

Significance
The PSD-95 family proteins serve as central scaffolds of excitatory synapses. Their expression levels dictate synaptic strength, and the functions of many synaptic organizing molecules are dependent on interactions with these proteins. Yet, it is unclear what guides PSD-95 into maturing synapses, which occurs postnatally and is required for proper synapse development. Here, we establish that the secreted protein LGI1 controls the functional incorporation of PSD-95 through interactions with the transmembrane protein ADAM22. This process occurs in a paracrine fashion, with LGI1 released from the pre- and postsynaptic cell able to modulate postsynaptic strength. Our data illustrate a previously undescribed level of synaptic organization, identifying a critical role for the LGI1–ADAM22 complex in controlling the function of PSD-95 itself and, in turn, normal synapse development.
Keywords: synaptic organization, synapse development, LGI1, ADAM22, PSD-95
Abstract
Synapse development is coordinated by a number of transmembrane and secreted proteins that come together to form synaptic organizing complexes. Whereas a variety of synaptogenic proteins have been characterized, much less is understood about the molecular networks that support the maintenance and functional maturation of nascent synapses. Here, we demonstrate that leucine-rich, glioma-inactivated protein 1 (LGI1), a secreted protein previously shown to modulate synaptic AMPA receptors, is a paracrine signal released from pre- and postsynaptic neurons that acts specifically through a disintegrin and metalloproteinase protein 22 (ADAM22) to set postsynaptic strength. We go on to describe a novel role for ADAM22 in maintaining excitatory synapses through PSD-95/Dlg1/zo-1 (PDZ) domain interactions. Finally, we show that in the absence of LGI1, the mature synapse scaffolding protein PSD-95, but not the immature synapse scaffolding protein SAP102, is unable to modulate synaptic transmission. These results indicate that LGI1 and ADAM22 form an essential synaptic organizing complex that coordinates the maturation of excitatory synapses by regulating the functional incorporation of PSD-95.
Proper development of synapses involves recruitment of proteins that establish presynaptic release sites and postsynaptic densities (PSDs), and later coordinate maturation, maintenance, and plasticity of the synapse. In the last decade, a number of transmembrane synaptic adhesion proteins and secreted proteins that initiate and modulate excitatory synapses—termed synaptic organizing proteins—have been identified (1, 2). Whereas the synaptogenic properties of many synaptic organizing proteins have been described in detail (2–11), much less is known about how synaptic organizing proteins regulate synapse maintenance and maturation (1).
A key component to the functional maturation of excitatory synapses is the recruitment of AMPA-type glutamate receptors (AMPARs) to the PSD, which is coordinated by PSD-95/Dlg1/zo-1 (PDZ) domain-containing scaffolding proteins (12). Specifically, one family of PDZ proteins, the membrane-associated guanylyl kinases (MAGUKs), is known to determine basal synaptic AMPAR content (13–16). Like synaptic AMPAR content, the expression of different MAGUKs is developmentally regulated; whereas synapse-associated protein 102 (SAP102) is expressed in the early postnatal period and is critical to the function of immature synapses, postsynaptic density proteins 93 and 95 (PSD-93 and PSD-95) are first expressed around postnatal day 10 (P10) and are required for proper function of mature synapses (14, 17). Mice lacking PSD-93 and PSD-95 have synapses with significantly reduced AMPAR content (14), indicating the shift in MAGUK expression is critical to synapse maturation. However, what guides the incorporation of PSD-93 and PSD-95 into developing synapses remains unknown.
We recently identified an instructive role for leucine-rich, glioma-inactivated protein 1 (LGI1) in regulating synaptic AMPAR content—application of LGI1 increases and loss of LGI1 decreases synaptic AMPAR localization (18, 19). LGI1 is a secreted protein that is localized to synapses, where it binds to the extracellular domain of the transmembrane a disintegrin and metalloproteinase proteins 11, 22, and 23 (ADAM11, ADAM22, and ADAM23) (19). Notably, LGI1 and ADAM22 are found in complex with the mature MAGUKs, PSD-93 and PSD-95, in vivo (18, 19), and their expression levels follow a similar timeline (17, 18, 20). However, little is known about the neuronal function of ADAM11, ADAM22, and ADAM23, and it is not clear which mediates the function of LGI1 at the synapse. Moreover, the source and destination of secreted LGI1 remains unstudied.
Here, we show that LGI1 released from pre- and postsynaptic cells acts in a paracrine fashion to regulate synaptic AMPAR content. We find that the function of LGI1 at the synapse is fully dependent on its interaction with ADAM22, the only ADAM in the LGI1 complex that contains a PDZ-binding motif. ADAM22, in turn, maintains excitatory synapses through PDZ domain interactions. Finally, we demonstrate that PSD-95, but not SAP102, requires LGI1 to function at synapses, indicating that the LGI1–ADAM22 complex directs synapse maturation by controlling the incorporation of the mature MAGUK PSD-95.
Results
LGI1 Expression Regulates Synaptic, but Not Surface, AMPAR Content.
Having previously identified a role for LGI1 in regulating synaptic AMPAR content, we were interested in determining the mechanism through which secreted LGI1 affects postsynaptic transmission. However, mice lacking LGI1 exhibit a lethal epilepsy phenotype, with seizures starting in the second postnatal week that are invariably fatal by postnatal week 3 (19, 21, 22). Thus, we turned to hippocampal slice culture, a system in which we are able to both investigate single-cell genetic manipulations and also bypass the potential confounding effects of repeated seizures in vivo (21, 23). Slice cultures were prepared from P6–P8 animals, before seizure onset in LGI1−/− mice, and recordings were made at day in vitro (DIV) 8 (Fig. 1A), a time at which LGI1 and other major synaptic proteins are robustly expressed (18–20).
Fig. 1.

Loss of LGI1 reduces synaptic, but not surface, AMPAR content. (A) Timeline of experimental preparation for hippocampal slice cultures and recordings (Bottom) compared with the LGI1−/− phenotype (Top). (B) AMPA/NMDA ratios recorded in slice cultures made from LGI1−/− mice are significantly reduced (P = 0.03, n = 15) compared with wild type. (Left) Sample traces of wild type (black) and LGI1−/− (gray). (Scale bar, 50 ms and 50 pA.) (Right) Bar graphs showing average AMPA/NMDA ratios ± SEM in wild type and LGI1−/−. (C) Paired-pulse stimulation reveals no significant difference (P = 0.46, n = 16 wild type, 17 LGI1−/−) in presynaptic release probability in LGI1−/− relative to wild type. (Left) Sample traces normalized to first-response amplitude in wild-type cell. (Scale bar, 40 ms.) (Right) Average ratio ± SEM (D) Miniature EPSC recordings in wild type and LGI1−/−. Loss of LGI1 results in a significant decrease in amplitude (P = 0.03, n = 21), but not frequency (P = 0.17, n = 21), of mEPSCs. Bar graphs of average amplitude and frequency of mEPSCs ± SEM, with sample traces for each condition shown above. [Scale bars, 25 ms (amplitude traces), 100 ms (frequency traces), and 10 pA.] (E) Somatic outside-out patch recordings in wild-type and LGI1−/− cells have similar glutamate-evoked currents (P = 0.92, n = 13). (Top) Sample traces of wild-type and LGI1−/− surface currents. (Scale bars, 1 s and 100 pA.) (Bottom) Average glutamate-evoked currents ± SEM.
We first set out to verify in slice cultures that, as in acute slices, LGI1 loss reduces synaptic AMPAR localization (19). Recording from CA1 neurons, we found the ratio of the AMPAR-mediated excitatory postsynaptic currents (EPSCs) to NMDA receptor (NMDAR)-mediated EPSCs (AMPA/NMDA ratio) was significantly decreased in LGI1−/− compared with wild-type littermate controls (Fig. 1B), confirming that the previously observed decrease in AMPA/NMDA ratio in acute slice is due to loss of LGI1 and not an effect of chronic seizures in vivo. Moreover, no difference was found in the paired-pulse ratio (PPR) between wild type and LGI1−/−, in line with our previous findings in acute slice (Fig. 1C).
We next sought to determine whether the change in AMPA/NMDA ratios observed after loss of LGI1 is due to the changing strength of individual synapses or to a decrease in the total number of AMPAR-containing synapses. Recording miniature EPSCs (mEPSCs) in wild-type versus LGI1−/− cells, we found a significant reduction in amplitude but not frequency (Fig. 1D), consistent with a deficit in synapse strength, not number. This was specifically due to decreased synaptic localization of AMPARs, as no difference was observed in glutamate-evoked currents recorded from LGI1−/− and wild-type neurons (Fig. 1E), indicating that receptor surface expression is normal in the absence of LGI1.
Previous research has shown that transgenic overexpression of an epilepsy-associated LGI1 mutant, which is thought to act as a dominant negative, increased synaptic transmission in the dentate gyrus (24). This finding suggests that LGI1 may play distinct roles in different regions of the hippocampus. To test if this is the case, we recorded EPSCs in dentate gyrus granule cells of LGI1−/− and wild-type littermates. We observed a decrease in AMPA/NMDA ratio similar in magnitude to our findings in acute and cultured slice CA1. Also similar to our findings in CA1, we did not see any change in paired-pulse ratio after loss of LGI1 (Fig. S1). Thus, loss of LGI1 impacts transmission similarly in the dentate gyrus and CA1 region of the hippocampus and is functionally distinct from mutant LGI1 overexpression.
Fig. S1.

Loss of LGI1 also results in decreased AMPAR-mediated transmission in dentate gyrus granule cells. (A) Loss of LGI1 leads to a significant decrease in AMPA/NMDA ratio in dentate gyrus (DG) granule cells (P = 0.02, n = 11 wild type, 8 LGI1−/−). (Left) Sample traces of wild-type (black) and LGI1−/− (gray) neurons recorded at −70 mV and +40 mV. (Scale bar, 50 ms and 50 pA.) (Right) Bar graphs showing average AMPA/NMDA ratios ± SEM in wild type and LGI1−/−. (B) Loss of LGI1 has no impact on paired-pulse ratio recorded in the dentate gyrus (P = 0.96, n = 9 wild type, 8 LGI1−/−). (Left) Sample traces normalized to first-response amplitude in wild-type cell. (Scale bar, 40 ms.) (Right) Average ratio ± SEM.
LGI1 Is a Paracrine Signal Released from Pre- and Postsynaptic Cells.
Next, we used molecular replacement strategies to dissect the mechanism by which LGI1 regulates postsynaptic AMPAR content, beginning by examining the impact of single-cell expression of LGI1 on synaptic transmission. LGI1−/− hippocampal slice cultures were sparsely transfected with LGI1 using biolistics, and simultaneous recordings were made from neighboring transfected and untransfected CA1 pyramidal cells in response to stimulation of stratum radiatum. Curiously, we observed no difference in AMPAR- or NMDAR-mediated EPSCs in the LGI1 expressing cell relative to the LGI1−/− control (Fig. 2A).
Fig. 2.

LGI1 is a paracrine signal derived from the pre- and postsynaptic cell. (A) AMPAR- and NMDAR-mediated EPSCs in LGI1−/− cells expressing LGI1 are the same as neighboring untransfected LGI1−/− cells (AMPAR: P = 0.17, n = 17; NMDAR: P = 0.37, n = 16). Scatterplots show individual dual recordings (open circles) and mean ± SEM (filled circle). (Insets) Sample traces of LGI1−/− (black) and LGI1−/− + LGI1 (green). (Scale bars, 50 ms and 50 pA.) (B) Recording set-up of naïve control cell in LGI1−/− slice and control cell near (either adjacent to or one cell body away from) an LGI1-transfected cell in LGI1−/− slice. (C) The AMPA/NMDA ratio of nearby control cells in LGI1−/− biolistically transfected slices is significantly higher than cells from naïve LGI1−/− slices (P = 0.03, n = 15). LGI1S473L expression, however, does not alter the AMPA/NMDA ratio of nearby cells relative to naïve LGI1−/− cells (P = 0.71, n = 13). Bar graphs show mean ratio ± SEM. (D) Viral injections of LGI1 in CA3 (presynaptic) and dentate gyrus (DG, nonsynaptic) were carried out to test the source of active LGI1. Recordings were made in CA1 pyramidal cells for both experiments. (E) Expression of LGI1 in CA3 significantly increases the AMPA/NMDA ratio of CA1 neurons relative to naïve LGI1−/− neurons (P = 0.05, n = 15 LGI1−/−, n = 16 CA3-expressing LGI1−/−); AMPA/NMDA ratios remain similar to naïve LGI1−/− when LGI1 is expressed in the DG (P = 0.82, n = 15 LGI1−/−, n = 20 DG-expressing LGI1−/−). Bar graphs show mean ratio ± SEM.
In our dual recording set-up, it is possible that LGI1 secreted from the transfected cell acts in a paracrine fashion, increasing receptor content in the neighboring LGI1−/− control cells and masking a change in EPSCs induced by LGI1 expression. To test if secreted LGI1 alters nearby cells, we compared the AMPA/NMDA ratios of untransfected LGI1−/− cells next to LGI1-transfected cells and LGI1−/− cells from untransfected slices (Fig. 2B). Consistent with LGI1 having paracrine activity, the AMPA/NMDA ratio of LGI1−/− cells near to transfected cells was rescued to wild-type levels and significantly increased relative to cells from the untransfected LGI1−/− slices (Fig. 2C).
LGI1 does not bind AMPARs directly (19), so it must work through a network of interactions to regulate synaptic strength. Based on previous biochemical data (18, 19), we hypothesized that LGI1 regulation of synaptic transmission is mediated by the transmembrane protein ADAM22, which directly binds LGI1 extracellularly and PSD-95 intracellularly. A recently identified missense mutation of LGI1 implicated in human autosomal dominant temporal lobe epilepsy (ADTLE), LG11S473L (25), is secreted but lacks the ability to bind ADAM22, although it can still bind ADAM23 (26). With the paracrine signal assay, we used this mutant to test whether the LGI1–ADAM22 interaction specifically is necessary for LGI1 function. Comparing AMPA/NMDA ratios of LGI1−/− cells near to LGI1S473L transfected cells and LGI1−/− cells from untransfected slices, we found that LGI1S473L is unable to rescue AMPA/NMDA ratios in neighboring neurons (Fig. 2C), indicating a requirement for ADAM22 interaction in LGI1 synaptic function. The impact of LGI1S473L mutation is not specific to inhibition of the paracrine signaling function, as LGI1−/− cells transfected with LGI1S473L show similar AMPAR- and NMDAR-meditated EPSCs as LGI1−/− cells (Fig. S2), indicating this mutant is also unable to rescue transmission cell autonomously.
Fig. S2.

Lack of cell-autonomous impact of LGI1S473L expression and localization of wild-type LGI1 expression in vivo. (A) Dual recordings were carried out in LGI1−/− and neighboring biolistically transfected LGI1S473L-expressing cells in LGI1−/− slices. (B) Expression of LGI1S473L does not alter AMPAR-mediated EPSCs (P = 0.46, n = 8). Scatterplots show individual dual recordings (open circles) and mean ± SEM (filled circle). (Scale bars, 50 ms and 25 pA.) (C) NMDAR-mediated EPSCs are also unchanged relative to LGI1−/− cells (P = 0.69, n = 7). (Scale bars, 50 ms and 50 pA.) (D) Diagram of Prox1 promoter-driven LGI1 tagged with FLAG and Hisx6 (–FH) designed for generating transgenic mice. (E) Dentate gyrus-specific expression of LGI1–FH was confirmed by Western blotting of microdissected brain regions of LGI1+/+ and +/−; Prox1–LGI1 (50 μg of total protein). Closed arrow, LGI1–FH; open arrow, endogenous LGI1; closed arrowhead, Prox1; DG, dentate gyrus; CA, CA1 plus CA3 regions; CTX, cortex. (F) Labeling of LGI1 in wild-type (LGI1+/+) and LGI1−/−; Prox1–LGI1 slices. Whereas LGI1 expression is uniform across the hippocampus in the wild-type slice, it is specifically localized to the molecular layer (site of granule cell dendrites) and mossy fiber axon tract in the LGI1−/−; Prox1–LGI1 slice. (Scale bar, 200 µm.)
LGI1 mediates a complex with pre- and postsynaptic components (19), although the source of the secreted protein remains unknown. Our previous experiment showed the expression of LGI1 in the postsynaptic and neighboring cells is sufficient to rescue AMPA/NMDA ratios. To test if presynaptic expression of LGI1 is sufficient to alter transmission, we used lentiviral injections in slice culture to specifically express LGI1 in CA3 neurons (Fig. 2D) and found that expression of LGI1 in CA3 rescues the AMPA/NMDA ratios of CA1 neurons to wild-type levels (Fig. 2E).
This finding could indicate that LGI1 is promiscuous in its ability to modulate synaptic transmission and can diffuse great distances from the site of secretion and impact far away synapses. To test this possibility, we again used viral expression of LGI1 in LGI1−/− slices, although this time in the dentate gyrus instead of CA3 (Fig. 2D). In slice culture, the somas of dentate gyrus granule cells and CA3 pyramidal neurons are roughly equidistant from CA1 neurons; however, unlike CA3 neurons, the axons and dendrites of granule cells never come in close contact with CA1. Recordings of AMPA/NMDA ratios in CA1 cells of slices with LGI1 expressed in the dentate gyrus were not significantly different from naïve LGI1−/− slices (Fig. 2E). Moreover, immunohistochemical analysis of transgenic mice that express LGI1 exclusively in dentate granule cells (LGI1−/−; Prox1–LGI1) shows specific labeling in the molecular layer (site of granule cell dendrites) and the mossy fiber axon tract, with little diffusion from these regions (Fig. S2). Together, these data suggest that secreted LGI1 only impacts synapses near the site of secretion. Because the dentate and CA3 cell bodies are equidistant from CA1 cells, and only CA3 expression of LGI1 can rescue transmission, these data indicate that LGI1 is likely released from axons and dendrites. However, we cannot rule out somatic secretion of LGI1, and it is possible that LGI1 is secreted from somas but is active only at synapses of the secreting cell or very nearby cells.
ADAM22 Is Required to Maintain Excitatory Synapses.
Our experiments indicate that LGI1 regulates synaptic AMPARs via an interaction with ADAM22, yet no research has been done on the role of ADAM22 at the synapse, and the way in which this interaction could modulate transmission remains unknown. Thus, we next sought to characterize the role of ADAM22 in synaptic transmission.
We began by examining the consequence of ADAM22 deletion on synaptic activity. Using biolistic transfection of Cre recombinase into slice cultures prepared from mice with the gene encoding ADAM22 flanked by loxP homologous recombination sites (ADAM22fl/fl), we were able to directly compare AMPAR- and NMDAR-mediated transmission in the presence and absence of ADAM22. Dual recordings from Cre-transfected, ADAM22-lacking (herein referred to as ADAM22−/−) neurons and neighboring untransfected, wild-type cells showed that loss of ADAM22 results in a significant decrease in both AMPAR- and NMDAR-mediated EPSCs (Fig. 3 A–C).
Fig. 3.
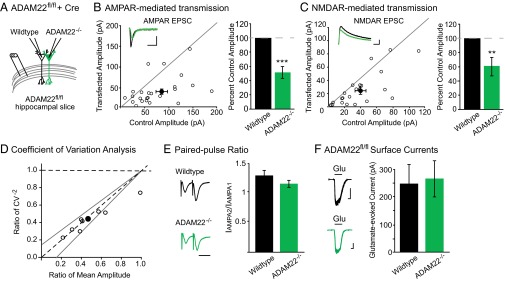
Loss of ADAM22 results in a decreased number of functional excitatory synapses. (A) Diagram of wild-type and ADAM22−/− dual recordings. (B) AMPAR- (P = 0.001, n = 23) and (C) NMDAR-mediated (P = 0.003, n = 19) EPSCs are significantly reduced after Cre-mediated deletion of ADAM22. Scatterplots show individual dual recordings (open circles) and mean ± SEM (filled circle). (Insets) Representative traces from a dual recording of a wild-type (black) and ADAM22−/− cell (green). (Scale bars, 50 ms and 50 pA.) Bar graphs show normalized mean EPSC amplitude ± SEM. (D) Coefficient of variation analysis of AMPAR-mediated EPSCs reveals a change in quantal content after loss of ADAM22. Scatterplots show individual dual recordings (open circles), mean (filled circle), y = line, and 95% confidence interval (dotted lines). (E) No difference in paired-pulse ratio was observed in ADAM22−/− cells (P = 0.27, n = 15 wild type, n = 14 ADAM22−/−). (Scale bar, 40 ms.) Bar graphs show mean ratio ± SEM. (F) Loss of ADAM22 does not alter surface currents in CA1 pyramidal cells (P = 0.79, n = 15 wild type, n = 12 ADAM22−/−). (Left) Representative traces of glutamate-evoked currents in outside-out patches pulled from wild-type (black) and ADAM22−/− (green) neurons. (Scale bars, 1 s and 100 pA.) Bar graphs show mean amplitude ± SEM.
The reduction in both AMPAR- and NMDAR-mediated currents after loss of ADAM22 could represent a decrease in the number of functional synapses, the probability of vesicle release, the receptor content at each synapse, or a combination of these changes. To distinguish among these possibilities, we carried out a coefficient of variation analysis in wild-type and ADAM22−/− neurons, which allows for determination of changes in quantal size and quantal content in our dual recording set-up. Reductions in quantal size result in reductions in variance relative to the change in mean EPSC amplitude, such that the normalized ratio of mean2/variance (CV−2) is unaffected and data points on a graph showing the relationship of mean versus CV−2 lie along the y = 1 line. Conversely, changes in quantal content result in correlated changes in CV−2, such that data points on a graph showing the relationship of mean versus CV−2 lie along the y = x line (27–29). Analysis of evoked AMPAR-mediated EPSCs from wild-type versus ADAM22−/− cells show correlated reductions in mean amplitude and CV−2, indicative of a reduction in quantal content after loss of ADAM22 (Fig. 3D).
A reduction in quantal content can reflect a decrease in the number of functional synapses or in release probability. To test whether release probability was reduced, we analyzed PPRs from wild-type and ADAM22−/− cells. No difference in PPR was observed, indicating that ADAM22 does not regulate probability of release (Fig. 3E). Thus, the reduction in quantal content observed after the loss of ADAM22 is due to a decrease in the number of excitatory synapses. Other synaptic protein complexes have been shown to act in formation and maintenance of dendritic spines, the anatomical site of excitatory synapses (30). However, no significant difference was found in the spine density between wild-type and ADAM22−/− neurons (Fig. S3), indicating that the decrease in excitatory synapses observed after loss of ADAM22 is not a result of a structural deficit but rather a reduction in functional excitatory synapses.
Fig. S3.

Loss of ADAM22 does not alter dendritic spine density. (A) Diagram showing region of apical dendrite where spine density was quantified and representative images of dendritic spines acquired from GFP- or Cre-expressing ADAM22fl/fl neurons. (Scale bar, 5 μm.) (B) No significant difference in spine density was found in the presence or absence of ADAM22 (P = 0.74, n = 8 wild type, 9 ADAM22−/−). Bar graphs show mean density ± SEM.
Although clustered at synapses, ADAM22 is spread diffusely along the surface of the neuron (18). Thus, it is possible that ADAM22 is required for the delivery or stabilization of AMPARs at the surface. However, glutamate-evoked currents recorded from outside-out patches of wild-type and ADAM22−/− cells are not different (Fig. 3F), demonstrating that ADAM22 is not required for proper surface localization of AMPARs. Together, these data indicate that ADAM22 is required for the maintenance of excitatory synaptic transmission. Loss of ADAM22 has a more severe effect on transmission than loss of LGI1, impacting both AMPAR and NMDAR-mediated EPSCs, suggesting that ADAM22 retains some function in the absence of LGI1. Supporting this conclusion, in the absence of LGI1, ADAM22 synaptic localization is significantly decreased, although a small portion (∼25%) remains at the synapse (26).
ADAM22 Function Is Dependent upon PDZ Interactions.
We next turned to a molecular replacement strategy to dissect the mechanism by which ADAM22 regulates excitatory synapse number. First, we tested whether expression of ADAM22 in ADAM22−/− neurons was sufficient to restore transmission. Dual recordings were carried out in untransfected, wild-type cells and cells expressing both Cre and ADAM22 in ADAM22fl/fl slice cultures. We found that coexpression of ADAM22 rescued both AMPAR- and NMDAR-mediated transmission to wild-type levels (Fig. 4 A and C).
Fig. 4.

ADAM22 lacking the PDZ-binding motif is unable to rescue synaptic transmission. (A) ADAM22 expression fully rescues the decrease in AMPAR- and NMDAR-mediated transmission observed in ADAM22−/− cells. Above, diagram of N-terminal protein domains, transmembrane domain (TM), and C-terminal PDZ-binding motif in wild-type ADAM22. Scatterplots show individual dual recordings (open circles) and mean ± SEM (filled circle). (Insets) Representative traces from a dual recording of a wild-type (black) and ADAM22−/− cell (green). (Scale bars, 50 ms and 25 pA.) (B) Expression of ADAM22 lacking the C-terminal PDZ-binding domain (ADAM22dC4, diagram above) is unable to rescue the AMPAR or NMDAR EPSC deficit in ADAM22−/− cells. (Scale bars, 50 ms and 25 pA.) (C) Summary bar graphs showing normalized mean ± SEM AMPAR and NMDAR EPSCs in wild type, Cre-, Cre + ADAM22-, and Cre + ADAM22dC4-expressing cells. Whereas wild-type ADAM22 is able to rescue both AMPAR- (P = 0.17, n = 23) and NDMAR-mediated EPSCs (P = 0.87, n = 20) to wild-type levels, expression of ADAM22dC4 fails to rescue AMPAR- (P = 0.006, n = 23) and NMDAR-mediated transmission (P = 0.009, n = 16).
ADAM22 contains a C-terminal PDZ-binding motif, a unique feature among the ADAM family proteins that bind LGI1 (18). To assess whether ADAM22 function depends on PDZ domain interactions, we tried rescuing ADAM22−/− neurons with an ADAM22 mutant lacking the PDZ-binding motif, ADAM22dC4. Both AMPAR- and NMDAR-mediated EPSCs were significantly reduced in cells expressing ADAM22dC4 (Fig. 4B), similar to ADAM22−/− neurons (Fig. 4C), indicating the function of ADAM22 at the synapse is completely dependent upon PDZ domain interactions. We considered that the C-terminal truncation of ADAM22 might result in improper processing of the ADAM22dC4 mutant, impacting surface localization and subsequent LGI1 binding. However, immunostaining of ADAM22dC4 revealed that it has normal surface localization and binds extracellular LGI1 (Fig. S4).
Fig. S4.
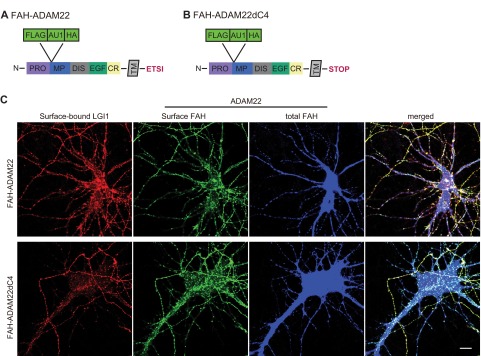
Normal surface localization and LGI1 binding of ADAM22dC4 mutant. FAH-tagged ADAM22 (A and B) and Hisx6-tagged LGI1 were expressed in dissociated hippocampal neurons. (C) Surface-bound LGI1 (red), surface-expressed ADAM22 (green), and total ADAM22 (blue) were labeled by anti-Hisx6, FAH, and HA antibodies, respectively. Wild-type ADAM22 is surface localized and binds extracellular LGI1. ADAM22 lacking the PDZ-binding motif (ADAM22dC4, Bottom) is also surface localized and able to bind extracellular LGI1. (Scale bar, 10 μm.)
PSD-95 Function Is Dependent on LGI1 Expression.
We have shown here that an LGI1 point mutant that cannot bind ADAM22 (LGI1 S473L) is unable to regulate transmission and that the function of ADAM22 requires PDZ interactions. Previous proteomic analysis indicates that the MAGUKs PSD-95 and PSD-93 are part of the LGI1–ADAM22 complex at synapses (19). Like LGI1, changing expression levels of either of these proteins modulates synaptic AMPAR localization (14). Moreover, loss of both PSD-95 and PSD-93 results in a decrease in the number of functional synapses without altering surface receptors or spine density (14), akin to the effect of ADAM22 deletion. Therefore, we hypothesized that the LGI1–ADAM22 complex modulates synaptic transmission by regulating the function of mature MAGUKs at the synapse.
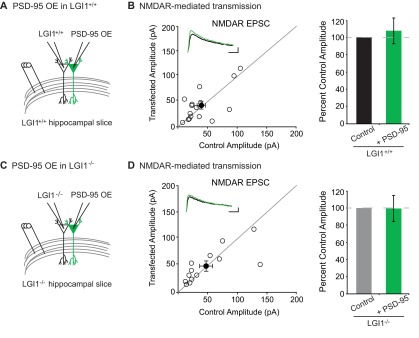
We first tested if LGI1 is required for overexpression of PSD-95 to increase AMPAR content by comparing the effect of overexpressing PSD-95 in wild-type and LGI1−/− slice cultures. As expected, overexpression of PSD-95 greatly increases AMPAR EPSCs in wild-type neurons (Fig. 5 A, B, and E). However, overexpression of PSD-95 in LGI1−/− cells had little effect, showing that LGI1 plays a critical role in mediating PSD-95 function (Fig. 5 C–E). Overexpression of PSD-95 has no effect on NMDAR EPSCs in wild-type or LGI1−/− neurons (Fig. S5). These data indicate that the dramatic increase in synaptic AMPARs normally observed after PSD-95 overexpression is dependent upon LGI1 expression.
Fig. 5.

LGI1 is required for PSD-95 potentiation of excitatory synapses. (A) Diagram depicting dual recording of wild-type and PSD-95–overexpressing cells. (B) Scatterplot of AMPAR EPSC amplitudes recorded from wild-type control and PSD-95–overexpressing cells. Representative traces of dual recordings are shown as Insets. (Scale bars, 50 ms and 50 pA.) (C) Diagram depicting dual recording of LGI1−/− and PSD-95–overexpressing cells. (D) Scatterplot of AMPAR EPSC amplitudes recorded from LGI1−/− control and LGI1−/− cells overexpressing PSD-95. (Scale bars, 50 ms and 25 pA.) (E) Summary bar graphs showing the normalized mean AMPAR EPSC ± SEM. Overexpression of PSD-95 in wild-type slice culture results in a threefold increase in AMPAR-mediated transmission (P = 0.0004, n = 17), whereas overexpression of PSD-95 in LGI1−/− slices has a slight, but significant, effect on AMPAR EPSCs (P = 0.03, n = 17).
Fig. S5.
PSD-95 overexpression does not alter NMDAR-mediated EPSCs. (A) Diagram depicting dual recording of wild-type and PSD-95 overexpressing cells. (B) Scatterplot of NMDAR EPSC amplitudes recorded from wild-type control and PSD-95–overexpressing cells. Representative traces of dual recordings are shown as Insets. (Scale bars, 50 ms and 50 pA.) Bar graphs show normalized mean NMDAR EPSC ± SEM. Overexpression of PSD-95 in wild-type slice culture has no effect on NMDAR EPSCs (P = 0.34, n = 16). (C) Diagram depicting dual recording of LGI1−/− and PSD-95–overexpressing cells. (D) Scatterplot of NMDAR EPSC amplitudes recorded from LGI1−/− control and LGI1−/− cells overexpressing PSD-95. (Scale bars, 50 ms and 25 pA.) Bar graphs show the mean NMDAR EPSC ± SEM. No effect was observed on NMDAR EPSCs in LGI1−/− neurons overexpressing PSD-95 (P = 0.59, n = 13).
LGI1 Regulates the Functional Incorporation of PSD-95 but Not SAP102.
We next sought to determine if the absence of LGI1 results in loss of endogenous function of PSD-95. To test this possibility, we examined the effect of decreased PSD-95 in wild-type and LGI1−/− slice cultures using a validated target sequence to knockdown PSD-95 expression (14). In agreement with previous findings (14), we found that reduced expression of PSD-95 results in a 50% reduction in AMPAR-mediated EPSCs. Strikingly, however, knockdown of PSD-95 in LGI1−/− cells had no effect on AMPAR-mediated EPSCs (Fig. 6 A–D). Knockdown of PSD-95 did not alter NMDAR-mediated EPSCs in wild-type or LGI1−/− neurons (Fig. S6). Thus, LGI1 expression is required for PSD-95 to function at the synapse. It is possible that PSD-95 is unable to function in the absence of LGI1 due to mislocalization. However, when we quantified the density and intensity of synaptic PSD-95 puncta in wild-type and LGI1−/− hippocampal slices, we find that loss of LGI1 does not alter the localization of PSD-95 (Fig. S7).
Fig. 6.
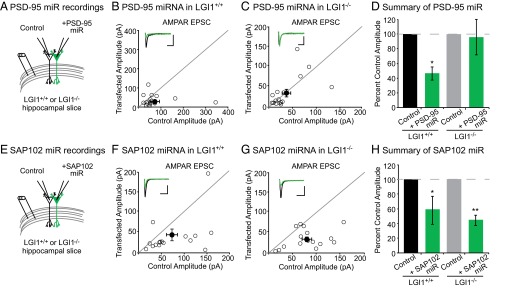
LGI1 is essential for PSD-95, but not SAP102, function. (A) Dual recordings of control and PSD-95 miRNA-expressing cells in LGI1+/+ or LGI1−/− slices. (B) Scatterplot of AMPAR EPSC amplitudes recorded from wild-type and PSD-95 miRNA-transfected cells. Representative traces of dual recordings are shown as Insets. (Scale bars, 50 ms and 50 pA.) (C) Scatterplot of AMPAR EPSC amplitudes recorded from LGI1−/− control and LGI1−/− cells transfected with PSD-95 miRNA. (Scale bars, 50 ms and 25 pA.) (D) Summary bar graphs showing normalized mean ± SEM of experiments in B and C. Reducing PSD-95 expression in wild-type slice culture results in a 50% reduction in AMPAR-mediated transmission (P = 0.03, n = 17). In LGI1−/− slices, reduction in PSD-95 expression has no effect on AMPAR-mediated transmission (P = 1.00, n = 16). (E) Dual recordings of control and SAP102 miRNA-expressing cells in LGI1+/+ or LGI1−/−. (F) Scatterplot of AMPAR EPSC amplitudes recorded from wild-type and SAP102 miRNA-transfected cells. Representative traces of dual recordings are shown as Insets. (Scale bars, 50 ms and 50 pA.) (G) Scatterplot of AMPAR EPSC amplitudes recorded from LGI1−/− control and LGI1−/− cells transfected with SAP102 miRNA. (Scale bars, 50 ms and 50 pA.) (H) Summary bar graphs showing the normalized mean ± SEM of experiments in F and G. Knockdown of SAP102 results in a significant reduction in AMPAR-mediated EPSCs in wild-type (P = 0.02, n = 16) and LGI1−/− (P = 0.002, n = 16) neurons.
Fig. S6.
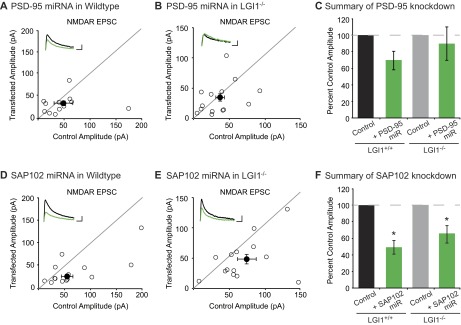
Reduction in SAP102, but not PSD-95, expression results in decreased NMDAR-mediated EPSCs in both wild-type and LGI1−/− neurons. (A) Scatterplot of NMDAR EPSC amplitudes recorded from wild-type and PSD-95 miRNA-transfected cells. Representative traces of dual recordings are shown as Insets. (Scale bars, 50 ms and 50 pA.) (B) Scatterplot of NMDAR EPSC amplitudes recorded from LGI1−/− control and LGI1−/− cells transfected with PSD-95 miRNA. (Scale bars, 50 ms and 25 pA.) (C) Summary bar graphs showing normalized mean NMDAR EPSC ± SEM of experiments in A and B. Reducing PSD-95 expression in wild-type slice culture results in a slight, but insignificant decrease in NMDAR EPSCs (P = 0.31, n = 13). In LGI1−/− slices, reduction in PSD-95 expression has no effect on NMDAR-mediated transmission (P = 0.67, n = 14). (D) Scatterplot of NMDAR EPSC amplitudes recorded from wild-type and SAP102 miRNA-transfected cells. Representative traces of dual recordings are shown as Insets. (Scale bars, 50 ms and 50 pA.) (E) Scatterplot of NMDAR EPSC amplitudes recorded from LGI1−/− control and LGI1−/− cells transfected with SAP102 miRNA. (Scale bars, 50 ms and 50 pA.) (F) Summary bar graphs showing the normalized mean NMDAR EPSC ± SEM of experiments in D and E. Knockdown of SAP102 results in a significant reduction in NMDAR-mediated EPSCs in wild-type (P = 0.02, n = 14) and LGI1−/− (P = 0.03, n = 14) neurons.
Fig. S7.
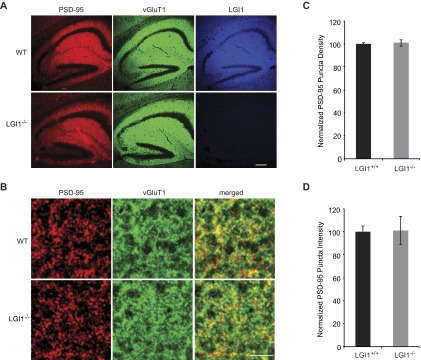
LGI1 is not required for proper localization of PSD-95. (A) PSD-95 (red), vGluT1 (green), and LGI1 (blue) were labeled in hippocampal slices from wild-type and LGI1−/− mice. (Scale bar, 200 μm.) (B) Higher magnification of images show colocalization or apposition of PSD-95 and vGluT1 at synapses, used to quantify synaptic PSD-95 puncta density and intensity. (Scale bar, 5 μm.) (C) Loss of LGI1 has no effect on the density of synaptic PSD-95 puncta counted in 10 × 10 μm regions of interest (ROIs) (P = 0.46, n = 27 ROIs). (D) Likewise, LGI1 expression does not alter the intensity of PSD-95 puncta (P = 0.60, n = 1,800 puncta).
A hallmark of the MAGUK family of scaffolding proteins is a developmental shift in the expression of individual family members—as the synapse matures, SAP102 is replaced by PSD-93 and PSD-95 (14, 17). Notably, the timeline of PSD-95/PSD-93 and LGI1 expression is similar (17, 18, 20), and, unlike PSD-95 and PSD-93, SAP102 was not found to interact with the LGI1–ADAM protein complex in vivo (18, 19). Thus, we wondered if in the absence of LGI1, SAP102 remains functional as a synaptic scaffold. Using a previously validated miRNA targeting SAP102 (14, 17), we assayed the impact of reduced SAP102 expression in wild type and LGI1−/−. In both wild-type and LGI1−/− neurons, we found that knockdown of SAP102 results in a significant decrease in AMPAR-mediated transmission (Fig. 6 E–H) as well as a reduction in NMDAR-mediated EPSCs (Fig. S6). Together our data indicate that in the absence of LGI1, synapses do not functionally incorporate PSD-95, leaving them in an immature state and limiting the strength of adult synapses.
Discussion
Here, we identify an essential role for the LGI1–ADAM22 complex in functional maturation of the synapse via molecular interaction with the scaffolding protein PSD-95. We show that LGI1 is secreted from pre- and postsynaptic cells and binds to nearby ADAM22 to control synaptic AMPAR content. Moreover, we reveal a novel role for ADAM22 in maintaining excitatory synapses through PDZ-dependent interactions. Finally, we show that in the absence of LGI1, PSD-95, although still targeted to synapses, is unable to function, leaving synapses in an immature state.
Previous work has shown that LGI1 binds to and regulates ADAM22 synaptic localization (26). LGI1 can also oligomerize—forming homodimers, -trimers, and -tetramers (19, 26)—giving it the ability to stabilize increasing numbers of ADAMs at the synapse (31). Together with our data, these findings support a model wherein ADAM22 works through PSD-95 to maintain mature synapses, and LGI1 binds to and incorporates more ADAM22 and, in turn, functional PSD-95 to increase the AMPAR-content at these synapses.
ADAM22 also interacts with the mature MAGUK PSD-93 at the synapse, which is found in the LGI1-mediated protein complex in vivo (19). Thus, it is possible that the LGI1–ADAM22 complex acts through both PSD-95 and PSD-93 to regulate synapse maturation. This finding is supported by the literature regarding the effects of changing MAGUK expression levels. Addition or removal of either PSD-95 or PSD-93 results in a corresponding change in synaptic AMPAR content (14, 15), similar to the effect of addition (18) or removal (19) of LGI1. However, loss of both PSD-95 and PSD-93 results in a decreased number of synapses (14), similar to what we observe after loss of ADAM22. Thus, when a small amount of ADAM22 is present, as is the case in LGI1−/− cells, some mature MAGUKs may be functional and synapses can be maintained—although, without LGI1 to oligomerize and incorporate more MAGUKs, these synapses remain weak. In the absence of ADAM22, however, no mature MAGUKs are able to function and thus excitatory synapses are not maintained.
What might account for the remaining synaptic ADAM22 in the absence of LGI1? Previous work has shown that both LGI2, implicated in canine epilepsy (32), and LGI4, found to regulate myelination in the peripheral nervous system (33), bind ADAM22 (32–35). Both family members are also found in CA1, although expressed at much lower overall levels than LGI1 (36). However, these proteins lack a unique insertion in the C-terminal epitempin (EPTP) domains, which is thought to contribute to the ability of LGI1 to oligomerize (31). It is possible, then, that in the absence of LGI1, LGI2 and LGI4 act to stabilize ADAMs at the synapse, but lack the ability to oligomerize and recruit more AMPARs, rendering the remaining synapses weaker.
Surprisingly, when we examined PSD-95 localization at the synapse, we found that it is similar in wild-type and LGI1−/− neurons. How might LGI1 regulate PSD-95 function, while not impacting its synaptic localization? Quantitative analysis of postsynaptic proteins indicates the MAGUK family proteins far outnumber NMDARs and AMPARs in the PSD (37). Thus, there may be other factors that control the function of PSD-95 once it has reached the synapse. Our data indicate the LGI1–ADAM22 complex is this factor. An enticing possibility is that LGI1 localizes PSD-95 into a functional microdomain within the synapse; it may be that the LGI1 creates a transsynaptic structure through its pre- and postsynaptic interactions (19) to guide PSD-95 into the functional synaptic space in line with the presynaptic active zone. It also may be that the LGI1–ADAM22 complex introduces a previously unknown protein modification that activates PSD-95. Future studies are required to determine the exact mechanism by which this complex regulates PSD-95 function.
Previous analysis of LGI1-interacting proteins revealed three candidate transmembrane proteins to mediate the LGI1 functional effect—ADAM11, ADAM22, and ADAM23 (19). Using an epilepsy-associated LGI1 mutant, S473L, we demonstrate that binding to ADAM22 is essential for LGI1 to regulate synaptic content. Consistent with these data, LGI1 is found bound to ADAM22 most often in vivo (19). Additionally, mice lacking ADAM11 do not exhibit a lethal epileptic phenotype (38), unlike LGI1-lacking or ADAM22-lacking mice (19, 21, 22, 39), suggesting that ADAM11 may not share a functional role with these proteins. In the future, it will be of interest to determine if ADAM11 and ADAM23 impact synaptic transmission as well and the nature of their interaction with LGI1.
Other research has suggested that, in addition to its role in synaptic transmission, LGI1 may be important during neuronal development. In previous reports, it was shown that LGI1 can act as an antagonist for myelin-based growth cone inhibition (20), can regulate early cerebellar cell development (40), and can increase neurite outgrowth in dissociated hippocampal neurons (41, 42). However, these developmental defects would not explain the specific decrease in synaptic AMPAR content in LGI1−/− mice we observe in the hippocampus. Moreover, acute application of LGI1 to hippocampal neurons increases synaptic AMPAR content (18). Together, these findings suggest that LGI1 may have independent roles in neural development and synapse maturation.
Recently, LGI1 has been identified as the target of antibodies made in the autoimmune disorder limbic encephalitis. Patients with limbic encephalitis (LE) develop adult-onset epilepsy and neuropsychiatric symptoms, such as memory deficits and psychosis (43). Research into the mechanism of action of autoantibodies has shown that antibodies isolated from LE patients target LGI1, disrupting the LGI1–ADAM22 interaction and reducing synaptic AMPARs (44). So, whereas it may be that the epilepsy phenotype related to mutations in LGI1 is a result of improper neurite development, it is certain that disruption of the LGI1–ADAM complex in the mature adult brain can also produce disease, indicating a critical function for this complex in synaptic maintenance as well as development.
Over the last decade, the importance of synaptic organizing complexes in proper development and maturation of excitatory synapses has begun to be appreciated. This work uncovers a novel role for the LGI–ADAM22 synaptic organizing complex in coordinating the function of PSD-95 at mature synapses. A deeper understanding of this complex is certain to contribute not only to our knowledge of the basic mechanisms of synaptic transmission but also to the dysfunctions that produce the devastating neurological disorders associated with this complex.
Materials and Methods
Mice.
All animal studies were performed according to the Institutional Animal Care and Use Committee guidelines at the University of California, San Francisco and the institutional guidelines at the National Institute for Physiological Sciences. LGI1−/− and ADAM22fl/fl mice were generated and genotyped as previously described (19, 33).
Slice Culture Preparation and Transfection.
Hippocampal slice cultures were prepared from 6- to 8-d-old mice as previously described (45). At 4 d in vitro, for overexpression experiments, or 1 d in vitro, for RNAi-mediated knockdown, slice cultures were transfected using a Helios Gene Gun (BioRad). For biolistic transfection, 50 μg total of each construct was coated on 1-μm-diameter gold particles, which were then coated onto PVC tubing and stored at 4 °C. For experiments where bullets were coated with two different constructs, coexpression was visually confirmed by using different fluorophores for each construct.
Slice Culture Viral Injections.
LGI1-expressing lentivirus was introduced into slice cultures using a microinjection technique. Virus was loaded into a glass pipette, and 2.3-nL injections were made into slices using a Micro4 microsyringe pump (WPI). Injection sites were visualized under 10× magnification and confirmed by visible depression of the slice culture after injection burst. For both CA3 and dentate gyrus, three injections were made into the region to ensure robust coverage.
Electrophysiology.
All datasets include recordings from at least seven hippocampal slices from three different animals. Recordings were made at DIV8, or DIV17–21 in ADAM22fl/fl to allow for complete turnover of existing ADAM22 after Cre-mediated deletion, using 3- to 4-MΩ glass electrodes filled with an internal solution consisting of 130 mM CsMeSO3, 8 mM NaCl, 10 mM Hepes, 4 mM Mg-ATP, 0.3 mM Na-GTP, 10 mM BAPTA-tetracesium, 5 mM QX314-Cl, and 0.1 mM spermine, pH 7.2 with CsOH. External perfusion medium consisted of 140 mM NaCl, 2.4 mM KCl, 1 mM NaH2PO4, 26.2 mM NaHCO3, 10 mM glucose, 4 mM CaCl2, 4 mM MgSO4, and 100 μM picrotoxin, saturated with 95% (vol/vol) O2 and 5% (vol/vol) CO2. Transfected pyramidal cells were identified using fluorescence microscopy. For experiments using two transfected constructs, both diffuse GFP and nuclear mCherry (tagged Cre) were confirmed. In all paired experiments, transfected and neighboring control neurons were recorded simultaneously.
A bipolar stimulating electrode was placed in stratum radiatum or stratum moleculare for synaptic recordings of CA1 pyramidal cells and dentate gyrus granule cells, respectively. After gaining whole-cell access, cells were held at −70 mV and stimulated for 5 min to allow for response stabilization. After this period, 20 trials were obtained at 0.2 Hz while holding the cells at −70 mV, followed by 20 trials at +40 mV. AMPA EPSCs were measured as the peak amplitude of the averaged traces recorded at −70 mV, and the NMDA EPSC was measured at +40 mV as the average amplitude of the current 100 ms after stimulation, at which the AMPA receptor-mediated EPSC had completely decayed. Series resistances typically ranged from 10 to 20 MΩ; a cell pair was discarded if the series resistance of either increased to >30 MΩ. For coefficient of variation analysis, 100 trials were acquired at −70 mV. Paired-pulse ratios were obtained by recording the responses to a pair of stimuli given 40 ms apart. The two resulting EPSCs were measured and the peak amplitude of the second EPSC divided by the peak amplitude of the first EPSC gives the paired-pulse ratio. Miniature excitatory postsynaptic currents were obtained in the presence of 1 μM tetrodotoxin (TTX) (Tocris Bioscience).
Outside-out patches were taken from CA1 pyramidal neurons by patching the cell bodies with 4- to 5-MΩ patch pipettes. After obtaining whole-cell access and clamping the cell to −70 mV, the patch pipette was slowly pulled away from the cell body until a gigaohm seal reformed. The tip of the pipette was perfused with Hepes-ACSF containing (in millimoles): 150 NaCl, 2.5 KCl, 10 Hepes, 10 glucose, 1 MgCl2, 2 CaCl2, 0.1 d-AP5, 0.1 picrotoxin, 0.1 cyclothiazide, and 0.5 μM TTX. To evoke glutamate currents, the solution was switched to Hepes-ACSF containing 1 mM l-glutamic acid. A ValveLink 8 (AutoMate Scientific) was used for fast perfusion of the control and glutamate containing Hepes-ACSF. Outside-out patches from experimental cells were interleaved with nontransfected control neurons to ensure the most consistent and direct comparisons.
Statistical Analysis.
Sample size (n) indicates number of cells, regions of interest, or puncta in each condition for unpaired data, and number of pairs for dual recordings. All statistical analyses were performed in Prism 5 (GraphPad). Dual recording statistics were calculated using a two-tailed Wilcoxon signed-rank test, whereas unpaired statistics were calculated using a two-tailed Wilcoxon–Mann–Whitney test. Statistics on spine density and PSD-95 puncta analysis also used a two-tailed Wilcoxon–Mann–Whitney test. Coefficient of variation was calculated as the square of the variance over the mean.
SI Materials and Methods
Experimental Constructs.
LGI1 and ADAM22 constructs were cloned as previously described (18). For biolistic expression, LGI1, ADAM22, and ADAM22 mutants were subcloned into the pCAGGS–IRES–GFP vector using PCR and In-Fusion HD Cloning system (Invitrogen). LGI1 used in lentivirus was subcloned after the IRES sequence in FUGW–GFP–IRES so that infected cells could be readily identified. PSD-95 used in overexpression experiments as well as PSD-95 and SAP102 miRNA target sequences were generated and validated in a previous publication (14). For GFP coexpression, the miRNA were subcloned into the 3′ UTR of the GFP in pCDNA6.2/EmGFP with the BLOCK-iT miRNA kit (Invitrogen) and then subcloned into the pCAGGS vector. To examine the cell-surface expression of ADAM22, the sequence encoding FAH tandem tag (FLAG–AU1–HA) was inserted between base pairs 696 and 697 of mouse ADAM22 (HM004095) as a synthetic DNA fragment 5′-GGAGGTGACTACAAGGATGACGACGACAAGGGAGGTGACACATACCGATACATAGGAGGTTACCCATACGATGTTCCAGATTACGCTGGAGGT-3′.
Lentivirus Production.
To create the LGI1 lentivirus, three T-75 flasks of rapidly dividing HEK293T cells (ATCC) were transfected with FUGW–GFP–IRES–LGI1 and the helper plasmids pVSV-G and psPAX2 using FuGENE HD (Promega). After 40 h of incubation, the supernatant was collected, filtered, and concentrated using PEG-It Virus Precipitation Solution (System Biosciences). The resulting pellet was resuspended in 400 μL Opti-Mem, flash frozen in liquid nitrogen, and stored at −80 °C.
Generation of Prox1–LGI1 Mice.
To specifically express LGI1 in the dentate gyrus granule cells, we took advantage of prospero homeobox 1 (Prox1) promoter [kindly suggested by Sebastian Jessberger (University of Zurich) and Ryuta Koyama (University of Tokyo)]. The cDNAs of rat LGI1 tagged with FLAG and Hisx6 (LGI1–FH) (19) and kanamycin-resistant gene were inserted at the locus of the start codon of a Prox1 gene in the Prox1-containing BAC clone (RP23-360I16) by Red/ET recombination system (gene bridges). This linearized modified BAC DNA was microinjected into BDF1 oocytes to produce transgenic mice (PhoenixBio). Genotyping was performed using PCR primers: 5′-GCTTGACCAGATTCATTGGCGACT-3′ and 5′-CTAATGGTGATGGTGATGATGACC-3′ for LGI1–FH. The LGI1–FH (Prox1–LGI1) transgenic mouse was crossbred with an LGI1+/− mouse. Obtained LGI1+/−; Prox1–LGI1 was crossed with LGI1+/− to obtain LGI1−/−; Prox1–LGI1.
Antibodies.
The following antibodies were used: a rabbit polyclonal antibody to LGI1 (Abcam ab30868), mouse monoclonal antibodies to PSD-95 (Thermo Scientific; MA1-046) and Hisx6 (WAKO 9F2), rat monoclonal antibody to HA (Roche 3F10), a guinea pig polyclonal antibody to vGluT1 (Millipore AB5905), and a human polyclonal antibody to LGI1 (44). A rabbit polyclonal antibody to an FAH tag was raised against GST–FAH (GGDYKDDDDKGGDTYRYIGGYPYDVPDYAGG; underlining corresponds to FLAG, AU1, and HA epitopes, respectively). The antibody was affinity purified on CNBr-activated Sepharose 4B (GE Healthcare) columns containing the immunizing antigen.
Western Blotting of Microdissected Hippocampal Regions.
LGI1+/+; Prox1–LGI1 and LGI1+/−; Prox1–LGI1 mice (1 mo, total three mice) were killed and their brains were removed. Horizontal brain slices (400 µm thickness, ∼8 slices from one animal) were taken with microslicer (DSK Pro7). Cortex and hippocampus were dissected and dentate gyrus and CA3 plus CA1 were further dissected from the hippocampus under the dissection microscope. Dissected pieces were collected, weighed, and lysed in 4 vol of homogenate buffer [10 mM Tris⋅HCl (pH 8.0), 2 mM EDTA, 320 mM sucrose, and 50 μg/mL PMSF] with a Dounce homogenizer. Protein concentration of lysates was measured and 50 µg of protein was subjected to SDS/PAGE and Western blotting with anti-LGI1 and -Prox1 antibodies.
Immunohistochemistry.
LGI1−/−; Prox1–LGI1 and littermate wild-type mice (P17) were anesthetized by pentobarbital (75 mg/kg, i.p.) and perfused with 4% (wt/vol) paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4). The brain was removed and immersed in the same fixative for 12 h at 4 °C and then cryoprotected in 20% (wt/vol) sucrose in PBS overnight at 4 °C. The 60-μm free-floating sections were cut on a cryostat (Leica CM1950). Sections were pretreated with 0.2 mg/mL pepsin (DAKO) for 10 min at 37 °C. Sections were blocked for 1 h in PBS containing 3% (wt/vol) normal goat and 0.3% Triton X-100 at room temperature and then incubated in the same buffer containing anti-LGI1 antibody (1 μg/mL) overnight at 4 °C. Endogenous peroxidase activity was inactivated by incubating brain sections in 1% H2O2 for 30 min. Immunohistochemical staining was performed with an avidin/biotin/peroxidase system (Vector Laboratories; ABC Elite) and DAB (Vector Laboratories). Images were taken using stereomicroscope equipped with a digital camera (Olympus DP20) with the same exposure time and camera settings.
For immunofluorescence analysis, LGI1−/− mice and their littermate controls (three animals for each) were killed (P17) and their brains were frozen in n-hexane cooled on dry ice and embedded in OCT compound. Then, the frozen sections (7 μm in thickness) were cut on a cryostat (CM1950; Leica) and fixed with acetone for 20 min on ice. Fixed sections were rehydrated and blocked for 1 h at room temperature in PBS containing 5% (wt/vol) donkey and goat serum and were incubated in the mixture of PSD-95, vGluT1, and LGI1 antibodies overnight at 4 °C, followed by Alexa 488–anti-guinea pig IgG, Cy3–anti-mouse IgG, and Alexa 647–anti-human IgG Fc secondary antibodies, respectively. Fluorescent images were acquired with a confocal laser scanning microscopy system (TCS SP5 II; Leica) equipped with a HCX PL APO 63×/1.40–0.60 oil immersion objective lens. Microscope control and all image analyses were performed with Leica LAS AF software, and images were acquired with the same laser power and detector settings. The number of PSD-95 clusters, which were adjacent to vGluT1, in randomly chosen 10 μm × 10 μm regions of CA1 stratum radiatum (nine regions per animal) was counted (threshold was set at 530 arbitrary units intensity). The mean fluorescence intensity of PSD-95 clusters in CA1 stratum radiatum (600 clusters per animal) was measured.
Immunocytochemical Analysis.
Cultured hippocampal neurons were obtained from E18 rat brains and seeded onto 12-mm coverslips (5 × 104 cells). The neurons (DIV10) were transfected with pCAGGS:ADAM22–FAH with pCAGGS:LGI1–Hisx6 by Lipofectamine 2000 (Life Technologies). At DIV17, cell-surface LGI1–Hisx6 and ADAM22–FAH were live labeled with anti-Hisx6 and FAH antibodies by incubating neurons in conditioned medium for 30 min at 37 °C. Neurons were then washed and fixed with methanol for 10 min at –30 °C and blocked with PBS containing 10 mg/mL BSA for 10 min on ice. Then, neurons were incubated with anti-HA antibody for total ADAM22–FAH staining, followed by Cy3-anti-mouse IgG, Alexa 488–anti-rabbit IgG and DyLight649–anti-rat IgG secondary antibodies. Fluorescent images were acquired with a confocal laser scanning microscopy system (TCS SP5 II; Leica) equipped with a HCX PL APO 63×/1.40–0.60 oil immersion objective lens.
Acute Slice Preparation.
For dentate gyrus granule cell recordings, acute slices were used to preserve the medial and lateral perforant paths for recordings. Transverse hippocampal slices (300 μm) were obtained from P14–P15 mice with a Leica vibratome in a high sucrose cutting solution containing (in millimoles): NaCl 50, KCl 2.5, CaCl2 0.5, MgCl2 7.0, NaH2PO4 1.0, NaHCO3 25, glucose 10, and sucrose 150. Slices were transferred to artificial cerebrospinal fluid (ACSF) containing (in millimoles) NaCl 119, KCl 2.5, NaHCO3 26.2, Na2PO4 1, glucose 11, CaCl2 2.5, MgCl2 1.3 in an incubating chamber, recovered at 35 °C for 30 min, then moved to room temperature for the duration of the experiment.
Spine Density Imaging.
Images were acquired using a Zeiss LSM 5 Pascal scanning confocal microscope at DIV17–21. ADAM22fl/fl slice cultures were biolistically transfected with GFP (control) or GFP and Cre-mCherry (ADAM22−/−). For quantification of spine density, 100 μm of the primary apical dendrite of transfected CA1 cells were imaged starting 100 μm away from the soma. To ensure bright enough fluorescence to visualize spines, a vector driving GFP expression under the strong hybrid CAG promoter (pCAGGS) was cotransfected with Cre. Spine density was quantified manually using ImageJ software.
Acknowledgments
We thank Dies Meijer for the ADAM22fl/fl mice; Kirsten Bjorgan, Manuel Cerpas, and Dan Qin for technical assistance; as well as the members of the R.A.N. laboratory for thoughtful comments and helpful suggestions. K.L.L. and A.J.G. were supported by National Science Foundation Graduate Research fellowships. Y.F. is supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) and the Ministry of Health, Labor, and Welfare. M.F. is supported by grants from the MEXT. R.A.N. is supported by the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511910112/-/DCSupplemental.
References
- 1.McMahon SA, Díaz E. Mechanisms of excitatory synapse maturation by trans-synaptic organizing complexes. Curr Opin Neurobiol. 2011;21(2):221–227. doi: 10.1016/j.conb.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siddiqui TJ, Craig AM. Synaptic organizing complexes. Curr Opin Neurobiol. 2011;21(1):132–143. doi: 10.1016/j.conb.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119(7):1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci USA. 2005;102(17):6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Wit J, et al. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64(6):799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ko J, Fuccillo MV, Malenka RC, Südhof TC. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009;64(6):791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linhoff MW, et al. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61(5):734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101(6):657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 9.DeNardo LA, de Wit J, Otto-Hitt S, Ghosh A. NGL-2 regulates input-specific synapse development in CA1 pyramidal neurons. Neuron. 2012;76(4):762–775. doi: 10.1016/j.neuron.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woo J, et al. Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nat Neurosci. 2009;12(4):428–437. doi: 10.1038/nn.2279. [DOI] [PubMed] [Google Scholar]
- 11.Kim S, et al. NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat Neurosci. 2006;9(10):1294–1301. doi: 10.1038/nn1763. [DOI] [PubMed] [Google Scholar]
- 12.Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5(10):771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 13.El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290(5495):1364–1368. [PubMed] [Google Scholar]
- 14.Elias GM, et al. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52(2):307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 15.Schnell E, et al. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99(21):13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, et al. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J Neurosci. 2011;31(17):6329–6338. doi: 10.1523/JNEUROSCI.5968-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sans N, et al. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20(3):1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukata Y, et al. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313(5794):1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- 19.Fukata Y, et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci USA. 2010;107(8):3799–3804. doi: 10.1073/pnas.0914537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas R, et al. LGI1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci. 2010;30(19):6607–6612. doi: 10.1523/JNEUROSCI.5147-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chabrol E, et al. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain. 2010;133(9):2749–2762. doi: 10.1093/brain/awq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu YE, et al. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet. 2010;19(9):1702–1711. doi: 10.1093/hmg/ddq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lynch MW, Rutecki PA, Sutula TP. The effects of seizures on the brain. Curr Opin Neurol. 1996;9(2):97–102. doi: 10.1097/00019052-199604000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Zhou YD, et al. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med. 2009;15(10):1208–1214. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berkovic SF, et al. LGI1 mutations in temporal lobe epilepsies. Neurology. 2004;62(7):1115–1119. doi: 10.1212/01.wnl.0000118213.94650.81. [DOI] [PubMed] [Google Scholar]
- 26.Yokoi N, et al. Chemical corrector treatment ameliorates increased seizure susceptibility in a mouse model of familial epilepsy. Nat Med. 2015;21(1):19–26. doi: 10.1038/nm.3759. [DOI] [PubMed] [Google Scholar]
- 27.Del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124(3):560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346(6280):177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- 29.Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346(6286):724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- 30.Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: Signalling functions at the synapse. Nat Rev Neurosci. 2007;8(3):206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leonardi E, et al. A computational model of the LGI1 protein suggests a common binding site for ADAM proteins. PLoS One. 2011;6(3):e18142. doi: 10.1371/journal.pone.0018142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seppälä EH, et al. LGI2 truncation causes a remitting focal epilepsy in dogs. PLoS Genet. 2011;7(7):e1002194. doi: 10.1371/journal.pgen.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ozkaynak E, et al. Adam22 is a major neuronal receptor for Lgi4-mediated Schwann cell signaling. J Neurosci. 2010;30(10):3857–3864. doi: 10.1523/JNEUROSCI.6287-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4(6):387–396. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishino J, Saunders TL, Sagane K, Morrison SJ. Lgi4 promotes the proliferation and differentiation of glial lineage cells throughout the developing peripheral nervous system. J Neurosci. 2010;30(45):15228–15240. doi: 10.1523/JNEUROSCI.2286-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herranz-Pérez V, Olucha-Bordonau FE, Morante-Redolat JM, Pérez-Tur J. Regional distribution of the leucine-rich glioma inactivated (LGI) gene family transcripts in the adult mouse brain. Brain Res. 2010;1307:177–194. doi: 10.1016/j.brainres.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: A more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi E, et al. Deficits in spatial learning and motor coordination in ADAM11-deficient mice. BMC Neurosci. 2006;7:19. doi: 10.1186/1471-2202-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sagane K, et al. Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci. 2005;6:33. doi: 10.1186/1471-2202-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie YJ, et al. Essential roles of leucine-rich glioma inactivated 1 in the development of embryonic and postnatal cerebellum. Sci Rep. 2015;5:7827. doi: 10.1038/srep07827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owuor K, et al. LGI1-associated epilepsy through altered ADAM23-dependent neuronal morphology. Mol Cell Neurosci. 2009;42(4):448–457. doi: 10.1016/j.mcn.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silva J, Sharma S, Cowell JK. Homozygous deletion of the LGI1 gene in mice leads to developmental abnormalities resulting in cortical dysplasia. Brain Pathol. 2014 doi: 10.1111/bpa.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai M, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: A case series. Lancet Neurol. 2010;9(8):776–785. doi: 10.1016/S1474-4422(10)70137-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohkawa T, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci. 2013;33(46):18161–18174. doi: 10.1523/JNEUROSCI.3506-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37(2):173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]