antigens were subjected to SDS-PAGE and blotted onto polyvinylidene fluoride (PVDF) membranes as previously described.30 PVDF membranes were blocked with 0.1% I-Block (Tropix, Bedford, MA, USA), incubated with mAb R53 for 1 h, and subsequently reacted with AP-conjugated goat anti-rabbit IgG at a dilution of 1:5000 for 30 min. The membranes were washed with the blocking buffer after each step. Western-light substrate was then applied to the membranes for 5 min. X-ray films were exposed to the dried membranes and developed using a Kodak processor.
Ab binding kinetics analysis
The binding kinetics of mAb R53 and five representative gp120s was studied using biolayer interferometry (BLI, ForteBio Octet QKe) in a 96-well format, as previously described.30 Protein A-coated tips (ProA sensor, ForteBio) were loaded with R53 at 10 µg/mL in the kinetics buffer (PBS/0.1% bovine serum albumin (BSA)/0.002% Tween 20). After capture, excess unbound antibodies were removed with a 1-min wash using the kinetics buffer to establish a new baseline signal. Then, the biosensor tips coated with mAb R53 were placed in wells containing the designated gp120 proteins at concentrations of 300, 100, 33.3, 11.1, 3.7, or 1.23 nM and allowed to associate in the kinetics buffer. Ab-gp120 association rates (on-rate, Kon) and dissociation rates (off-rate, Koff) were measured in the kinetics buffer. The sensorgrams were corrected with the blank reference and the binding curves were generated by global and local fitting with ForteBio Data Analysis software package 7.0 using a 1:1 binding model when R2 values were greater than 0.95. The affinity constant KD values (in molar units) were calculated using off-rates (Koff)/on-rates (Kon). To regenerate the Protein A sensor for reuse, the sensor was placed in wells containing regenerating buffer (500 mM phosphoric acid) as necessary.
Competition binding assays
The sheep gp120 C5-specific mAb, D3724 (Aalto Bio Reagents, Dublin, Ireland) was used to coat ELISA plates at 2 µg/mL. The plates were washed five times and blocked with PBS containing 4% whey and 5% powdered milk, and gp120 proteins were then added to these pre-coated plates. After the five washes, the competitor rabbit mAbs or sCD4 were diluted in the blocking buffer and added to the plates at three-fold serial dilutions, starting at 500 µg/mL. After a 30-min incubation, 10 µL of Ig-CD4 at 0.1 µg/mL was added to the wells. Bound Ig-CD4 was detected as described above in the ELISA assay.
Virus capture assay
Virus capture assays were performed as previously described.31 Pseudovirions expressing the JR-FL Env and vesicular stomatitis virus (VSV) G protein were produced with the pSG3ΔENV backbone in 293T cells. Plates were coated with 50 μL of mAb at 5 μg/mL for 1 h at room temperature and then blocked in PBS with 3% BSA overnight at 4 °C. Hybridoma supernatants of mAb cell cultures were incubated with pseudoviruses for 1 h before the mixtures were added to the ELISA wells, and incubated for 3 h at room temperature. Plates were washed five times with sterile PBS, overlaid with 10 000 TZM-bl cells per well and incubated for 48 h at 37 °C. Luciferase activity was determined according to the manufacturer's instructions (Promega, Madison, WI, USA). The competition percentage was reported as the reduction of the luciferase signal compared to a serum negative control.
Fab production and purification
Details of the production and antigenicity characterization of rabbit mAb R53 have been published.30 The Fab fragment of R53 was prepared by papain digestion as previously described.32 Briefly, IgG was mixed with papain (Worthington, Lakewood, NJ, USA) at a 20:1 ratio in 100 mM Tris (pH 6.8) containing 1 mM cysteine hydrochloride and 4 mM EDTA. The mixture was incubated for 1 h at 37 °C and the reaction was stopped by the addition of 10 mM iodoacetamide. The Fab fragment was separated from the Fc fragment and undigested IgG using a Protein A column and was further purified by size exclusion chromatography. The concentration of the Fab fragment used for crystallization was approximately 10 mg/mL.
Crystallization, data collection, structure determination and refinement
The C4 peptide was synthesized by Biomatik (Wilmington, DE, USA) and was dissolved in water to a concentration of 10 mg/mL. Preliminary crystals of Fab in complex with peptide, or Fab alone, were obtained by robotic screening using the vapor diffusion hanging drop method. Well-diffracted crystals of R53 Fab in complex with the peptide were obtained in a well solution of 24% polyethylene glycol 6000, 0.1 M citric acid, pH 5.0, and 0.02% NaN3. Crystals of R53 Fab alone were obtained in a well solution of 14% polyethylene glycol 8000, 0.1 M HEPES, pH 7.5, and 8% ethylene glycol. Crystals of Fab R53/epitope complex and Fab R53 alone were first soaked in the mother liquor with additional 20% glycerol (v/v) and 20% ethylene glycol, respectively, before being placed in the X-ray beam. X-ray diffraction data of the crystals of Fab R53/epitope complex were collected at beam line X6A, National Synchrotron Light Source, Brookhaven National Laboratory. Data of the crystals of R53 Fab alone were collected at the synchrotron beamline GM/CA CAT at the Advanced Photo Source (APS), Argonne National Laboratory. All data sets were processed using the HKL 2000 software package,33 and structures were determined by molecular replacement, using as the starting model the structure of another rabbit mAb R56 that we had previously determined (Protein Data Bank (PDB) ID 4JO1). Cycles of refinement for each model were carried out in COOT and Phenix.34,35 The final structural analysis was carried out using ICM, and the figures were generated using Pymol and ICM.36,37
Results
Immunological characterization of rabbit mAb R53
Rabbit mAb R53 is one of the mAbs that we recently generated from a New Zealand white rabbit that was immunized with JR-FL DNA prime, followed by a homologous protein boost, as previously reported.30 The location of its epitope was mapped to the C4 region of gp120 using a 15-aa overlapping peptide array, and a virus capture assay was used to identify whether the epitope of R53 overlapped functionally with those of CD4-binding sites (CD4bs) mAbs (Table 1). CD4bs mAb b12, V3-specific mAb 3074, and glycan-specific antibody 2G12 were used as competition targets. Supernatant from the R53 hybridoma was able to prevent 56% of the virus binding to mAb b12, suggesting that R53 indeed targets the CD4bs region, but not as effectively as the CD4bs mAbs. In contrast, it could not compete against either anti-V3 mAb 3074 or anti-glycan mAb 2G12, further supporting its specific competition against b12. Because previously described mAbs targeting the C4 region were found to block CD4 binding of gp120,11,26 we determined the binding specificity of R53 using a competition ELISA (Figure 1A). R53, sCD4 and R56 were used to compete with Ig-CD4 for binding to JR-FL gp120. Here, R56 is a rabbit mAb that was isolated from the same rabbit as that of R53, and recognizes the V3 crown region of gp120.30,38 As expected, R53 could inhibit Ig-CD4 binding, although it is not as effective as the positive control sCD4. As a negative control, R56 had no impact on Ig-CD4 binding of gp120. These results confirm that R53 can block CD4 binding to gp120.
Table 1. The inhibition percentages of R53 to outcompete binding of mAbs to a JR-FL and VSV-G pseudotyped virus.
| Competing mAb | Hybridoma cell line ID | ||
|---|---|---|---|
| 53 | 56 | 15 | |
| b12 | 56% | 34% | 24% |
| 3074 | 21% | 83% | 17% |
| 2G12 | 39% | 22% | 11% |
Figure 1.
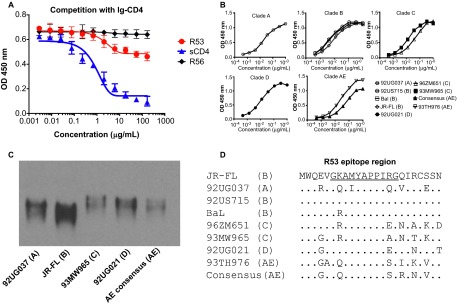
Broad reaction of R53 with gp120s of diverse clades. (A) Competition assay of R53 with Ig-CD4. The soluble CD4 (sCD4) and anti-V3 mAb R56 were used as positive and negative controls, respectively. Results shown represent the mean levels of two independent experiments and error bars indicate the standard deviations. (B) R53 bound nine gp120s from clades A, B, C, D, and AE, as revealed by ELISA. (C) Western blot analysis showed that five selected gp120 proteins from different clades were recognized by R53. (D) A protein sequence alignment of R53 epitope region of the gp120s used in panel B and C. The R53 epitope is underlined in the JR-FL sequence.
To determine the binding breadth of R53 for HIV-1 gp120 from different clades, nine representative gp120s were tested: 92UG037.8 from clade A; 92US715, BaL, and JR-FL from clade B; 96ZM651 and 93MW965 from clade C; 92US021 from clade D; and 93TH976.17 and AE consensus from clade AE. R53 was able to recognize all of the Envs that were tested in the ELISA (Figure 1B). This result was verified by a Western blot analysis, which demonstrated that the five representative gp120s from different clades were recognized by R53 under denaturing conditions (Figure 1C). Binding kinetics of R53 with five selective gp120s were further determined using a ForteBio instrument (Figure 2). R53 was shown to have high binding affinities to all five gp120s tested, with KD values ranging from 1.79 nM to 3.98 nM (Figure 2 and Table 2). A sequence alignment with the R53 epitope region revealed that it is highly conserved among clades A, B, C, D, and AE (Figure 1D).
Figure 2.
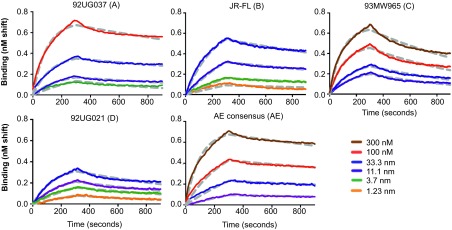
Binding kinetics of R53 and gp120s. The binding kinetics of R53 to 92UG037 (clade A), JR-FL (clade B), 93MW965 (clade C), 92UG021 (clade D), and AE consensus (clade AE) were measured using ForteBio at different concentrations of the ligand. The brown, red, blue, purple, green, and orange lines represent 300 nM, 100 nM, 33.3 nM, 11.1 nM, 3.7 nM, and 1.23 nM of gp120 proteins, respectively. Dashed gray lines represent the theoretical fitting curves. The binding data are summarized in Table 2.
Table 2. R53 binding kinetics with five representative gp120 proteins.
| gp120 proteins (clade) | KD (M) | Kon (M−1s−1) | Koff (s−1) |
|---|---|---|---|
| 92UG037 (A) | 1.54E-09 | 3.41E+05 | 5.24E-04 |
| JR-FL (B)* | 1.96E-09 | 1.57E+05 | 3.09E-04 |
| 93MW965 (C) | 3.98E-09 | 3.04E+05 | 1.21E-03 |
| 92UG021 (D) | 1.79E-09 | 3.56E+05 | 6.38E-04 |
| AE consensus (AE) | 2.49E-09 | 3.91E+05 | 9.73E-04 |
Published in Ref. 30.
Crystal structure of the Fab/epitope complex of rabbit mAb R53
We determined the crystal structure of the R53 antigen-binding fragment (Fab) in complex with a peptide (VGKAMYAPPIRGQIR, residues 430 to 444 in HXB2 numbering). The complex structure of the R53 Fab/epitope was solved by molecular replacement and refined to a resolution of 2.3 Å with an Rwork/Rfree of 16.7%/22.3% (Figure 3 and Table 3). The crystals grew in the orthorhombic space group P212121 with two Fab/epitope complexes in the asymmetric unit. The two complexes are highly similar (RMSD = 0.55 Å); we thus chose only one for description here. We numbered the residues following the Kabat et al.'s convention,39 with the light and heavy chains preceded by “L” and “H”, respectively, and the residues of the epitope by a “P”. Although a 15-mer peptide was used in the crystallization, only 11 residues, i.e., GKAMYAPPIRG (residues 431 to 441), were observed in the electron density map. We also determined the structure of the Fab alone (Table 3); superposition of the two Fab structures showed that the R53 Fab appears to undergo a minimal conformational change upon epitope binding (RMSD = 0.77 Å). R53 has a typical rabbit antibody inter-domain disulfide bond between residues 80 and 170 of the light chain, which places a constraint on the variability of its elbow angle.38
Figure 3.

Structure of the Fab R53/epitope complex. (A) A ribbon representation of Fab R53 in complex with its epitope in a front view. The light chain, heavy chain and the R53 epitope are colored cyan, green, and magenta, respectively (a coloring scheme maintained throughout the manuscript, except where otherwise indicated). (B) A side view of the complex. (C) A top view of the R53/epitope complex looking at the antigen-binding site. The CDR regions are labeled and colored differently from the rest of the Fab. (D) Electrostatic surface potentials of R53, with red as the negatively charged and blue as positively charged regions. The inset is the sequence of the peptide used in crystallization, with the magenta region indicating residues visualized in the crystal structure.
Table 3. Crystallization and refinement statistics.
| Fab R53/epitope | Fab R53 | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | C2 |
| Cell dimensions | ||
| a, b, c (Å) | 72.28, 84.83, 167.24 | 105.43, 78.55, 68.48 |
| α, β, γ (°) | 90, 90, 90 | 90, 92.25, 90 |
| Resolution (Å) | 2.26 (2.30–2.26) | 1.63 (1.66–1.63) |
| Rmerge | 16.5 (48.3) | 5.6 (44.0) |
| I/σI | 17.9 (3.7) | 21.5 (2.3) |
| Completeness (%) | 99.1 (97.5) | 95.4 (79.0) |
| Redundancy | 6.5 (6.1) | 3.9 (3.1) |
| Refinement | ||
| Resolution (Å) | 44.1–2.3 | 31.5–1.6 |
| Number of reflections | 45616 | 66081 |
| Rwork/Rfree | 16.7/22.3 | 19.5/22.8 |
| Number of atoms | ||
| Protein | 6632 | 3251 |
| Solvent | 988 | 624 |
| B-factors | ||
| Protein | 19.8 | 29.9 |
| Solvent | 25.4 | 40.6 |
| Root-mean-square deviations | ||
| Bond lengths (Å) | 0.008 | 0.007 |
| Bond angles (°) | 1.073 | 1.096 |
Values in parentheses are for the highest resolution shell.
The epitope of R53 (431GKAMYAPPIRG441) does not form a regular secondary structure, but it is shaped like a stretched spring, lying across the top of the light and heavy chains (Figure 3). Its N-terminus sits in a groove formed by the heavy chain, while the C-terminus straddles on a saddle formed by the R53 light chain residue AspL30, on one side hanging the side chain of ArgP440 and on the other side the side chain of IleP439. Electrostatic potential analysis indicates that the antigen-binding site of R53 is negatively charged (Figure 3D), complementing the positively charged epitope that harbors two positively charged residues, LysP432 and ArgP440. All complementarity-determining regions (CDRs) are involved in antigen binding, except for the second CDR loop of the light chain (CDR L2).
Antigen–antibody interaction of R53
The antigen–antibody interaction of R53 involves extensive hydrophobic contacts, with ∼730 Å2 of buried surface area, and multiple potential hydrogen bonds between the antibody and the epitope. The epitope of R53 is comprised of six hydrophobic residues, including AlaP433, MetP434, AlaP436, ProP437, ProP438 and IleP439, and they are involved in several hydrophobic interactions (Figure 4). Interestingly, the second proline residue in the epitope, ProP438, has barely any contact with R53 (less than 5 Å2 of calculated contact area). The epitope residue TyrP435 has the largest contact with the antibody; its side chain stacks in parallel with the backbone of SerL95A of CDR L3. There are also hydrogen bonds between the epitope backbone and the antibody: for example, TyrL32 and TyrL92 of the light chain with the backbone of the epitope residues ProP437 and IleP439, respectively; the backbone amide of AlaP433 with the side chain of heavy chain residue AspH51B; the backbone of MetP434 with the amide of TyrH97; and the OH group of the epitope residue TyrP435 with the side chain of the heavy chain GluH55 (Figure 4).
Figure 4.
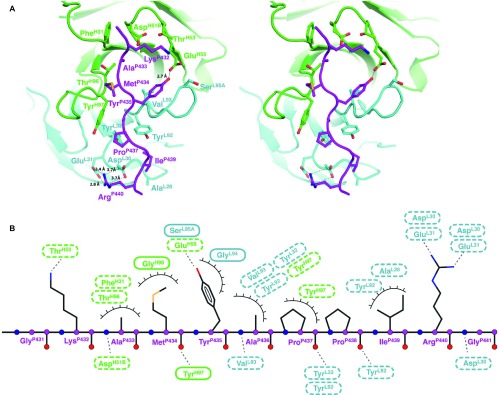
Details of the epitope binding in the Fab R53/epitope complex. (A) A stereo view of the antigen-binding site. The side chains of the key residues involved in hydrogen bonding and van der Waals interactions (with contact areas greater than 10 Å2) are shown in sticks. Note that (i) three tyrosines and four negatively charged residues in R53 play a key role in binding the epitope, (ii) C4 residue ArgP440 interacts with two acidic residues AspL30 and GluL31 from the light chain, and (iii) the side chain of C4 residue TyrP435 engages in van der Waals stacking with the antibody backbone, and its hydroxyl group forms a hydrogen bond with the side chain of GluH56 of the heavy chain. (B) A schematic of the antigen–antibody interaction. Hydrogen-binding interactions are indicated by dashed lines between the residues, whereas van der Waals contacts are indicated by eyelashes. Residues in solid ovals contribute to the interaction by their main chain atoms, and those in dashed ovals contribute to the interaction by their side chain atoms.
The epitope of R53 contains no acidic residues but has two basic residues, LysP432 and ArgP440. The side chain of LysP432 is located in an acidic environment that is formed by AspH51B and GluH55, while the side chain of ArgP440 forms salt bridges with two heavy chain residues, AspL30 and GluL31, of R53 (Figure 4). The R53-contacting residues, AM/IYAPPI, are highly conserved among clades A, B, C, D, and AE (Figure 1D).
Spatial location of the R53 epitope in gp120 structures
After an examination of all currently available gp120 structures in the RCSB PDB, we found that the region of the R53 epitope can form four distinct conformations (Figure 5): (i) the CD4-bound (represented by the first gp120 core structure with PDB ID 1GC1. The majority of the gp120 structures, including the unbound core structures, belong to this group), (ii) the mAb b12-bound (PDB ID 2NY7), (iii) the mAb F105-bound (PDB ID 3HI1), and (iv) the mAb b13-bound conformations (PDB ID 3IDX).13,27,28,29 In the CD4-bound conformation, this region extends from beta strand 21 of the bridging sheet to loop F13. In the b12-bound gp120, the conformation of this region is similar to that of the CD4-bound gp120. However, the bridging sheet is not completely formed due to the dislocation of the two strands (strands 2 and 3) from the inner domain.29 In the b13-bound and F105-bound gp120s, this region, as well as the region of strand 20, forms a coil. Superimposing the R53 epitope with the C4 region in these gp120 structures showed that the epitope is only available in the b13-bound conformation, i.e., R53 binding of the epitope does not clash with the rest of the gp120 molecule. In a recently reported work, structures of the stabilized BG505 SOSIP.664 trimer,2,7,8 the epitope region of R53 is covered under the variable loops V1V2 and V3, and is thus completely buried in the interior of the trimer (Figure 5E).
Figure 5.

Locations and conformations of the R53 epitope region in gp120 structures. The structure of the R53 epitope, together with Fab R53 (only the Fv region is shown), was superimposed onto the epitope region of gp120 structures with distinct C4 conformations, including the CD4-bound (A), b12-bound (B), F105-bound (C), and b13-bound (D) gp120s.11,25,26,27 The R53 epitope is colored magenta, while gp120 is colored gray. Clashes between R53 and gp120s are indicated. The R53 epitope is accessible (without clashes between the antibody and gp120) only in the b13-bound conformation of gp120. (E) The location of the R53 epitope in the recent published structure of BG505 SOSIP.664 trimer. The R53 epitope region, located underneath V1V2/V3, is colored magenta, while the rest of the three gp120s is colored gray, yellow, and orange, respectively.
Discussion
We have structurally defined a linear C4 epitope in complex with a broadly reactive gp120-specific mAb to understand the antigen–antibody interactions in a region of gp120 that serves as a target for many previously reported CD4 blocking mAbs, for which structural information was previously unavailable. The epitope is located at the C-terminus of the bridging sheet and loop F. This region plays critical roles in gp120 functions and thus is highly conserved. Residues TyrP435, ProP438, IleP439, ArgP440, and GlyP441 in the R53 epitope region are critical for co-receptor binding.6,40 This region is also highly immunogenic, as indicated by several mAbs that were reported in the literature. However, most of the antibodies that target this region are not neutralizing or have only weak neutralization activities,23,25,30,41,42 suggesting that this region is masked in the gp120 trimer.
Our data provide a mechanistic understanding of how this region is masked. In the pre-fusion gp120/gp41 complex trimer, this region is buried by V1V2 and V3 loops that lie on top of it; thus, this region is sequestered from interactions with antibodies. In the CD4-bound state, this region is located on the surface of the gp120 monomer (not shown). However, in this conformation the epitope of R53 is not available for mAb binding (Figure 5A), as the side chains of amino acids, such as TyrP435 that binds R53, are buried and face the core of the molecule. Thus, the high immunogenicity of this region may be derived from the side chains that are not accessible in the CD4-bound state. Therefore, our structure can explain the mechanism by which this family of mAbs blocks the binding of CD4, i.e., as illustrated in Figure 5A, the CD4-bound conformation of gp120 is not compatible with R53 binding. In other words, the binding of the CD4-blocking mAbs will prevent the correct formation of the CD4 binding site, thus preventing CD4 binding to gp120.
RCSB PDB accession numbers for the crystal structures
Atomic coordinates for the R53 Fab/epitope complex and the Fab alone have been deposited in the RCSB PDB with the accession codes 4ZTO and 4ZTP, respectively.
Acknowledgments
This work is supported in part by NIH (grant NOs AI082274, AI082676, AI065250, and AI100151). We thank staff members of the beamlines for their help with X-ray diffraction data collection. The X6A beam line is funded by the National Institute of General Medical Sciences under agreement NO GM-0080. The National Synchrotron Light Source, Brookhaven National Laboratory is supported by the US Department of Energy under contract NO DE-AC02-98CH10886. GM/CA at APS has been funded in whole or in part with federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of the APS was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract NO DE-AC02-06CH11357.
References
- Kwong PD, Doyle ML, Casper DJ, et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature. 2002;420:678–682. doi: 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- Pancera M, Zhou T, Druz A, et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Burke V, Totrov M, et al. Conserved structural elements in the V3 crown of HIV-1 gp120. Nat Struct Mol Biol. 2010;17:955–961. doi: 10.1038/nsmb.1861. [DOI] [PubMed] [Google Scholar]
- Killikelly A, Zhang HT, Spurrier B, et al. Thermodynamic signatures of the antigen binding site of mAb 447-52D targeting the third variable region of HIV-1 gp120. Biochemistry. 2013;52:6249–6257. doi: 10.1021/bi400645e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao HX, Bonsignori M, Alam SM, et al. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity. 2013;38:176–186. doi: 10.1016/j.immuni.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lam SN, Acharya P, et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science. 2007;317:1930–1934. doi: 10.1126/science.1145373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP, Cupo A, Sok D, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D, Julien JP, de Val N, et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrow S, Hahn BH, Shaw GM, Gallo RC, Wong-Staal F, Wolf H. Computer-assisted analysis of envelope protein sequences of seven human immunodeficiency virus isolates: prediction of antigenic epitopes in conserved and variable regions. J Virol. 1987;61:570–578. doi: 10.1128/jvi.61.2.570-578.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner L, Fisher A, Jagodzinski LL, et al. Complete nucleotide sequences of functional clones of the AIDS virus. AIDS Res Hum Retroviruses. 1987;3:57–69. doi: 10.1089/aid.1987.3.57. [DOI] [PubMed] [Google Scholar]
- Lasky LA, Nakamura G, Smith DH, et al. Delineation of a region of the human immunodeficiency virus type 1 gp120 glycoprotein critical for interaction with the CD4 receptor. Cell. 1987;50:975–985. doi: 10.1016/0092-8674(87)90524-1. [DOI] [PubMed] [Google Scholar]
- Rizzuto C, Sodroski J. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res Hum Retroviruses. 2000;16:741–749. doi: 10.1089/088922200308747. [DOI] [PubMed] [Google Scholar]
- Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto CD, Wyatt R, Hernandez-Ramos N, et al. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science. 1998;280:1949–1953. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- Moore JP, Thali M, Jameson BA, et al. Immunochemical analysis of the gp120 surface glycoprotein of human immunodeficiency virus type 1: probing the structure of the C4 and V4 domains and the interaction of the C4 domain with the V3 loop. J Virol. 1993;67:4785–4796. doi: 10.1128/jvi.67.8.4785-4796.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt R, Thali M, Tilley S, et al. Relationship of the human immunodeficiency virus type 1 gp120 third variable loop to a component of the CD4 binding site in the fourth conserved region. J Virol. 1992;66:6997–7004. doi: 10.1128/jvi.66.12.6997-7004.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NG, Seillier-Moiseiwitsch F, Ahn J, Walker JM, Swanstrom R. Variability in the human immunodeficiency virus type 1 gp120 Env protein linked to phenotype-associated changes in the V3 loop. J Virol. 2002;76:3852–3864. doi: 10.1128/JVI.76.8.3852-3864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cease KB, Margalit H, Cornette JL, et al. Helper T-cell antigenic site identification in the acquired immunodeficiency syndrome virus gp120 envelope protein and induction of immunity in mice to the native protein using a 16-residue synthetic peptide. Proc Natl Acad Sci USA. 1987;84:4249–4253. doi: 10.1073/pnas.84.12.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berzofsky JA, Bensussan A, Cease KB, et al. Antigenic peptides recognized by T lymphocytes from AIDS viral envelope-immune humans. Nature. 1988;334:706–708. doi: 10.1038/334706a0. [DOI] [PubMed] [Google Scholar]
- Morrow WJ, Williams WM, Whalley AS, et al. Synthetic peptides from a conserved region of gp120 induce broadly reactive anti-HIV responses. Immunology. 1992;75:557–564. [PMC free article] [PubMed] [Google Scholar]
- Li H, Chien PC, Jr, Tuen M, et al. Identification of an N-linked glycosylation in the C4 region of HIV-1 envelope gp120 that is critical for recognition of neighboring CD4 T cell epitopes. J Immunol. 2008;180:4011–4021. doi: 10.4049/jimmunol.180.6.4011. [DOI] [PubMed] [Google Scholar]
- McKeating JA, Moore JP, Ferguson M, et al. Monoclonal antibodies to the C4 region of human immunodeficiency virus type 1 gp120: use in topological analysis of a CD4 binding site. AIDS Res Hum Retroviruses. 1992;8:451–459. doi: 10.1089/aid.1992.8.451. [DOI] [PubMed] [Google Scholar]
- Coussens PM, Tieber VL, Mehigh CS, Marcus M. Identification of a novel transcription factor, ACF, in cultured avian fibroblast cells that interacts with a Marek's disease virus late gene promoter. Virology. 1991;185:80–89. doi: 10.1016/0042-6822(91)90756-2. [DOI] [PubMed] [Google Scholar]
- Sun NC, Ho DD, Sun CR, et al. Generation and characterization of monoclonal antibodies to the putative CD4-binding domain of human immunodeficiency virus type 1 gp120. J Virol. 1989;63:3579–3585. doi: 10.1128/jvi.63.9.3579-3585.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell J, Moore JP, Dean CJ, Klasse PJ, Weiss RA, McKeating JA. Rat monoclonal antibodies to nonoverlapping epitopes of human immunodeficiency virus type 1 gp120 block CD4 binding in vitro. Virology. 1991;185:72–79. doi: 10.1016/0042-6822(91)90755-z. [DOI] [PubMed] [Google Scholar]
- Nakamura GR, Byrn R, Wilkes DM, et al. Strain specificity and binding affinity requirements of neutralizing monoclonal antibodies to the C4 domain of gp120 from human immunodeficiency virus type 1. J Virol. 1993;67:6179–6191. doi: 10.1128/jvi.67.10.6179-6191.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YD, Finzi A, Wu X, et al. Unliganded HIV-1 gp120 core structures assume the CD4-bound conformation with regulation by quaternary interactions and variable loops. Proc Natl Acad Sci USA. 2012;109:5663–5668. doi: 10.1073/pnas.1112391109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Kwon YD, Zhou T, et al. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science. 2009;326:1123–1127. doi: 10.1126/science.1175868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Xu L, Dey B, et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature. 2007;445:732–737. doi: 10.1038/nature05580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Vaine M, Wallace A, et al. A novel rabbit monoclonal antibody platform to dissect the diverse repertoire of antibody epitopes for HIV-1 Env immunogen design. J Virol. 2013;87:10232–10243. doi: 10.1128/JVI.00837-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaine M, Wang S, Crooks ET, et al. Improved induction of antibodies against key neutralizing epitopes by human immunodeficiency virus type 1 gp120 DNA prime-protein boost vaccination compared to gp120 protein-only vaccination. J Virol. 2008;82:7369–7378. doi: 10.1128/JVI.00562-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke V, Williams C, Sukumaran M, et al. Structural basis of the cross-reactivity of genetically related human anti-HIV-1 mAbs: implications for design of V3-based immunogens. Structure. 2009;17:1538–1546. doi: 10.1016/j.str.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Carter CW, Sweet R. Macromolecular Crystallography, Part A, Vol. 276. New York; Academic Press; 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, Hung LW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Abagyan RA, Totrov M, Kuznetsov D. ICM – A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- DeLano WL. The PyMOL User's Manual. Palo Alto, CA; DeLano Scientific; 2002. [Google Scholar]
- Pan R, Sampson JM, Chen Y, et al. Rabbit anti-HIV-1 monoclonal antibodies raised by immunization can mimic the antigen-binding modes of antibodies derived from HIV-1-infected humans. J Virol. 2013;87:10221–10231. doi: 10.1128/JVI.00843-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat EA, Wu TT, Perry HM, Gottesman KS. Sequences of Proteins of Immunological Interest, 5th edn. Bethesda, MD; National Institutes of Health; 1991. [Google Scholar]
- Xiang SH, Wang L, Abreu M, et al. Epitope mapping and characterization of a novel CD4-induced human monoclonal antibody capable of neutralizing primary HIV-1 strains. Virology. 2003;315:124–134. doi: 10.1016/s0042-6822(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Nakamura GR, Byrn R, Rosenthal K, et al. Monoclonal antibodies to the extracellular domain of HIV-1IIIB gp160 that neutralize infectivity, block binding to CD4, and react with diverse isolates. AIDS Res Hum Retroviruses. 1992;8:1875–1885. doi: 10.1089/aid.1992.8.1875. [DOI] [PubMed] [Google Scholar]
- Kelker HC, Itri VR, Valentine FT. A strategy for eliciting antibodies against cryptic, conserved, conformationally dependent epitopes of HIV envelope glycoprotein. PLoS One. 2010;5:e8555. doi: 10.1371/journal.pone.0008555. [DOI] [PMC free article] [PubMed] [Google Scholar]