

Abstract
Although many Archaea have AMP-Acs (acetyl-coenzyme A synthetase) and ADP-Acs, the extant methanogenic genus Methanosarcina is the only identified Archaeal genus that can utilize acetate via acetate kinase (Ack) and phosphotransacetylase (Pta). Despite the importance of ack as the potential urkinase in the ASKHA phosphotransferase superfamily, an origin hypothesis does not exist for the acetate kinase in Bacteria, Archaea, or Eukarya. Here we demonstrate that Archaeal AMP-Acs and ADP-Acs contain paralogous ATPase motifs previously identified in Ack, which demonstrate a novel relation between these proteins in Archaea. The identification of ATPase motif conservation and resulting structural features in AMP- and ADP-acetyl-CoA synthetase proteins in this study expand the ASKHA superfamily to include acetyl-CoA synthetase. Additional phylogenetic analysis showed that Pta and MaeB sequences had a common ancestor, and that the Pta lineage within the halophilc archaea was an ancestral lineage. These results suggested that divergence of a duplicated maeB within an ancient halophilic, archaeal lineage formed a putative pta ancestor. These results provide a potential scenario for the establishment of the Ack/Pta pathway and provide novel insight into the evolution of acetate metabolism for all three domains of life.
The simplest alkane, one of the most abundant organic compounds on Earth, and the main component of natural gas, methane, has been a byproduct of microbial metabolism for over 3.46 Byr1. Methane production, carried out by methanogenic Archaea (methanogens), is an essential part of the global carbon cycle. Methanogens convert microbial metabolites into methane thereby allowing the bacterial breakdown of complex organic carbon compounds that would otherwise be thermodynamically unfavorable due to build-up of hydrogen and/or other metabolites2. Methanogens contain enzymes capable of producing methane via CO2 and H2 (hydrogenotrophic methanogenesis), one-carbon compounds (e.g., formate, methanol, methylamines and methylthiols) (methylotrophic methanogenesis) and/or acetate (aceticlastic methanogenesis)3. Acetate, a major microbial intermediate in anaerobic environments, can account for approximately two-thirds of the methane produced in the biosphere4,5,6,7.
The ability to utilize and/or excrete acetate to balance carbon and electron flow is wide-spread across the three domains of life, and this observation suggests that acetate utilization evolved early. The “acetate switch” refers to a metabolic capacity for cells to either excrete or assimilate acetate based upon the availability of nutrients8. This ability provides an adaptive advantage in dynamic environments by allowing rapid growth in the presence of abundant nutrients and enhanced survival when nutrients are depleted8. The proteins AMP acetyl-coenzyme A synthetase (AMP-Acs), ADP acetyl-coenzyme A synthetase (ADP-Acs) and a pathway involving acetate kinase (Ack) and phosphotransacetylase (Pta) are central to the acetate switch and catalyze the overall process of acetate production or acetate utilization8.
The Ack/Pta pathway is completely reversible and functions optimally at high concentrations of acetate that allows an adaptive advantage in dynamic environments where overflow metabolism occurs in the presence of abundant nutrients9,10. Although many Archaea have AMP-acs and ADP-acs, the extant methanogenic genus Methanosarcina is the only identified Archaeal genus with the Ack/Pta pathway and is the only known lineage to have all three genes (i.e., ack, AMP-acs, and ADP-acs). The absence of the Ack/Pta pathway in all other known Archaea indicates this pathway either evolved within an ancient archaeal genome or was horizontally transferred from another domain. Based upon the available sequences at the time, previous research suggested both ack and pta were provided to an ancient methanogen through horizontal gene transfer from Bacteria11. Although there is not an origin hypothesis for ack (or pta) in Bacteria, an alternative hypothesis has not been proposed because ack or pta homologs have not been identified in any other Archaea. Here we demonstrate that Archaeal AMP-Acs and ADP-Acs contain paralogous ATPase motifs previously identified in Ack, revealing novel Ack homologs in Archaea. Furthermore, a Pta homolog with presumptive active site residues was identified within halophilic Archaea (e.g., Halobacteriales) that have been previously shown to exchange DNA with phylogenetically disparate relatives12,13. These observations provide a possible scenario for the transfer of pta to an ancient halophilic methanogen that possessed ack and the establishment of the Ack/Pta pathway. The presented results coincide with the evolution of the first protein kinase as Ack that was previously postulated to be the urkinase and the most ancient protein in the large ASKHA (acetate and sugar kinases/Hsc70/actin) superfamily of phosphotransferases that share an ATPase domain through gene duplication and divergence14,15. Recent work indicated that a brief period of the Archaeon eon coincided with rapid diversification of bacterial lineages that gave rise to 27% of modern gene families as a consequence of duplication and divergence of genes involved in cellular metabolism16. The evolution of ack and the development of the ASKHA superfamily may have contributed to the early metabolic expansion in biology identified in the Archaeon time period.
Results
ATPase Motif Conservation
The presented results demonstrate that AMP-Acs, ADP-Acs and Ack share motif and active site features (Fig. 1A). The active site of AMP-Acs, located between the two domains of the protein, is formed by core motifs that include a serine and glycine-rich loop (termed region A3), a sequence that contains a threonine-glutamate dipeptide with an invariant glutamate residue (A5), a DX6GXR motif with an invariant glycine (A8), and a region that contains a lysine residue completely conserved in all AMP-forming enzymes (A10)17,18. The ADP-Acs active site has not been well defined due to a lack of protein structures but active site and motif inferences have been previously reported based on a close relationship to succinyl-CoA synthetase (SuccCoA) proteins19,20,21. The α- and β-subunits of ADP-Acs and SuccCoA are often either fused or closely associated ensuring that these subunits are transcribed and work in tandem20,22,23. The ADP-Acs and SuccCoA α−subunits contain a motif similar to the A3 motif in AMP-Acs and an active site dependent on the phosphorylation of a conserved histidine and glutamate residue that aligns with the A5 region in AMP-Acs (Fig. 1A)19. Similar to AMP-Acs and ADP-Acs, Ack is a two domain protein with an initial serine and glycine rich motif (termed Phosphate 1)14,24. The second motif (Connect 1) aligns with A5 in AMP-Acs, with an invariant negatively charged glutamate in place of aspartate in both ADP-Acs and AMP-Acs. An active site histidine residue is completely conserved in Ack, ADP-Acs, SuccCoA and several AMP-Acs20,25,26. In addition, the phosphate 2 motif and the adenosine motif in Ack share homology and secondary structural features with the A8 and A10 motifs from AMP-Acs respectively (Fig. 1A)17,24.
Figure 1.

(A) Methanosarcina AMP-Acs, ADP-Acs and Ack multiple alignment highlighting the shared motifs. The previously identified motifs in Ack and AMP-Acs are above the alignment along with the secondary structure of Ack. Residues are colored according to ClustalX and dashes indicate gaps59. The secondary structure of AMP-Acs is labeled below the alignment. (B) Structural comparisons between Ack and AMP-Acs with previously identified motifs highlighted and color coded25,26.
A HMM-HMM comparison27 with a full alignment (Supp. Fig. 1) identified significant homology between the AMP-Acs, ADP-Acs, SuccCoA synthetase, butyrate kinase (Buk), and Ack sequences. HHPred28 predicted Ack, AMP-Acs and ADP-Acs as homologs with high confidence based on structures and folds (E-values < 1E-12) according to both HMM-HMM global and local multiple alignment comparisons. The three dimensional structures of AMP-Acs, ADP-Acs and Ack indicate that all three proteins consist of two domains17,20,29. The two domains join together forming the active site cleft with an ATP-binding site at the bottom, a feature shared by all proteins in the ASKHA superfamily. This structural feature indicates all of the proteins in the ASKHA superfamily were likely formed through gene duplication and divergence because the structure is the result of complex internal core folds due to motif conservation involved with ATP binding15,30. Previous identification of these structural features in Ack suggested this protein was the ancestral protein of the ASKHA superfamily and potentially the first kinase protein that evolved (Urkinase)14,30. The structural comparisons in Fig. 1B provided the ability to calculate the root-mean-square distance (RMSD) values for the motifs shared between AMP-Acs and Ack (Table 1). The identification of ATPase motif conservation and resulting structural features in AMP- and ADP-acetyl-CoA synthetase proteins in this study expand the ASKHA superfamily to include acetyl-CoA synthetase (Fig. 1).
Table 1. Comparison between previously identified Ack and AMP-Acs motifs with resulting RMSD values8,15.
| Ack Motif | AMP-Acs Motif | RMSD Value |
|---|---|---|
| Phosphate 1 | A3 | 1.82 |
| Connect 1 | A5 | 3.72 |
| Phosphate 2 | A8 | 2.18 |
| Adenosine | A10 | 1.19 |
| Connect 2 | C-terminus | 2.76 |
Phylogenetic Relationships
A phylogenetic comparison of Ack, AMP-Acs, ADP-Acs and SuccCoA shows the evolutionary relationship of the proteins (Fig. 2). Branch lengths indicate a large number of substitutions between Ack and both ADP-Acs and AMP-Acs. Buk and Ack appear to share a common ancestor, and this result supports previous research that suggested Buk evolved from an ancient Ack14. The results demonstrate homology for AMP-Acs, ADP-Acs and Ack that suggests these acetate-metabolizing proteins share a common ancestor.
Figure 2. Phylogenetic analysis of Buk, Ack, SuccCoA, ADP-Acs and AMP-Acs using BEAST.

The posterior probabilities label each node and branch lengths are scaled to expected substitutions per site. The yellow box denotes Methanosarcina Ack sequences, the green box denotes Buk sequences, the red box denotes ADP-Acs sequences, and the blue box denoted AMP-Acs sequences.
Genome Synteny Investigations
Methanosarcina spp. are the only identified Archaea with Ack, and this observation suggests ack was either horizontally transferred or could have evolved within the archaeal genomes11. Genome synteny can be maintained for hundreds of millions of years, therefore genes of close phylogenetic relatives in the same loci are frequently orthologous31. Genome synteny comparisons suggest ack in Methanosarcina is located in the same locus as the gene encoding the ADP-acs α subunit in the close phylogenetic relatives M. mahii and M. evestigatum (Fig. 3). Furthermore, ack in Methanosarcina spp. is located near the ADP-acs β-subunit while in most other Archaeal genomes, including M. mahii and M. zhilinae, the genes encoding the α- and β-subunits of ADP-Acs are either fused or closely associated22,23. As demonstrated in Fig. 3, the ack and ADP-acs (β−subunit) genes are in the same locus (separated by 1 or 2 genes) in the Methanosarcina mazei and Methanosarcina acetivorans genomes. M. mazei and M. acetivorans contain a sole gene encoding the ADP-acs α−subunit (gi20904723 and gi19919192) in a gene locus different from the halophilic methanogens and separate from the gene encoding the β-subunit of ADP-Acs and these results support a possible scenario for the evolution of ack (see discussion).
Figure 3. Genome alignment between Methanosarcina and halophilic methanogens.
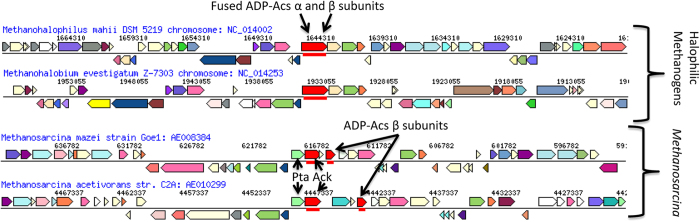
Halophilic methanogens fused ADP-Acs a and b subunits (red) exhibit synteny to ack (red) and the ADP-Acs b subunit (red) in Methanosarcina mazei and Methanosarcina acetivorans. The pta (light green) is closely associated with ack within all sequenced Methanosarcina genomes. The color shading indicates homology shared between the genomes.
Evolution of a Catalytic Pta in Halobacteriales
Pta reversibly exchanges CoA and phosphate groups between acetyl-CoA and acetyl-P, maintaining a balance between biosynthesis and energy generation32,33. The presented analyses identified two different Pta isoforms (Pta and MaeB) in Halobacteriales not previously described in Archaea (Fig. 4). MaeB consists of a fusion between the malic enzyme and Pta that has been characterized in Bacteria34. Results suggest the homologous Pta portion of MaeB enables better metabolic modulation of malic enzyme but lacks Pta activity34. Many Halobacteriales contain duplicate maeB genes providing the possibility for one copy to diverge. Halococcus spp. contain maeB as well as a presumptive pta that contains the conserved catalytic residues35; however, further work is needed to confirm Pta activity. Phylogenetic analysis showed that Pta and MaeB sequences had a common ancestor, and that the Pta lineage within the halophilc archaea was an ancestral lineage to the bacterial and Methanosarcina sequences (Fig. 4). These results suggest that divergence of a duplicated maeB within the ancient Halococcus lineage could have formed a putative pta ancestor. A conserved Pta residue (R310) was present in all Halobacteriales MaeB but absent in bacterial MaeB analyzed in this report (Fig. 5)35. The haloarchaeal Pta sequences were more closely related to those of Methanosarcina and Bacteria, and further work is needed to discern the relationship and significance of the homologous regions of archeal Pta and MaeB sequences.
Figure 4. Phylogenetic relationships of archaeal and bacterial Pta and Ack sequences via BEAST analysis.

The yellow box denotes Methanosarcina Pta sequences, the green box denotes haloarchaeal Pta sequences, and the red box denotes MaeB sequences.
Figure 5. Alignment highlighting the shared active site residues between archaeal and bacterial MaeB and Pta.

Active site residues are in black boxes. Sequences are colored according to the ClustalX color scheme and dashed indicate gaps in the alignment.
Discussion
Our results provide a novel link between AMP-Acs, ADP-Acs and Ack through duplication and divergence of shared motifs involved in ATP binding. The major high-transfer potential molecule of the cell, ATP, is critical for all extant life on Earth that is currently known. It is suggested that deep within hydrothermal vents, AMP-Acs evolved the ability to bind ATP through bio-mimicry of the mineral greigite36. The notion of divergent evolution for the Ack has been previously postulated, and the ancestral ATPase has evolved into a diverse set of proteins across all three domains with a myriad of functions (e.g., actin, hexokinase, hsp70)8. Bork et al. argued it unlikely that the rather particular and complicated fold of the ATPase domain would arise twice as result of evolutionary pressure11; and therefore, the relationship is likely a result of divergent evolution. Providing a relationship between AMP-Acs, ADP-Acs, and Ack allows deeper insight into the evolution of the ASKHA superfamily and an ATPase domain common to proteins in all three domains of life, including actin in eukaryotic cells14,15.
Genome synteny results suggest a gene encoding an ADP-Acs α subunit could have duplicated and diverged into a gene which encodes Ack within an ancient Archaeon. The ADP-Acs α subunit from halophilic methanogens has features including a two domain structure, a glutamate residue that aligns with the magnesium binding aspartate in Ack, and a conserved C-terminus glycine residue that was predicted to be part of the ancient Ack by other researchers14,15. In addition, the evolution of a Pta in Halobacteriales could have allowed horizontal gene transfer of a duplicated pta to an ancient halophilic methanogen with ack and the establishment of the first Ack/Pta pathway. This scenario is supported by research that indicates halophilic methanogens and Halobacteriales engage in genetic exchange similar to conjugation in Bacteria37,38.
The high identity of the archaeal Ack and Pta to homologs in several Eukarya and many Bacteria suggests a single ancient origin of the Ack/Pta pathway39. The presence of biogenic methane in approximately 3.5-Gyr-old fluid inclusions, when the oceans were 1.6 times saltier than today, indicate halophilic methane production and halophiles evolved early1,40. An early reliance on sodium may have promoted co-localization of halophilic methanogens that possessed the presumptive pta and ack genes with ancient Bacteria that relied on F-type and/or V-type ATPases for energy production40. Phylogenetic analysis suggests pta and ack in Methanosarcina are most closely related to those in different Clostridium species11. The ack and pta in Clostridium are in a locus different from the gene cluster for the ancient Wood-Ljungdahl pathway41 and in the same orientation as the homologs observed in Methanosarcina which implicates horizontal gene transfer of both genes in one transfer event between the ancient Methanosarcina and Clostridium. The presented evolutionary relationship between ADP-Acs, AMP-Acs, and Ack provides an evolutionary path for the development of Ack that was then shared between Methanosarcina and Clostridium. The extensive distribution of ack throughout Bacteria and Eukarya suggests Ack contributed to the evolutionary fitness of many organisms across all domains of life11.
Today, many organisms take advantage of the reversibility of the Ack/Pta pathway to conserve energy and maintain steady-state levels of free CoA in the cell42. This flexibility contributes greatly to cell survival when alternating between copiotrophic and oligotrophic conditions often experienced in situ8. Thermodynamic constraints of acetate activation suggest Ack/Pta is suboptimal for acetate consumers at low acetate concentrations, while these organisms would have an increased energetic yield in high acetate environments. Conversely for acetate producing organisms, Ack/Pta provides a thermodynamic gradient which is functional under the largest range of acetate concentrations whereas AMP-Acs is often considered irreversible. During direct resource competition, increased rate or alternative substrate utilization can provide a major competitive advantage43.
Research indicates that a brief period of the Archaean eon coincided with rapid diversification of bacterial lineages that gave rise to 27% of major modern gene families, and functional analyses of the genes originating during this expansion show likely involvement in electron-transport and respiratory pathways16. The expansion could have contributed to the wide distribution of the Ack/Pta pathway in many bacterial lineages and the evolution of the ASKHA superfamily14. Additionally, eukaryotes have incorporated Ack into at least three different metabolic pathways including the Ack/Pta pathway, the Ack/Xfp pathway and a pathway involving ADP-Acs and Ack44. Ack has been observed to be widespread in fungi with putative ack sequences identified in three of the four fungal phyla including the Chytridiomycota, the earliest branch of true fungi44. Although the evolution of the Ack/Pta pathway may have greatly expanded the diversity of life on Earth, the enhanced methane production provided by the pathway (i.e., acetoclastic methanogenesis) under certain environmental conditions may have greatly decreased diversity at least once.
Five major extinction events have been identified that greatly diminished the diversity of life on Earth45. Previous research suggests the largest extinction, the end-Permian extinction that decimated up to 95% of shell-bearing marine species and 80% of land animals, was the result of a large release of biogenic methane46,47. The presented results with additional genome sequences suggest that a Ack/Pta pathway could have been acquired or evolved within a methanogenic genome. Thus, the potential for a large methane release was in place long before the end-Permian extinction, suggesting a change in environmental conditions could have promoted methanogenic microorganisms. Recent research indicates that the ancient combustion of coal through an interaction with basalt sills of the Siberian Traps released trace metals that created stimulatory conditions for methane production and instigated the end-Permian extinction47,48,49,50. Today, atmospheric methane concentrations have nearly tripled since pre-industrial times and this increase has been partly attributed to coal combustion51,52.
The presented data offers the first evolutionary relationships between acs and ack as well as mae and pta and has implications for the development of protein kinases and the Ack/Pta pathway. The presented results reveal novel motif, active site, and structural conservation between AMP-Acs, ADP-Acs and Ack that suggest the proteins are related through gene duplication and divergence. A possible scenario is suggested in which the first Ack could have evolved from a duplicated ADP-Acs α-subunit in an ancestral Archaeon. A pta homolog is conserved in all the Halobacteriales genomes sequenced to date, and the close relation of Methanosarcina spp. to halophilic methanogens suggests the pta gene could have been acquired via horizontal gene transfer from ancient Halobacteriales that existed in the same environment. This event greatly impacted the cycling of carbon via a metabolic node that coordinates cellular catabolism and anabolism, and likely was a turning point in the evolutionary history of microbial life and carbon cycling on a global scale. For example, access to the acetate switch would have allowed greater energy conservation that likely played a role in early cell development53. An increased understanding of the Ack/Pta pathway origins and the historical impacts on the biosphere will provide insight into the evolution and diversification of biological function in diverse microbial communities.
Methods
Sequence Alignment
Amino acid sequences: L. ferriphilum Ack gi406774110 A. capsulatum Ack gi506377611, C. parvum Ack gi193086734, A. xylosoxidans Ack gi310762969, A. laidlawii Ack gi161985465, A. butzleri Ack gi526474464, B. hyodysenteriae Ack gi225619127, F. nodosum Ack gi154153381, B. adolescentis Ack gi740659093, A. colombiense Ack gi293617041, I. album Ack gi383801907, M. barkeri Ack gi73669326, M. acetivorans Ack gi20092406, M. mazei Ack gi17907861, C. cellulolyticum Ack gi506405859, A. fermentans Ack gi502703469, D. thermophilum Ack gi501541912, C. exile Ack gi381363692, E. minutum Ack gi501383324, G. bemidjiensis Ack gi197088018, C. nitroreducens Ack gi312939484, D. indicum Ack gi503271010, F. succinogenes Ack gi261372347, E. coli Ack gi15802843, S. negevensis Ack gi503709960, C. akajimensis Ack gi293614294, M. hydrothermalis Ack gi328451244, G. aurantiaca Ack gi226090818, A. arabaticum Buk gi302391242, S. smaragdinae Buk gi301637019, Clostridium sp. Buk gi357173877, G. thermoglucosidasius Buk gi336234763, M. zhillinae ADP-Acs gi335930152, M. evestigatum ADP-Acs gi298675798, M. mahii ADP-Acs gi294496302, M. psychrophilus ADP-Acs gi410670387, M. hollandica ADP-Acs gi505136916, M. ruminatium ADP-Acs gi502721385, H. mukohataei ADP-Acs gi506243470, H. hispanica ADP-Acs gi503806362, N. gregoryi ADP-Acs gi491747964, Salinarchaeum sp. ADP-Acs gi510882650, H. jeotgali ADP-Acs gi495691923, H. borinquense ADP-Acs gi313126866, M. igneus ADP-Acs gi503565020, Methanocaldococcus sp. ADP-Acs gi502746162, M. fervens ADP-Acs gi506272139, M. fervidus ADP-Acs gi312137221, A. fulgidus ADP-Acs gi668356014, T. kodakarensis SuccCoA gi57160139, Pyrococcus sp. SucSoA gi389853130, A. saccharovorans ADP-Acs gi302349008, M. acetivorans ADP-Acs gi19917192, M. mazei Tuc01 ADP-Acs gi452208966, M. mazei ADP-Acs gi21226460, M. barkeri AMP-Acs gi73669665, M. acetivorans AMP-Acs gi20091733, M. mazei Tuc1 AMP-Acs gi452211690, C. methylpentosum AMP-Acs gi493399067, C. cellulolyticum AMP-Acs gi220928894, C. akajimensis AMP-Acs gi754108219, B. australis AMP-Acs gi505211453, G. kilaueensis AMP-Acs gi752573627, A. capsulatum AMP-Acs gi225791282, M. thermophila AMP-Acs gi116666161, M. harundinacae AMP-Acs gi386001446, M. concilii AMP-Acs gi330506788, A. ferrooxidans AMP-Acs gi753964065, A. colombiense AMP-Acs gi502814190, C. calidirosea AMP-Acs gi512725278, A. xylosoxidans AMP-Acs gi503158389, M. mazei Pta gi452099000, M. acetivorans Pta gi19917662, C. cellulolyticum Pta gi220929552, C. saccharolyticus Pta gi346722880, D. metallireducens Pta gi493768054, T. celere Pta gi517490922, C. akajimensis Pta gi502808194, F. succinogenes Pta gi504358929, T. halophilus Pta gi503890526, B. fragilis Pta gi53711769, D. indicum Pta gi503271009, I. album Pta gi383801098, Fusobacterium sp. Pta gi696260554, A. fermentans Pta gi502703540, B. hyodysenteriae Pta gi501921082, G. auranitica Pta gi226090483, H. morrhuae Pta gi490155740 H. thailandensis Pta gi495013868, I. album MaeB gi504373553, D. indicum MaeB gi503270289, D. desulfuricans MaeB gi502772806, D. baarsil MaeB gi760132925, H. thailandensis MaeB gi495015987, H. hamelinensis MaeB gi445789056, H. mukohataei MaeB gi257170109 H. borinquense MaeB gi313125645, B. fragilis MaeB gi695330578, F. succinogenes MaeB gi504358448, E. coli MaeB gi378259043 were obtained from the National Center for Biotechnology Information (NCBI) genomic database. The sequences were trimmed and aligned with Mafft using a default setting followed by the settings E-INS-I and finally L-INS-I54,55. The sequences were manually adjusted based on secondary structures provided by Jpred56. Sequences were colored according to ClustalX with dashes indicating gaps57. Using JGI-IMG Usearch and Nearest Neighbor functions, a gene synteny map was generated for Methanosarcina Ack (gi17907861)58,59. In an iterative fashion we investigated the synteny in related halophilic methanogens.
HHPred Analysis
Using HHPred28 (Open source 2.0) a homology detection and structure predictor by HMM-HMM comparison 6 sequences (2AMP-Acs, 2ADP-Acs, 2Ack) from our alignment were analyzed by HHPred. Using the HHBlits module at default parameters in global alignment mode these sequences were compared to the PDB70 HMM database28. The structural comparison results indicated ADP-Acs, AMP-Acs, Ack and Buk were homologous at a probability of 100% with e-values of 9.1E-82, 8.4E-63, 1.3E-23, 1E-12, respectively. This structural evidence supports homology at the superfamily and fold level. Using default parameters but different databases such as CDD, Interpro andPfam28, the outcome resulted with lower e-values and better probability due to the availability of more sequenced data.
Pymol Analysis
The structure AMP-Acs M. acetivorans (3ETC_A) and Ack M. thermophila (1g99) were downloaded from the protein database (PDB)28. All structures were trimmed to represent the multiple alignment. The motifs which form the ATPase cleft were identified with Pymol. (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC).
Phylogenetic Analysis
A Bayesian BEASTv1.8.060 tree was created from the manually adjusted sequence alignment using a 10 million step chain with 20% burn-in; run under the WAG amino acid substitution model61 and rate heterogeneity among sites (gamma distribution with 8 categories) with relaxed exponential clock model and coalescent constant population tree prior. Four Constraint priors were applied for the Buk, Ack, AMP-Acs and ADP-Acs groups from a preliminary UPGMA tree generated from Molecular Evolutionary Genetics Analysis (MEGA)62 (data not shown); a randomized tree was used as the starting tree input.
Additional Information
How to cite this article: Barnhart, E. P. et al. Potential Role of Acetyl-CoA Synthetase (acs) and Malate Dehydrogenase (mae) in the Evolution of the Acetate Switch in Bacteria and Archaea. Sci. Rep. 5, 12498; doi: 10.1038/srep12498 (2015).
Supplementary Material
Acknowledgments
This material by ENIGMA- Ecosystems and Networks Integrated with Genes and Molecular Assemblies (http://enigma.lbl.gov), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Biological & Environmental Research under contract number DE-AC02-05CH11231. We thank many students from Dr. Marcie McClure’s Molecular Evolution and Genomic Analysis Lab for their insightful comments and guidance as well as Hannah Schweitzer for helpful discussions. E.P.B. was also supported by the U.S. Geological Survey Energy Program and the DOE-ZERT Program under grant No. DE-FC26-04NT42262. Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the US Government.
Footnotes
Author Contributions E.P.B. and M.W.F. developed hypotheses, and E.P.B., K.J., S.C. and M.A.M. performed bioinformatic analyses. E.P.B., K.A.H., M.A.M. and M.W.F. refined hypotheses and wrote and revised the manuscript.
References
- Ueno Y., Yamada K., Yoshida N., Maruyama S. & Isozaki Y. Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era. Nature 440, 516–519 (2006). [DOI] [PubMed] [Google Scholar]
- Schink B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 61, 262–280 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinder S. H. in Methanogenesis ecology physiology biochemistry and genetics (Ferry J. G. ) 128–206 (Chapman & Hall, 1993). [Google Scholar]
- Ferry J. G. Methane from acetate. J. Bacteriol. 174, 5489–5495 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetten M. S. M., Stams A. J. M. & Zehnder A. J. B. Methanogenesis from acetate: a comparison of the acetate metabolism in Methanothrix soehngenii and Methanosarcina spp. FEMS Microbiol. Rev. 88, 181–197 (1992). [Google Scholar]
- Ferry J. G. How to make a living by exhaling methane. Ann. Rev. Microbiol. 64, 453–473 (2010). [DOI] [PubMed] [Google Scholar]
- Kotsyurbenko O. R. et al. Acetoclastic and hydrogenotrophic methane production and methanogenic populations in an acidic West-Siberian peat bog. Environ. Microbiol. 6, 1159–1173 (2004). [DOI] [PubMed] [Google Scholar]
- Wolfe A. J. The acetate switch. Microbiol. Mol. Biol. Rev. 69, 12–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min H. & Zinder S. H. Kinetics of acetate utilization by two thermophilic acetotrophic methanogens: Methanosarcina sp. Strain CALS-1 and Methanothrix sp. Strain CALS-1. Appl. Environ. Microbiol. 55, 488–491 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri G. N., Altman E., Sangurdekar D. P., Khodursky A. B. & Eiteman M. A. Overflow metabolism in Escherichia coli during steady-state growth: Transcriptional regulation and effect of the redox ratio. Appl. Environ. Microbiol. 72, 3653–3661 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier G. P. & Gogarten J. P. Evolution of acetoclastic methanogenesis in Methanosarcina via horizontal gene transfer from cellulolytic Clostridia. J. Bacteriol. 190, 1124–1127 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaere M. Z. et al. High level of intergenera gene exchange shapes the evolution of haloarchaea in an isolated Antarctic lake. Proc. Nat. Acad. Sci. USA 110, 16939–16944 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan N. R. et al. Evidence from phylogenetic and genome fingerprinting analyses suggests rapidly changing variation in Halorubrum and Haloarcula populations. Front. Microbiol. 5, 143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss K. A. et al. Urkinase: structure of acetate kinase, a member of the ASKHA superfamily of phosphotransferases. J. Bacteriol. 183, 3536 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P., Sander C. & Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc. Nat. Acad. Sci. USA 89, 7290–7294 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- David L. A. & Alm E. J. Rapid evolutionary innovation during an Archaean genetic expansion. Nature 469, 93–96 (2011). [DOI] [PubMed] [Google Scholar]
- Gulick A. M., Starai V. J., Horswill A. R., Homick K. M. & Escalante-Semerena J. C. The 1.75 A crystal structure of acetyl-CoA synthetase bound to adenosine-5’-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873 (2003). [DOI] [PubMed] [Google Scholar]
- Starai V. J., Celic I., Cole R. N., Boeke J. D. & Escalante-Semerena J. C. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 (2002). [DOI] [PubMed] [Google Scholar]
- Bräsen C., Schmidt M., Grötzinger J. & Schönheit P. Reaction mechanism and structural model of ADP-forming Acetyl-CoA synthetase from the hyperthermophilic archaeon Pyrococcus furiosus: evidence for a second active site histidine residue. J. Biol. Chem. 283, 15409–15418 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez L. B., Galperin M. Y. & Müller M. Acetyl-CoA synthetase from the amitochondriate eukaryote Giardia lamblia belongs to the newly recognized superfamily of acyl-CoA synthetases (Nucleoside diphosphate-forming). J. Biol. Chem. 275, 5794–5803 (2000). [DOI] [PubMed] [Google Scholar]
- Musfeldt M., Selig M. & Schönheit P. Acetyl coenzyme A synthetase (ADP forming) from the hyperthermophilic Archaeon Pyrococcus furiosus: identification, cloning, separate expression of the encoding genes, acdAI and acdBI, in Escherichia coli, and in vitro reconstitution of the active heterotetrameric enzyme for its recombinant subunits. J. Bacteriol. 181, 5885–5888 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräsen C. & Schönheit P. Unusual ADP-forming acetyl-coenzyme A synthetases from the mesophilic halophilic euryarchaeon Haloarcula marismortui and from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. Arch. Microbiol. 182, 277–287 (2004). [DOI] [PubMed] [Google Scholar]
- Bapteste É., Brochier C. & Boucher Y. Higher-level classification of the Archaea: evolution of methanogenesis and methanogens. Archaea 1, 353–363 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker T. M. et al. Crystal structures of acetate kinases from the eukaryotic pathogens Entamoeba histolytica and Cryptococcus neoformans. J. Structural Biol. 181, 185–189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram-Smith C. The role of histidines in the acetate kinase from Methanosarcina thermophila. J. Biol. Chem. 275, 33765–33770 (2000). [DOI] [PubMed] [Google Scholar]
- Fraser M. E., James M. N., Bridger W. A. & Wolodko W. T. A detailed structural description of Escherichia coli succinyl-CoA synthetase. J. Mol. Biol. 285, 1633–1653 (1999). [DOI] [PubMed] [Google Scholar]
- Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960 (2005). [DOI] [PubMed] [Google Scholar]
- Söding J., Biegert A. & Lupas A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittori S., Savithri H. S. & Murthy M. R. Structural and mechanistic investigations on Salmonella typhimurium acetate kinase (AckA): identification of a putative ligand binding pocket at the dimeric interface. BMC Structural Biol. 12, 24 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheek S., Zhang H. & Grishin N. V. Sequence and structure classification of kinases. J. Mol. Biol. 320, 855–881 (2002). [DOI] [PubMed] [Google Scholar]
- Ghedin E. et al. Gene synteny and evolution of genome architecture in trypanosomatids. Mol. Biochemical Parasitol. 134, 183–191 (2004). [DOI] [PubMed] [Google Scholar]
- Lawrence S. H. & Ferry J. G. Steady-state kinetic analysis of phosphotransacetylase from Methanosarcina thermophila. J. Bacteriol. 188, 1155–1158 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mansi M., Cozzone A. J., Shiloach J. & Eikmanns B. J. Control of carbon flux through enzymes of central and intermediary metabolism during growth of Escherichia coli on acetate. Curr. Opin. Microbiol. 9, 173–179 (2006). [DOI] [PubMed] [Google Scholar]
- Bologna F. P., Andreo C. S. & Drincovich M. F. Escerichia coli malic enzymes: two isoforms with substantial differences in kinetic properties, metabolic regulation, and structure. J. Bacteriol. 189, 5937–5946 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence S. H., Luther K. B., Schindelin H. & Ferry J. G. Structural and functional studies suggest a catalytic mechanism for the phosphotransacetylase from Methanosarcina thermophila. J. Bacteriol. 188, 1143–1154 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell M. J. & Martin W. The rocky roots of the acetyl-CoA pathway. Trends Biochemical Sci. 29, 358–363 (2010). [DOI] [PubMed] [Google Scholar]
- Papke R. T., Koenig J. E., Rodríguez-Valera F. & Doolittle W. F. Frequent recombination in a saltern population of Halorubrum. Science 306, 1928–1929 (2004). [DOI] [PubMed] [Google Scholar]
- Allers T. & Mevarech M. Archaeal genetics-the third way. Nature Rev. Genetics 6, 58–73 (2005). [DOI] [PubMed] [Google Scholar]
- Ferry J. G. Acetate kinase and phosphotransacetylase. Meth. Enzymol. 494, 219–231 (2011). [DOI] [PubMed] [Google Scholar]
- Mulkidjanian A. Y., Galperin M. Y., Makarova K. S., Wolf Y. I. & Koonin E. V. Evolutionary primacy of sodium bioenergetics. Biol. Direct 3, 13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poehlein A. et al. An ancient pathway combining carbon dioxide fixation with the generation and utilization of a sodium ion gradient for ATP synthesis. PLoS One 7, e33439 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai V. J. & Escalante-Semerena J. C. Acetyl-coenzyme A synthetase (AMP forming). Cellular Mol. Life Sci. 61, 2020–2030 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer T., Schuster S. & Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science 292, 504–507 (2001). [DOI] [PubMed] [Google Scholar]
- Ingram-Smith C., Martin S. R. & Smith K. S. Acetate kinase: not just a bacterial enzyme. Trends Microbiol. 14, 249–253 (2006). [DOI] [PubMed] [Google Scholar]
- Rohde R. A. & Muller R. A. Cycles in fossil diversity. Nature 434, 208–210 (2005). [DOI] [PubMed] [Google Scholar]
- Knoll A. H., Bambach R. K., Payne J. L., Pruss S. & Fischer W. W. Paleophysiology and end-Permian mass extinction. Earth Planetary Sci. Lett. 256, 295–313 (2007). [Google Scholar]
- Rothman D. et al. Methanogenic burst in the end-Permian carbon cycle. Proc. Natl. Acad. Sci. USA 15, 5462–5467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden D. E. & Sleep N. H. Explosive eruption of coal and basalt and the end-Permian mass extinction. Proc. Natl. Acad. Sci. USA 109, 59–62 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass J. B. & Orphan V. J. Trace metal requirements for microbial enzymes involved in the production and consumption of methane and nitrous oxide. Frontiers Microbiol. 3, 61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M. S., Basha S., Joshi H. V. & Jha B. Evaluation of the emission characteristics of trace metals from coal and fuel oil fired power plants and their fate during combustion. J. Haz. Mat. 123, 242–249 (2005). [DOI] [PubMed] [Google Scholar]
- Bousquet P. et al. Contribution of anthropogenic and natural sources to atmospheric methane variability. Nature 443, 439–443 (2006). [DOI] [PubMed] [Google Scholar]
- Kirschke S. et al. Three decades of global methane sources and sinks. Nature Geosci. 6, 813–823 (2013). [Google Scholar]
- Ferry J. G. & House C. H. The stepwise evolution of early life driven by energy conservation. Mol. Biol. Evol. 23, 1286–1292 (2006). [DOI] [PubMed] [Google Scholar]
- Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M. & Barton G. J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. & Standley D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C., Barber J. D. & Barton G. J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F. & Higgins D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz V. M. et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Database 40, 115–122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- Drummond A. J. & Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S., Li P. & Goldman N. Molecular phylogenetics: State-of-the-art methods for looking into the past. Trends Genetics 17, 262–272 (2001). [DOI] [PubMed] [Google Scholar]
- Tamura K. et al. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. 113, 1530–1534 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.