

Abstract
Introduction
Malignant gliomas (MGs) represent the most common primary brain tumors in adults, the most deadly of which is grade IV glioblastoma. Patients with glioblastoma undergoing current standard-of-care therapy have a median survival of 12 – 15 months.
Areas covered
Over the past 25 years, there have been modest advancements in the treatment of MGs. Assessment of therapeutic responses has continued to evolve to account for the increasing number of agents being tested in the clinic. Currently approved therapies for primary tumors have been extended for use in the setting of recurrent disease with modest efficacy. Agents initially approved for recurrent gliomas have begun to demonstrate efficacy against de novo tumors but will ultimately need to be evaluated in future studies for scheduling, timing and dosing relative to chemotherapy.
Expert opinion
Screening and identification of tumor-specific mutations is critical for the advancement of effective therapy that is both safe and precise for the patient. Two unique antigens found in glioblastoma are currently being employed as targets for immunotherapeutic vaccines, one of which has advanced to Phase III testing. Whole genome sequencing of MGs has yielded two other novel mutations that offer great promise for the development of molecular inhibitors.
Keywords: antiangiogenic agents, EGFR variant III, glioblastoma, isocitrate dehydrogenase, malignant glioma, temozolomide, tumor-specific antigen
1. Malignant glioma: prognosis and assessment of therapeutic response
Malignant gliomas (MGs) comprise the most common primary brain tumors in the adult population. The histologic subtypes of malignant glial neoplasms range from anaplastic astrocytoma (AA) to the most deadly WHO Grade IV glioblastoma (GBM) [1]. Over the past 25 years, only modest advancements in the treatment of GBM tumors have been reached. Current therapies are predominantly for palliative end points rather than curative, although some treatment modalities have been shown to extend survival. Without any therapy, GBM patients uniformly die within 3 months. Patients undergoing current standard-of-care therapy, including surgical resection, radiation therapy (RT) and chemotherapy, have a median survival of 12 – 15 months, with < 25% of patients surviving up to 2 years and fewer than 10% surviving up to 5 years [1-3]. In order to properly assess clinical responses to therapy or disease progression in patients with MGs, medical centers require an assessment of both initial responses to treatment as well as subsequent evidence of progressive disease. This approach has traditionally utilized the Macdonald criteria, which rely upon alterations in two-dimensional tumor measurements with contrast-enhanced computed tomography or MRI [4]. Revised criteria have been proposed by the Response Assessment in Neuro-Oncology (RANO) group to address inaccuracies in assessing patients with pseudoprogression or in assessing progressive disease in patients with non-enhancing lesions [5].
Despite these revisions, current radiographic response criteria for progressive disease following surgical resection are limited in their ability to detect changes surrounding postoperative resection cavities [6]. This difficulty arises in the inherent biology of these gliomas, which may contain large cysts or resection cavities, serve as a reservoir for postoperative blood products that create false-positive MRI signal changes and possess irregular shapes with satellite lesions and small amounts of postoperative residual rim enhancement that are difficult to quantify. In some circumstances, these cavities can collapse, dramatically altering the size and configuration of these irregular enhancing areas. The Response Evaluation Criteria In Solid Tumors criteria are not recommended for evaluating changes to resection cavities, as these criteria consider all lesions that are either < 1 cm or cystic to be unmeasurable. To address these limitations, Kanaly et al. have implemented an algorithm to quantify enhancing tumor volume changes despite resection cavity collapse and can detect enhancing tumor even when it is obscured by intrinsically bright T1 images, such as subacute hemorrhage in a resection cavity [7]. This approach holds a strong advantage over relying on experienced operators to manually outline the tumor volume and then perform the analysis, which brings forth a considerable amount of intra- and inter-operator variability.
Important measures of treatment efficacy in Phase II and III clinical trials for patients with GBM include overall survival (OS), radiographic response, and the duration of any treatment effect, or progression-free survival (PFS). Although OS is considered the gold standard clinical end point, it does not directly measure the impact of a specific regimen because of confounding factors, including standard-of-care and salvage therapy. As a consequence, both radiographic response rate and PFS are valuable end points when attempting to isolate the relative efficacy of a given treatment and to understand the nature of on-study progression [8]. These surrogate measures of tumor burden, however, have well-documented limitations, including the potential for variability, the likelihood of false-positive signals and the discordance in radio-graphic interpretation between observers [9]. Methodologies and techniques that are used to determine tumor response and progression thus continue to evolve, with the goal of minimizing inherent biases and improving accuracy. Neuro-oncologists have also included additional measures such as more informative neurologic examinations and the requirement for steroid therapy in response assessments to strengthen their value. The continued refinement of response assessments is particularly important in the context of an increasing number of agents that are being evaluated in patients with MGs.
With the advent of information about the oncogenic process and molecular expression of these tumors, clinicians and scientists have undertaken endeavors to more precisely target these tumors as monotherapies or in conjunction with current standard-of-care therapy. The following review encompasses an explanation of current standard-of-care treatments as well as the most promising targeted therapies suitable for clinical scalability. Each section of the review includes a reporting strategy of the most recently performed clinical studies, with both positive and negative results, that have been identified in the Cochrane Database, ClinicalTrials.gov and the EU Clinical Trials Register. To eliminate common reporting biases (publication, citation and outcome reporting biases), we performed a comprehensive search of clinical studies using currently approved and investigational agents that were both recently shared at scientific meetings and that report conflicting results compared to findings in similar studies, highlighting the need for well-controlled and randomized design.
2. Current FDA-approved therapies for primary and recurrent GBM
The current regimen for treatment of primary GBM tumors is surgical resection [10] in combination with RT and chemotherapy. To date, the US FDA has approved only a select few therapies for primary GBM tumors, which include nitrosoureas (lomustine and carmustine) and temozolomide (TMZ). Oral lomustine received approval in 1976 [11], and intravenous carmustine received approval in 1977 [12] for use as single agents or in combination with other approved chemotherapeutic agents in patients with primary or metastatic brain tumors who had already underwent surgery or RT [13]. Carmustine wafers are synthetic biodegradable polymers impregnated with carmustine. This product was first approved in 1996 for the treatment of recurrent GBM as an adjunct to surgery and was subsequently approved in 2003 for first-line treatment of high-grade MGs as an adjunct to surgery and radiation [14,15]. While treated patients demonstrated a longer OS compared to placebo controls, approval was based on a reduction in systemic toxicity using this locally applied therapy. In a Phase III trial, 240 newly diagnosed adults undergoing resection of any type of MG were randomly assigned to placement of up to eight carmustine wafers or a placebo, followed by standard RT. Patients receiving the carmustine polymer had only a modest 2-month increase in median survival, although statistically significant (13.9 vs 11.6 months). When the analysis was restricted to patients specifically with GBM tumors, the difference in survival was not statistically significant. Moreover, toxicities with carmustine polymers were similar to the placebo with an additional increase in the incidence of cerebrospinal fluid leakage and intracranial hypertension compared to placebo [16].
2.1 Re-irradiation therapy for primary and recurrent disease
Essentially, all patients with GBM recur after initial therapy, and the majority of patients do not survive beyond 1 year after a diagnosis of recurrent disease (1-year survival following recurrence is approximately 20 – 25%) [17,18]. The current difficulty in offering efficacious and durable treatments has opened up a new area of research for the treatment of MGs. Presently, a number of salvage approaches have been introduced. The first type of salvage therapy is stereotactic radiosurgery (SRS), which is applied as a single dose. With this approach, it is possible to deliver very high doses to small target volumes, while sparing surrounding healthy tissues [19]. The largest available prospective cohort study on SRS determined the efficacy of SRS as a salvage treatment in patients with recurrent MG. A total of 114 patients were included in the analysis, and median OS from the time of diagnosis was 37.5 months for patients with grade III gliomas and 23 months for patients with GBM. The median PFS following SRS was 8.6 months for patients with grade III gliomas and 4.6 months for patients with GBM. A significant survival benefit of SRS as salvage treatment could be shown in patients with recurrent GBM compared to a historical control group (23 vs 12 months; p < 0.0001), but there was no significant difference in patients with recurrent grade III gliomas (37.5 vs 26 months; p = 0.789) [20]. And so, SRS for recurrent glioma is possible, but with higher tumor volumes the risk of side effects increases.
Fractionated stereotactic radiotherapy (FSRT) is another noninvasive precision RT technique. FSRT comprises first obtaining the required therapeutic dose, which is then divided into a number of fractions. By exploiting the radiobiological advantage of fractionation, the risk of side effects to normal tissue can be minimized over time. FSRT can be applied safely for very small target volumes as an alternative to SRS; moreover, for bulky tumors, FSRT can also be performed safely and effectively without the high risk of side effects associated with SRS in such tumors [19]. Cho et al. reported patients receiving FSRT had comparable survival to SRS patients and lower risk of late complications despite having poorer pretreatment prognostic factors. Investigators concluded that FSRT may be a better option for patients with larger tumors or tumors in eloquent structures [21]. Patel et al. also conducted a prospective study comparing salvage re-irradiation with SRS and with FSRT for recurrent GBM. Median OS was not significantly different between the two therapies, with OS following SRS extending to 8.5 months compared to 7.4 months following FSRT (p = 0.81). Of note, patients who responded to either treatment had statistically improved survival compared to non-responders, with a median survival of 15.8 versus 7.3 months (p < 0.05) [22]. One of the largest trials to date was performed by Fokas et al. on 53 patients with recurrent GBM who were re-irradiated using hypofractionated stereotactic radiotherapy (HFSRT). At the time of recurrence, a median total dose of 30 gray (Gy) was delivered in median fractions of 3 Gy/day. After HFSRT, the median survival was 9 months, and the 1-year PFS was 22%. The median OS from initial diagnosis was 27 months [23]. Thus, FSRT seems to represent a safe and feasible option as a treatment for recurrent MG, even for larger tumors and shows adequate efficacy in a number of clinical studies.
2.2 Antiangiogenic treatment for recurrent MG
Antiangiogenic agents target abnormal tumor vasculature, but several aspects of their mechanism of action are incompletely understood. The classic hypothesis is that antiangiogenic therapy, through vessel pruning and reduced blood perfusion, starves the tumor of oxygen and essential nutrients, halting the tumor's uncontrolled growth [24]. However, a logical consequence of diminished tumor blood perfusion following antiangiogenic therapy might be reduced delivery of concurrent chemotherapy. In fact, bevacizumab rapidly reduced blood perfusion in a small study of lung cancer patients, resulting in a decreased influx rate of concurrent docetaxel [25]. However, the relationship between antiangiogenic therapies and chemotherapy delivery is complex and has varied depending on underlying patient characteristics, different tumor profiles or class and dose of antiangiogenic treatments [26]. As a result, bevacizumab and other antiangiogenic agents are being evaluated for use against GBM tumors (Figure 1).
Figure 1. Molecular targets of antiangiogenic agents in GBM.
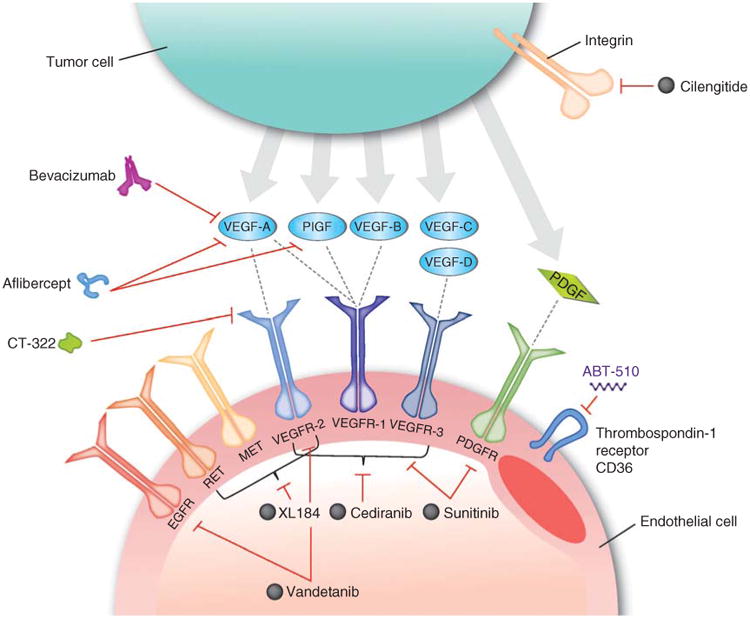
Cilengitide is a cyclic peptide that binds to and inhibits the activities of the α(v)β(3) and α(v)β(5) integrins. Bevacizumab is a humanized monoclonal IgG1 antibody that binds to and inhibits VEGF-A. Aflibercept is a fusion protein that binds all isoforms of VEGF-A, as well as PlGF. Cediranib, sunitinib, vandetanib, XL184 and CT-322 are multireceptor tyrosine kinase inhibitors. ABT-510 is a nonapeptide that targets the thrombospondin-1 receptor CD36.
Reproduced with permission from [43].
EGFR: Epidermal growth factor receptor; GBM: Glioblastoma; PDGFR: Platelet-derived growth factor receptor; PlGF: Placental growth factor; VEGF-A: Vascular endothelial growth factor A; VEGFR: Vascular endothelial growth factor receptor.
Currently, bevacizumab is the only standard therapy for recurrent GBM or WHO grade III MG. In 2009, the FDA granted accelerated approval for bevacizumab in 2009 as a single agent for patients with GBM with progressive disease following prior RT and TMZ therapy [27]. Bevacizumab is a recombinant humanized monoclonal IgG1 antibody that selectively binds to and neutralizes the biologic activity of human VEGF by interfering with the binding of VEGF to its receptors, fms-like tyrosine kinase-1 and kinase insert domain receptor on the surface of endothelial cells.
The outcomes of patients with recurrent GBM treated with bevacizumab have been evaluated in several studies [17,28-31]. Reardon et al. performed a retrospective analysis of outcomes among patients with recurrent GBM who were treated with bevacizumab in combination with either irinotecan, daily TMZ, etoposide, bortezomib or erlotinib [32]. The OS for recurring patients without any treatment (n = 41), recurring patients treated without bevacizumab (n = 44) and recurring patients treated with bevacizumab (n = 55) were 1.5, 4.0 and 5.9 months, respectively (hazard ratio, HR = 0.64, p = 0.04). The PFS of recurring patients in the non-bevacizumab-treated (n = 44) and bevacizumab-treated (n = 55) groups were 1.6 and 2.8 months, respectively (HR = 0.64, p < 0.0001). It is important to note that these results stemmed from a pooled analysis of five consecutive single-arm, Phase II studies. The authors attempted to adjust comparisons of outcome associated with bevacizumab and non-bevacizumab therapy for potential confounding factors, including factors related to treatment selection. Eligibility criteria (histopathological confirmation of grade IV MG, recurrent disease following TMZ therapy and Karnofsky Performance Status ≥ 60) were matched across the studies. Additionally, recurring patients in this study showed favorable prognostic features, including young age and good performance status. Therefore, it should be noted that results from this retrospective analysis may not be applicable to the overall recurrent GBM population. With regard to quality-of-life assessment, these analyses did not assess patient function or quality of life, while receiving therapy after bevacizumab progression. Given the overall poor outcome of GBM patients after progression on bevacizumab, future studies to evaluate therapeutic interventions for such patients should prioritize assessment of these two parameters.
In the recurrent setting, there is a clear and urgent need for sufficiently powered, prospective, well-controlled studies to address whether there is a PFS or OS benefit with bevacizumab for patients with recurrent MGs. Using the Cochrane Database and clinical trial registries (ClinicalTrials.gov and EU Clinical Trials Register), we performed a search for recent clinical studies evaluating these parameters. These studies are summarized in Table 1. One recent well-controlled example is the Dutch BELOB study [33]. In this three-arm, multicenter randomized Phase II study, patients were assigned to either bevacizumab alone, bevacizumab and lomustine, or lomustine alone. A total of 148 eligible patients were enrolled. Importantly, the prognostic factors of patients were balanced across the groups. Results, so far, have shown that patients receiving lomustine in combination with bevacizumab have a longer 6-month PFS (50%, median PFS = 11 months) compared to those receiving bevacizumab (18%, median PFS = 3 months) or lomustine alone (11%, median PFS = 2 months).
Table 1. Recent clinical trials employing antiangiogenic therapy for newly diagnosed and recurrent GBM.
| Clinical trial identifier | Eligibility criteria | Study end point(s) | Treatment groups | |
|---|---|---|---|---|
| Primary | ||||
| Bevacizumab | NCT00943826 (AVAglio) [41] | Newly diagnosed, histologically confirmed GBM Stable or decreasing steroid dose within 5 days prior to randomization |
Primary: PFS and OS Secondary: 1- and 2-year survival rate, quality of life, adverse events |
Arm I: Bevacizumab 10 mg/kg + RT + TMZ 75 mg/m2 Arm II: RT + TMZ 75 mg/m2 |
| NCT00884741 (RTOG 0825) [40] | Newly diagnosed, histologically confirmed GBM with supratentorial component (partial or complete resection) No recurrent or multifocal malignant glioma No prior TMZ or bevacizumab |
Primary: PFS and OS (from randomization) Secondary: Treatment-related toxicity, molecular profile |
Arm I: RT + TMZ Arm II: RT + TMZ + bevacizumab |
|
| NCT00967330 (GLARIUS) [42] | Histologically confirmed GBM No previous chemotherapy or RT for GBM Non-methylated MGMT promoter |
Primary: 6-month PFS Secondary: OS, response rate, time to treatment failure, adverse events, quality of life |
Arm I: Bevacizumab 10 mg/kg every 2 wks + irinotecan 125 mg/m2 every 2 wks + TMZ 75 mg/m2 Arm II: TMZ 75 mg/m2 |
|
| Cediranib | NCT00662506 [26] | Histologically confirmed newly diagnosed GBM Scheduled to receive standard post-surgical RT + TMZ |
Primary: Safety profile and optimal dosing of cediranib during TMZ (Phase I) PFS (Phase II) Secondary: MRI parameters, blood biomarkers, tumor biomarkers |
Arm I: Cediranib + TMZ (dose-limiting toxicity 15 mg, 20 mg, 30 mg) |
| NCT01062425 | Histologically confirmed newly diagnosed GBM with supratentorial component No recurrent or multifocal malignant glioma |
Primary: 6-month PFS Secondary: PFS and OS (from randomization), treatment-related toxicity |
Arm I: Cediranib + RT + TMZ Arm II: RT + TMZ |
|
| Cilengitide | NCT00689221 (CENTRIC) [48] | Newly diagnosed, histologically confirmed supratentorial GBM Methylated MGMT promoter |
Primary: OS (from time of randomization) Secondary: PFS, pharmacokinetics, quality-of-life assessment and safety and tolerability |
Arm I: Cilengitide + RT + TMZ ARM II: RT + TMZ |
| 2004-004849-18 (EMD121974-010) [47] | Newly diagnosed, histologically confirmed supratentorial GBM Stable or decreasing dose of steroids for ≥ 8 days |
Primary: 6-month PFS Secondary: Response rate, OS, 1-year survival rate, median time to progression, pharmacokinetics of cilengitide + TMZ, safety and tolerability |
Arm I: Cilengitide + RT + TMZ Arm II: RT + TMZ |
|
| NCT00813943 (CORE) [49] | Newly diagnosed, histologically confirmed supratentorial GBM Non-methylated MGMT promoter |
Primary: OS (from time of randomization) Secondary: PFS (from randomization), pharmacokinetics, adverse events |
Arm I: Cilengitide (twice weekly) + TMZ + RT Arm II: Cilengitide (five times weekly) + TMZ + RT Arm III: TMZ + RT |
|
| Recurrent | ||||
| Bevacizumab | NTR1929 (BELOB) [33] | Histologically confirmed GBM First relapse after prior treatment with standard RT/TMZ No prior treatment with nitrosoureas or VEGF-R signaling inhibitors |
Primary: 9-month OS Secondary: Response rate, median PFS and OS, 6- and 12-month PFS, quality of life |
Arm I: Bevacizumab 10 mg/kg every 2 wks Arm II: Bevacizumab 10 mg/kg every 2 weeks + 110 mg/m2 lomustine every 6 wks Arm III: Lomustine 110 mg/m2 every 6 wks |
| ACTRN12610000915055 (CABARET) [34] | Histologically confirmed GBM Prior treatment with standard RT/TMZ Recurrent/progressive disease confirmed by surgical resection or MRI |
Primary: PFS Secondary: OS, Response rate, MMSE cognitive function, quality of life, corticosteroid dose, toxicity, time to treatment failure |
Arm I: Bevacizumab every 2 wks until disease progression Arm II: Bevacizumab every 2 wks + carboplatin every 4 wks until disease progression |
|
| Cediranib | NCT00305656 [36] | Histologically confirmed GBM Contrast-enhancing tumor ≥ 1 cm in longest diameter |
Primary: Rate of 6-month PFS Secondary: Response rate, OS, toxicity profile |
Arm I: Cediranib once daily on days 1 – 28 |
| NCT00777153 (REGAL) [37] | Histologically confirmed recurrent GBM Receive one prior chemotherapy with TMZ |
Primary: PFS Secondary: OS (from randomization), response rate, progression-free rate at 6 months, steroid-free days |
Arm I: Cediranib 30 mg Arm II: Cediranib 20 mg + lomustine Arm III: Lomustine alone |
|
| NCT01310855 (DORIC) | Histologically or cytologically confirmed GBM No other prior treatment for GBM except Gliadel or steroids Recurrent or progressive disease after standard therapy |
Primary: PFS (from randomization) Secondary: OS (from randomization), response rate, progression-free rate at 6 months, steroid use |
Arm I: Cediranib 30 mg + gefitinib 500 mg Arm II: Cediranib alone |
|
| Cilengitide | NCT00093964 (EMD 121974-009) [93] | Recurrent or progressive GBM following surgery or biopsy RT and one previous regimen of systemic chemotherapy Solid contrast-enhancing lesion ∼ 1 cm in any dimension within 2 weeks prior to the first dose of cilengitide |
Primary: Rate of 6-month PFS Secondary: Response rate, time to disease progression, survival time, safety, tolerability and pharmacokinetics |
Arm I: Cilengitide 500 mg twice weekly Arm II: Cilengitide 2,000 mg twice weekly |
GBM: Glioblastoma; kg: Kilograms; m2: Meters squared; mg: Milligrams; MGMT: O(6)-methylguanine-DNA methyltransferase; OS: Overall survival; PFS: Progression-free survival; RT: Radiation therapy; RTOG: Radiation therapy oncology group; TMZ: Temozolomide; wks = Weeks.
Another study assessing the therapeutic benefits between using bevacizumab as a monotherapy or combination therapy is the CABARET study [34], which was a sequential stratified two-part randomized Phase II study. The primary objective was to determine the effect of bevacizumab plus carboplatin versus bevacizumab alone for 6-month PFS using the modified RANO criteria. The second stratification included randomizing patients who had progressed but were able to continue treatment to continue or cease bevacizumab. Secondary end points included response rate, cognitive function, quality of life, toxicity and OS. From the 122 patients enrolled, the 6-month PFS was 26% (combination) versus 24% (monotherapy) (HR = 0.96, 95% confidence interval [CI] [0.66, 1.39], p = 0.82). Median OS between the two cohorts was 6.9 versus 6.4 months (HR = 1.08, 95% CI [0.74, 1.59], p = 0.68). Ongoing follow-up of patients on bevacizumab beyond progression, and novel secondary and exploratory end points are not yet available.
A fairly recent antiangiogenic agent, cediranib, is an orally available pan-VEGFR tyrosine kinase inhibitor. Cediranib has a sub-nanomolar half maximal inhibitory concentration for VEGF receptors with additional activity against c-Kit and lower potency against platelet-derived growth factor [35]. In a prior Phase II study of cediranib (45 mg/d) for patients with recurrent GBM, 8 of 30 subjects (27%) achieved a partial radiographic response using Macdonald criteria [36]. A recent multi-centre, randomized double-blind Phase II study (DORIC) is comparing cediranib with and without gefitinib in patients with recurrent GBM. The trial has completed recruitment and is ongoing in the follow-up period. Importantly, results for PFS will be stratified for prognostic factors for GBM, including O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation and isocitrate dehydrogenase enzymes 1/2 (IDH-1/IDH-2) mutations.
A recent international, Phase III, randomized, partially blinded, placebo-controlled study (REGAL) was conducted to investigate the efficacy of cediranib as a monotherapy and in combination with lomustine, versus lomustine alone in patients with recurrent GBM [37]. The primary end point of PFS was not significantly different for either cediranib alone (HR = 1.05; 95% CI [0.74, 1.50]; p = 0.9) or cediranib in combination with lomustine (HR = 0.76; 95% CI [0.53, 1.08]; p = 0.16) versus lomustine. These results show a lack of synergism between chemotherapy and antiangiogenic therapy, which is quite different from the positive results seen with the BELOB trial. Although cediranib monotherapy or in combination with lomustine did not improve PFS compared with lomustine alone in REGAL, preclinical models suggest synergistic activity of anti-VEGF therapy in combination with radiation, owing to the ability of these agents to normalize tumor vessels. On the basis of these observations, cediranib in combination with chemoradiation therapy is being studied in Phase II trials in the newly diagnosed GBM population [26] (NCT00662506; NCT01062425).
2.3 Antiangiogenic treatment for primary MG
Antiangiogenic therapy has shown improved clinical outcomes in certain tumor types other than MGs [38,39]. We employed the aforementioned reporting strategy for recent clinical studies evaluating antiangiogenic therapies for newly diagnosed disease. These studies are summarized in Table 1. Bevacizumab treatment for patients with newly diagnosed GBM was recently tested in two Phase III clinical trials. In the Radiation Therapy Oncology Group (RTOG) 0825 study, 637 patients were randomized to TMZ-based chemoradiation with or without bevacizumab. Patients in the bevacizumab arm started the agent 4 weeks into the RT protocol and continued for 6 – 12 cycles of maintenance therapy. Primary end points of these trials were to evaluate OS and PFS. Results showed a median OS of 15.7 months with the addition of bevacizumab compared to 16.1 months without the treatment. PFS improved slightly in the bevacizumab arm (10.7 vs 7.3 months, p = 0.004), but the difference did not meet the pre-determined level of statistical significance (p = 0.002) [40]. The European trial (AVAglio trial) involved 921 patients and revealed a modest improvement in PFS (10.6 months in the bevacizumab arm vs 6.2 months in the placebo arm) but virtually identical OS (16.8 months vs 16.7 months, respectively) [41]. Furthermore, the AVAglio trial has reported an improvement of quality of life, while the RTOG 0825 study did not, and even suggested a negative impact on neurocognitive functions.
The failure to detect any benefit from antiangiogenic therapy across these two trials stems from an incomplete understanding of the mechanisms of action for these agents, in particular, their impact on blood flow and oxygenation status of the tumor (vessel ‘pruning’ vs normalization). Batchelor et al. investigated this disparity in treatment efficacy and found that clinical outcomes of newly diagnosed GBM patients to chemoradiation with and without cediranib were dependent on improved perfusion, which only occurred in a subset of patients in cediranib-containing regimens [26]. Moreover, an increase in perfusion was associated with improved tumor oxygenation status as well as with pharmacodynamic biomarkers, such as changes in plasma placenta growth factor and soluble VEGFR2. Finally, treatment resistance was associated with elevated plasma IL-8 and soluble VEGFR1 post-therapy. Therefore, these tumor perfusion changes following antiangiogenic therapy may distinguish responders versus non-responders early in the course of therapy, which can ultimately provide new insight into the selection of GBM patients most likely to benefit from anti-VEGF treatments.
Secondly, the interactions between antiangiogenic therapy and chemotherapy in many of these prospective trials do not take into consideration the underlying contributions of epigenetic profiles in GBM tumors. For instance, the GLARIUS trial [42], focused on evaluating bevacizumab and irinotecan combinatorial therapy in newly diagnosed GBMs with a non-methylated MGMT gene. A total of 182 GBM patients with non-methylated MGMT received standard RT and were then randomly assigned to receive bevacizumab and irinotecan (116 patients) or TMZ alone (54 patients). With 6-month PFS as the primary end point of the study, patients receiving the combination therapy had a significantly prolonged PFS compared to those receiving TMZ alone (9.74 vs 5.99 months for TMZ; HR = 0.30, 95% CI [0.19, 0.48]; p < 0.0001). Even the secondary end point of OS showed that the combination therapy in non-methylated patients was significantly longer compared to the TMZ arm (16.6 vs 14.8 months for TMZ; HR 0.60, 95% CI [0.37, 0.96]; p = 0.031).
Several lines of evidence suggest that integrin antagonists, including cilengitide, may have enhanced antitumor benefit when administered in combinatorial therapeutic regimens [43]. Integrins are critically involved in many tumor-promoting activities, such as proliferation, survival, invasion and angiogenesis. Therefore, effective integrin inhibition may enhance other therapeutics targeting regulators of these processes [44]. In addition, recent evidence suggests that integrin inhibitors may potentiate the activity of cytotoxic agents [45,46].
For these reasons, a trial combining cilengitide with RT and TMZ for newly diagnosed GBM patients was recently performed [47]. Fifty-two patients received cilengitide 500 mg twice weekly during RT with daily TMZ and then during six post-RT monthly TMZ cycles. With a median follow-up time of 14 months, the 6-month PFS and 1-year OS rates were 69 and 67%, respectively. In comparison, patients treated with the same regimen without cilengitide have 6-month PFS and 1-year OS rates of 54 and 62%, respectively [3]. Furthermore, patients treated on the cilengitide study whose tumors lacked MGMT expression had a particularly favorable outcome.
These observations were evaluated in a recent multicenter, randomized controlled Phase III study (CENTRIC) comparing RT plus TMZ versus the same regimen plus cilengitide in newly diagnosed GBM patients with methylated MGMT promoters [48]. Median OS was 26.3 months in both arms (HR = 1.02; 95% CI [0.81 – 1.29]; p = 0.86), and median PFS was 13.5 months in the cilengitide arm and 10.7 months in the control arm (HR = 0.93; 95% CI [0.76 – 1.14]; p = 0.48). A similar study (CORE study, NABTT 0306) evaluated clinical outcomes in 112 newly diagnosed patients [49]. The median OS was 19.7 months for all patients, 17.4 months for the patients in the 500-mg dose group, 20.8 months for patients in the 2000-mg dose group, 30 months for patients who had methylated MGMT status and 17.4 months for patients who had non-methylated MGMT status. For patients aged ≤ 70 years, the median survival and survival at 24 months were superior to what was observed in the EORTC trial [3] (20.7 vs 14.6 months and 41 vs 27%, respectively; p = 0.008).
Although the safety and efficacy of combining antiangiogenic agents with chemotherapy has been documented in the recurrent setting, the ideal chemotherapy partner has yet to be identified by prospective, randomized trials. Moreover, the scheduling, timing and dosing of antiangiogenic agents relative to chemotherapy also remains to be defined and should be a focus of future studies. As the field progresses towards patient-specific approaches, gene expression studies and other correlative analyses are needed to assess the safety and efficacy of antiangiogenic therapies on the basis of the molecular pathophysiology of the disease. These antiangiogenic agents are expected to play a significant role in the treatment of GBM in the future, and it is hoped that the consideration of molecular profiling will further improve target selection.
2.4 Cytotoxic therapy with TMZ for MG
TMZ given concurrently with RT or as an adjuvant after RT has become the standard of care for patients with GBM. This drug was granted accelerated approval in 1999 based on durable objective responses in patients with AA refractory to a nitrosourea and procarbazine, two chemotherapeutic alkylating agents [50]. Full approval for TMZ was granted in 2005 after confirmation of significantly improved OS was observed in a randomized trial of patients with newly diagnosed GBM [3].
TMZ has also demonstrated activity in recurrent gliomas. In a Phase II trial in patients with recurrent GBM, the objective response rate was only 8%. However, an additional 45% of patients displayed disease stabilization, suggesting that 53% of patients experienced a minor clinical benefit with TMZ treatment [1]. The 6-month PFS for TMZ-treated patients was 18%, and the 6-month OS was 46% [51]. In a large randomized Phase II trial in patients with recurrent GBM, the efficacy of TMZ was compared with that of procarbazine [52]. In this study, the 6-month PFS was 21% for patients treated with TMZ compared with only 8% for patients treated with procarbazine (p < 0.008). In all these trials, TMZ was administered at a dose of 150 – 200 mg/m2/d for 5 days, with cycles beginning every 28 days. Treatment with TMZ is usually well tolerated, with grade 3 and 4 thrombocytopenia and neutropenia occurring in fewer than 10% of patients [1].
2.5 Dose-intensified TMZ administration
TMZ has been shown to produce a survival benefit in patients with GBM and has become a routine part of standard-of-care therapy. As such, more aggressive, dose-intensified (DI) regimens are now being evaluated [53,54]. Recent advances have been made in the treatment of GBM utilizing a common side effect of serial TMZ administration, which is profound lymphopenia, to enhance cancer vaccine efficacy against MGs. The profound lymphopenia induced by therapeutic TMZ would be expected to limit the induction of functional immune responses induced by cellular vaccines. In our previous clinical trial (ACT II), our group evaluated the effectiveness of a peptide vaccine directed against the unique EGFRvIII mutation in combination with standard-of-care TMZ [55]. The EGFRvIII vaccine was given in coordination with concurrent daily TMZ in monthly cycles after completion of radiation. Patients were enrolled sequentially into two groups based on the dose of TMZ, standard (STD) or DI. Patients in group A received TMZ at a dose of 200 mg/m2 for 5 days of a 28-day cycle (STD) and those in group B received TMZ at a dose of 100 mg/m2 for 21 days of a 28-day cycle (DI). Patients were vaccinated on day 21 of each cycle until progression. Patients enrolled in ACT II vaccinated in coordination with monthly cycles of TMZ had a median PFS of 15.2 versus 6.3 months for historical controls (p = 0.024) and a median OS of 23.6 versus 15 months for historical controls (p = 0.019) matched for entry criteria and known prognostic factors. The significant finding of our study is that both humoral and cellular vaccine-induced immune responses were unexpectedly enhanced by a DI TMZ that induced more profound and more persistent lymphopenia than the STD TMZ regimen [56]. Although counterintuitive, this is consistent with preclinical studies [57,58] and findings after adoptive T-cell transfer [59] that lymphopenic states can induce reactive homeostatic proliferation of the immune cell compartment and enhance antitumor immune responses [60].
Although TMZ is the most proven effective chemotherapeutic agent for these tumors, the prognosis for patients treated with surgery, RT and TMZ still remains poor, with survival just over 14 months in high-performance status patients [3]. Two prominent reasons for TMZ failure is that a large percentage of tumors are resistant to the cytotoxic effects of the TMZ-induced DNA lesion O(6)-methylguanine due to elevated expression of the repair protein MGMT [61] or a defect in the mismatch repair pathway [62]. Overcoming resistance mediated by MGMT is being explored by incorporating adjunctive therapy with competitive inhibitors of MGMT, such as O6-benzyl-guanine [63].
3. Bypassing and direct manipulation of the blood–brain barrier
The most important factor affecting the delivery of any drug to the brain and to brain tumors is the transport of the agent across the blood–brain barrier (BBB) and the blood–tumor barrier (BTB). The effectiveness of delivering agents across the BBB and BTB is also influenced by the regional blood flow and drug pharmacokinetic profile. Strategies to increase drug delivery to brain tumors have included intra-arterial drug administration, disruption of the BBB by hyperosmolar solutions or biomolecules, direct intratumoral injection of free drug, or the use of drug embedded in a controlled-release, biodegradable matrix delivery system. Even if the BBB is overcome, drug access to tumor cells may be hindered by increased intercapillary distances, greater interstitial pressure, lower microvascular pressure and the uptake of drug by surrounding normal brain tissue (referred to as the ‘sink effect’) [64].
Direct delivery of a drug into the tumor or a postoperative tumor resection cavity is a promising alternative to maximize local drug concentrations, while minimizing systemic effects. Two techniques have been used to directly administer drug to the tumor: slow-release systems and direct infusion. Slow-release carrier systems are controlled-release methods employing various carrier systems permit constant drug delivery into the tumor, while protecting the unreleased drug from metabolism. The most extensively evaluated slow-release system is the carmustine polymer wafer, which has been approved for use in patients with MGs [15].
Convection enhanced delivery (CED) is a form of direct intratumoral infusion with various chemotherapeutic drugs. This method has been demonstrated in several clinical trials [65,66]. Current direct drug administration has utilized CED through surgically implanted catheters. CED optimizes the delivery of antitumor agents to the tumor using a positive-pressure infusion to control drug distribution by adjusting the infusion rate and volume [67]. The potential utility of CED with small molecule chemotherapy agents was illustrated by a report of 15 patients with recurrent MG who were treated with CED of paclitaxel [68]. There were five complete and six partial responses. Complications included chemical meningitis, infections and transient neurologic deterioration, the latter which was initially thought to be due to peritumoral edema. CED may be particularly useful for the delivery of large molecules [69]. As an example, CED has been used to deliver cintredekin besudotox, a conjugate of human IL-13 with pseudomonas exotoxin [70]. MGs express the IL-13 receptor on the cell surface, and this conjugate is used to deliver locally high concentrations of the pseudomonas exotoxin. A Phase III trial to assess the efficacy of this approach in patients with GBM upon first relapse showed no improvement in survival as compared to treatment with carmustine wafers [71].
4. Expert opinion: more precise therapies via targeting of tumor-specific antigens
Therapeutic efficacy in treating MGs with current standard-of-care therapies ultimately has proven to be short-lived. Treatment failure can be attributed to a variety of factors, including the high-grade invasiveness of MGs at the time of diagnosis, increased tumor resistance to RT, the impracticality of optimal surgical resection and the comparative intolerance of the normal brain for cytotoxic therapies. Therefore, identification of unique tumor mutations is critical for the advancement of targeted therapy. Tumor-specific antigens (TSAs) are unique mutations that stem from random somatic point mutations induced by physical or chemical carcinogens. Targeting of TSAs in MGs ensures safety to the patient due to the inherent high specificity of these antigens that are expressed solely in the tumor tissue. Consequently, TSAs elicit a response clinically more effective than that of any amplified or overexpressed self-antigens, while reducing the risk of any autoimmune reactions. A summary of current TSAs identified in MGs is delineated in Table 2.
Table 2. Conserved and tumor-specific antigens identified in malignant gliomas.
| Protein | Mutation | Function |
|---|---|---|
| EGFR [72] | EGFRvIII | Constitutively activated form of EGFR, promotes cell proliferation, inhibits apoptosis, resistance to radiation and chemotherapy |
| IDH1 [79] | R132H | Central metabolism, oxidative decarboxylation of isocitrate to 2-oxoglutarate |
| TERT [87] | C250T and C228T | Somatic mutations that maintain telomere length by prolonged activation of telomerase |
| CMV pp65 [74-76] | Nuclear localization in infected cells; modulating/evading the host cell immune response during HCMV infections | |
| CMV IE1 [74-76] | Regulates transcription of viral and host genes, driving viral replication |
CMV: Cytomegalovirus; EGFR: Epidermal growth factor receptor;
IDH 1: Isocitrate dehydrogenase enzymes 1; IE: Immediate early 1;
TERT: Telomerase reverse transcriptase.
The EGFRvIII mutation is currently one of the most prominent examples of directly targeting a TSA in malignant brain tumors. The EGFR gene is amplified in up to 50% and overexpressed in over 90% of GBM specimens. Originally, it was believed that the impact of EGFR on neoplastic processes was merely a result of the corresponding gene amplification, or acquired increase in copy number. It has now been well-characterized that the malignant profile of many tumors, including GBM, is a result of aberrant, overexpressed forms of the EGFR gene. The mutated form of the receptor harbors a constant deletion within the extracellular domain and serves as a tumor-specific neoantigen that is not expressed in normal tissue [72]. Results from the ACT II study [55] can offer important insights into the in vivo dynamics of targeting the EGFRvIII mutation. Vaccination with the EGFRvIII peptide spanning the mutated EGFR region resulted in prolonged PFS and OS in newly diagnosed patients compared to a historically matched cohort for eligibility criteria and prognostic factors (median OS and PFS of 23.6 and 15.2 months, respectively, compared to matched cohort median OS and PFS of 15 and 6.3 months, respectively). The EGFRvIII vaccine is now being tested in an international Phase III trial.
Since the advent of exploiting the EGFR mutation, there has been extensive investigation into the targeting of human Cytomegalovirus (CMV) in patients with GBM. CMV has been shown to be reactivated in GBM tumors but not surrounding normal brain [73,74], owing to the specificity of this potential target. Over 90% of GBMs express CMV proteins, including pp65 and immediate early 1, which have been shown to promote glioma progression [75,76]. Over the past few years, there has been a vast amount of experience with both the safety and efficacy in targeting CMV [77,78] proteins. Thus, the unique and specific expression of CMV proteins in GBM tumors provides a second viable TSA for targeted therapy.
4.1 Novel TSAs in MGs: IDH1 and telomerase reverse transcriptase
Using genome-wide sequencing, tumor-specific mutations in MGs have been discovered exclusively the active sites of IDH1 and IDH2 [79,80], an evolutionarily conserved enzyme essential to cell function [81]. Investigators found that 50 – 80% of low-grade gliomas carried mutations of IDH1 or IDH2. Later studies showed that 5% of primary GBMs and 60 – 90% of secondary GBMs express mutant IDH proteins. As such, mutant IDH enzymes acquire a gain-of-function mutation and reduce α-KG to an oncometabolite, 2-hydroxyglutarate (2-HG) [82]. Overexpression of these mutated IDH enzymes also induces histone and DNA hyper-methylation and blocks cellular differentiation. Greater than 90% of all IDH1 mutations occur from a substitution of histidine for arginine resulting in the highly conserved and tumor-specific mutation, IDH1R132H [83]. Immunohistochemical analysis has revealed that IDH1R132H is homogeneously expressed in all tumor cells, including single infiltrating tumor cells [84] but is absent in normal cells [80,84]. The high frequency, specificity and homogeneous expression of the IDH1 mutation thus make it an ideal target for therapeutic intervention.
These unique IDH mutations offer the potential for targeted therapy with molecular inhibitors, lowering levels of the 2-HG metabolite and thus halting tumor growth. This approach would benefit the treatment of low-grade gliomas for which there is a current lack of precise and highly effective therapy. In two recent studies, IDH1 and IDH2 inhibitors were selective for targeting the cancer-mutated IDH enzymes. Wang et al. inhibited the mutated IDH2 enzyme in leukemia cells, slowing cell proliferation and inducing differentiation [85]. Rohle et al. used the IDH1 inhibitor to slow proliferation of GBM cells, induce demethylation of histones and enhance astroglial differentiation [86]. These preclinical results have exciting implications for clinical translation. For example, a mutated IDH inhibitor with low toxicity might halt the growth of MGs dependent on the increasing activity of 2-HG or even delay progression of low-grade MGs to high-grade tumors.
Recently, screening of > 1000 tumor samples spanning 60 histologic types revealed that point mutations in the promoter of the telomerase reverse transcriptase (TERT) gene, which in turn increase telomerase expression, could subdivide gliomas into tumors with high and low frequencies of this mutation. Several MG subtypes were found to be high expressers of TERT promoter mutations, 83% of which were primary GBM tumors [87]. The recent identification of TERT mutations holds much promise in leveraging the expression and presentation of this antigen as a targeted therapy. Furthermore, TERT mutations provide a biomarker that may be indicative of clinical prognoses for MGs.
Microarray expression profiling of MGs has identified molecular subtypes as well as genes associated with tumor grade, progression and patient survival [88-90]. While MGs such as GBM and AA continue to be defined by histological criteria, several reports demonstrate that expression profiles can better predict outcome [91,92]. Given the possibility that molecularly distinct tumors may exhibit different clinical responses, a greater understanding of molecularly defined subsets rather than histologic subtypes of tumors may be a better tool for the development of more effective therapies.
Expert Opinion.
Malignant glioma: prognosis and assessment of therapeutic response
Current FDA-approved therapies for primary and recurrent GBM
Bypassing and direct manipulation of the blood–brain barrier
Expert opinion: more precise therapies via targeting of tumor-specific antigens
Article highlights.
Assessment of treatment responses for malignant gliomas (MGs) has undergone a recent shift from two-dimensional radiographic measurements to novel algorithmic-based modalities to assess intricate alterations in postoperative resection cavities.
Current standard-of-care therapies such as temozolomide and radiation therapy have been extended into treatment for recurrent disease with dose-intensified and re-irradiation treatment regimens.
A review of recent clinical trials evaluating antiangiogenic and molecular inhibitor therapy for MGs in the primary and recurrent setting is included.
Efforts to overcome the inherent difficulty in drug delivery to intracranial tumors have focused on slow-release carrier systems and direct infusion with convection-enhanced delivery.
Screening and identification of tumor-specific mutations in MGs has yielded two promising targets for sensitive targeting of glioblastoma tumors.
The recent discovery of novel tumor-specific mutations in MGs offers promising avenues for the development of safer, more precise therapies.
This box summarizes key points contained in the article.
Footnotes
Declaration of interest: JH Sampson has patents related to technology discussed in this manuscript and receives consulting fees and licensing fees from Celldex Therapeutics, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1•.Stupp R, Dietrich PY, Ostermann Kraljevic S, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20:1375–82. doi: 10.1200/JCO.2002.20.5.1375. Large Phase II trial estimating safety and efficacy of concomitant radiation with novel temozolomide (TMZ) therapy followed by adjuvant TMZ therapy for patients with newly diagnosed glioblastoma (GBM) [DOI] [PubMed] [Google Scholar]
- 2.Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long-term survivors with malignant astrocytomas. Ann Neurol. 1990;28:818–22. doi: 10.1002/ana.410280614. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–80. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 5.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–72. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 6.Mehta AI, Kanaly CW, Friedman AH, et al. Monitoring radiographic brain tumor progression. Toxins (Basel) 2011;3:191–200. doi: 10.3390/toxins3030191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Kanaly CW, Ding D, Mehta AI, et al. A novel method for volumetric MRI response assessment of enhancing brain tumors. PLoS One. 2011;6:1–8. doi: 10.1371/journal.pone.0016031. A novel method for volumetric MRI response assessment of enhancing brain tumors that addresses limitations of traditional response assessment criteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Bent MJ, Afra D, de Witte O, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet. 2005;366:985–90. doi: 10.1016/S0140-6736(05)67070-5. [DOI] [PubMed] [Google Scholar]
- 9.Provenzale JM, Mancini MC. Assessment of intra-observer variability in measurement of high-grade brain tumors. J Neurooncol. 2012;108:477–83. doi: 10.1007/s11060-012-0843-2. [DOI] [PubMed] [Google Scholar]
- 10.Stummer W, Pichlmeier U, Meinel T, et al. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7:392–401. doi: 10.1016/S1470-2045(06)70665-9. [DOI] [PubMed] [Google Scholar]
- 11.Parney IF, Chang SM. Current chemotherapy for glioblastoma. Cancer J. 2003;9:149–56. doi: 10.1097/00130404-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Walker MD, Alexander E, Jr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49:333–43. doi: 10.3171/jns.1978.49.3.0333. [DOI] [PubMed] [Google Scholar]
- 13••.Walker MD, Green SB, Byar DP, et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med. 1980;303:1323–9. doi: 10.1056/NEJM198012043032303. Large-scale randomized clinical trial comparing radiotherapy and nitrosoureas for the treatment of malignant glioma (MG) following surgical resection. [DOI] [PubMed] [Google Scholar]
- 14••.Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. Lancet. 1995;345:1008–12. doi: 10.1016/s0140-6736(95)90755-6. High-impact placebo-controlled study evaluating safety and therapeutic response with intraoperative slow-release biodegradable polymers of chemotherapy for the treatment of recurrent MGs. [DOI] [PubMed] [Google Scholar]
- 15.Valtonen S, Timonen U, Toivanen P, et al. Interstitial chemotherapy with carmustine-loaded polymers for high-grade gliomas: a randomized double-blind study. Neurosurg. 1997;41:44–9. doi: 10.1097/00006123-199707000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 18.Chamberlain MC. Bevacizumab for the treatment of recurrent glioblastoma. Clin Med Insights Oncol. 2011;5:117–29. doi: 10.4137/CMO.S7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Combs SE, Debus J, Schulz-Ertner D. Radiotherapeutic alternatives for previously irradiated recurrent gliomas. BMC Cancer. 2007;7:167. doi: 10.1186/1471-2407-7-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong DS, Lee JI, Park K, et al. Efficacy of stereotactic radiosurgery as a salvage treatment for recurrent malignant gliomas. Cancer. 2008;112:2046–51. doi: 10.1002/cncr.23402. [DOI] [PubMed] [Google Scholar]
- 21.Cho KH, Hall WA, Gerbi BJ, et al. Single dose versus fractionated stereotactic radiotherapy for recurrent high-grade gliomas. Int J Radiat Oncol Biol Phys. 1999;45:1133–41. doi: 10.1016/s0360-3016(99)00336-3. [DOI] [PubMed] [Google Scholar]
- 22.Patel M, Siddiqui F, Jin JY, et al. Salvage reirradiation for recurrent glioblastoma with radiosurgery: radiographic response and improved survival. J Neurooncol. 2009;92:185–91. doi: 10.1007/s11060-008-9752-9. [DOI] [PubMed] [Google Scholar]
- 23•.Fokas E, Wacker U, Gross MW, et al. Hypofractionated stereotactic reirradiation of recurrent glioblastomas : a beneficial treatment option after high-dose radiotherapy? Strahlenther Onkol. 2009;185:235–40. doi: 10.1007/s00066-009-1753-x. One of the largest clinical trials evaluating responses in patients with recurrent GBM using re-irradiation with hypofractionated stereotactic radiotherapy and implicates this therapy as a viable option for recurrent disease. [DOI] [PubMed] [Google Scholar]
- 24.Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg. 1972;175:409–16. doi: 10.1097/00000658-197203000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van der Veldt AA, Lubberink M, Bahce I, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21:82–91. doi: 10.1016/j.ccr.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 26.Batchelor TT, Gerstner ER, Emblem KE, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci USA. 2013;110:19059–64. doi: 10.1073/pnas.1318022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14:1131–8. doi: 10.1634/theoncologist.2009-0121. [DOI] [PubMed] [Google Scholar]
- 28.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Metronomic chemotherapy with daily, oral etoposide plus bevacizumab for recurrent malignant glioma: a phase II study. Br J Cancer. 2009;101:1986–94. doi: 10.1038/sj.bjc.6605412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740–5. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vredenburgh JJ, Desjardins A, Herndon JE, II, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 31.Vredenburgh JJ, Desjardins A, Herndon JE, II, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–9. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 32.Reardon DA, Herndon JE, II, Peters KB, et al. Bevacizumab continuation beyond initial bevacizumab progression among recurrent glioblastoma patients. Br J Cancer. 2012;107:1481–7. doi: 10.1038/bjc.2012.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taal W, Oosterkamp HM, Walenkamp AME, et al. A randomized phase II study of bevacizumab versus bevacizumab plus lomustine versus lomustine single agent in recurrent glioblastoma: the Dutch BELOB study. J Clin Oncol. 2013;31(Suppl) abstract 2001. [Google Scholar]
- 34.Maree Field K, Simes J, Wheeler H, et al. A randomized phase II study of carboplatin and bevacizumab in recurrent glioblastoma multiforme (CABARET) J Clin Oncol. 2013;31(Suppl) abstract 2017. [Google Scholar]
- 35.Wedge SR, Kendrew J, Hennequin LF, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 36.Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–23. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–18. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 39.Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–34. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gilbert MR, Dignam J, Won M, et al. RTOG 0825: Phase III double-blind placebo-controlled trial evaluating bevacizumab (Bev) in patients (Pts) with newly diagnosed glioblastoma (GBM) J Clin Oncol. 2013;31(Suppl) abstract 1. [Google Scholar]
- 41.Henriksson R, Bottomley A, Mason W, et al. Progression-free survival (PFS) and health-related quality of life (HRQoL) in AVAglio, a phase III study of bevacizumab (Bv), temozolomide (T), and radiotherapy (RT) in newly diagnosed glioblastoma (GBM) J Clin Oncol. 2013;31(Suppl) abstract 2005. [Google Scholar]
- 42.Herrlinger H, Schaefer N, Steinbach JP, et al. Bevacizumab, irinotecan, and radiotherapy versus standard temozolomide and radiotherapy in newly diagnosed, MGMT-nonmethylated glioblastoma patients: First results from the randomized multicenter GLARIUS trial. J Clin Oncol. 2013;31(Suppl) abstract LBA2000. [Google Scholar]
- 43.Beal K, Abrey LE, Gutin PH. Antiangiogenic agents in the treatment of recurrent or newly diagnosed glioblastoma: analysis of single-agent and combined modality approaches. Radiat Oncol. 2011;6:1–15. doi: 10.1186/1748-717X-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eskens FA, Dumez H, Hoekstra R, et al. Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer. 2003;39:917–26. doi: 10.1016/s0959-8049(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 45.Abdollahi A, Griggs DW, Zieher H, et al. Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin Cancer Res. 2005;11:6270–9. doi: 10.1158/1078-0432.CCR-04-1223. [DOI] [PubMed] [Google Scholar]
- 46.Albert JM, Cao C, Geng L, et al. Integrin alpha v beta 3 antagonist Cilengitide enhances efficacy of radiotherapy in endothelial cell and non-small-cell lung cancer models. Int J Radiat Oncol Biol Phys. 2006;65:1536–43. doi: 10.1016/j.ijrobp.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 47.Stupp R, Hegi ME, Neyns B, et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:2712–18. doi: 10.1200/JCO.2009.26.6650. [DOI] [PubMed] [Google Scholar]
- 48.Stupp R, Hegi ME, Gorlia T, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma and methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter: Key results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study. J Clin Oncol. 2013;31(Suppl) abstract LBA2009. [Google Scholar]
- 49.Nabors LB, Mikkelsen T, Hegi ME, et al. A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306) Cancer. 2012;118:5601–7. doi: 10.1002/cncr.27585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newlands ES, Stevens MF, Wedge SR, et al. Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat Rev. 1997;23:35–61. doi: 10.1016/s0305-7372(97)90019-0. [DOI] [PubMed] [Google Scholar]
- 51.Brada M, Hoang-Xuan K, Rampling R, et al. Multicenter phase II trial of temozolomide in patients with glioblastoma multiforme at first relapse. Ann Oncol. 2001;12:259–66. doi: 10.1023/a:1008382516636. [DOI] [PubMed] [Google Scholar]
- 52.Yung WK, Albright RE, Olson J, et al. A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83:588–93. doi: 10.1054/bjoc.2000.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brock CS, Newlands ES, Wedge SR, et al. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res. 1998;58:4363–7. [PubMed] [Google Scholar]
- 54.Tolcher AW, Gerson SL, Denis L, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer. 2003;88:1004–11. doi: 10.1038/sj.bjc.6600827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55•.Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13:324–33. doi: 10.1093/neuonc/noq157. Influential clinical study demonstrating that chemotherapy-induced lymphopenia is actually beneficial for vaccine responses by fostering enhanced homeostatic proliferation of immune cells in patients with GBM following treatment with dose-intensified TMZ therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56•.Heimberger AB, Sampson JH. The PEPvIII-KLH (CDX-110) vaccine in glioblastoma multiforme patients. Expert Opin Biol Ther. 2009;9:1087–98. doi: 10.1517/14712590903124346. High impact study elucidating the importance of effectively targeting tumor-specific mutations in GBM, specifically that of the EGFRvIII mutation with an informative review on developing vaccines specific for this mutation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asavaroengchai W, Kotera Y, Mule JJ. Tumor lysate-pulsed dendritic cells can elicit an effective antitumor immune response during early lymphoid recovery. Proc Natl Acad Sci USA. 2002;99:931–6. doi: 10.1073/pnas.022634999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dummer W, Niethammer AG, Baccala R, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–92. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dudley ME, Wunderlich JR, Yang JC, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–51. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 62.Friedman HS, McLendon RE, Kerby T, et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin Oncol. 1998;16:3851–7. doi: 10.1200/JCO.1998.16.12.3851. [DOI] [PubMed] [Google Scholar]
- 63.Quinn JA, Desjardins A, Weingart J, et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005;23:7178–87. doi: 10.1200/JCO.2005.06.502. [DOI] [PubMed] [Google Scholar]
- 64.Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990;9:253–66. doi: 10.1007/BF00046364. [DOI] [PubMed] [Google Scholar]
- 65.Voulgaris S, Partheni M, Karamouzis M, et al. Intratumoral doxorubicin in patients with malignant brain gliomas. Am J Clin Oncol. 2002;25:60–4. doi: 10.1097/00000421-200202000-00013. [DOI] [PubMed] [Google Scholar]
- 66.Patchell RA, Regine WF, Ashton P, et al. A phase I trial of continuously infused intratumoral bleomycin for the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2002;60:37–42. doi: 10.1023/a:1020291229317. [DOI] [PubMed] [Google Scholar]
- 67.Lopez KA, Waziri AE, Canoll PD, Bruce JN. Convection-enhanced delivery in the treatment of malignant glioma. Neurol Res. 2006;28:542–8. doi: 10.1179/016164106X116836. [DOI] [PubMed] [Google Scholar]
- 68.Lidar Z, Mardor Y, Jonas T, et al. Convection-enhanced delivery of paclitaxel for the treatment of recurrent malignant glioma: a phase I/II clinical study. J Neurosurg. 2004;100:472–9. doi: 10.3171/jns.2004.100.3.0472. [DOI] [PubMed] [Google Scholar]
- 69.Sampson JH, Akabani G, Friedman AH, et al. Comparison of intratumoral bolus injection and convection-enhanced delivery of radiolabeled antitenascin monoclonal antibodies. Neurosurg Focus. 2006;20:E14. doi: 10.3171/foc.2006.20.4.9. [DOI] [PubMed] [Google Scholar]
- 70.Kunwar S, Prados MD, Chang SM, et al. Direct intracerebral delivery of cintredekin besudotox (IL13-PE38QQR)in recurrent malignant glioma: a report by the Cintredekin Besudotox Intraparenchymal Study Group. J Clin Oncol. 2007;25:837–44. doi: 10.1200/JCO.2006.08.1117. [DOI] [PubMed] [Google Scholar]
- 71.Kunwar S, Chang S, Westphal M, et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010;12:871–81. doi: 10.1093/neuonc/nop054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72••.Ekstrand AJ, James CD, Cavenee WK, et al. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991;51:2164–72. In depth analysis of genes amplified and altered for the EGFR, transforming growth factor-α, and epidermal growth factor and their expression in human gliomas in vivo. [PubMed] [Google Scholar]
- 73.Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Engl J Med. 2008;359:539–41. doi: 10.1056/NEJMc0804818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitchell DA, Xie W, Schmittling R, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008;10:10–18. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cobbs CS, Harkins L, Samanta M, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62:3347–50. [PubMed] [Google Scholar]
- 76.Lucas KG, Bao L, Bruggeman R, et al. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neurooncol. 2011;103:231–8. doi: 10.1007/s11060-010-0383-6. [DOI] [PubMed] [Google Scholar]
- 77.Walter EA, Greenberg PD, Gilbert MJ, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–44. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 78.Numazaki K, Ikehata M, Yanai S, et al. Adoptive immunotherapy for interstitial pneumonia associated with cytomegalovirus infection. Clin Infect Dis. 1997;25:1246–7. doi: 10.1086/516959. [DOI] [PubMed] [Google Scholar]
- 79••.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. Paramount study using integrated genomic analysis of GBM tumors that identified recurrent mutations in the active site of the isocitrate dehydrogenase enzymes 1 (IDH1) enzyme that were associated with secondary GBM tumors as well as a more favorable prognosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kato Y, Jin G, Kuan CT, et al. A monoclonal antibody IMab-1 specifically recognizes IDH1R132H, the most common glioma-derived mutation. Biochem Biophys Res Commun. 2009;390:547–51. doi: 10.1016/j.bbrc.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Merritt TJ, Kuczynski C, Sezgin E, et al. Quantifying interactions within the NADP(H) enzyme network in Drosophila melanogaster. Genetics. 2009;182:565–74. doi: 10.1534/genetics.109.100677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82••.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:739–44. doi: 10.1038/nature08617. Critical study demonstrating that tumor-associated IDH1 somatic mutations result in a gain of function that causes the accumulation of the metabolite 2-Hydroxyglutarate, which holds implications in driving oncogenesis for MGs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174:1149–53. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Capper D, Weissert S, Balss J, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20:245–54. doi: 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85•.Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–6. doi: 10.1126/science.1234769. Proof-of-concept study employing a molecular inhibitor of the IDH2 enzyme that holds the capacity to induce differentiation of malignant cells. [DOI] [PubMed] [Google Scholar]
- 86•.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–30. doi: 10.1126/science.1236062. Significant study using high-throughput screening to develop a novel molecular inhibitor of the mutant R132H-IDH1 enzyme and promote the differentiation of glioma cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87••.Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6. doi: 10.1073/pnas.1303607110. Novel identification of telomerase reverse transcriptase promoter mutations in MGs and a subset of tumors derived from cells with low rates of self-renewal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88•.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. Paradigm-shifting analysis to identify molecular subclasses of high-grade glioma that more reliably predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. [DOI] [PubMed] [Google Scholar]
- 89.Godard S, Getz G, Delorenzi M, et al. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res. 2003;63:6613–25. [PubMed] [Google Scholar]
- 90.Rickman DS, Bobek MP, Misek DE, et al. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001;61:6885–91. [PubMed] [Google Scholar]
- 91.Freije WA, Castro-Vargas FE, Fang Z, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64:6503–10. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 92.Nutt CL, Mani DR, Betensky RA, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003;63:1602–7. [PubMed] [Google Scholar]
- 93.Reardon DA, Fink KL, Mikkelsen T, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610–17. doi: 10.1200/JCO.2008.16.7510. [DOI] [PubMed] [Google Scholar]