

Abstract
Background
Dickeya zeae is a bacterial species that infects monocotyledons and dicotyledons. Two antibiotic-like phytotoxins named zeamine and zeamine II were reported to play an important role in rice seed germination, and two genes associated with zeamines production, i.e., zmsA and zmsK, have been thoroughly characterized. However, other virulence factors and its molecular mechanisms of host specificity and pathogenesis are hardly known.
Results
The complete genome of D. zeae strain EC1 isolated from diseased rice plants was sequenced, annotated, and compared with the genomes of other Dickeya spp.. The pathogen contains a chromosome of 4,532,364 bp with 4,154 predicted protein-coding genes. Comparative genomics analysis indicates that D. zeae EC1 is most co-linear with D. chrysanthemi Ech1591, most conserved with D. zeae Ech586 and least similar to D. paradisiaca Ech703. Substantial genomic rearrangement was revealed by comparing EC1 with Ech586 and Ech703. Most virulence genes were well-conserved in Dickeya strains except Ech703. Significantly, the zms gene cluster involved in biosynthesis of zeamines, which were shown previously as key virulence determinants, is present in D. zeae strains isolated from rice, and some D. solani strains, but absent in other Dickeya species and the D. zeae strains isolated from other plants or sources. In addition, a DNA fragment containing 9 genes associated with fatty acid biosynthesis was found inserted in the fli gene cluster encoding flagellar biosynthesis of strain EC1 and other two rice isolates but not in other strains. This gene cluster shares a high protein similarity to the fatty acid genes from Pantoea ananatis.
Conlusion
Our findings delineate the genetic background of D. zeae EC1, which infects both dicotyledons and monocotyledons, and suggest that D. zeae strains isolated from rice could be grouped into a distinct pathovar, i.e., D. zeae subsp. oryzae. In addition, the results of this study also unveiled that the zms gene cluster presented in the genomes of D. zeae rice isolates and D. solani strains, and the fatty acid genes inserted in the fli gene cluster of strain EC1 were likely derived from horizontal gene transfer during later stage of bacterial evolution.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1545-x) contains supplementary material, which is available to authorized users.
Background
Dickeya zeae is the major pathogen responsible for the maize stalk rot and rice foot rot diseases, and has the ability to infect both monocotyledons and dicotyledons. Other members of Dickeya are D. chrysanthemi, D. dianthicola, D. dieffenbachiae, D. paradisiaca, D. dadantii and D. solani [1,2]. A recent recA-based molecular evolution and genetic diversity study showed that D. zeae is least related to other Dickeya species and even within D. zeae, there are many sequence variants (sequevars) among the isolates from different host plants [3], suggesting a long history of D. zeae adaptation and evolution in the processes of pathogen-host interactions.
The symptoms caused by Dickeya infection include soft rot as well as wilts resulting from vascular invasions. Extensive studies on the Dickeya pathogens which infect dicotyledon crops and ornamental plants led to identify a range of virulence factors including extracellular enzymes, phospholipase, iron metabolism, siderophores, indigoidine pigment, and type III secretion system, collectively contributing to the bacterial infections [4]. In contrast, genetic analysis and biochemical characterization of the pathogenic mechanisms of D. zeae were initiated only in recent years [5-8], following the outbreak of the rice root rot disease in China [9]. These studies led to identification of a new family of phytotoxins, i.e., zeamine and zeamine II [5,6], which appear to act as the key virulence determinants of the pathogen as mutation of the zeamine synthase genes zmsA and zmsK resulted in partial or almost complete loss of virulence on rice seeds germination [6,7]. The regulatory mechanisms that govern virulence gene expression are largely unknown, except that an acyl-homoserine lactone (AHL) quorum sensing (QS) system was shown to be involved in the regulation of certain virulence traits in D. zeae, including bacterial motility and biofilm development [8].
Up till now, four complete Dickeya bacterial genome sequences have become publically available, including D. paradisiaca Ech703 isolated from Solanum tuberosum [10] (sequence release date: June 26, 2009), D. chrysanthemi Ech1591 causing maize and Zea mays soft rot [10] (sequence release date: July 7, 2009), D. zeae Ech586 isolated from Philodendron [10] (sequence release date: December 11, 2009), and D. dadantii 3937 isolated from Saintpaulia ionantha [11] (sequence release date: September 16, 2010). Additionally, a range of partial genome sequences of Dickeya species have also been presented, including four D. danthicola strains (GBBC 2039, NCPPB 3534, NCPPB 453 and IPO 980), six D. solani strains (GBBC 2040, MK10, MK16, IPO 2222, Ds0432-1, and 3337) [12-14], three D. chrysanthemi strains (NCPPB 402, NCPPB 516 and NCPPB 3533), three D. dadantii strains (NCPPB 898, NCPPB 2976 and NCPPB 3537), one D. paradisiaca strain NCPPB 2511, eight D. zeae strains (ZJU1202 [15] and DZ2Q [16] isolated from diseased rice, CSL RW192 and MK19 isolated from river water, NCPPB 2538 isolated from maize, NCPPB 3531 and NCPPB 3532 isolated from potato, and MS1 isolated from banana [17]) and other five unassigned Dickeya spp. (NCPPB 569, NCPPB 3274, CSL RW240, DW0440 and MK17) [18]. These genome sequences may provide a good opportunity to study bacterial evolution and identify the genes contributing to host-range determination.
In this study, a complete genome sequence of D. zeae strain EC1, isolated from rice plants, was obtained using the Illumina next-generation sequencing technology coupling with polymerase chain reaction (PCR) method for gap closure. The genome sequence was annotated and compared with the representative genome sequences of other Dickeya species with a special focus on the virulence determinants and potential regulatory mechanisms. Substantial genomic rearrangements were revealed by comparing the genome sequence of D. zeae EC1 with other Dickeya isolates. Furthermore, we found that the gene cluster encoding for phytotoxin zeamine biosynthesis is conserved only in the D. zeae strains isolated from rice plants and the D. solani strains isolated from potato.
Results and discussion
General genomic features of D. zeae EC1
Assembly of D. zeae EC1 genome sequencing data resulted in 3 scaffolds with 3430.072 kb, 1294.767 kb and 1.821 kb in length, respectively, with 148× coverage in average. For finishing, the gaps were closed by custom primer walks or PCR amplification, followed by sequencing. Hence, the EC1 genome consists of a circular chromosome with 4,532,364 bps in size with no apparent autonomous plasmids. The average GC content of the whole genome is 53.43%. Coding sequences account for about 85.77% of the genome with GC content of 54.57%. The genome contains 4,154 open reading frames (ORFs) predicted to encode polypeptides with 935 bps in average length. The GC content in the intergenic region is 46.55%. An origin of replication designated as dnaA boxes was identified between the deduced gene for the 50S ribosomal protein L34 and the gyrB locus encoding DnaA, DnaN and RecF. The first nucleotide of the dnaA start codon was assigned as the base pair 1 of the chromosome (Table 1, Figure 1).
Table 1.
Genomic features of D. zeae EC1 and other Dickeya spp
| Features | EC1 | Ech1591 | Ech586 | 3937 | Ech703 |
|---|---|---|---|---|---|
| Size (bp) | 4,532,364 | 4,813,854 | 4,818,394 | 4,922,802 | 4,679,450 |
| GC content (%) | 53.43 | 54.5 | 53.6 | 56.3 | 55 |
| CDS | |||||
| Predicted no. of CDS | 4,154 | 4,367 | 4,318 | 4,687 | 4,136 |
| Function assigned | 3,880 | 4,163 | 4,144 | 4,543 | 3,970 |
| Function unknown | 274 | 204 | 174 | 144 | 166 |
| Ribosomal RNA operons | 7 | 7 | 7 | 7 | 7 |
| Transfer RNA | 88 | 74 | 77 | 75 | 74 |
| sRNA | 10 | 3 | 3 | 14 | 4 |
| Transposases | 65 | 55 | 11 | 50 | 13 |
Figure 1.

Circular chromosome map of D. zeae strain EC1. The distribution of genes is shown on the two outer rings. The next circle (black) indicates the GC content and the central circle (green/purple) shows GC-skew value. The window size of the GC content and GC-skew is 100 nt.
Seven rRNA operons were found in the chromosome of EC1 with 3 on the positive strand and 4 on the negative strand. Both chains contain an unusual rRNA operon with a 16S-23S-23S-5S organization, and another unusual one with 16S-23S-5S-5S organization locates on the negative chain, differing from the common 16S-23S-5S organization pattern. Similar unusual rRNA operons were also found in some other bacterial species, e.g. a 16S-23S-5S-5S gene cluster was found in Erwinia pyrifoliae Ep1/96, E. tasmaniensis Et1/99 [19]. The occurrence of the unusual organization of rRNAs within the chromosome was interpreted as the result of a chromosomal recombination event [19]. Altogether, 88 tRNA genes which recognize 20 codons were found in the genome of D. zeae EC1.
Genome similarity of D. zeae EC1 and other Dickeya bacteria
The complete genomic sequences of several different Dickeya species including D. chrysanthemi Ech1591 (GenBank: CP001655.1; 4,813,854 bp), D. zeae Ech586 (GenBank: CP001836.1; 4,818,394 bp), D. dadantii 3937 (GenBank: CP002038.1; 4,922,802 bp) [11] and D. paradisiaca Ech703 (GenBank: CP001654.1; 4,679,450 bp) have been determined recently. In comparison, D. zeae EC1 has the smallest chromosome with 4,532,364 bp nucleotides with 4,154 ORFs, whereas D. dadantii 3937 possesses the largest genome with 4,922,802 bp nucleotides containing 4,687 ORFs (Table 1). The GC content of D. zeae EC1 genome is lowest (53.43%), similar to that of D. zeae Ech586 (53.6%) (Table 1). In addition, D. zeae EC1 contains substantially more tRNAs and transposases than any other bacterial species, suggesting a complex history of DNA transposition and recombination in the process of genome evolution [20]. Consistent with the above features, D. zeae EC1 has more ORFs encoding peptides with function unknown than any other Dickeya species with complete genome sequence (Table 1).
Phylogenetic relationships of D. zeae EC1 with other Dickeya species were assessed by performing Multilocus Sequence Analysis (MLSA) using four housekeeping genes (atpD, dnaX, gyrB and recA), and the concatenated data set of the four genes was constructed with Pectobacterium atrosepticum SCR1043 as an outgroup. The numbers of Dickeya species used in this phylogeny assessment were varied from 35 to 38 depending on the availability of the housekeeping gene sequences indicated above. The phylogenic analysis results showed that the three D. zeae strains isolated from diseased rice plants (EC1, ZJU1202 and DZ2Q) are located in the same branch, and share the same clade with the D. zeae isolates collected from other plants or sources (Additional file 1A, B, C, D). The same results also showed that among the four Dickeya strains with complete genome sequences released, EC1 was most homologous to D. zeae Ech586 and then D. chrysanthemi Ech1591 (Additional file 1A, B and C).
To further evaluate the evolutionary relationships among the five Dickeya species with available complete genome sequences, the sequence of D. zeae EC1 was aligned with those of other four species by using BLAST program, respectively. The results showed that D. zeae EC1 is best co-linearly matched with D. chrysanthemi Ech1591 with a few rearrangement events (reverse match; blue) (Figure 2A), similarly, a few inversed fragments were observed between EC1 and D. dadantii 3937 (Figure 2B), while the alignment between EC1 and D. zeae Ech586 showed a big inversion with the largest amounts of homologous DNA fragments (Figure 2C). Intriguingly, the genes adjacent to the borders of this inversion are W909_RS06895 and W909_13075, encoding an integrase IntB (WP_016943617.1) and a transposase (AJC68444.1), respectively, and the sequence between them is highly similar to that in Ech586 (Dd586_2634 ~ Dd586_1449) with reverse orientation, suggesting that the inversion might be due to homologous recombination between these two genes. The entire length of the D. paradisiaca Ech703 genome was least matched with D. zeae EC1 with numerous gene content dissimilarities (Figure 2D). The distant relationship of D. zeae EC1 with other four complete Dickeya species indicated by the genomic collinearity analysis (Figure 2A-D) is supported by the estimated numbers of shared genes through OrthoMCL comparative analyses (only 1835 proteins in total, Figure 3A). The number of the shared genes between D. zeae EC1 and D. chrysanthemi Ech1591, D. zeae Ech586 and D. dadantii 3937 was 2213, 3208 and 2192, respectively (Figure 3A). Agreeable with the genomic collinearity analysis shown in Figure 2, the most dissimilar species D. paradisiaca Ech703 shared only 961 genes with D. zeae EC1, and 935, 964 and 940 with D. chrysanthemi Ech1591, D. zeae Ech586, and D. dadantii 3937, respectively.
Figure 2.
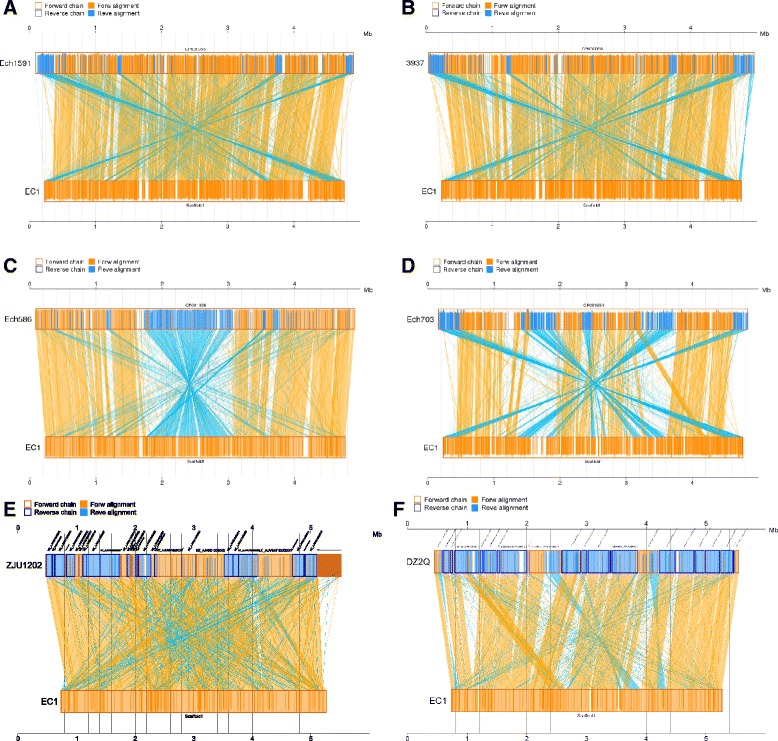
Nucleic acid colinearity of EC1 vs. Ech1591 (A), 3937 (B), Ech586 (C), Ech703 (D), ZJU1202 (E) and DZ2Q (F), respectively. The sequence of EC1 is ordered according to that of the reference bacterium based on Mummer 3.22 (http://mummer.sourceforge.net/). Then upper and following axes of linear synteny graph are constructed after the same proportion of size reduction in length of both sequences (parameter: b, 200; c, 65; extend: l, 20). According to BLAST results, each pair nucleic acid sequence of two alignments is marked in the coordinate diagram according to its position information after the same proportion of size reduction. Orange lines connecting homologous regions present in the same orientation while the blues present inverted orientation.
Figure 3.
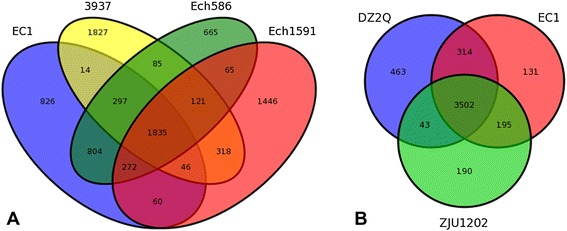
Venn diagrams for the deduced proteins of EC1, Ech1591, 3937, and Ech586 (A), and the rice strains D. zeae EC1, ZJU1202 and DZ2Q (B). Values were calculated by OrthoMCL clustering analyses using the following parameters: P-value Cut-off = 1 × 10−5; Identity Cut-off = 90%; Percent Match Cut-off = 80. The overlapping sections indicate shared numbers of deduced proteins.
To identify the genes specific to D. zeae EC1, we compared its genome sequence to the released complete genome sequences of D. chrysanthemi Ech1591, D. zeae Ech586, D. paradisiaca Ech703 and D. dadantii 3937, which unveiled 826 unique proteins located only in the EC1 genome (Figure 3A). For example, the chromosome of EC1 harbors several large specific regions, including the region from 1418747 nt to 1469974 nt, involved in the biosynthesis of zeamine phytotoxins, containing two characterized zeamine synthase genes zmsA and zmsK [6,7], the region from 1551137 nt to 1566214 nt, encoding different peptide products including three HTH-type transcriptional regulators (AJC65834.1, AJC65839.1 and AJC65846.1), three transporter proteins (AJC65835.1, AJC65838.1, and AJC65845.1), an acetyltransferase (AJC65836.1), a phosphoserine phosphatase (AJC65837.1), an acetaldehyde dehydrogenase (AJC65840.1), a transposase IS4 (AJC64674.1), a chemotaxis protein CheX (AJC65842.1), and five hypothetical proteins (AJC68379.1, AJC68380.1, AJC68381.1, AJC65843.1 and AJC68382.1) .
Taken together, the above analyses show that D. zeae EC1 is most co-linear with D. chrysanthemi Ech1591, most conserved with D. zeae Ech586 and least similar to D. paradisiaca Ech703. The findings highlight substantial divergence among Dickeya species. For paving ways to understand the genetic basis of pathogenicity and host specificity, we specifically compared the similarity and divergence of the genes encoding virulence determinants and regulatory mechanisms in the following sections.
In addition, we compared the genome sequences of three D. zeae strains isolated from rice plants. Synteny analysis showed that EC1 isolated from Guangdong Province of China was 99.962% identity to strain ZJU1202, which was also from Guangdong Province of China [15], and 95.863% identity to strain DZ2Q from Italy [16] (Figure 2E, 2 F), and the number of the shared genes among these three rice pathogen strains is up to 3502, only 131 genes are unique in strain EC1 (Figure 3B), among which, 95.4% (125 out of 131) encode hypothetical proteins, and the 6 genes with known functions share high similarities with other Dickeya strains (Additional file 2), 4 of which encode tranposases (457403 ~ 457603 nt, AJC65190.1, AJC65197.1, and AJC65071.1), 1 encode citrate lyase acyl carrier protein CitD (AJC65916.1), and the remaining encodes a PilT plasmid maintenance protein pirin (AJC64970.1), respectively. Given the high level of genome sequence similarity and the fact that the genomes of strains ZJU1202 and DZ2Q have only been partially sequenced with numerous gaps, which hinders the accurate comparison, we believe that the common genes shared by these three strains could be substantially higher than the currently obtained 3502.
Cell wall-degrading extracellular enzymes and proteases
Extracellular enzymes, including pectinases, polygalacturonases, cellulases and proteases, are one of the key virulence factors during bacterial pathogenesis when infecting host plants to cause soft rot symptoms. Among these enzymes, pectate lyases (Pel) and other pectinases such as pectin methylesterases (Pem) and pectin lyases (Pnl) of D. dadantii 3937 have been shown to play a major role in the virulence and tissue maceration [21]. The genome of EC1 contains a total of 14 genes encoding pectin degradation enzymes, including pnl, pelN, pelL, pelI, pelA, pelE, pelD, pelC, pelB, and pelZ encoding pectate lyases, pelW and pelX encoding pectate disaccharide-lyases, paeY encoding a pectin acetylesterase, and pemA encoding a pectin methylesterase (Table 2). These pectin degradation genes are highly conserved in various Dickeya species, except that pnl is absent in D. zeae Ech586 and D. chrysanthemi Ech1591, and pelI and pelD are not included in the D. paradisiaca Ech703 genome (Table 2). The pehK and pehX genes encoding polygalacturonases are also conserved in Dickeya spp. except that pehX has two copies in D. chrysanthemi Ech1591 (pehW and pehX), and three copies in D. dadantii 3937 (pehV, pehW and pehX) (Table 2).
Table 2.
Homolog of cell wall-degrading enzyme genes in D. zeae EC1 and other Dickeya spp
*AJC65107.1, AJC66160.1, AJC66869.1, AJC67145.1, AJC67288.1, AJC67289.1, AJC67290.1, AJC67296.1, AJC67297.1, and AJC67298.1 are pectate lyases, AJC66425.1 and AJC68275.1 are pectate disaccharide-lyases, AJC67291.1 is a pectin acetylesterase, and AJC67292.1is a pectin methylesterase.
Similarly, ten genes in EC1 are involved in cellulose degradation, including 2 endoglucanase encoding genes celY and celZ, 7 beta-glucosidase encoding genes bglA, bgxA, bglB, nagZ, bglC, bglD and celH, and an alpha-glucosidase encoding gene lfaA. These genes, associated with oligosaccharide degradation, are conserved in Dickeya stains, except that bglC and bglD were absent in D. zeae Ech586, and bglA and lfaA were not found in D. paradisiaca Ech703 (Table 2).
The prtGDEFBCX cluster (W909_09760 ~ 09795) encoding four proteases and three protease secretion associated proteins is located on the negative strand of EC1 chromosome. Among them, the four protease encoding genes, i.e., prtG, B, C and X, encode serralysin homologs sharing about 59.8% similarity at amino acid level, and the prtD, E and F encode the type I secretion system (T1SS) components PrtDEF (AJC66336.1, AJC66336.1, and AJC66336.1). In addition, a proteinase inhibitor encoding gene inh is located between prtG and prtD. Alignment with other Dickeya species showed that prtG is absent in Ech1591, while the whole prt gene cluster is lost in the genome of D. paradisiaca Ech703 (Table 2).
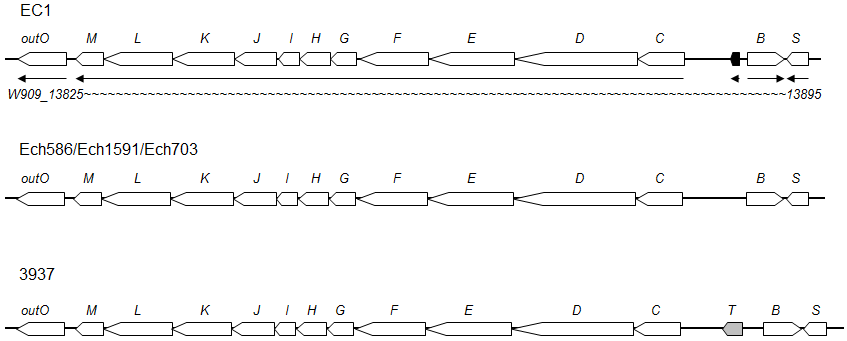
Type II secretion system
The type II secretion system (T2SS) is used by diverse gram-negative bacteria to translocate extracellular proteins across the outer membrane. In D. dadantii 3937, the T2SS is encoded by the out genes and stt genes. The Out system allows the secretion of several proteins including most pectinases while the Stt system only presents to the outer part of the outer membrane, such as the pectin lyases encoded by the adjacent pnl gene [22,23]. D. zeae EC1 chromosome contains a highly conserved T2SS gene cluster (outSBCDEFGHIJKLMO; W909_13825 ~ 13895) (Additional file 3), covering 13.814 kb with 15 ORFs, but no stt genes. The gene cluster shares a high similarity with that of D. zeae Ech586 (coverage 100%, identity 94%), D. chrysanthemi Ech1591 (coverage 90%, identity 87%), D. dadantii 3937 (coverage 89%, identity 85%), and D. paradisiaca Ech703 (coverage 77%, identity 77%) at nucleic acid level. The out gene cluster is highly conserved among Dickeya spp. except that D. zeae EC1 harbors an extra predicted gene designated as W909_13885 encoding a hypothetical protein (AJC68452.1) at the upstream of outC, and that D. dadantii 3937 contains an additional outT gene (Additional file 3).
Type III secretion system encoded by the hrp gene cluster
The hypersensitive response and pathogenicity (Hrp) type III secretion systems (T3SS) were established as pathogenicity factors in many phytopathogenic bacteria [24,25]. Similarly, the hrp genes in Dickeya spp. have also been reported to play an important role in their pathogenesis and interaction with host plants [26-28]. In the genome of D. zeae EC1, a large hrp gene cluster spanning 24.8 kb was identified, which is composed of 27 genes and arranged in three transcriptional units. Alignment of this gene cluster with the counterparts in other Dickeya species showed that this hrp gene cluster is absent in D. paradisiaca Ech703, but present in other three species, i.e., D. chrysanthemi Ech1591, D. zeae Ech586, and D. dadantii 3937 with certain variations (Figure 4). Among the strains containing the hrp gene cluster, the only variation was found at the adjacent regions of the plcA gene, which encodes an extracellular phospholipase [29]. The plcA gene is located between hrpE and hrpF genes, in D. zeae EC1, there is no additional genes, while in Ech586, Dd586_1900 (encoding a hypothetical protein) and Dd586_1899 (encoding a lytic murein transglycosylase) are located at the upstream, and Dd586_1902 and Dd586_1903 (both encoding hypothetical proteins) are located at the downstream (Figure 4). Similarly, Dd1591_1903 and Dd1591_1902 (encoding a HrpE/YscL family type III secretion apparatus protein) are located at the upstream in D. chrysanthemi Ech1591, and in D. dadantii 3937, three ORFs encoding two hypothetical proteins and a membrane-bound lytic murein transglycosylase MltB, respectively are located at the upstream (Figure 4, additional file 4). However, the biological significance of these extra genes surrounding plcA has not yet been investigated.
Figure 4.

Genetic organization of the hrp cluster in Dickeya spp. The varied genes are indicated by filled arrows. Symbol:  , hrp response elicitor gene;
, hrp response elicitor gene;  , regulator gene;
, regulator gene;  , T3SS structural gene;
, T3SS structural gene;  , hypothetical gene;
, hypothetical gene;  , extracellular phospholipase gene plcA. The hrp genes are fully conserved except the genes adjacent to plcA.
, extracellular phospholipase gene plcA. The hrp genes are fully conserved except the genes adjacent to plcA.
Some of these T3SS genes of the Dickeya species have been characterized in details. Among them, hrpXY encode a two-component system (TCS), which is required for the activation of the hrp gene expression in D. dadantii 3937 [30]. After phosphorylation, HrpY binds to the hrpS promoter and activates the expression of hrpS, which encodes a sigma (54)-dependent enhancer-binding protein HrpS. In turn, HrpS binds to the sigma factor homolog hrpL, and consequently activates the genes encoding T3SS structural proteins such as HrcJ and HrpA and secreted effectors including hrpN [30]. In D. dadantii EC16, the Hrp system appears to contribute to an early stage of pathogenesis, whereas in D. dadantii 3937 mutation of hrpN, hrpG, and hrcC resulted in substantially reduced lesion formation and bacterial growth in African violet leaves [26,27]. In addition to playing a role in plant–microbe interactions, the D. dadantii T3SS is also required for the formation of bacterial aggregates at the air-liquid interface [30,31]. Given that most key hrp genes are highly conserved in D. zeae EC1, it is highly possible that the T3SS in D. zeae EC1 could also play certain roles in the bacterial virulence and physiology, which awaits further characterizations.
Type IV secretion system
Type IV secretion system (T4SS) is unique from the other secretion systems in its ability to translocate nucleic acids in addition to proteins from donor to recipient cells [32]. It functions in conjugation, pathogenicity and DNA release/uptake [32,33]. In the EC1 genome, T4SS gene cluster covers 9.742 kb, harboring 11 T4SS-core genes (W909_12990 ~ 13040) encoding VirB1 ~ 11 (AJC66938.1 ~ AJC66948.1) (Additional file 5). VirB1 forms bores hole in peptidoglycan layer allowing T4SS complex assembly to occur, while previous studies indicated that VirB2 and VirB5 proteins constitute an extracellular pilus of T4SS, virB4 and virB11 encode ATPases providing energy and motor force for macromolecular secretion, architecture assembly and substrate pumping, and VirB6, VirB8, and VirB10 form a membrane channel encompassing both membranes, and the periplasmic protein VirB9 in complex with the short lipoprotein VirB7 could be part of this structure [32,34,35]. This T4SS gene cluster is highly conserved in D. chrysanthemi Ech1591 (coverage 100%, identity 97%), D. zeae Ech586 (coverage 99%, identity 96%), and D. dadantii 3937 (coverage 100%, identity 96%), but not found in D. paradisiaca Ech703.
Type VI secretion system
The type VI secretion system (T6SS) was identified recently and initially thought to take part in bacterial pathogenicity and host colonization [36]. Subsequent studies showed that T6SS functions in various biological processes, such as mediating cooperative or competitive interactions between bacteria and eukaryotes, and bacterial biofilm formation [37-41]. T6SS is typically encoded by clusters of 12 to 20 genes, with a minimum of 13 genes for production of a functional apparatus [42]. In Dickeya genus, D. zeae Ech586 and D. chrysanthemi Ech1591 were shown to possess an identical T6SS loci consisting of 17 genes [43], but the biological function has not yet been determined. In this study, a gene cluster encoding T6SS was found in D. zeae EC1, spanning 44.774 kb with 37 ORFs designated from W909_06460 to W909_06640. In this gene cluster, in addition to the 17 T6SS genes described previously [43], there are 20 additional ORFs inserted between W909_06465 (vgrG encoding Valine-glycine repeat G) and W909_06570 (impB) genes (Figure 5). These inserted genes encode a PAAR repeat-containing protein phospholipase A1 PldA (AJC65737.1), two ankyrins (AJC65738.1 and AJC65739.1), four YD repeat-containing protein RhsAs (AJC65742.1, AJC65744.1, W909_06510 and AJC65746.1), two plasmid stabilization system protein ParEs (AJC65749.1 and AJC65750.1), a peptidase M20 (AJC65751.1) and 10 hypothetical proteins (AJC65740.1, AJC65741.1, AJC65743.1, AJC68373.1, AJC68374.1, AJC65745.1, AJC65747.1, AJC65748.1, W909_06545, and AJC68375.1), amongst, AJC68373.1 and AJC68374.1 were not found in any other Dickeya strains. The 17 core T6SS genes are highly conserved in D. dadantii 3937, D. zeae Ech586 and D. chrysanthemi Ech1591, but substantial variations were found in the additional genes inserted between vgrG and impB (Figure 5). Similar to the cases of T3SS and T4SS, the T6SS gene cluster is totally absent in the genome of D. paradisiaca Ech703.
Figure 5.
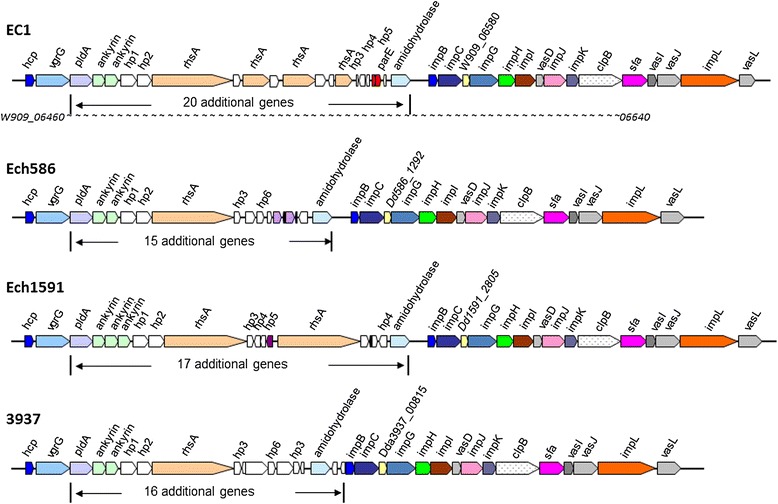
Genetic organization of the T6SS gene cluster in Dickeya spp. The variable region is indicated under the gene cluster. Colored ORF indicates the genes with known function, and the same color represents the same biological function. The gene encoding hypothetical protein is indicated by open ORF.
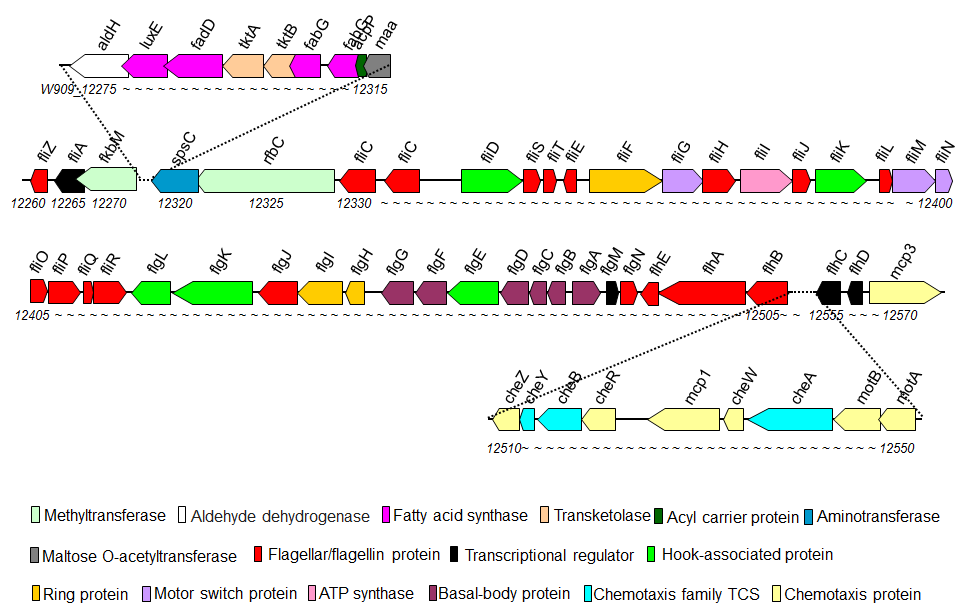
Flagellar and chemotaxis genes
One set of gene cluster encoding flagellar biosynthesis and chemotaxis proteins was found in the genome of D. zeae EC1 (W909_12260 ~ 12570), covering 63.371 kb in length. The gene cluster encodes 40 proteins (FliZ, FliA, 2 FliCs, FliD ~ T, FlgA ~ N, and FlhA ~ E) involved in flagellar biosynthesis, 10 chemotaxis-associated proteins (CheZ, CheY, CheB, CheR, MCP1, CheW, CheA, MotB, MotA, and MCP3) (Additional file 6), two methyltransferases (AJC66805.1 and AJC66816.1), an aminotransferase (AJC66815.1), and 9 fatty acid biosynthesis proteins (W909_12275 ~ 12315) (Additional file 7). The gene cluster closely resembles that found in D. zeae Ech586 (coverage 83%, identity 93%), D. chrysanthemi Ech1591 (coverage 82%, identity 83%), D. dadantii 3937 (coverage 83%, identity 84%) and D. paradisiaca Ech703 (coverage 49%, identity 77%) in nucleotide sequence, except that D. zeae EC1 contains a big inserted fragment designated as W909_12275 ~ 12315 encoding fatty acid biosynthesis (Additional file 6; Additional file 7). A blast search found that these D. zeae EC1 specific genes encoding an acyl-CoA reductase (AJC66806.1), a long-chain-fatty-acid--luciferin-component ligase LuxE (AJC66807.1), a long-chain acyl-CoA synthetase FadD (AJC66808.1), two transketolases TktA (AJC66809.1) and TktB (AJC66810.1), and two 3-oxoacyl-[acyl-carrier protein] reductases FabG (AJC66811.1 and AJC66812.1), an acyl carrier protein (AJC66813.1) and a maltose O-acetyltransferase (AJC66814.1), respectively, which share 47-70% protein similarity to their homologs in Pantoea ananatis and seem unlikely associated with the flagellar biogenesis. This fragment was also found between the fragellar genes in the rice strains DZZ2Q and ZJU1202 in same gene organization, but absent in other sequenced D. zeae strains. The flagellar genes are not clustered in the genome of D. paradisiaca Ech703, indicating again its distant evolution relationship with the other four Dickeya strains.
Unlike some other Enterobacteriaceae, which encode more than one type of flagella and flagellin [44], Dickeya spp. encodes only one type of flagella and one type of flagellin. Flagellar proteins are generally responsible for cell motility and intracellular trafficking, secretion and vesicular transport, while the chemotaxis proteins are responsible for cell motility and signal transduction. In D. dadantii 3937, mutation of fliA encoding a sigma factor abolished the bacterial motility, significantly reduced Pel production and the bacterial attachment to plant tissues, demonstrating that FliA is a positive regulator of many traits associated with virulence [44]. Mutation of chemotactic genes (cheW, cheB, cheY and cheZ) caused substantial reduction in swimming motility and decreased the bacterial virulence against Arabidopsis and potato [45]. The roles of flagellar and chemotaxis systems in D. zeae infection, especially in pathogen-monocotyledon host interactions, remain to be investigated.
Quorum-sensing systems
Many Gram-negative bacterial pathogens utilize the luxI/luxR quorum sensing system to regulate the expression of virulence genes. Typically, luxI encodes a synthase for production of acylhomoserine lactone (AHL) family quorum sensing signals, and luxR encodes an AHL signal receptor. Upon interaction with AHL signal, LuxR becomes an active transcription factor and hence modulating the expression of virulence genes. Our previous study showed that D. zeae EC1 produces an AHL family quorum sensing signal, i.e., N-(3-oxo-hexanoyl)-homoserine lactone (OHHL), which is encoded by the luxI homolog expI [8]. Mutation of expI resulted in the increased bacterial cell motility and decreased biofilm formation [8]. A Blast search of the EC1 genome revealed only one copy of expI (W909_00485) and also one well-conserved luxR homolog (W909_00480) designated as expR. ExpR is highly conserved in D. zeae Ech586 (97%), 3937 (91%), and D. chrysanthemi Ech1591 (91%), and D. paradisiaca Ech703 (64%). Interestingly, ExpI is absent in D. paradisiaca Ech703 (Additional file 8).
AI-2 produced by the S-ribosylhomocysteine lyase LuxS represents another type of conversed QS system involved in bacterial interspecies communication [46]. In Dickeya spp., evidence suggests that AI-2 production was switched off by indole-3-acetic acid (IAA) pathway under tryptophan control [47], but the biological significance of tryptophan modulation in planta and the role of AI-2 in pathogenesis remains unclear. A luxS gene homolog (W909_04510) encoding a S-ribosylhomocysteine lyase was also identified in D. zeae EC1, sharing about 79% identity to the luxS gene of Serratia marcescens ATCC 274 [48], and about 79% - 93% identity to its homologs in Ech586, Ech1591 and Ech703 at nucleic acid level, respectively.
Interestingly, in the upstream of the expI/expR genes (W909_00480 ~ 00485), the vfm gene cluster encoding the biosynthesis of a novel QS signal was found highly conserved in D. zeae EC1 and other four Dickeya species and strains used in this study (70% to 98% identity at nucleic acid level, additional file 9, Additional file 10). The gene cluster was originally identified in D. dadantii 3937, and is associated with the regulation of virulence factor production and pathogenesis [49]. The biological significance of various QS systems in D. zeae EC1 and other Dickaya species remains to be further investigated.
Zeamine synthesis gene cluster
D. zeae EC1 produces at least two polyamino phytotoxins and antibiotics, i.e., zeamine and zeamine II, which were shown to be the major virulence determinants [6]. The genes zmsA and zmsK, which respectively encode a multidomain polyketide synthase and an non-ribosomal peptide synthase containing only a condensation domain, are involved in zeamines biosynthesis and play a key role in the bacterial virulence [6,7]. Interestingly, zeamines have also been identified from a bacterial isolate of Serratia plymuthica RVH1 and a gene cluster containing 23 ORFs was proposed as the zeamine biosynthetic gene (zmn) cluster [50]. As the genome and the zmn sequences of S. plymuthica RVH1 are not publically available, we used the same gene cluster from S. plymuthica AS12 for comparison analysis. We found that most of the zmn genes of S. plymuthica are conserved in the D. zeae EC1 genome (W909_06685 ~ 06770) except a few genes including zmn1-4 and zmn23 (Figure 6, additional file 11). Moreover, as expected, the two sets of zeamine biosynthetic genes from D. zeae EC1 and S. plymuthica AS12 share a high degree of similarity with amino acids identity ranging from 50-92% (Additional file 11). S. plymuthica is an uncommon cause of human infection, but the role of zeamines in its pathogenesis has not yet been determined
Figure 6.
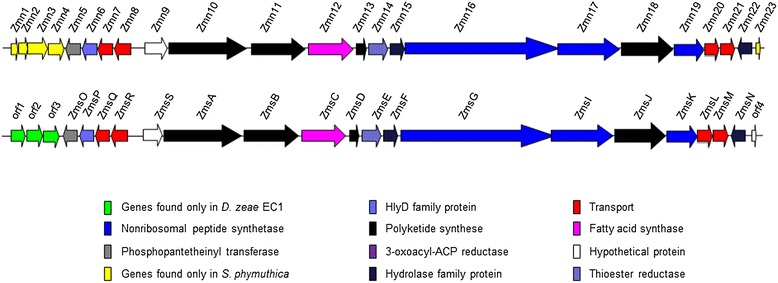
Genetic organization of zeamine biosynthetic gene cluster in S. phymuthica AS12 (top), D. zeae EC1 and D. solani D s0432-1 (bottom).
The zms gene cluster of D. zeae EC1 spans 51,169 bp, and includes 18 ORFs ranging from zmsO to zmsN (Figure 6). Surprisingly, the zms gene cluster is not conserved in the four Dickeya strains used in this study. Alignments of the nucleic acid sequence of the zms gene cluster showed that only few sporadic fragments in the cluster are present in the four whole-genome sequenced Dickeya spp., but surprisingly, it shares 69.35% similarity (95% coverage, 73% identity) with that in D. solani strains, such as 3337, D s0432-1, MK10, MK16, M005 and GBCC. The zeamine biosynthetic genes from D. zeae EC1 and D. solani D s0432-1 share a high degree of similarity with amino acids identity ranging from 59-94% (Additional file 11). We have also searched the partial genome sequences of other nine D. zeae strains, and found surprisingly that this zms gene cluster is only fully or almost fully conserved in the rice pathogens ZJU1202 [15] and DZ2Q [16] with 100% and 99% identity at nucleotide level, but absent in other D. zeae strains isolated from river water, maize, potato and banana, respectively [17,18]. Noticeably, the GC content of this cluster from D. zeae EC1 is 48.77%, significantly lower than the genomic GC contents of strains EC1 (53.43%), Ech1591 (54.5%), Ech586 (53.6%), Ech703 (55%) and 3937 (56.3%), suggesting that this zeamine biosynthesis gene cluster was acquired through horizontal gene transfer from other organisms at a relatively later stage of bacterial evolution, and seemingly associated with host specificity.
Conclusion
In this study, we present a complete genome sequence of D. zeae strain EC1 isolated from rice plants, which together with the other four released complete genome sequences of Dickeya species, i.e., D. chrysanthemi Ech1591, D. zeae Ech586, D. dadantii 3937, and D. paradisiaca Ech703, allows detailed genomic comparison to study bacterial evolution and identify clues associated with pathogenesis and host specificity. Synteny analysis of D. zeae EC1 and the other four released Dickeya spp. showed that the genome sequences of D. zeae strain EC1 is most co-linear with D. chrysanthemi Ech1591, most conserved with D. zeae Ech586 and least similar to D. paradisiaca Ech703. This is also evident from the overall similarity of the virulence genes conserved in the Dickeya strains used in this study. Several groups of virulence genes, such as protease genes (Table 2), T1SS gene cluster prtDEF, T3SS gene cluster (Figure 4), T4SS gene cluster (Additional file 5) and T6SS gene cluster (Figure 5) are highly or fully conserved in D. zeae EC1 and D. zeae Ech586 but absent in the genome of D. paradisiaca Ech703.
Except for D. paradisiaca Ech703, most virulence genes are well-conserved in other four Dickeya pathogens. However, we also noted some variations in certain virulence determinants, for example, the T3SS (hrp) and T6SS gene clusters. Given the important role of these two virulence determinants in host-pathogen interactions, it would be interesting to determine the impact of such variations on bacterial virulence and host-specificity. Importantly, a range of unique genes associated with virulence were identified in D. zeae strain EC1, such as zeamine synthesis gene cluster, 4 hypothetical proteins in the T6SS gene cluster (AJC65743.1, AJC68373.1, AJC68374.1 and AJC65745.1), and the 9 fatty acid biosynthesis proteins (AJC66806.1 ~ AJC66814.1) in the flagella biosynthesis cluster (Additional file 7). Among them, the genes encoding zeamine biosynthesis have been shown critical for D. zeae EC1 to establish infections in rice [6,7]. Intriguingly, the zms gene cluster was found in genomes of the six sequenced D. solani strains and strains of S. plymuthica RVH1 and AS12, suggesting that this cluster is possibly derived from a same organism by horizontal gene transfer during bacterial evolution. Given that the high similarity of the proteins encoded by the fragment inserted in the flagellar cluster (W909_12275 ~ 12315)) to P. ananatis (Additional file 7), we were tempted to speculate that this gene fragment may be transferred from an unknown organism or similar organisms to D. zeae EC1, P. ananatis strains AJ13355 (isolated from soil in Japan) and LMG20103 (isolated from Eucalyptus blight and dieback in South Africa).
In the last few years, up to 8 D. zeae isolates have been partially sequenced. Most intriguingly, sequence alignment showed that the zms gene cluster was only found in the D. zeae strains EC1, ZJU1202 and DZ2Q infecting rice plants [8,15,16], but not in other D. zeae strains from environment or other hosts, such as CSL RW192 and MK19 isolated from river water, NCPPB 2538 isolated from maize [51], Ech586 isolated from Philodendron [10], NCPPB 3531 and NCPPB 3532 isolated from potato, and MS1 isolated from banana [17,18]. Phylogenetic analysis of D. zeae strains indicates that these D. zeae strains are closely related. In particular, three rice isolates were found in the same evolutionary clade (Additional file 1). Taken together, our data suggests that the remaining D. zeae strains can be at least divided into two pathovars with one infecting rice and the other infecting other crops. We proposed to reclassify these rice strains as D. zeae subsp. oryzae.
In summary, comparison of the genome information of D. zeae EC1 with the closely related Dickeya species provides new insights for the conservation and evolution processes of virulence determinants in these important bacterial pathogens. The overall similarity in extracellular enzymes and hrp and T6SS systems of EC1 and other Dickeya pathogens seems to explain well why EC1 could also infect dicotyledons, and the unique genes (W909_06510 and W909_06520) in T6SS gene clusters might hold the keys to decipher the mechanisms of EC1 in infecting monocotyledons. Experimental dissection of the roles of these strain-specific genes and variations identified in this study shall reveal novel insights into the molecular mechanisms that specify host ranges and pathogenesis.
Methods
Whole-genome sequencing
Genomic DNA was extracted from D. zeae EC1 grown at 28°C in YEB medium [8] using MasterPure DNA purification kit (EPICENTRE Biotechnologies). The complete genome sequence was determined by the Beijing Genome Institute (Shenjun, China) using Solexa technology. A shotgun paired-end library with a fragment size between 150 to 500 bp and a long jumping distance mate-pair library with an insert-size between 2 to 10 kb were constructed. The two libraries were sequenced, and reads were trimmed on quality and length. In total, 1,664,407 high-quality filtered sequence reads were generated, with an average coverage equivalent to 148×. Sequence assembly was carried out using the SOAP software (http://soap.genomics.org.cn) 7 with a read length of 0.5 and a similarity of 0.8. Seven contigs were generated and joined into 3 scaffolds using pair-end information, with 3430.072 kb, 1294.767 kb and 1.821 kb in length, respectively. The in silico finishing of some gaps was carried out by mapping. We used the borders of the gaps as anchor and retrieved the reads in both orientations to perform a new de novo assembly on the gaps. The mapped reads were collected and used for de novo local assembling (read length of 0.5 and similarity of 0.8). Other gaps were closed by PCR amplification followed by DNA sequencing. The genome sequence data has been deposited in NCBI database with accession number CP006929.1.
Gene prediction and annotation
Genes were predicted using the coding sequence (CDS) prediction program Glimmer 3.0 [52]. Amino acid genome comparison was performed by bi-directional BLASTp (2.2.21) sequence alignment of translated ORFs in the Nr, Nt, SwissProt, COG (release: 20090331), and KEGG databases with a 10−5 e-value threshold. The best hit was filtered using a 50% identity cut-off value. Genes were considered as strain-specific if identity of the encoded protein was lower than 80% on the similar portion between them. In addition, ORFs with a size less than 110 bp were cut-off by Glimmer 3.0. Transposon sequences were searched using the RepeatMasker and RepeatProteinMasker softwares. Tandem repeat sequences were predicted by Tandem repeat finder (TRF) and aligned in the known transposon sequence database. rRNAs were found in rRNA database or predicted by rRNAmmer [53], tRNAscan [54] was used to predict the tRNA regions and analyze the tRNA secondary structures, and Rfam was used to analyze miRNA, sRNA and snRNA.
Phylogenetic analysis
Relationships of EC1 with other isolates of Dickeya were determined from multilocus sequence analysis (MLSA) on four housekeeping genes including atpD, dnaX, gyrB and recA, which were always used for the identification of Dickeya spp.[1,55]. AtpD, gyrB and recA gene sequences of 38 Dickeya strains, and dnaX gene sequences of 35 Dickeya strains were compared with sequences available in the public database (http://www.ncbi.nlm.nih.gov/). All sequences for the atpD (642 nucleotides), dnaX (536 nucleotides), gyrB (745 nucleotides) and recA (425 nucleotides) genes were edited and aligned using the ClustalW 1.6 software with parameters as: Gap Opening Penalty of 15, and Gap Extension Penalty of 6.66 for Pairwise and Multiple Alignment; Transition Weight of 0.5; and Delay Divergent Cut-off of 30%. Trees obtained with the concatenated data set of the four genes were constructed with Pectobacterium atrosepticum SCRI1043 as an outgroup. Method of Neighbor-joining was used for analysis, and 1,000 bootstrap replicates were included in a heuristic search, with a random tree and the tree bisection-reconnection branch-swapping algorithm. The percent variation was calculated by comparing all isolates to the nearest relative.
Genome comparisons
According to the phylogenetic analysis, we selected the four closely relative species with released whole genomes including D. chrysanthemi Ech1591 (formerly named D. zeae, GenBank accession number CP001655.1), D. zeae Ech586 (formerly named D. dadantii, CP001836, GCF_000025065), D. dadantii 3937 (CP002038) and D. paradisiaca Ech703 (formerly named D. dadantii, CP001654) [10], and another two D. zeae rice strains including ZJU1202 (GCF_000264075) [15] and DZ2Q (GCF_000404105) [16] for genome comparisons. The sequence of EC1 is ordered according to that of the reference bacterium based on Mummer 3.22 (http://mummer.sourceforge.net/). Then the upper and following axes of linear synteny graph are constructed after the same proportion of size reduction in length of both sequences (parameter: b, 200; c, 65; extend: l, 20). According to BLAST results, each pair nucleic acid sequence of two alignments is marked in the coordinate diagram according to its position information after the same proportion of size reduction.
To identify the set of common genes and the set of genes unique to strain EC1, OrthoMCL clustering analyses were performed with the following parameters: P-value Cut-off = 1 × 10−5; Identity Cut-off = 90%; Percent Match Cut-off = 80.
Ethics statement
All the experiments were conducted according to the experiment security regulations of South China Agricultural University (SCAU), and approved by the biosafety committee in SCAU.
Availability of supporting data
The data sets supporting the results of this article are included within the article and its additional files.
Acknowledgements
We are gratefully acknowledged for grants supported by the National Basic Research Program of China (973 Program, grant number 2015CB150600), the National Natural Science Foundation of China (grant numbers 31270170, 31201479 and 31470236) and the specialized research fund for the doctoral program of higher education of china (20124404120031).
Additional files
Phylogenetic tree based on the concatenated nucleotide sequences of the atpD (A), dnaX (B), gyrB (C) and recA (D) housekeeping genes using maximum likelihood method. The tree was constructed with P. atrosepticum SCRI1043 as outgroup, and generated with 1000 bootstrap replicates.
Characteristics of the unique proteins with known function in D. zeae EC1 compared with other two D. zeae rice strains.
{kind=link}
Physical map of type II secretion system in Dickeya spp. Arrows denote putative transcriptional units. Open arrow indicates conserved ORF. Filled arrow indicates inserted genes.
Similarity and variations of Hrp Proteins in Dickeya spp.
{kind=link}
Genetic organization of T4SS genes in Dickeya spp.
{kind=link}
Physical map of flagellar associate genes and chemotaxis associate genes in D. zeae EC1. Arrows denote putative transcriptional units. The dashed line on the top indicates the 9 fatty acid biosynthesis genes ranging from W909_12275 to W909_12315, and the dashed line on the bottom indicates the 9 chemotaxis-associated genes ranging from W909_12510 to W909_12550.
Characteristics of the fatty acid genes inserted in the fli gene cluster of D. zeae EC1.
Conservation of the common quorum sensing related proteins in Dickeya spp* . *The data in brackets indicates the number of amino acids in peptide.
The Vfm homologs in Dickeya spp*. *The sequences of vfm gene cluster are available at the asap database: http://asap.ahabs.wisc.edu/asap/ASAP1.htm. The data in brackets indicates the number of amino acids in peptide.
{kind=link}
Vfm quorum sensing genes in Dickeya spp.
Homology analysis of the zms genes of D. zeae EC1, S. plymuthica AS12 and D. solani D s0432-1. *zmn23 is predicted to encode a NifA subfamily transcriptional regulator, and its homolog in the genomes of S. plymuthica AS12 (CP002774.1), AS9 (CP002773.1) and AS13 (CP002775.1) (NCBI accession No.: AEF52351.1, AEF47399.1 and AEG30058.1), encoding a product of 684 aa, is located far from the zeamine biosynthesis gene cluster.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JN Zhou contributed to manual annotation of the genome, drafted and coordinated the manuscript, YY Cheng, SY Liu, MF Lv, LS Liao, YF Chen and YF Gu helped to extracted the DNA and analyzed the data, YY Xiong contributed to performed the phylogenetic analysis, ZD Jiang and LH Zhang conceived the study and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jianuan Zhou, Email: jianuanzhou@scau.edu.cn.
Yingying Cheng, Email: jinlingmeng@163.com.
Mingfa Lv, Email: mingfalv2010@163.com.
Lisheng Liao, Email: lishengliao@outlook.com.
Yufan Chen, Email: chenyufanscau@gmail.com.
Yanfang Gu, Email: yfgu12@163.com.
Shiyin Liu, Email: l.liu19@hotmail.com.
Zide Jiang, Email: zdjiang@scau.edu.cn.
Yuanyan Xiong, Email: mody0911@gmail.com.
Lianhui Zhang, Email: lhzhang01@scau.edu.cn.
References
- 1.van der Wolf JMNE, Kowalewska MJ, Saddler GS, Parkinson N, Elphinstone JG, Pritchard L. Dickeya solani sp. nov., a pectinolytic plant-pathogenic bacterium isolated from potato (Solanum tuberosum) Int J Syst Evol Microbiol. 2014;64(3):768–774. doi: 10.1099/ijs.0.052944-0. [DOI] [PubMed] [Google Scholar]
- 2.Samson R, Legendre JB, Christen R, Fischer-Le Saux M, Achouak W, Gardan L. Transfer of Pectobacterium chrysanthemi (Burkholder et al. 1953) Brenner et al. 1973 and Brenneria paradisiaca to the genus Dickeya gen. nov as Dickeya chrysanthemi comb. nov and Dickeya paradisiaca comb. nov and delineation of four novel species, Dickeya dadantii sp. nov., Dickeya dianthicola sp. nov., Dickeya dieffenbachiae sp. nov and Dickeya zeae sp. nov. Int J Syst Evol Microbiol. 2005;55:1415–1427. doi: 10.1099/ijs.0.02791-0. [DOI] [PubMed] [Google Scholar]
- 3.Parkinson N, Stead D, Bew J, Heeney J, Tsror L, Elphinstone J. Dickeya species relatedness and clade structure determined by comparison of recA sequences. Int J Syst Evol Microbiol. 2009;59:2388–2393. doi: 10.1099/ijs.0.009258-0. [DOI] [PubMed] [Google Scholar]
- 4.Charkowski ABC, Condemine G, Expert D, Franza T, Hayes C, Hugouvieux-Cotte-Pattat N, et al. The role of secretion systems and small molecules in soft-rot Enterobacteriaceae pathogenicity. Annu Rev Phytopathol. 2012;50:425–449. doi: 10.1146/annurev-phyto-081211-173013. [DOI] [PubMed] [Google Scholar]
- 5.Wu J, Zhang HB, Xu JL, Cox RJ, Simpson TJ, Zhang LH. C-13 Labeling reveals multiple amination reactions in the biosynthesis of a novel polyketide polyamine antibiotic zeamine from Dickeya zeae. Chem Commun. 2010;46(2):333–335. doi: 10.1039/B916307G. [DOI] [PubMed] [Google Scholar]
- 6.Zhou JN, Zhang HB, Wu J, Liu QG, Xi PG, Lee J, et al. A novel multidomain polyketide synthase is essential for zeamine production and the virulence of Dickeya zeae. Mol Plant-Microbe Interact. 2011;24(10):1156–1164. doi: 10.1094/MPMI-04-11-0087. [DOI] [PubMed] [Google Scholar]
- 7.Cheng YY, Liu XL, An SW, Chang CQ, Zou YQ, Huang LH, et al. A nonribosomal peptide synthase containing a stand-alone condensation domain is essential for phytotoxin zeamine biosynthesis. Mol Plant-Microbe Interact. 2013;26(11):1294–1301. doi: 10.1094/MPMI-04-13-0098-R. [DOI] [PubMed] [Google Scholar]
- 8.Hussain MBBM, Zhang HB, Xu JL, Liu QG, Jiang ZD, Zhang LH. The acyl-homoserine lactone-type quorum-sensing system modulates cell motility and virulence of Erwinia chrysanthemi pv. zeae. J Bacteriol. 2008;190(3):1045–1053. doi: 10.1128/JB.01472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu QG, Wang ZZ. Infection characteristics of Erwinia chrysanthemi pv. zeae on rice. J South China Agric Univ. 2004;25(3):55–57. [Google Scholar]
- 10.Pritchard LHS, Saddler GS, Parkinson NM, Bertrand V, Elphinstone JG. Detection of phytopathogens of the genus Dickeya using a PCR primer prediction pipeline for draft bacterial genome sequences. Plant Pathol. 2012;62:587–596. doi: 10.1111/j.1365-3059.2012.02678.x. [DOI] [Google Scholar]
- 11.Glasner JD, Yang CH, Reverchon S, Hugouvieux-Cotte-Pattat N, Condemine G, Bohin JP, et al. Genome Sequence of the Plant-Pathogenic Bacterium Dickeya dadantii 3937. J Bacteriol. 2011;193(8):2076–2077. doi: 10.1128/JB.01513-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pritchard L, Humphris S, Baeyen S, Maes M, Van Vaerenbergh J, Elphinstone J, Saddler G, Toth I: Draft Genome Sequences of Four Dickeya dianthicola and Four Dickeya solani Strains. Genome Announcements 2013;1(4):e00087-12 [DOI] [PMC free article] [PubMed]
- 13.Garlant L, Koskinen P, Rouhiainen L, Laine P, Paulin L, Auvinen P, et al. Genome sequence of Dickeya solani, a New soft Rot pathogen of potato, suggests its emergence May Be related to a novel combination of Non-ribosomal peptide/polyketide synthetase clusters. Diversity. 2013;5:824–842. doi: 10.3390/d5040824. [DOI] [Google Scholar]
- 14.Pédron J, Mondy S, Raoul des Essarts Y, Van Gijsegem F, Faure D’. Genomic and metabolic comparison with Dickeya dadantii 3937 reveals the emerging Dickeya solani potato pathogen to display distinctive metabolic activities and T5SS/T6SS-related toxin repertoire. BMC Genomics. 2014;15:283. [DOI] [PMC free article] [PubMed]
- 15.Li B, Shi Y, Ibrahim M, Liu H, Shan C, Wang Y, et al. Genome sequence of the rice pathogen Dickeya zeae strain ZJU1202. J Bacteriol. 2012;194(16):4452–4453. doi: 10.1128/JB.00819-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertani I, Passos da Silva D, Abbruscato P, Piffanelli P, Venturi V: Draft Genome Sequence of the Plant Pathogen Dickeya zeae DZ2Q, Isolated from Rice in Italy. Genome Announcements 2013;1(6):e00905-13. [DOI] [PMC free article] [PubMed]
- 17.Zhang JX, Lin BR, Shen HF, Pu XM: Genome Sequence of the Banana Pathogen Dickeya zeae Strain MS1, Which Causes Bacterial Soft Rot. Genome Announcements 2013;1(3):e00317-13. [DOI] [PMC free article] [PubMed]
- 18.Pritchard L, Humphris S, Saddler GS, Elphinstone JG, Pirhonen M, Toth IK: Draft genome sequences of 17 isolates of the plant pathogenic bacterium dickeya. Genome Announcements 2013;1(6):e00978-13. [DOI] [PMC free article] [PubMed]
- 19.Kube M, Migdoll AM, Gehring I, Heitmann K, Mayer Y, Kuhl H, Knaust F, Geider K, Reinhardt R: Genome comparison of the epiphytic bacteria Erwinia billingiae and E. tasmaniensis with the pear pathogen E. pyrifoliae. BMC Genomics 2010;11:393. [DOI] [PMC free article] [PubMed]
- 20.Aziz RK, Breitbart M, Edwards RA. Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res. 2010;38(13):4207–4217. doi: 10.1093/nar/gkq140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hugouvieux-Cotte-Pattat N, Condemine G, Nasser W, Reverchon S. Regulation of pectinolysis in Erwinia chrysanthemi. Annu Rev Microbiol. 1996;50:213–257. doi: 10.1146/annurev.micro.50.1.213. [DOI] [PubMed] [Google Scholar]
- 22.Lindeberg M, Boyd CM, Keen NT, Collmer A. External loops at the C terminus of Erwinia chrysanthemi pectate lyase C are required for species-specific secretion through the out type II pathway. J Bacteriol. 1998;180(6):1431–1437. doi: 10.1128/jb.180.6.1431-1437.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrandez YCG. Novel mechanism of outer membrane targeting of proteins in gram negative bacteria. Mol Microbiol. 2008;69:1349–1357. doi: 10.1111/j.1365-2958.2008.06366.x. [DOI] [PubMed] [Google Scholar]
- 24.Xiao Y, Lu Y, Heu S, Hutcheson SW. Organization and environmental regulation of the Pseudomonas syringae pv. syringae 61 hrp cluster. J Bacteriol. 1992;174(6):1734–1741. doi: 10.1128/jb.174.6.1734-1741.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindgren PB. The role of hrp genes during plant-bacterial interactions. Annu Rev Phytopathol. 1997;35:129–152. doi: 10.1146/annurev.phyto.35.1.129. [DOI] [PubMed] [Google Scholar]
- 26.Bauer DW, Bogdanove AJ, Beer SV, Collmer A. Erwinia chrysanthemi hrp genes and their involvement in soft rot pathogenesis and elicitation of the hypersensitive response. Mol Plant-Microbe Interact. 1994;7(5):573–581. doi: 10.1094/MPMI-7-0573. [DOI] [PubMed] [Google Scholar]
- 27.Yang CH, Gavilanes-Ruiz M, Okinaka Y, Vedel R, Berthuy I, Boccara M, et al. hrp genes of Erwinia chrysanthemi 3937 are important virulence factors. Mol Plant-Microbe Interact. 2002;15(5):472–480. doi: 10.1094/MPMI.2002.15.5.472. [DOI] [PubMed] [Google Scholar]
- 28.Rojas CM, Ham JH, Schechter LM, Kim JF, Beer SV, Collmer A. The Erwinia chrysanthemi EC16 hrp/hrc gene cluster encodes an active hrp type III secretion system that is flanked by virulence genes functionally unrelated to the hrp system. Mol Plant-Microbe Interact. 2004;17(6):644–653. doi: 10.1094/MPMI.2004.17.6.644. [DOI] [PubMed] [Google Scholar]
- 29.Keen NT, Ridgway D, Boyd C. Cloning and characterization of a phospholipase gene from Erwinia chrysanthemi EC16. Mol Microbiol. 1992;6(2):179–187. doi: 10.1111/j.1365-2958.1992.tb01999.x. [DOI] [PubMed] [Google Scholar]
- 30.Yap MN, Yang CH, Barak JD, Jahn CE, Charkowski AO. The Erwinia chrysanthemi type III secretion system is required for multicellular behavior. J Bacteriol. 2005;187(2):639–648. doi: 10.1128/JB.187.2.639-648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yap MN, Rojas CM, Yang CH, Charkowski AO. Harpin mediates cell aggregation in Erwinia chrysanthemi 3937. J Bacteriol. 2006;188(6):2280–2284. doi: 10.1128/JB.188.6.2280-2284.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fronzes R, Christie PJ, Waksman G. The structural biology of type IV secretion systems. Nat Rev Microbiol. 2009;7(10):703–714. doi: 10.1038/nrmicro2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alvarez-Martinez CE, Christie PJ. Biological Diversity of Prokaryotic Type IV Secretion Systems. Microbiol Mol Biol Rev. 2009;73(4):775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chandran V, Fronzes R, Duquerroy S, Cronin N, Navaza J, Waksman G. Structure of the outer membrane complex of a type IV secretion system. Nature. 2009;462(7276):1011–U1066. doi: 10.1038/nature08588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fronzes R, Schafer E, Wang L, Saibil HR, Orlova EV, Waksman G. Structure of a type IV secretion system core complex. Science. 2009;323(5911):266–268. doi: 10.1126/science.1166101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mougous JD, Cuff ME, Raunser S, Shen A, Zhou M, Gifford CA, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312(5779):1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aschtgen MS, Bernard CS, De Bentzmann S, Lloubes R, Cascales E. SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative escherichia coli. J Bacteriol. 2008;190(22):7523–7531. doi: 10.1128/JB.00945-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hood RD, Singh P, Hsu F, Guevener T, Carl MA, Trinidad RRS, et al. A type VI secretion system of Pseudomonas aeruginosa targets, a toxin to bacteria. Cell Host Microbe. 2010;7(1):25–37. doi: 10.1016/j.chom.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jani AJ, Cotter PA. Type VI secretion: Not just for pathogenesis anymore. Cell Host Microbe. 2010;8(1):2–6. doi: 10.1016/j.chom.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarz S, West TE, Boyer F, Chiang WC, Carl MA, Hood RD, Rohmer L, Tolker-Nielsen T, Skerrett SJ, Mougous JD: Burkholderia Type VI Secretion Systems Have Distinct Roles in Eukaryotic and Bacterial Cell Interactions. Plos Pathogens 2010;6(8):e1001068. [DOI] [PMC free article] [PubMed]
- 41.Zheng J, Ho B, Mekalanos JJ: Genetic Analysis of Anti-Amoebae and Anti-Bacterial Activities of the Type VI Secretion System in Vibrio cholerae. Plos One 2011;6(8):e23876. [DOI] [PMC free article] [PubMed]
- 42.Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I: Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from available microbial genomic resources? BMC Genomics 2009;10–104 [DOI] [PMC free article] [PubMed]
- 43.Sarris PF, Trantas EA, Skandalis N, Tampakaki AP, Kapanidou M, Kokkinidis M, Panopoulos NJ: Phytobacterial Type VI Secretion System - Gene Distribution, Phylogeny, Structure and Biological Functions; 2012;53–84
- 44.Jahn CE, Willis DK, Charkowski AO. The flagellar sigma factor FliA is required for Dickeya dadantii virulence. Mol Plant-Microbe Interact. 2008;21(11):1431–1442. doi: 10.1094/MPMI-21-11-1431. [DOI] [PubMed] [Google Scholar]
- 45.Antunez-Lamas M, Cabrera-Ordonez E, Lopez-Solanilla E, Raposo R, Trelles-Salazar O, Rodriguez-Moreno A, et al. Role of motility and chemotaxis in the pathogenesis of Dickeya dadantii 3937 (ex Erwinia chrysanthemi 3937) Microbiology-Sgm. 2009;155:434–442. doi: 10.1099/mic.0.022244-0. [DOI] [PubMed] [Google Scholar]
- 46.Vendeville A, Winzer K, Heurlier K, Tang CM, Hardie KR. Making ‘sense’ of metabolism: Autoinducer-2, LuxS and pathogenic bacteria. Nat Rev Microbiol. 2005;3(5):383–396. doi: 10.1038/nrmicro1146. [DOI] [PubMed] [Google Scholar]
- 47.Crepin A, Barbey C, Beury-Cirou A, Helias V, Taupin L, Reverchon S, Nasser W, Faure D, Dufour A, Orange N, Feuilloley M, Heurlier K, Burini JF, Latour X: Quorum Sensing Signaling Molecules Produced by Reference and Emerging Soft-Rot Bacteria (Dickeya and Pectobacterium spp.). Plos One 2012;7(4):e35176. [DOI] [PMC free article] [PubMed]
- 48.Coulthurst SJ, Kurz CL, Salmond GPC. LuxS mutants of Serratia defective in autoinducer-2-dependent ‘quorum sensing’ show strain-dependent impacts on virulence and production of carbapenem and prodigiosin. Microbiology-Sgm. 2004;150:1901–1910. doi: 10.1099/mic.0.26946-0. [DOI] [PubMed] [Google Scholar]
- 49.Nasser W, Dorel C, Wawrzyniak J, Van Gijsegem F, Groleau MC, Deziel E, et al. Vfm a new quorum sensing system controls the virulence of Dickeya dadantii. Environ Microbiol. 2013;15(3):865–880. doi: 10.1111/1462-2920.12049. [DOI] [PubMed] [Google Scholar]
- 50.Masschelein J, Mattheus W, Gao LJ, Moons P, Van Houdt R, Uytterhoeven B, Lamberigts C, Lescrinier E, Rozenski J, Herdewijn P, Aertsen A, Michiels C,Lavigne R: A PKS/NRPS/FAS Hybrid Gene Cluster from Serratia plymuthica RVH1 Encoding the Biosynthesis of Three Broad Spectrum, Zeamine-Related Antibiotics. Plos One 2013;8(1):e54143. [DOI] [PMC free article] [PubMed]
- 51.Ma B, Hibbing ME, Kim HS, Reedy RM, Yedidia I, Breuer J, et al. Host range and molecular Phylogenies of the soft rot enterobacterial genera Pectobacterium and dickeya. Phytopathology. 2007;97(9):1150–1163. doi: 10.1094/PHYTO-97-9-1150. [DOI] [PubMed] [Google Scholar]
- 52.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23(6):673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lowe TM, Eddy SR. TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brady CL CI, Denman S, Venter SN, Rodríguez-Palenzuela P, Coutinho TA, De Vos P: Proposal to reclassify Brenneria quercina (Hildebrand and Schroth 1967) Hauben et al. 1999 into a new genus, Lonsdalea gen. nov., as Lonsdalea quercina comb. nov., descriptions of Lonsdalea quercina subsp. quercina comb. nov., Lonsdalea quercina subsp. iberica subsp. nov. and Lonsdalea quercina subsp. britannica subsp. nov., emendation of the description of the genus Brenneria, reclassification of Dickeya dieffenbachiae as Dickeya dadantii subsp. dieffenbachiae comb. nov., and emendation of the description of Dickeya dadantii. Int J Systemat Evol Microbioly 2012, 62(7):1592–1602 [DOI] [PubMed]