

Abstract
An extremely drug resistant mutant of HIV-1 protease (PR) bearing 20 mutations (PR20) has been studied with two potent antiviral investigational inhibitors. GRL-5010A and GRL-4410A were designed to introduce hydrogen bond interactions with the flexible flaps of the PR by incorporating gem-difluorines and alkoxy, respectively, at the C4 position of the bis-THF of darunavir. PR20 provides an excellent model for high level resistance, since clinical inhibitors are >1000-fold less active on PR20 than on wild-type enzyme. GRL-5010A and GRL-4410A show inhibition constants of 4.3 ± 7.0 and 1.7 ± 1.8 nM, respectively, for PR20, compared to the binding affinity of 41 ± 1 nM measured for darunavir. Crystal structures of PR20 in complexes with the two inhibitors confirmed the new hydrogen bond interactions with Gly 48 in the flap of the enzyme. The two new compounds are more effective than darunavir in inhibiting mature PR20 and show promise for further development of antiviral agents targeting highly resistant PR mutants.
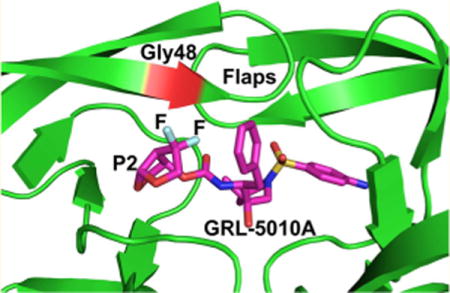
INTRODUCTION
Drug resistance is a major obstacle in the treatment of HIV and AIDS.1 New infections by resistant virus can effectively limit the therapeutic options and increase the risk of treatment failure. Transmission rates of drug resistant strains vary from moderate to high in different regions of the world.2–5 Patients can also acquire drug resistance in the course of treatment, and the risk steadily increases with duration of treatment.6,7 In addition, prevention strategies employing antiviral drugs can increase the prevalence of drug resistant strains.8,9 Tens of millions of people worldwide will undergo antiviral drug therapy for many years, which has the potential to increase the occurrence of drug resistant strains. These factors increase the urgency for the design and development of inhibitors effective against the drug resistant strains.
Previously, we characterized a highly evolved multidrug resistant strain of HIV-1 protease (PR20), derived from a clinical isolate, with 20 mutations in comparison to the wild-type protease (PR). PR20 is extremely resistant to clinically available HIV protease inhibitors.10,11 In addition, the autoprocessing of PR20, which releases the mature protein, is not inhibited by any of the clinical drugs, while darunavir and saquinavir inhibit the autoprocessing of wild-type PR.10 Structural studies show that PR20 forms fewer hydrogen bond interactions than PR does with a substrate analog bound in the active site cavity.11 The extremely drug resistant mutant PR20 exemplifies a prototype to aid the design of new inhibitors retaining high potency against highly resistant mutants. Recently, we determined the crystal structure of PR20 in complex with GRL-0519A, a potent antiviral inhibitor of HIV-1 PR. GRL-0519A is derived from darunavir with three tetrahydrofuran (THF) rings instead of the bis-THF group of darunavir. GRL-0519A exhibits ∼10-fold higher potency than darunavir against multidrug resistant HIV-1 clinical strains.12 However, GRL-0519A and darunavir show similar binding affinity for PR20.13
HIV-1 protease inhibitor GRL-4410A was derived by the incorporation of an alkoxy substituent at the C4-position of bis-THF in darunavir. The bis-THF group is designated as the P2 position in darunavir from the standard nomenclature for protease substrates, where cleavage occurs between amino acid residues P1 and P1′.14 Structural studies have shown that the bis-THF group is involved in a network of hydrogen bonding interactions with the main chain atoms of HIV-1 protease.15 The C4-alkoxy addition at the bis-THF in GRL-4410A forms new interactions with the main chain atoms of Gly 48 in the flaps of PR.16 GRL-4410A displays excellent inhibition of PR activity with a Ki of 2.9 pM. Further, GRL-4410A has impressive antiviral efficacy as determined by the MTT assay with IC50 value of 2.4 nM. Thus, GRL-4410A is an extremely potent antiviral inhibitor of wild-type PR and a good candidate to study against PR20.
Similar to GRL-4410A, the recently reported HIV-1 protease inhibitor GRL-5010A was also derived from darunavir and designed with substitutions at the C4 position of bis-THF group. GRL-5010A has gem-difluorines at the C4 position of bis-THF. Halogen bonding has been shown to play an important role in molecular recognition and assembly.17 In recent years, halogen interactions in protein–ligand complexes have generated increased interest.18,19 The presence of fluorine in the compound often leads to increased lipid solubility, thereby enhancing absorption and transport of drugs.20 Fluorine containing compounds are used in the therapeutic management of central nervous system (CNS) diseases, including psychoses, anxiety, depression, and obsessive-compulsive disorder, due to the improved absorption and transport across the blood–brain barrier.20 The formation of an HIV reservoir in the CNS poses a critical threat in AIDS therapy, and improving the drug concentration in the CNS may be required to eliminate the viral sanctuaries. Furthermore, the presence of viral particles in the CNS leads to HIV-1 associated neurocognitive disorders (HAND), possibly due to poor CNS penetration of current antiviral drugs.21 The fluorine containing compound GRL-5010A was shown to have better blood–brain barrier penetration than darunavir.22,23 Also, the two fluorines in GRL-5010A form interactions with the main chain atoms of flap residue Gly 48 in wild-type PR.23 GRL-5010A is a potent inhibitor of wild-type PR with a Ki of 5.8 pM and inhibits viral replication with an EC50 of 0.003 μM.22 GRL-5010A also showed significantly better inhibition of the replication of seven tested multidrug resistant strains compared to saquinavir, lopinavir, atazanavir, amprenavir, and darunavir, with EC50 values in the range of 0.002–0.02 μM.22
In order to discover potent inhibitors for the extremely drug resistant PR20, we have solved the crystal structures of PR20 in complex with inhibitors GRL-4410A and GRL-5010A. We have analyzed their crystal structures in comparison to the corresponding PR complexes and relative inhibition values. The current structural analysis provides valuable insight for improved design of inhibitors against extremely drug resistant variants like PR20.
RESULTS
Inhibition of PR20 and TFR-PR20 by GRL-4410A and GRL-5010A
GRL-5010A has similar antiviral potency as darunavir against multidrug resistant variants harboring 9–14 mutations associated with drug resistance (DRMs) as measured by the EC50 values.22 GRL-5010A also exhibits moderate antiviral activity against a highly darunavir resistant strain of HIV-1 selected in the laboratory.22 Both GRL-4410A and GRL-5010A exhibit nanomolar inhibition of the proteolytic activity of PR20. The Ki value of GRL-4410A for PR20 is 4.3 ± 7.0 nM, while that of GRL-5010A is 1.7 ± 1.8 nM. The inhibitors, GRL-4410A and GRL-5010A, were reported to have picomolar inhibition values of 2.9 pM16 and 5.8 pM,23 respectively, for wild-type PR. Thus, inhibition of the highly resistant PR20 by GRL-4410A is 1500-fold weaker than for wild-type PR, while GRL-5010A exhibits 300-fold weaker inhibition of PR20 in comparison to wild-type enzyme. However, GRL-4410A has ∼10-fold lower Ki for the inhibition of PR20 relative to darunavir, which has a ligand dissociation constant (KL) of 41 nM for PR20 as measured by isothermal titration calorimetry (ITC).10 Similarly, for GRL-5010A, the relative Ki value is about 2-fold lower for PR20 in comparison to the binding affinity of darunavir. Thus, the two new inhibitors are an order of magnitude better than darunavir for the extremely drug resistant mutant PR20.
The autocatalytic cleavage of the PR from the Gag-Pol precursor polyprotein is a critical step in the maturation of HIV.24,25 The cleavage at the N-terminus of the PR in its precursor is concomitant with formation of stable dimer with very low dimer dissociation constant and consequently the appearance of mature like catalytic activity. The autoprocessing of a model precursor comprising the transframe region (TFR) and PR (TFR-PR) is inhibited most effectively by darunavir, saquinavir, and atazanavir relative to other clinical protease inhibitors. In contrast, the autoprocessing of TFR-PR20 is uninhibited by any of the clinical inhibitors up to a concentration of 150 μM.10 The results of the dose–response study indicate that both GRL-4410A and GRL-5010A inhibit the autoprocessing of the wild-type precursor TFR-PR with about the same IC50 value of 1–2 μM (see Supporting Information Figure S1, panels A and B and references). In spite of an order of magnitude better inhibition of mature PR20 by GRL-4410A and GRL-5010A, however, no significant improvement was observed in the inhibition of the TFR-PR20 precursor in tests up to 100 μM concentration (Figure S1, panels C and D). This suggests that molecular interactions defining the binding affinity between the inhibitor and the mature PR20 are likely to be different from those for the TFR-PR20 precursor undergoing autoprocessing.
Overall Structure
The X-ray crystal structures were determined of PR20 in complex with the potent HIV-1 protease inhibitors GRL-4410A (Figure 1A) and GRL-5010A (Figure 1B) at resolutions of 1.35 and 1.30 Å, respectively. Both of the crystal structures were solved in the P61 space group with one PR20 dimer numbered 1–99 and 1′–99′ in the asymmetric unit (Figure 1C). The two structures were refined to R-factors of 15.7% and 15.5%, respectively, as shown in the crystallographic statistics in Table 1. All 20 protease mutations and the two inhibitors (Figure 1A and Figure 1B) are unambiguously visible in the electron density maps. In both structures, the inhibitor molecule is bound at the active site in two mutually exclusive orientations related by a 180° rotation and the relative occupancy of 0.5. The two monomers of the PR20/4410 and PR20/5010 dimer structures are very similar to each other with an rms deviation of 0.36 and 0.34 Å, respectively. The low values for mean temperature factors of 20.18 Å2 (PR20/4410) and 20.16 Å2 (PR20/5010) for all protein and inhibitor atoms indicate the structures are well ordered, which is important for precise comparison of atomic positions.
Figure 1.
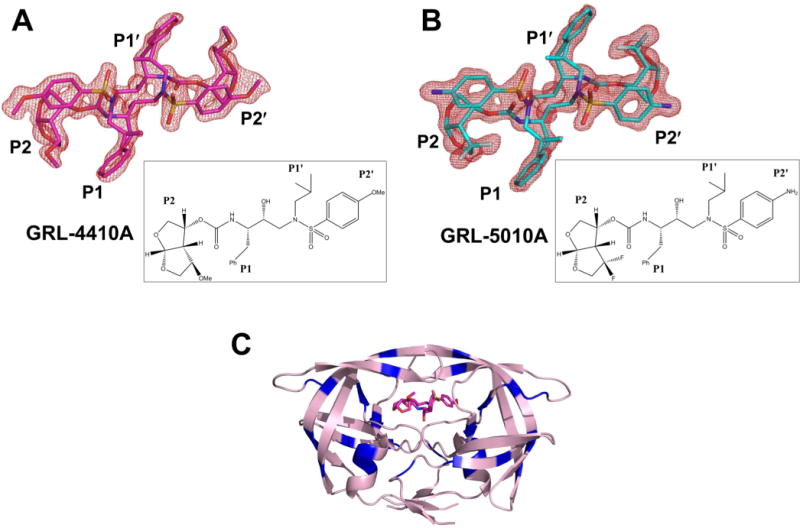
Structures of PR20 dimer with GRL-4410A and GRL-5010A: Fo − Fc omit maps contoured at 3σ level for (A) GRL-4410A and (B) GRL-5010A. The chemical structure of the inhibitor is shown in the inset. (C) Overall structure of PR20 dimer with the bound GRL-4410A colored by atom type in stick representation. Sites of the 20 mutations on HIV-1 PR20 (pink cartoon representation) are colored blue.
Table 1.
Crystallographic Data Collection and Refinement Statistics
| PR20 complex
|
||
|---|---|---|
| PR20/4410 | PR20/5010 | |
| space group | P61 | P61 |
| cell dimensions | ||
| a (Å) | 60.35 | 60.30 |
| b (Å) | 60.35 | 60.30 |
| c (Å) | 85.78 | 86.14 |
| resolution range (Å) | 50.0–1.35 | 50.0–1.30 |
| unique reflections | 38558 | 41329 |
| redundancy | 5.9 (3.2) | 5.9 (4.5) |
| completeness | 99.1 (92.4)a | 94.6 (73.8) |
| ⟨I/σ(I)⟩ | 24.5 (2.3) | 28.5 (3.4) |
| Rsym (%) | 6.5 (47.2) | 5.2 (40.9) |
| refinement resolution range (Å) | 50–1.35 | 50.0–1.30 |
| Rcryst (%) | 15.7 | 15.5 |
| Rfree (%) | 20.0 | 21.0 |
| no. of solvent molecules | 176 | 175 |
| average B-factor (Å2) | ||
| main chain | 16.4 | 16.6 |
| side chain | 21.4 | 21.2 |
| inhibitor | 12.7 | 11.6 |
| solvent | 35.6 | 33.9 |
| rms deviation from ideality | ||
| bond length (Å) | 0.03 | 0.03 |
| angle (deg) | 2.7 | 2.8 |
Values in parentheses are given for the highest resolution shell.
Comparison of PR20/5010 with PR/5010
GRL-5010A was designed from the scaffold of the clinical protease inhibitor darunavir with substitution of two fluorines at the C4 position of bis-THF to form strong interactions with flap residue Gly 48. GRL-5010A was shown to have significantly higher blood–brain barrier permeability than darunavir.23 To better understand the molecular mechanism of PR20 inhibition by GRL-5010A, we compared the crystal structure of PR20/5010 with the wild-type PR/5010 complex (PDB code 4U8W).23 The two dimer structures can be superposed with an RMS deviation of 0.99 Å for 198 Cα atoms. The two monomers of PR20/5010 complex are more similar to the corresponding monomers of PR/5010 and can be superposed with an rms deviation of 0.87, 0.92, 0.87, and 0.88 Å for four different combinations of monomers in the two dimers. Since both of the monomers are similar, the conformation and interactions henceforth will be discussed for one monomer unless there are differences in the second monomer. In PR20/5010 complex, residues in both flaps (44–54 and 45′–54′) are split into two alternative conformations. Also, the residues 59–64 at the base of the flaps are seen in two alternative conformations. In particular, residues Asp 60 and Gln 61 have their main chain carbonyl oxygens flipped in the two conformations (Figure 2A). The corresponding residues 71–74 in the adjacent β strand are also split into two alternative conformations. Only the side chain of Gln 61 in these two strands has an interaction with a symmetry-related molecule. Although crystal contacts cannot be completely ruled out, it is most likely that the presence of alternative conformations for residues in the β strands at the base of flaps is due to the binding of inhibitor GRL-5010A in two alternative, mutually exclusive conformations. The existence of alternative conformations for so many residues in the flaps, the base of the flaps, and adjacent strands is unusual for crystal structures of HIV PR.
Figure 2.
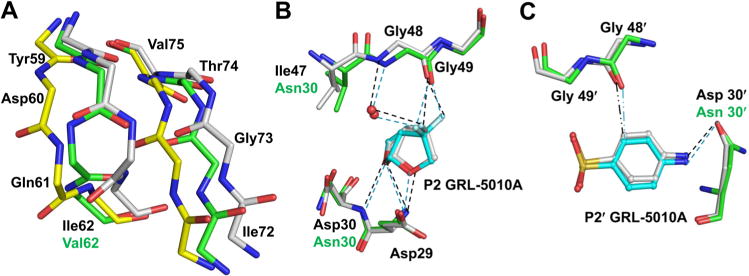
Polar interactions of PR20 with GRL-5010A. (A) The residues 59–64 at the base of the flaps were observed in two alternative conformations with the peptides of Asp 60 and Gln 61 flipped in one conformation. The corresponding residues in the adjacent strand comprising Val 71–Thr 74 also show two alternative conformations. The two conformations of the PR20 main chain atoms are colored by element type with green and yellow carbons, respectively, while the wild-type PR main chain atoms are shown with gray carbons. The side chains of PR20 have been removed for the sake of clarity. (B) The P2 gem-difluorines of GRL-5010A form strong interactions with the carbonyl of Gly 48 in the flaps of PR20 in addition to the water meditated hydrogen bond with the amide of Gly 48. In this and subsequent figures, PR20 and wild-type PR residues are colored by element type with green and white carbons, respectively. GRL-5010A is shown in ball and stick representation with cyan carbons. (C) The P2′ aniline group of GRL-5010A forms a C–H⋯O interaction (··–) with the carbonyl oxygen of Gly 48 in the flap. The P2′group of GRL-5010A also forms a hydrogen bond with the Asn 30 side chain of the active site mutation D30N in PR20.
Mutation D30N in the active site cavity of PR20/5010 complex retains the hydrogen bond observed between Asp 30 and aminophenyl moiety of the inhibitor in the corresponding wild-type complex despite the loss of charge in the mutation. The two mutations I47V and I84V in the S2/S2′ subsites of PR20 maintain the hydrophobic interaction observed between wild-type residues and the inhibitor in the PR/5010 complex despite the smaller side chains. All the hydrogen bond interactions between the inhibitor GRL-5010A and the main chain of PR, like the hydrogen bond interaction between the urethane NH and carbonyl oxygen of Gly 27, are conserved in the mutant PR20/5010 complex. However, the PR20/5010 structure shows differences in the hydrogen bond interactions between the central hydroxyl group of the inhibitor and the two catalytic aspartates in the dimer in comparison to the wild type PR/5010 as described in a later section. In the wild type complex of PR/5010, both of the fluorine atoms on the bis-THF moiety form short interactions with the carbonyl oxygen of flap residue Gly 48. Similar interactions are also observed in the PR20/5010 complex (Figure 2B). Similar fluorine interactions with carbonyl oxygen have been reported for various protein structures.26,27 In addition, one of the two fluorines forms a water mediated interaction with the amide nitrogen of Gly 48 in both the PR/5010 and PR20/5010 complexes. The carbonyl oxygen of Gly 48 in the flaps forms a C–H⋯O interaction with the P2′ aniline moiety of GRL-5010A in both PR/5010 and PR20/5010 complexes (Figure 2C). Thus, GRL-5010A forms strong fluorine mediated interactions with the flaps of both PR and PR20. Further, the water mediated interaction and the C–H⋯O interaction between the flap and GRL-5010A are conserved in both PR and PR20. The inhibitor preserves all of its major interactions with PR when bound to the PR20 mutant, with the marked exception of those between the central hydroxyl and the catalytic Asp 25/25′.
Comparison of PR20/4410 with PR/4410
GRL-4410A is a substituted bis-THF-containing inhibitor derived from the clinical inhibitor darunavir. To understand the molecular mechanism of inhibition of PR20 by GRL-4410A, the structure of PR20/4410 was compared with that of wild-type PR/4410. The two dimer structures can be superposed with an rms deviation of 0.91 Å for 198 Cα atoms. The two monomers of PR20/4410 complex are a little closer to those of PR/4410 and can be superposed with rms deviations of 0.78, 0.82, 0.81, and 0.84 Å for the four different combinations of monomers A and B in the two dimers. The dimer structures are very similar for PR20/4410 complex and PR20/5010 and superpose with an rms deviation of 0.35 Å. Significant differences with more than 1 Å in rms deviation occur in the hinge loop and 60’s loop when PR20/4410 is compared with the wild-type PR/4410 complex. Most of the residues at the base of the flap have a single conformation (Figure 3A), unlike the two alternative conformations in the PR20/5010 complex. Asp 60 and Gln 61 also do not exhibit the flipped conformation of the carbonyl oxygens seen in the PR20/5010 complex. The entire flap region from Met 46 to Phe 53 is split into two alternative conformations in both PR20/4410 and PR/4410 complexes. This indicates that the alternative conformations of the flap are likely due to the two conformations of the bound inhibitor. Mutated residue I47V, one of the mutations that expand the S2/S2′ pockets in PR20, lies within the split flap region, while mutation I54L is adjacent to it. The mutated residue Asn 30 in PR20 and Asp 30 in wild type PR form similar van der Waals interactions with the P2 group of GRL-4410A (Figure 3B). Despite their smaller side chains, the mutations that increase the space in the S2/S2′ pockets, I47V and I84V, form similar hydrophobic interactions with the P2 and P2′ groups of the inhibitor in PR20/4410 and the wild-type PR/4410 complex, albeit in lower numbers for the mutant. Inhibitor GRL-4410A was designed to form interactions with the PR flap though the methoxy group on the bis-THF moiety.16 The C–H⋯O interaction observed between the carbonyl oxygen of Gly 48 and the methoxy methyl of P2 group in the wild type PR/4410 complex also occurs in the mutant PR20/4410 complex (Figure 3B). In addition, the methoxy oxygen of the P2 group forms a water mediated hydrogen bond with the amide nitrogen of Gly 48 in PR20/4410 similar to that of the wild-type PR/4410 complex. The D30N mutation in PR20/4410 retains the hydrogen bond and CH⋯O interaction with the oxygen of the P2′ methoxy group of the inhibitor seen in the wild-type PR/4410 complex (Figure 3C). The carbonyl oxygen of flap residue Gly 48 forms a C–H⋯O interaction with the P2′ aniline group in both PR20/4410 and wild-type PR/4410 complexes (Figure 3C). Other important hydrogen bond interactions between the ligand and protease main chain residues observed in the wild-type PR/4410 complex are retained in the PR20/4410 structure. However, the central hydroxyl group of GRL-4410A exhibits lengthened interactions with one of the catalytic Asp 25/25′ in both orientations, as discussed in a later section. Thus, GRL-4410A maintains its signature interactions with the flap that are observed in both the wild-type PR and PR20 complexes. Further, the inhibitor retains most of its interactions with the mutated residues D30N, I47V, and I84V in the active site cavity of PR20.
Figure 3.
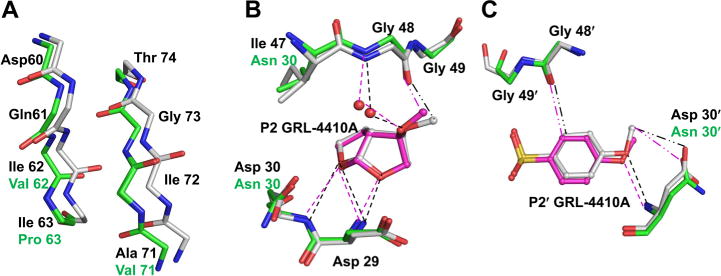
Polar interactions of PR20 with GRL-4410A. (A) The residues at the base of the flaps in the PR20/4410 complex show deviations of more than 1 Å in comparison to PR/4410. PR20 is shown with green carbons and wild type PR with gray carbons. (B) The P2 substituted bis-THF of GRL-4410A forms a C–H⋯O interaction with the main chain carbonyl oxygen of flap residue Gly 48. The methoxy oxygen of the P2 moiety forms water mediated hydrogen bonds with the amide NH of Gly 48 similar to those in the PR complex. GRL-4410A is shown in ball and stick representation with magenta carbons. The P2′ group of GRL-4410A forms a C–H⋯O interaction with the carbonyl oxygen of Gly 48. The methoxy group of P2′ forms hydrogen bond and C–H⋯O interactions (··–) with the main chain amide and the side chain of mutated Asn 30 in the PR20/4410 complex.
Expanded S2/S2′ Pocket Elongates Interactions with the Second Catalytic Asp in PR20
The catalytically active homodimer of HIV protease facilitates binding of many inhibitors in two orientations related by a rotation of 180°. The central hydroxyl group of the inhibitors forms hydrogen bond interactions with the side chain oxygen atoms of the two catalytic Asp 25 and 25′ residues. In both the wild-type PR/4410 and PR/5010 complexes, the central hydroxyl group makes strong interactions with both the catalytic Asp residues. In PR/4410, the hydroxyl of the inhibitor has hydrogen bond interactions of lengths 2.5, 2.7 Å and lengths 2.9, 3.0 Å with Asp 25 and Asp 25′, respectively (Figure 4). The second orientation of the inhibitor has similar or even lower values. The corresponding distances for PR/5010 are almost identical at 2.5, 2.6 Å and at 2.8, 3.1 Å. However, in both PR20/4410 and PR20/5010 complexes, the central hydroxyl group forms strong interactions with one Asp and relatively longer interactions with one of the carboxylate oxygen of the other. In PR20/4410, the inhibitor hydroxyl forms short hydrogen bonds of 2.4 and 2.7 Å with Asp 25, while longer 3.0 and 3.4 Å interactions are observed with Asp 25′. The hydroxyl group from the second inhibitor orientation forms similar shorter and longer interactions with Asp 25′ and Asp 25, respectively. For PR20/5010, the distances for strong interactions with Asp25 were 2.5 and 2.7 Å for Asp 25, while longer interactions of 3.0 and 3.3 Å were seen for Asp 25′. Similar interactions were observed for the second inhibitor orientation with Asp 25′ and Asp 25, respectively. The separations between the hydroxyl groups in the two orientations of the inhibitor are 1.2 and 1.1 Å for the wild-type PR/4410 and PR/5010 complexes, respectively, while the corresponding values for both of the PR20 complexes are 1.7 Å. Thus, it is apparent that the 0.5 Å or more increase in the distance between the hydroxyl groups of the two orientations of inhibitor positions the hydroxyl asymmetrically closer to one of the catalytic aspartate residues in the dimer of PR20 compared to the wild type PR. Comparison with PR20 in complex with darunavir, which shares the chemical scaffold with GRL-4410A and GRL-5010A, shows that the central hydroxyl of darunavir in one orientation makes shorter interactions (2.7, 2.7 Å and 2.8, 3.0 Å) with both the Asp 25 and Asp 25′ residues. The second orientation forms short (2.4, 2.5 Å) and relatively long interactions (2.9, 3.3 Å) with Asp 25′ and Asp 25, respectively. The distance between central hydroxyl in the two orientations is 1.2 Å, which is similar to the separations seen in the wild-type complexes of PR/darunavir, PR/4410 and PR/5010.
Figure 4.
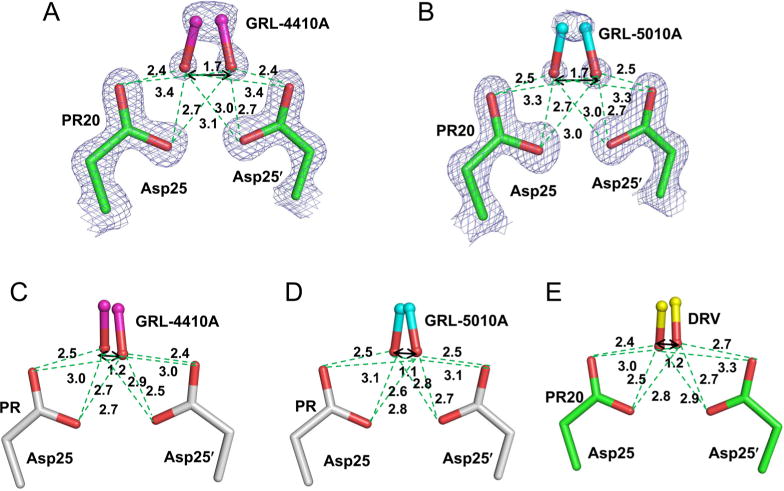
Hydrogen bonds between the central hydroxyl group of the two inhibitor conformations and the catalytic Asp25 and Asp25′ in the complex of (A) PR20/4410, (B) PR20/5010, (C) PR/4410, (D) PR/5010, and (E) PR20/darunavir. Inhibitor atoms of GRL-4410A, GRL-5010A, and darunavir are shown in ball and stick representation with magenta, cyan, and yellow carbons, respectively. PR20 is shown with green carbons and wild type PR with gray carbons. The PR20/4410 and PR20/5010 complexes are shown with 2Fo − Fc maps contoured at 1.7σ and 1.9σ, respectively.
DISCUSSION AND CONCLUSIONS
Drug resistant HIV protease poses a major challenge in the treatment of HIV and AIDS. Previously, we characterized a highly evolved multidrug resistant HIV protease from a clinical isolate (PR20), which is resistant to all tested clinical drugs.11 PR20 has more than 3 orders of magnitude lower affinity for clinical inhibitors than does wild-type PR.10 In addition, the autocatalytic cleavage of the PR20-containing precursor that results in mature enzyme is not inhibited by any clinical inhibitors, while darunavir and saquinavir effectively inhibit the autoprocessing of wild-type precursors.10 The crystallographic analyses of PR20 revealed several important structural features that correlate with its diminished affinity for inhibitors.11 Mutations in and around the active site expanded the S2/S2′ pockets of PR20. Also, mutations in the hinge loop of PR20 contribute to increased flexibility of its flaps. Structural studies of PR20 in complex with darunavir and saquinavir have suggested two strategies for the design of successful inhibitors for PR20-like resistant variants. The first strategy is to design inhibitors with bigger P2 groups that better fit into the expanded S2 pocket of PR20. The second strategy is to design inhibitors that form stronger interactions with the flaps of PR20.
In the quest to identify promising inhibitors of PR20, we recently studied novel protease inhibitors with P1′-pyrrolidi-none (GRL-02031A)28 and P2-tris-THF (GRL-0519A).12 GRL-02031A has an enhanced P1′ group and GRL-0519A has a larger P2 group, which parallels the design strategy to fill the expanded S2 pocket of PR20. Both these inhibitors showed added interactions with wild-type PR, together with excellent Ki values and antiviral efficacy against both wild-type HIV-1 and other resistant variants.12,28 However, GRL-02031A had weaker affinity for PR20, since the modified P1′ group forms poorer interactions with PR20 than with wild-type PR.13 GRL-0519A with three THF rings in the P2 group fills the S2 pocket well, but its binding affinity for PR20 is essentially identical to that of darunavir.13 In addition, like darunavir, neither GRL-02031A nor GRL-0519A inhibits the autoprocessing of PR20-containing precursors, which is essential for viral replication.
Here, we investigated the strategy of adding interactions with the flap residues of PR20 for the new antiviral inhibitors GRL-4410A and GRL-5010A. Drug resistant mutations and polymorphisms are known to alter the flap conformation and flexibility, which can be overcome by inhibitors forming strong interactions with the flaps.29–31 Both of the inhibitors GRL-4410A and GRL-5010A exhibit improved interactions with the flaps of wild-type PR relative to those of darunavir and are remarkably potent against the wild-type PR with Ki values of 2.9 and 5.8 pM, respectively, comparable to the affinity of 5 pM measured for darunavir. Furthermore, the fluorine substituted GRL-5010A has 6.5-fold higher apparent permeability coefficient (Papp), which is a measure of penetration efficiency of the drug across the blood–brain barrier, than darunavir.22 Viral particles hidden in the central nervous system (CNS) are a major problem in the treatment of HIV, and improving the drug concentration in CNS may help reduce the incidence of HIV-1 associated dementia and other CNS disorders.32,33 The current structures show that both of these inhibitors form direct, as well as water-mediated, interactions with the flap residue Gly 48 of PR20, similar to those of the wild-type PR. However, in both PR20/4410 and PR20/5010 complexes, the distance between the central hydroxyl groups of the two conformation of the inhibitor lengthens by more than 0.5 Å when compared with the corresponding complexes with wild-type PR. In both the PR20 complexes, the inhibitors form interactions with the flaps in the S2 subsite, while the P2′ moieties with aniline in GRL-4410A and methoxy in GRL-5010A shift by 0.5 Å to maintain the van der Waals interactions with the smaller I84V side chain in the expanded S2′ subsite of PR20. The two orientations of inhibitor fit in the expanded S2/S2′ pocket of PR20 in a manner that increases the distance between the central hydroxyl groups in comparison to those seen in the wild-type PR complexes and the PR20/DRV complex. Both GRL-4410A and GRL-5010A exhibit improved inhibition for PR20 over darunavir, which is consistent with their additional interactions with the flaps. In future designs, increasing the size of the P2′ moiety may be necessary to fill the S2′ pocket, in addition to the S2 pocket, in order to effectively inhibit mutants like PR20 that expand these pockets.11 Also, substitution of other flap interacting groups may further improve the potency of future inhibitors against PR20. Though the Ki values of GRL-4410A and GRL-5010A for PR20 are 1000-fold weaker than those for wild-type enzyme, they offer promising leads in the quest to find inhibitors for extremely resistant mutants represented by PR20. Clearly, further studies and new designs of inhibitors are needed to obtain potent inhibitors of highly resistant PR.
EXPERIMENTAL SECTION
Construction, Expression, and Purification of PR20
A synthetic protease gene coding the 99 amino acid protein derived from the sequence of a clinical isolate,34 termed PR20, was cloned between the Nde1 and BamH1 sites of pET11a vector (Novagen, San Diego, CA) and transformed into E. coli BL-21(DE3; Stratagene). Constructions of vectors for the expression of wild-type PR and precursor constructs, TFR-PR and TFR-PR20, have been reported previously.10 Protein expression, purification, and folding were performed as described before.35
Kinetic Assays
PR20 was diluted in reaction buffer (100 mM MES, pH 5.6, 400 mM sodium chloride, 1 mM EDTA, and 5% glycerol) for kinetic assays. Inhibitors were dissolved and diluted to various concentrations in DMSO. The assay was prepared by mixing 10 μL of diluted enzyme, 98 μL of reaction buffer, and 2 μL of inhibitor. The mixture was incubated at 37 °C for 5 min, then 90 μL of fluorogenic substrate, Abz-Thr-Ile-Nle-p-nitro-Phe-GlnArg-NH2, where Abz is anthranilic acid and Nle is norleucine (Bachem, King of Prussia, PA, USA), with the sequence derived from the p2/NC cleavage site of the Gag polyprotein, was added to initialize the reaction. The reaction was monitored over 5 min at wavelengths of 340 and 420 nm for excitation and emission in POLARstar OPTIMA microplate reader. The data analysis was performed with Sigmaplot 9.0 (SPSS Inc., Chicago, IL, USA). The reaction rate at different substrate concentrations was fitted in Michaelis–Menten equation to obtain Km and kcat values. The kcat/Km is determined from the equation (A/B) ± [(B2a2 + A2b2)1/2]/B2, where A is kcat, a is kcat error, B is Km, and b is error in Km. The IC50 values were obtained through inhibitor dose–response curve. The Ki values were calculated from the equation Ki = (IC50 − [E]/2)/(1 + [S]/Km), where [E] and [S] are the PR and substrate concentrations, respectively.
Comparison of Inhibition of N-Terminal Autoprocessing in E. coli
Inhibition of N-terminal autoprocessing of wild-type precursor (TFR-PR) and corresponding mutant precursor (TFR-PR20) by GRL-4410A and GRL-5010A was assessed by growing the cultures in the presence of increasing concentrations of inhibitor provided in the medium. The expressed protein was partially purified and analyzed by SDS–PAGE as described and compared to the inhibition of autoprocessing of TFR-PR and TFR-PR20 by darunavir.10,13
Crystallization, X-ray Data Collection, and Structure Determination
The PR20 was complexed with GRL-4410A and GRL-5010A at 1:7 molar ratios and crystallized by hanging drop vapor diffusion at room temperature. The crystals of PR20/5010 were obtained by mixing 1 μL of protein complex at 5 mg/mL with 1 μL of 1.15 M sodium chloride, 0.1 M sodium acetate, pH 5.5, and 0.22 μL of yittrium chloride. PR20/4410 was crystallized from a drop containing 1 μL of protein complex at 5 mg/mL with 1 μL of 1.2 M sodium chloride, 0.1 M sodium acetate, pH 5.5, and 0.22 μL of yittrium chloride. The crystals were cryocooled in a cryoprotectant composed of the respective mother liquor in addition to 30% glycerol. Diffraction data were collected at 100 K on beamline 22-ID (SER-CAT) at the Advanced Photon Source, Argonne National Laboratory. The data were integrated and scaled with HKL2000.36
The crystal structures were determined by molecular replacement using PHASER.37,38 The previously published structure of PR20/DRV structure (3UCB)11 was selected as the initial model for the two PR20 complexes. The PR20 complexes were refined using REFMAC 5.2.39 The X-ray diffraction data for the PR20 complexes were initially scaled in P6122 and successfully solved with a PR20 monomer and half the inhibitor molecule in the asymmetric unit. However, after refinement of both structures, the entire flap region of the monomer and the inhibitor were found to have two alternative conformations. To rule out the possibility that averaging of the two monomers from the PR20 dimer was the cause of the alternative conformations, the X-ray data were reprocessed in the lower symmetry space group of P61. In P61 a noncrystallographic 2-fold replaces the crystallographic 2-fold in P6122, resulting in an asymmetric unit containing the PR20 dimer with clearly resolved alternative conformations and better interpretation of the interactions between PR20 and inhibitor. Hence, both structures of PR20 were refined in the P61 space group.
The inhibitors were fitted into unambiguous electron density. The models were subjected to several rounds of refinement in REFMAC39 and model building with Coot.40 Solvent molecules were inserted at stereochemically reasonable positions using 2Fo − Fc and Fo − Fc maps at 1σ and 3σ levels, respectively. Molecular figures were prepared with PyMOL (http://www.pymol.org).
Supplementary Material
Acknowledgments
The research was supported in part by the National Institutes of Health Grants GM062920 (I.T.W.), GM053386 (A.K.G.) and Intramural Research Program of the NIDDK, National Institutes of Health. We thank Annie Aniana for technical assistance. This research was authored, in whole or in part, by National Institutes of Health staff. Data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22ID at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html.
ABBREVIATIONS
- HIV
human immunodeficiency virus
- AIDS
acquired immunodeficiency syndrome
- PR
protease
- THF
tetrahydrofuran
- CNS
central nervous system
- ITC
isothermal titration calorimetry
- DRM
drug resistance mutation
- DRV
darunavir
- rms
root mean square
- EDTA
ethylenediaminetetraacetic acid
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
Footnotes
Supporting Information
Autoprocessing assay of wild-type TFR-PR and TFR-PR20 precursors by GRL-4410A and GRL-5010A and a csv file of molecular formula strings. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00474.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 4YE3 for PR20/4410 and 4YHQ for PR20/5010.
Notes
The authors declare no competing financial interest.
References
- 1.Pennings PS. HIV drug resistance: Problems and perspectives. Infect Dis Rep. 2013;5:21–25. doi: 10.4081/idr.2013.s1.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleyn TJ, Liedtke MD, Harrison DL, Lockhart SM, Salvaggio MR, Ripley TL, Rathbun RC. Incidence of transmitted antiretroviral drug resistance in treatment-naive HIV-1-infected persons in a large south central united states clinic. Ann Pharmacother. 2014;48:470–475. doi: 10.1177/1060028013519246. [DOI] [PubMed] [Google Scholar]
- 3.Loubet P, Charpentier C, Visseaux B, Nuta C, Adu E, Chapplain JM, Baysah M, Walters-Doe S, Tattevin P, Peytavin G, Yazdanpanah Y, Descamps D. Prevalence of HIV-1 transmitted drug resistance in liberia. AIDS Res Hum Retroviruses. 2014;30:863–866. doi: 10.1089/AID.2014.0089. [DOI] [PubMed] [Google Scholar]
- 4.Hamers RL, Wallis CL, Kityo C, Siwale M, Mandaliya K, Conradie F, Botes ME, Wellington M, Osibogun A, Sigaloff KC, Nankya I, Schuurman R, Wit FW, Stevens WS, van Vugt M, de Wit TF. HIV-1 drug resistance in antiretroviral-naive individuals in Sub-Saharan Africa after rollout of antiretroviral therapy: A multicentre observational study. Lancet Infect Dis. 2011;11:750–759. doi: 10.1016/S1473-3099(11)70149-9. [DOI] [PubMed] [Google Scholar]
- 5.Sigaloff KC, Mandaliya K, Hamers RL, Otieno F, Jao IM, Lyagoba F, Magambo B, Kapaata A, Ndembi N, Rinke de Wit TF. High prevalence of transmitted antiretroviral drug resistance among newly HIV type 1 diagnosed adults in Mombasa, Kenya. AIDS Res Hum Retroviruses. 2012;28:833–837. doi: 10.1089/AID.2011.0348. [DOI] [PubMed] [Google Scholar]
- 6.Stadeli KM, Richman DD. Rates of emergence of hiv drug resistance in resource-limited settings: A systematic review. Antiviral Ther. 2013;18:115–123. doi: 10.3851/IMP2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips AN, Dunn D, Sabin C, Pozniak A, Matthias R, Geretti AM, Clarke J, Churchill D, Williams I, Hill T, Green H, Porter K, Scullard G, Johnson M, Easterbrook P, Gilson R, Fisher M, Loveday C, Gazzard B, Pillay D. Long term probability of detection of HIV-1 drug resistance after starting antiretroviral therapy in routine clinical practice. AIDS. 2005;19:487–494. doi: 10.1097/01.aids.0000162337.58557.3d. [DOI] [PubMed] [Google Scholar]
- 8.Heneine W, Kashuba A. HIV prevention by oral preexposure prophylaxis. Cold Spring Harbor Perspect Med. 2012;2:1–13. doi: 10.1101/cshperspect.a007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Supervie V, Garcia-Lerma JG, Heneine W, Blower S. HIV, transmitted drug resistance, and the paradox of preexposure prophylaxis. Proc Natl Acad Sci USA. 2010;107:12381–12386. doi: 10.1073/pnas.1006061107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Louis JM, Aniana A, Weber IT, Sayer JM. Inhibition of autoprocessing of natural variants and multidrug resistant mutant precursors of HIV-1 protease by clinical inhibitors. Proc Natl Acad Sci USA. 2011;108:9072–9077. doi: 10.1073/pnas.1102278108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agniswamy J, Shen CH, Aniana A, Sayer JM, Louis JM, Weber IT. HIV-1 protease with 20 mutations exhibits extreme resistance to clinical inhibitors through coordinated structural rearrangements. Biochemistry. 2012;51:2819–2828. doi: 10.1021/bi2018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SG, Amano M, Mitsuya H. Probing multidrug-resistance and protein-ligand interactions with oxatricyclic designed ligands in HIV-1 protease inhibitors. ChemMedChem. 2010;5:1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agniswamy J, Shen CH, Wang YF, Ghosh AK, Rao KV, Xu CX, Sayer JM, Louis JM, Weber IT. Extreme multidrug resistant HIV-1 protease with 20 mutations is resistant to novel protease inhibitors with p1′-pyrrolidinone or p2-tris-tetrahydrofuran. J Med Chem. 2013;56:4017–4027. doi: 10.1021/jm400231v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh AK, Martyr CD, Steffey M, Wang YF, Agniswamy J, Amano M, Weber IT, Mitsuya H. Design of substituted bis-tetrahydrofuran (bis-THF)-derived potent HIV-1 protease inhibitors, protein-ligand X-ray structure, and convenient syntheses of bis-THF and substituted bis-THF ligands. ACS Med Chem Lett. 2011;2:298–302. doi: 10.1021/ml100289m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metrangolo P, Neukirch H, Pilati T, Resnati G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc Chem Res. 2005;38:386–395. doi: 10.1021/ar0400995. [DOI] [PubMed] [Google Scholar]
- 18.Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X, Jiang H, Zhu W. Halogen bonding—a novel interaction for rational drug design? J Med Chem. 2009;52:2854–2862. doi: 10.1021/jm9000133. [DOI] [PubMed] [Google Scholar]
- 19.Hernandes MZ, Cavalcanti SM, Moreira DR, de Azevedo Junior WF, Leite AC. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr Drug Targets. 2010;11:303–314. doi: 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- 20.Filler R, Saha R. Fluorine in medicinal chemistry: A century of progress and a 60-year retrospective of selected highlights. Future Med Chem. 2009;1:777–791. doi: 10.4155/fmc.09.65. [DOI] [PubMed] [Google Scholar]
- 21.Langford D, Marquie-Beck J, de Almeida S, Lazzaretto D, Letendre S, Grant I, McCutchan JA, Masliah E, Ellis RJ. Relationship of antiretroviral treatment to postmortem brain tissue viral load in human immunodeficiency virus-infected patients. J Neurovirol. 2006;12:100–107. doi: 10.1080/13550280600713932. [DOI] [PubMed] [Google Scholar]
- 22.Salcedo Gomez PM, Amano M, Yashchuk S, Mizuno A, Das D, Ghosh AK, Mitsuya H. Grl-04810 and grl-05010, difluoride-containing nonpeptidic HIV-1 protease inhibitors (pis) that inhibit the replication of multi-pi-resistant HIV-1 in vitro and possess favorable lipophilicity that may allow blood-brain barrier penetration. Antimicrob Agents Chemother. 2013;57:6110–6121. doi: 10.1128/AAC.01420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK, Yashchuk S, Mizuno A, Chakraborty N, Agniswamy J, Wang YF, Aoki M, Gomez PM, Amano M, Weber IT, Mitsuya H. Design of gem-difluoro-bis-tetrahydrofuran as p2 ligand for HIV-1 protease inhibitors to improve brain penetration: Synthesis, X-ray studies, and biological evaluation. ChemMedChem. 2015;10:107–115. doi: 10.1002/cmdc.201402358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Louis JM, Ishima R, Torchia DA, Weber IT. HIV-1 protease: Structure, dynamics, and inhibition. Adv Pharmacol. 2007;55:261–298. doi: 10.1016/S1054-3589(07)55008-8. [DOI] [PubMed] [Google Scholar]
- 25.Louis JM, Weber IT, Tozser J, Clore GM, Gronenborn AM. HIV-1 protease: Maturation, enzyme specificity, and drug resistance. Adv Pharmacol. 2000;49:111–146. doi: 10.1016/s1054-3589(00)49025-3. [DOI] [PubMed] [Google Scholar]
- 26.Zhou P, Zou J, Tian F, Shang Z. Fluorine bonding—How does it work in protein-ligand interactions? J Chem Inf Model. 2009;49:2344–2355. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]
- 27.Vulpetti A, Schiering N, Dalvit C. Combined use of computational chemistry, NMR screening, and X-ray crystallography for identification and characterization of fluorophilic protein environments. Proteins. 2010;78:3281–3291. doi: 10.1002/prot.22836. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh AK, Leshchenko-Yashchuk S, Anderson DD, Baldridge A, Noetzel M, Miller HB, Tie Y, Wang YF, Koh Y, Weber IT, Mitsuya H. Design of HIV-1 protease inhibitors with pyrrolidinones and oxazolidinones as novel p1′-ligands to enhance backbone-binding interactions with protease: Synthesis, biological evaluation, and protein-ligand X-ray studies. J Med Chem. 2009;52:3902–3914. doi: 10.1021/jm900303m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, Kovalevsky AY, Louis JM, Boross PI, Wang YF, Harrison RW, Weber IT. Mechanism of drug resistance revealed by the crystal structure of the unliganded HIV-1 protease with f53l mutation. J Mol Biol. 2006;358:1191–1199. doi: 10.1016/j.jmb.2006.02.076. [DOI] [PubMed] [Google Scholar]
- 30.Kear JL, Blackburn ME, Veloro AM, Dunn BM, Fanucci GE. Subtype polymorphisms among HIV-1 protease variants confer altered flap conformations and flexibility. J Am Chem Soc. 2009;131:14650–14651. doi: 10.1021/ja907088a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coman RM, Robbins AH, Goodenow MM, Dunn BM, McKenna R. High-resolution structure of unbound human immunodeficiency virus 1 subtype c protease: Implications of flap dynamics and drug resistance. Acta Crystallogr, Sect D: Biol Crystallogr. 2008;64:754–763. doi: 10.1107/S090744490801278X. [DOI] [PubMed] [Google Scholar]
- 32.McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- 33.Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 34.Dierynck I, De Wit M, Gustin E, Keuleers I, Vandersmissen J, Hallenberger S, Hertogs K. Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J Virol. 2007;81:13845–13851. doi: 10.1128/JVI.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sayer JM, Agniswamy J, Weber IT, Louis JM. Autocatalytic maturation, physical/chemical properties, and crystal structure of group n HIV-1 protease: Relevance to drug resistance. Protein Sci. 2010;19:2055–2072. doi: 10.1002/pro.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.