

Abstract
Background
A long juvenile period between germination and flowering is a common characteristic among fruit trees, including Malus hupehensis (Pamp.) Rehd., which is an apple rootstock widely used in China. microRNAs (miRNAs) play an important role in the regulation of phase transition and reproductive growth processes.
Results
M. hupehensis RNA libraries, one adult and one juvenile phase, were constructed using tree leaves and underwent high-throughput sequencing. We identified 42 known miRNA families and 172 novel miRNAs. We also identified 127 targets for 25 known miRNA families and 168 targets for 35 unique novel miRNAs using degradome sequencing. The identified miRNA targets were categorized into 58 biological processes, and the 123 targets of known miRNAs were associated with phase transition processes. The KEGG analysis revealed that these targets were involved in starch and sucrose metabolism, and plant hormone signal transduction. Expression profiling of miRNAs and their targets indicated multiple regulatory functions in the phase transition. The higher expression level of mdm-miR156 and lower expression level of mdm-miR172 in the juvenile phase leaves implied that these two small miRNAs regulated the phase transition. mdm-miR160 and miRNA393, which regulate genes involved in auxin signal transduction, could also be involved in controlling this process. The identification of known and novel miRNAs and their targets provides new information on this regulatory process in M. hupehensis, which will contribute to the understanding of miRNA functions during growth, phase transition and reproduction in woody fruit trees.
Conclusions
The combination of sRNA and degradome sequencing can be used to better illustrate the profiling of hormone-regulated miRNAs and miRNA targets involving complex regulatory networks, which will contribute to the understanding of miRNA functions during growth, phase transition and reproductive growth in perennial woody fruit trees.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-1125) contains supplementary material, which is available to authorized users.
Keywords: Phase change, Malus hupehensis, Small RNA, miRNA targets, Expression profile, Degradome
Background
Fruit trees, like most perennial woody plants, have a long juvenile phase before flowering and fruiting [1–3]. Studies of the mechanisms involved in juvenile development and the juvenile to adult phase transition are vital for shortening the juvenile phase and accelerating the breeding of economically beneficial traits in woody plants, especially in apple trees. Rootstock breeding is an important aspect of fruit breeding, since many apple trees are propagated by grafting a scion on a rootstock, such as T337 and M26. Shortening the rootstock’s juvenile phase is necessary for early flowering and fruiting of several scion varieties [4]. Presently, China produces more apples than any other country in the world and up to 90% of the rootstocks are bred using seed propagation [5]. Malus hupehensis, which originated in Pingyi, Shandong Province, is an important apple rootstock that undergoes apomixis at a rate greater than 95% [4].
Many species have long juvenile vegetative phases. This may be associated with the biological processes involved in hormone synthesis and metabolism, carbohydrate synthesis and metabolism, and photosynthesis [1, 6]. It has been reported that carbohydrates play an important role in the reproductive development of olive trees, [7] and that the SQUAMOSA promoter binding protein (SBP)-box genes SPL10, SPL11 and SPL2 control morphological changes in Arabidopsis reproduction [8]. Transgenic experiments in Populus trichocarpa and Arabidopsis, indicate that the terminal flower 1 (TFL1) gene expression level affects the transition from the vegetative to reproductive phase [9, 10]. Additionally, other genes, including the MADS-box family of transcription factor (TF) genes in citrus, are involved in the juvenile to adult transition [11]. These control mechanisms represent a preliminary understanding of the molecular base of the transition from juvenile to adult phase in plants.
MicroRNAs (miRNAs) play an important role in the regulating the juvenile to adult phase transition in annual plants [12, 13]. For example, in Arabidopsis thaliana, high levels of miR156 reduced the expression levels of SPL TFs, which activated SUPPRESSOR of CONSTANS 1 (SOC1), LEAFY (LFY), AGAMOUS-LIKE 42 (AGL42), FRUITFULL (FUL) and APETALA1 (AP1) genes that regulate the transition from juvenile to adult phase [14]. miR156 acts in several pathways that control different aspects of vegetative development and play an important role in the juvenile phase [12]. miR172 down-regulates GLOSSY15 expression, which promotes the vegetative phase change in maize [15]. In perennial woody species, related studies on the molecular mechanisms of phase change have been performed [16, 17]. The overexpression of miR156 in transgenic Populus × canadensis reduced the expression of miR156-targeted SPL genes and miR172, and drastically prolonged the juvenile phase [1]. The increase in SsmiR156 and decrease in SsmiR172 during plant rejuvenation showed that these two miRNAs affect phase transition in Sequoia sempervirens[18].
Plant hormones are known to play an important role in the juvenile to adult phase transition and in plant flowering [19]. Currently, the miRNA-mediated regulation of plant growth, development and flowering through phytohormone crosstalk and other developmental processes mainly involves six classes of phytohormones, auxin (AUX), cytokinin (CK), abscisic acid (ABA), gibberellic acid (GA), ethylene (ET) and jasmonic acid (JA) [20]. It was reported that GA accelerates flowering through the degradation of transcription repressors, DELLAs, and that DELLAs directly bind to miRNA156-targeted TFs (SPL family members), which promote flowering by activating miR172 and MADS-box genes [21].
We determine if miRNAs are involved in hormone regulation during the transition from vegetative to reproductive growth in apple trees. Additionally, we investigated the complex hormonal and miRNA-mediated regulatory networks in which the miRNAs associated with phase transition control plant growth, development and the transition to flowering.
Methods
Plant material and RNA isolation
Leaf samples from the Apple Demonstration Nursery of Yangling Modern Agriculture Technology Park (Northwest Agriculture & Forestry University), Shaanxi Province of China (34° 52′ N, 108° 7′ E), were collected directly into liquid nitrogen. In June, ‘Adult phase’ (A) leaves were collected as mixed samples from the tops of 18 6-year-old M. hupehensis (Pamp.) Rehd. trees and ‘Juvenile phase’ (J) leaves were collected from the base. Since M. hupehensis has the ability to undergo apomixis at a rate greater than 95%, their growth is highly synchronous. Roots, stems, flowers and fruits were also collected in the same manner at the same time. The samples were stored in a −80°C freezer until used (Figure 1). Additionally, leaf samples were collected from the tops of M. hupehensis of different ages (1-, 2-, 3-, 4-, 5- and 6-year-old trees) (Figure 1C). Two leaf samples, A and J, were used for small RNA and degradome sequencing, and the samples were used for qRT-PCR to verify the expression patterns of miRNA and their targets (Figure 1C,D). Total RNA was isolated from each sample by a modified method [22].
Figure 1.

Morphology of adult and juvenile trees in Malus hupehensis . Adult phase at the tree top and juvenile phase at the tree base (A); Adult tree that can flower and juvenile tree that cannot flower (B); Trees of different ages; and (C) Adult leaves at the tree top and juvenile leaves at the tree base (D).
Construction of small RNA and degradome libraries
Small RNA construction and deep sequencing of the A and J leaf samples was carried out by the Beijing Genomics Institute (BGI) (Shenzhen, China) using an Illumina Genome Analyzer [23]. Total RNA extractions from A and J leaves were performed using the RNeasyPlant Mini Kit (Qiagen, Hilden, Germany) and collected into A and J RNA pools. Briefly, 18–30-nt gel fragments were selected and then ligated to a pair of adapters at the 5′- and 3′-ends using T4 RNA ligase. The small RNAs with the adapters were transcribed into cDNA using Super-Script II Reverse Transcriptase (Invitrogen, Shanghai), and then the cDNA products were amplified by polymerase chain reaction (PCR). Finally, the purified PCR products were directly sequenced using Solexa sequencing technology (BGI). The leaf total RNA from the J sample was also used for degradome sequencing and library construction as previously described [24, 25], as well as for miRNA target identification.
Bioinformatics analyses of sequencing data
Raw reads produced using an Illumina 1G Genome Analyzer at BGI were processed into clean full-length reads by removing the low-quality reads (e.g., those larger than 30 nt or smaller than 18 nt, those with between 30 and 50 adapter contaminants, those with polyA sequences and those without insert tags) by a data-cleaning pipeline. All high-quality sequences were queried using NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank) databases and Rfam databases (http://www.sanger.ac.uk/resources/databases/rfam.html), and used for further analyses. The small RNA tags were annotated with rRNA, scRNA, snoRNA, snRNA and tRNA using the tag2annotation software developed by BGI. To map every unique small RNA to only one annotation, we followed the following priority rule: rRNAetc (GenBank > Rfam) > known miRNA > repeat > exon > intron3 (Shenzhen, China).
Identification of known miRNAs and prediction of novel miRNAs in M. hupehensis (Pamp.)Rehd
To identify known miRNAs in M. hupehensis, the miRNA categories were mapped to the reference genome Malus domestica in miRBase 18.0 (http://www.mirbase.org) with the criterion that sequences in the small RNA libraries (A and J) have less than two mismatches and more than 16 matches without gaps. miRNAs that could not be annotated were used to predict novel miRNA using the software Mireap (http://sourceforge.net/projects/mireap/) developed by the BGI. Additionally, the characteristic structures of miRNA precursors, including hairpins, secondary structures, Dicer cleavage sites and the minimum free energy, were used to predict novel miRNAs with the MIREAP pipeline (https://sourceforge.net/projects/mireap/). The criteria included hairpin miRNAs that can fold into necessary secondary structures and mature miRNAs that are present in one arm of the hairpin precursors. Additionally, the free energy of hybridization must be lower than or equal to −18 kcal/mol, and the mature miRNA strand and its complementary strand (miRNA*) must contain 2-nt 3' overhangs.
Target prediction and identification
We identified targets by degradome sequencing [26]. Briefly, we matched the degraded fragments to the apple genome (Malus × domestica Borkh.) and removed ncRNAs, as well as polyN fragments, in the samples to reduce interference. We then used PairFinder software developed by the BGI degradome group to predict potential mRNA-miRNA pairings (Additional file 1). To predict potential functions of the putative miRNA targets in various biological processes, molecular functions and cellular components we used gene ontology (GO) categories (http://www.geneontology.org/) to classify the identified target genes [27]. Additionally, the KEGG database (fttp://fttp.genome.jp/pub/kegg/pathway/) was used for KEGG pathway analyses.
qRT-PCR validation of miRNAs and their targets
cDNAs of miRNAs and targets were generated from 2 μg of total RNAs of 24 M. hupehensis samples (leaf tissue at 3, 4, 5, 6, 7 and 8 months, and the top leaves of 1-, 2-, 3-, 4-, 5- and 6-year-old trees, as well as roots, stems, flowers and fruit in June) using miRcute miRNA cDNA (Tiangen, Beijing) and PrimeScript™ RT reagent Kit with gDNA Eraser (Takara) (Figure 1). qRT-PCR was performed using a miRNA qPCR Detection Kit (SYBR Green) with 10 μl of 2X miRcute miRNA premix with ROX and SYBR green (Tiangen), and 0.4 μM of forward and reverse primers in a 20-μl system for the expression of miRNAs. PCR was also performed using SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) with 10 μl of 2X SYBR® Premix Ex Taq II, and 0.8 μl of forward and reverse primers in a 20-μl system to determine the expression of the targets (Takara). The reactions were incubated in a Bio-Rad (iCycler iQ5) for 30 s at 95°C, followed by 40 cycles of 5 s at 95°C and 35 s at 60°C, followed by 81 cycles for the melt curve. Each reaction was performed in three replicates. All primers used in the qRT-PCR experiments are listed in Additional file 2.
Leaf morphology characteristics and hormone contents
The juvenile leaves from the bases of the trees and the adult leaves from the tops of the trees were used to measure and calculate the length, width, area and dry weight (Figure 1A and D). The methods of hormone extraction and determination in leaves were carried out as previously described [28, 29].
Results
Construction and sequencing of small RNA and degradome libraries
To determine responsive sRNAs in the juvenile and adult vegetative phases, A and J miRNA libraries were constructed and sequenced A total of 29,945,580 raw reads were generated by the high-throughput Illumina HiSeq. 2000 Sequencing System, with 16,316,909 and 13,628,671 reads from the A and J libraries, respectively (Table 1). After processing primary reads, 16,220,576 (99.74%) and 13,547,321 (99.68%) total clean reads were selected from the A and J libraries, respectively. We also constructed a degradome library using total RNA from the J sample (Additional file 3). The size distributions of the reads in the A and J libraries were quite similar, but there was at least 50% more 21-nt length reads in the juvenile library compared with the adult library (Figure 2). The length of the sRNA varied from 18 to 28 nt in the samples, and 21-, 23- and 24-nt small RNAs formed the major population with 24 nt being the most dominant, which is similar to the results obtained from most tested plants, including A. thaliana and Brassica juncea[30–32]. The sequencing data have been deposited in NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/Traces/sra_sub/sub.cgi). And accession number was SRP048848. Meanwhile, related data have been already deposited in Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63373). And accession number was GSE63373.
Table 1.
Raw and clean read statistics of small RNAs isolated from Malus hupehensis leaves
| A | J | |||
|---|---|---|---|---|
| Count | % | Count | ||
| total_reads | 16316909 | total_reads | 16316909 | |
| high_quality | 16262057 | 100% | high_quality | 16262057 |
| 3′adapter_null | 7741 | 0.05% | 3′adapter_null | 7741 |
| insert_null | 2554 | 0.02% | insert_null | 2554 |
| 5′adapter_contaminants | 25157 | 0.15% | 5′adapter_contaminants | 25157 |
| smaller_than_18nt | 5041 | 0.03% | smaller_than_18nt | 5041 |
| polyA | 988 | 0.01% | polyA | 988 |
| clean_reads | 16220576 | 99.74% | clean_reads | 16220576 |
Figure 2.
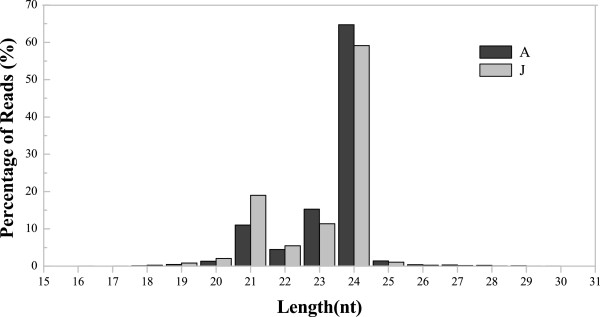
Length distribution of small RNAs from adult and juvenile Malus hupehensis leaves. The length distributions of high-quality sequences were obtained from A and J libraries. The distributions of the total reads are shown as percentages. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base.
The sRNA reads were grouped based on their identities (4,912,555 for A and 3,652,984 for J, respectively) as determined by mapping them to the domesticated apple’s genome (Malus × domestica Borkh.) using SOAP2 software [33]. Approximately, a half and a third of the unique sRNA sequences for A and J matched this genome, respectively (Table 2). The reads were categorized into different classes of sRNAs, including rRNA, miRNA, snRNA, snoRNA and repeats, by matching them with the domesticated apple’s genome in the Rfam (http://www.sanger.ac.uk/resources/databases/rfam.html) and GenBank (http://www.ncbi.nlm.nih.gov/genbank) databases using the tag2annotation software. The repeats, miRNAs and rRNAs formed the major population, and repeats were the most dominant class of sRNA; however, the majority of sRNAs remained unannotated. For almost every type of sRNA, a number of unique sRNAs were shared between the A and J libraries (Figure 3).
Table 2.
Mapping statistics of small RNAs isolated from Malus hupehensis leaves
| Libraries | Unique sRNAs | Percentage(%) | Total sRNAs | Percentage(%) |
|---|---|---|---|---|
| A | 4912555 | 47.83 | 5754465 | 19.33 |
| J | 3652984 | 35.57 | 4095344 | 13.76 |
Figure 3.
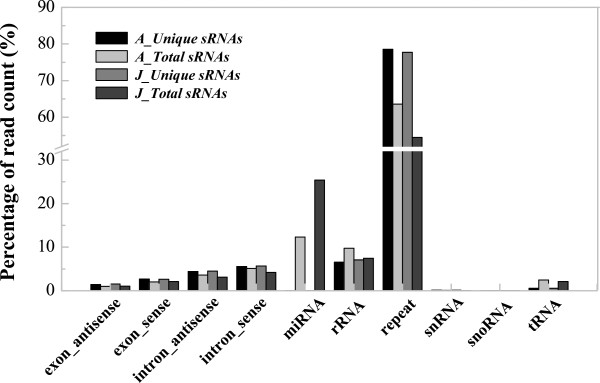
Classification of unique small RNA reads from adult and juvenile Malus hupehensis leaves. The reads were categorized into 10 different classes of small RNAs with their distributions as percentages. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base.
Known miRNA and expression levels
To identify the known miRNAs in M. hupehensis (Pamp.) Rehd., the sRNAs in the two libraries were queried using BLASTN to known mature plant miRNAs of M. domestica in the miRBase 18.0 (http://www.mirbase.org) and plant miRNA (http://bioinformatics.cau.edu.cn/PMRD) databases [25]. A total of 207 known miRNAs belonging to 42 miRNA families were identified (Additional file 4). Different sequences are cloned into libraries at different frequencies, so the read numbers of different miRNA species may be biased by the methods of miRNAs library construction. This potential problem can be addressed by using a new method of miRNAs library construction, increasing the sample size or improving the accuracy of the data analysis and comparisons (Additional file 4). The number of members within different miRNA families varied significantly. A majority of the 42 known miRNA families had several members, and five families, mdm-miR156, mdm-miR171, mdm-miR172, mdm-miR167 and mdm-miR399, had 31, 15, 15, 10 and 10 members, respectively. Seven of the known miRNA families, mdm-miR1511, mdm-miR391, mdm-miR7125, mdm-miR7126, mdm-miR7128, mdm-miR827 and mdm-miR858, had only one member (Figure 4).
Figure 4.
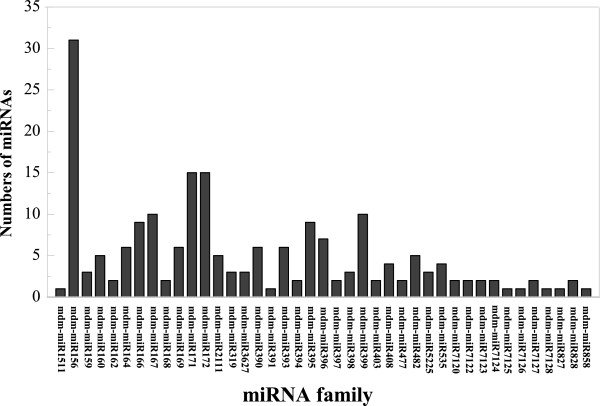
Numbers of identified miRNAs in known miRNA families in Malus hupehensis.
The expression levels of known miRNAs were determined by their sequence count frequencies in the A and J libraries. Some miRNAs showed library-specific expression patterns and the frequency of members from the same or different miRNA families varied greatly in each library, ranging from one to 446. Additionally, the expression levels of some miRNAs, including mdm-miR159, 164, 172, 319, 477 and 827, were found at very low levels, with read counts ranging from 0 to 10 between the A and J libraries. Others, including mdm-miR156, 166, 167, 168, 408 and 391, had high expression levels, with read counts that reached more than 10,000 in each library (Additional file 4).
Analyzing known miRNA expression levels between the A and J libraries revealed that the 17 known miRNA families (mdm-miR156, 172, 398, 397, 7125, 408, 160, 7124, 393, 3627, 5225, 396, 858, 535, 162, 2118 and 7120) were differentially expressed (Figure 5). Among these, the expression levels of mdm-miR156 and 11 other miRNA family members, mdm-miR160, 7124, 393, 3627, 5225, 162, 2118, 7120, 396, 858 and 535, in the J library were significantly higher than in the A library (Figure 5A,B). However, the expression levels of mdm-miR172 and four other miRNA family members, mdm-miR398, 397, 7125 and 408, were significantly higher in the A library than in the J library (Figure 5C,D). We also found that mdm-miR1511, 159, 164, 166, 167, 168, 171, 390, 191, 395, 403, 483, 7121, 7122, 7123 and 7126 showed no significant differences in expression levels between the A and J libraries (Additional file 4). The different expression patterns of known miRNAs in the A and J libraries may reflect a divergence in their potential biological functions during the phase transition from vegetative growth to reproductive growth.
Figure 5.
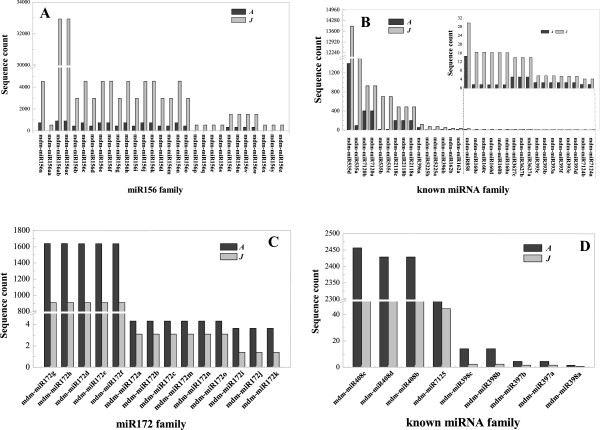
Differentially expressed known miRNA families from libraries of adult and juvenile Malus hupehensis leaves. The differentially expressed known miRNAs are shown for the A and J libraries. The expression levels are given as sequence counts. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. miR156 family numbers (A) and other known miRNA family numbers (B) expressed higher in J than in A; miR172 family numbers (C) and other known miRNA family numbers (D) expressed higher in A than in J.
Putative novel miRNA in M. hupehensis
The reference genome sequences of the domesticated apple (Malus × domestica Borkh.) were used to predict potential novel miRNAs. In this study, several miRNA characteristics, including the miRNA precursor’s hairpin structure, which was predicted by the software Mireap (http://sourceforge.net/projects/mireap/), the Dicer cleavage site and the minimum free energy of the unannotated sRNA tags, which could be mapped to the genome, were used to identify putative novel miRNAs. We also used 10 reads per million as a cutoff to eliminate miRNAs with low expression levels. We identified 172 putative unique M. hupehensis (Pamp.) Rehd. miRNAs in the A and J sRNA libraries (Additional file 5). The lengths of the predicted novel miRNA hairpin structures ranged from 61 to 242 nt. We also found that a majority of identified novel miRNA sequences were at the 5′-ends of the hairpins rather than the 3′-ends (Additional file 5).
Of the 172 novel miRNAs, 31 were highly expressed in at least one library (more than 100 reads per million), and novel_miR22, novel_miR276 and novel_miR275 were the most highly expressed novel miRNAs (Additional file 6). When comparing the expression levels of these novel miRNAs between the A and J libraries, some of the putative novel miRNAs showed distinctive expression profiles. For example, 42 were expressed only in the J library, including novel_miR479, 485, and 388 (Figure 6A). The expression of another 48, including novel_miR127, 144, 204, 312, 275, 254 and 282, were detected only in the A library (Figure 6C). Additionally, 21 novel miRNAs, including novel_miR116, 302, 320, 149, 123, 207 and 262, were expressed significantly higher in the J library than in the A library (Figure 6B); however, the expression levels of novel_miR82, 249, 27, 326, 292 and 326 were significantly lower in the J library than in the A library (Figure 6D). We also found that another 52 novel miRNAs, including novel_miR343, 253, 133, 273, 128 130, 260, 17, 244 and 290, were similarly expressed in both the A and J libraries (Additional file 5). In summary, our results showed that the known and novel miRNAs presented highly diverse expression patterns between the libraries, indicating that they may play different roles in phase-associated biological processes.
Figure 6.
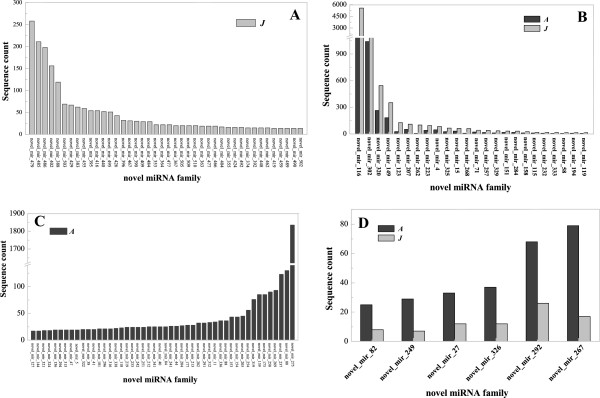
The differentially expressed novel miRNAs from libraries of adult and juvenile Malus hupehensis leaves. The differentially expressed novel miRNAs are shown for the A and J libraries. The expression levels are given as sequence counts. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. Novel miRNA family numbers expressed in J (A) and expressed higher in J than in A (B); novel miRNA family numbers expressed in A (C) and expressed higher in A than in J (D).
Targets of known and novel miRNAs
To explore the functions of the identified miRNAs, known and novel, in diverse biological processes and obtain insights into the mechanisms of the juvenile to adult transition in apple trees, we identified their targets by degradome sequencing (Tables 3 and 4; Additional file 1). In total, 127 targets of 25 known miRNA families, including mdm-miR156, mdm-miR159, mdm-miR166 and mdm-miR172, were detected in our library (Table 3; Additional file 6). The distributions of known miRNAs and their target cleavage sites are shown in an Additional file 6. We also detected 168 unique targets of 35 unique novel miRNAs (Table 4; Additional file 7). The distributions of novel miRNAs and their target cleavage sites are shown in an Additional file 7. In most cases, a single miRNA regulated multiple targets. For example, mdm-miR156 regulated 15 genes, including the SBP domain, SPL2, SPL9 and acyl-CoA synthetase5. However, in some cases, single genes were targeted by several miRNAs. The MYB domain protein 65 was targeted by both mdm-miR159 and mdm-miR319, and the autoinhibited Ca2 + −ATPase was regulated by both mdm-miR858 and mdm-miR3627. The known miRNA targets included some TFs, including SPL2 (mdm-miR156), SPL9 (mdm-miR156), ARF16 (mdm-miR160) and MYB5 (mdm-miR858), and others contained several regulatory proteins, including the LETM1-LIKE protein (mdm-miR162), AUX signaling F-box 2 protein (mdm-miR393) and the AT hook motif DNA-binding family protein (mdm-miR3627) (Table 3). We also identified several novel miRNA targets, including some regulatory proteins and some TFs. For example, novel_miR329, novel_miR157 and novel_miR413 targeted genes encoding a C2H2-like zinc finger protein, the MYB domain protein105 and a SBP family protein, respectively. Additionally, novel_miR169 targeted the RING/U-box and integrase-type DNA-binding superfamily proteins (Table 4).
Table 3.
Potential targets of the identified known miRNAs from Malus hupehensis leaves by degradome analysis
| miRNA | Target protein | Target function | At Locus | Accession ID |
|---|---|---|---|---|
| mdm-miR156 | Squamosa promoter-binding protein-like | transcription factor | AT1G69170.1 | MDP0000119458 ;MDP0000778465;MDP0000589558 |
| (SBP domain) transcription factor family protein | MDP0000142582; MDP0000146640; MDP0000171877 | |||
| MDP0000176265 ;MDP0000193702; MDP0000210138 | ||||
| MDP0000246046 ;MDP0000263766 | ||||
| mdm-miR156 | squamosa promoter binding protein-like 2(SPL2) | transcription factor | AT5G43270.1 | MDP0000155354 |
| mdm-miR156 | squamosa promoter binding protein-like 9(SPL9) | transcription factor | AT2G42200.1 | MDP0000297978;MDP0000322647 |
| mdm-miR156 | acyl-CoA synthetase 5 | AT1G62940.1 | MDP0000249364 | |
| mdm-miR159 | myb domain protein 65 | transcription factor | AT3G11440.1 | MDP0000147309 |
| mdm-miR160 | auxin response factor 16 | transcription factor | AT4G30080.1 | MDP0000131481; MDP0000221322; MDP0000750392 |
| mdm-miR160 | auxin response factor 17 | transcription factor | AT1G77850.1 | MDP0000232116; MDP0000256621 |
| mdm-miR160 | Co-chaperone GrpE family protein | AT1G36390.1 | MDP0000273491 | |
| mdm-miR162 | LETM1-LIKE protein | AT3G11560.2 | MDP0000187512 | |
| mdm-miR164 | NAC domain containing protein 1 | AT1G56010.2 | MDP0000298182; MDP0000528658 | |
| mdm-miR166 | homeobox gene 8 | AT4G32880.1 | MDP0000005879; MDP0000126553 | |
| mdm-miR166 | Homeobox-leucine zipper family protein/ lipid- | AT2G34710.1 | MDP0000050082;MDP0000943529 | |
| binding START domain-containing protein | AT5G60690.1 | MDP0000236500; MDP0000242861; MDP0000426630 | ||
| AT1G52150.3 | MDP0000251484 | |||
| AT1G52150.1 | MDP0000313059 | |||
| mdm-miR167 | auxin response factor 6 | transcription factor | AT1G30330.2 | MDP0000319957; MDP0000550049 |
| AT1G30330.1 | MDP0000153538 | |||
| mdm-miR167 | auxin response factor 8 | transcription factor | AT5G37020.1 | MDP0000137461; MDP0000232417 |
| mdm-miR167 | auxin response factor 19 | transcription factor | AT1G19220.1 | MDP0000268306 |
| mdm-miR168 | Stabilizer of iron transporter SufD/ Polynucleotidyl transferase | AT1G48410.1 | MDP0000069525; MDP0000161046; MDP0000305971 | |
| mdm-miR169 | nuclear factor Y, subunit A7 | transcription factor | AT1G30500.2 | MDP0000164531; MDP0000183865 |
| AT1G30500.1 | MDP0000279028 | |||
| mdm-miR169 | nuclear factor Y, subunit A9 | transcription factor | AT3G20910.1 | MDP0000146933 |
| mdm-miR169 | translocation protein-related | AT5G12840.1 | MDP0000296077 | |
| mdm-miR171 | GRAS family transcription factor | transcription factor | AT4G00150.1 | MDP0000151144 ;MDP0000275704 ;MDP0000274120 |
| AT2G45160.1 | MDP0000784909 | |||
| mdm-miR172 | related to AP2.7 | transcription factor | AT2G28550.3 | MDP0000163645; MDP0000181606; MDP0000200319; |
| MDP0000296716 | ||||
| mdm-miR172 | Integrase-type DNA-binding superfamily protein | transcription factor | AT4G36920.1 | MDP0000137561 |
| mdm-miR319 | myb domain protein 65 | transcription factor | AT3G11440.1 | MDP0000147309 |
| mdm-miR3627 | AT hook motif DNA-binding family protein | AT2G45850.1 | MDP0000133746;MDP0000231744 | |
| mdm-miR3627 | autoinhibited Ca2+ − ATPase, isoform 8 | AT5G57110.1 | MDP0000258197 | |
| mdm-miR390 | exocyst subunit exo70 family protein H7 | AT5G59730.1 | MDP0000145463 | |
| mdm-miR391 | Transmembrane proteins 14C | AT3G43520.1 | MDP0000207199; MDP0000244081 | |
| mdm-miR393 | F-box/RNI-like superfamily protein | AT3G62980.1 | MDP0000125975; MDP0000498419 | |
| mdm-miR393 | auxin signaling F-box 2 | AT3G26810.1 | MDP0000203334; MDP0000469943 | |
| mdm-miR393 | auxin signaling F-box 3 | AT1G12820.1 | MDP0000268652 | |
| mdm-miR395 | ATP sulfurylase 1 | AT3G22890.1 | MDP0000263161 | |
| mdm-miR398 | DC1 domain-containing protein | AT1G60420.1 | MDP0000152817 ;MDP0000308890 | |
| mdm-miR398 | GroES-like zinc-binding dehydrogenase family protein | AT5G43940.1 | MDP0000193167 | |
| mdm-miR5225 | autoinhibited Ca2+ − ATPase, isoform 8 | AT5G57110.1 | MDP0000258197 | |
| mdm-miR535 | Papain family cysteine protease(RD19) | AT4G39090.1 | MDP0000189200 | |
| mdm-miR858 | myb domain protein 3 | transcription factor | AT1G22640.1 | MDP0000184538 |
| mdm-miR858 | myb domain protein 4 | transcription factor | AT4G38620.1 | MDP0000031172 |
| mdm-miR858 | myb domain protein 5(MYB5) | transcription factor | AT3G13540.1 | MDP0000133817; MDP0000143276; |
| MDP0000226215;MDP0000253904 | ||||
| mdm-miR858 | myb domain protein 7 | transcription factor | AT2G16720.1 | MDP0000210851 |
| mdm-miR858 | myb domain protein 12 | transcription factor | AT2G47460.1 | MDP0000140609 ;MDP0000887107 |
| mdm-miR858 | myb domain protein 66 | AT5G14750.1 | MDP0000124555 | |
| mdm-miR858 | Duplicated homeodomain-like superfamily protein | AT5G35550.1 | MDP0000318013; MDP0000437717 | |
| mdm-miR858 | high response to osmotic stress 10 | AT1G35515.1 | MDP0000931057 |
Table 4.
Potential targets of the identified novel miRNAs from Malus hupehensis leaves by degradome analysis
| miRNA | Target protein | Target function | At Locus | Accession ID |
|---|---|---|---|---|
| novel_mir_106 | Late embryogenesis abundant (LEA) hydroxyproline rich glycoprotein family | AT2G46150.1 | MDP0000405151 | |
| novel_mir_108 | RNA-binding (RRM/RBD/RNP motifs) family protein | AT2G44710.1 | MDP0000242219;MDP0000303270 | |
| novel_mir_11 | Flavin-containing monooxygenase family protein | AT1G48910.1 | MDP0000138851;MDP0000208234 | |
| novel_mir_157 | myb domain protein 105 | transcription factor | AT1G69560.1 | MDP0000136541; MDP0000146675 |
| novel_mir_116 | autoinhibited Ca2+ − ATPase, isoform 8 | AT5G57110.1 | MDP0000258197 | |
| novel_mir_156 | phosphate transporter 1;4 | AT2G38940.1 | MDP0000141330; MDP0000523104 | |
| novel_mir_156 | phosphate transporter 1;7 | AT3G54700.1 | MDP0000746621 | |
| novel_mir_160 | Transmembrane proteins 14C | AT3G43520.1 | MDP0000207199; MDP0000244081 | |
| novel_mir_169 | RING/U-box superfamily protein | AT3G05200.1 | MDP0000909888 | |
| novel_mir_169 | Integrase-type DNA-binding superfamily protein | AT3G23230.1 | MDP0000930655 | |
| novel_mir_207 | phytosulfokine 4 precursor | AT3G49780.1 | MDP0000145144 ;MDP0000509438; MDP0000824044 | |
| novel_mir_261 | cell elongation protein / dwarf1 / diminuto (dim) | AT3G19820.1 | MDP0000278275; MDP0000682675 | |
| novel_mir_262 | nuclear factor Y, subunit A9 | transcription factor | AT3G20910.1 | MDP0000146933 |
| novel_mir_262 | nuclear factor Y, subunit A7 | transcription factor | AT1G30500.2 | MDP0000164531 ;MDP0000183865 |
| AT1G30500.1 | MDP0000279028 | |||
| novel_mir_262 | nuclear factor Y, subunit A1 | transcription factor | AT5G12840.1 | MDP0000296077 |
| novel_mir_268 | nuclear factor Y, subunit A7 | transcription factor | AT1G30500.2 | MDP0000164531 ;MDP0000183865 |
| AT1G30500.1 | MDP0000279028 | |||
| novel_mir_27 | plasmodesmata-located protein 2 | AT1G04520.1 | MDP0000412849 | |
| novel_mir_301 | phospholipid/glycerol acyltransferase family protein | AT1G32200.1 | MDP0000171689 ;MDP0000532750 | |
| novel_mir_335 | SOS3-interacting protein 4 | AT2G30360.1 | MDP0000127732; MDP0000146449 | |
| novel_mir_351 | response regulator 2 | AT4G16110.1 | MDP0000228719 | |
| novel_mir_37 | Translation initiation factor IF6 | AT3G55620.1 | MDP0000247249 | |
| novel_mir_374 | S-adenosyl-l-homocysteine (SAH) hydrolase 2 | AT3G23810.1 | MDP0000212365; MDP0000679173 | |
| novel_mir_378 | ATP binding; valine-tRNA ligases; nucleotide binding; aminoacyl-tRNA ligases | AT5G16715.1 | MDP0000155593; MDP0000238240 | |
| novel_mir_384 | S-adenosyl-l-homocysteine (SAH) hydrolase 2 | AT3G23810.1 | MDP0000212365; MDP0000679173 | |
| novel_mir_392 | nuclear factor Y, subunit A9 | transcription factor | AT3G20910.1 | MDP0000146933 |
| novel_mir_392 | nuclear factor Y, subunit A7 | transcription factor | AT1G30500.2 | MDP0000164531; MDP0000183865 |
| AT1G30500.1 | MDP0000279028 | |||
| novel_mir_392 | nuclear factor Y, subunit A1 | transcription factor | AT5G12840.1 | MDP0000296077 |
| novel_mir_422 | related to AP2 4 | transcription factor | AT1G78080.1 | MDP0000401140; MDP0000633218 |
| novel_mir_446 | S-methyl-5-thioribose kinase | AT1G49820.1 | MDP0000148984; MDP0000278395; MDP0000234656 | |
| novel_mir_477 | aldehyde dehydrogenase 2B4 | AT3G48000.1 | MDP0000159395; MDP0000221713 | |
| novel_mir_477 | 5\′-3\′ exonuclease family protein | AT1G34380.2 | MDP0000259472 | |
| novel_mir_477 | OPC-8:0 CoA ligase1 | AT1G20510.1 | MDP0000716496 | |
| novel_mir_486 | phospholipase D delta | AT4G35790.2 | MDP0000125742 | |
| novel_mir_486 | zinc knuckle (CCHC-type) family protein | AT5G43630.1 | MDP0000147872; MDP0000196131 | |
| novel_mir_492 | BRI1-associated receptor kinase | AT4G33430.1 | MDP0000287771; MDP0000291093 ;MDP0000309283 | |
| novel_mir_495 | ENTH/VHS/GAT family protein | AT1G06210.1 | MDP0000320808 | |
| novel_mir_506 | GDSL-like Lipase/Acylhydrolase superfamily protein | AT3G26430.1 | MDP0000182713 | |
| novel_mir_89 | NB-ARC domain-containing disease resistance protein | AT3G07040.1 | MDP0000137113; MDP0000142444 ;MDP0000249156 | |
| MDP0000662922; MDP0000242361 | ||||
| novel_mir_89 | NB-ARC domain-containing disease resistance protein | AT3G14470.1 | MDP0000206335; MDP0000241462 ;MDP0000243301 | |
| novel_mir_89 | LRR and NB-ARC domains-containing disease resistance protein | AT3G14460.1 | MDP0000196621 | |
| novel_mir_413 | Squamosa promoter-binding protein-like (SBP domain) transcription factor family protein | transcription factor | AT1G69170.1 | MDP0000119458; MDP0000146640; MDP0000171877 |
| MDP0000193702; MDP0000246046 ;MDP0000589558 | ||||
| MDP0000778465 | ||||
| AT5G50670.1 | MDP0000142582; MDP0000176265; MDP0000210138 | |||
| MDP0000263766 | ||||
| novel_mir_329 | myb domain protein 105 | transcription factor | AT1G69560.1 | MDP0000146675 |
| novel_mir_329 | C2H2-like zinc finger protein | AT1G75710.1 | MDP0000179049 | |
| novel_mir_329 | Frigida-like protein | AT5G27220.1 | MDP0000179887 | |
| novel_mir_329 | RNA-binding protein | AT2G43970.1 | MDP0000266270 |
GO and KEGG analyses of the degradome predicted target genes
A total of 58 GO terms from various biological processes were identified (Additional file 8). A total of 123 known miRNA targets were associated with processes such as the growth, regulation of developmental processes, hormone-mediated signaling pathways, and organ, flower and reproductive developmental processes, which are thought to be associated with the juvenile to adult transition. To better understand their biological functions, we also identified 44 GO terms for the predicted targets of the putative novel miRNAs (Additional file 9). They were associated with plant growth and development.
The GO analysis also revealed that the known miRNAs’ potential targets were associated with the biological processes of the juvenile to adult transition (Table 5). For example, the targets of mdm-miR156 and mdm-miR160 were associated with growth and flower development in plants. The targets of mdm-miR160 and mdm-miR393 were associated with AUX- and hormone-mediated signaling pathways, which play important roles in the reproductive growth of plants.
Table 5.
GO analyses showing that miRNAs from Malus hupehensis leaves potentially target the juvenile to adult transition-related biological processes
| miRNAs | GO biological process | GO ID | P value | Targets | Target number |
|---|---|---|---|---|---|
| 156 ,160,162,398 | growth | GO:0040007 | 0.0005 | MDP0000146640,MDP0000155354,MDP0000131481,MDP0000221322; | 8 |
| MDP0000256621, MDP0000187512, MDP0000308890,MDP0000193167 | |||||
| 166,167,393 | developmental maturation | GO:0021700 | 2.60E-20 | MDP0000005879; MDP0000126553,MDP0000319957,MDP0000137461 | 7 |
| MDP0000125975,MDP0000203334,MDP0000268652 | |||||
| 162,166,398 | regulation of developmental growth | GO:0048638 | 1.20E-12 | MDP0000187512,MDP0000943529,MDP0000426630,MDP0000308890 | 5 |
| MDP0000193196 | |||||
| 156, 162,166 | developmental growth | GO:0048589 | 1.10E-09 | MDP0000119458,MDP0000155354,MDP0000249364,MDP0000187512 | 5 |
| MDP0000005879 | |||||
| 160,164,167,393 | auxin mediated signaling pathway | GO:0009734 | 1.90E-18 | MDP0000131481,MDP0000221322,MDP0000256621,MDP0000298182; | 8 |
| MDP0000528658,MDP0000319957,MDP0000125975,MDP0000203334 | |||||
| 159,160, 393 | hormone-mediated signaling pathway | GO:0009755 | 5.10E-10 | MDP0000147309,MDP0000131481,MDP0000221322,MDP0000125975, | 6 |
| MDP0000203334,MDP0000268652 | |||||
| 159,160,399,858 | response to abscisic acid stimulus | GO:0009737 | 3.0E-10 | MDP0000147309,MDP0000131481,MDP0000256621,MDP0000143276 | 6 |
| MDP0000166425,MDP0000318013 | |||||
| 164,166,167,393 | response to auxin stimulus | GO:0009733 | 3.10E-11 | MDP0000298182,MDP0000005879,MDP0000319957,MDP0000125975 | 6 |
| MDP0000203334,MDP0000268652 | |||||
| 156,169,172,393 | flower development | GO:0009908 | 6.30E-22 | MDP0000146640,MDP0000155354,MDP0000297978,MDP0000164531, | 9 |
| MDP0000296716,MDP0000137561,MDP0000125975,MDP0000203334 | |||||
| MDP0000268652 | |||||
| 160,166,167 | floral organ development | GO:0048437 | 2.00E-23 | MDP0000131481,MDP0000221322,MDP0000256621,MDP0000153538 | 5 |
| MDP0000268306 | |||||
| 156,393,858 | androecium development | GO:0048466 | 1.60E-10 | MDP0000146640,MDP0000155354,MDP0000297978,MDP0000249364, | 14 |
| MDP0000125975,MDP0000203334,MDP0000268652,MDP0000143276 | |||||
| MDP0000140609.MDP0000931057 | |||||
| 156,393,858 | stamen development | GO:0048443 | 1.60E-10 | MDP0000146640,MDP0000155354,MDP0000297978,MDP0000249364, | 14 |
| MDP0000125975,MDP0000203334,MDP0000268652,MDP0000143276 | |||||
| MDP0000140609.MDP0000931057 | |||||
| 172 | specification of floral organ identity | GO:0010093 | 6.90E-06 | MDP0000163645,MDP0000181606, MDP0000200319,MDP0000296716 | 5 |
| MDP0000137561 | |||||
| 156 | anther development | GO:0048653 | 2.40E-05 | MDP0000146640; MDP0000171877,MDP0000155354,MDP0000297978 | 6 |
| MDP0000322647,MDP0000249364 | |||||
| 172 | floral organ formation | GO:0048449 | 0.00099 | MDP0000163645,MDP0000181606, MDP0000200319,MDP0000296716 | 5 |
| MDP0000137561 | |||||
| 172 | floral organ morphogenesis | GO:0048444 | 0.0038 | MDP0000163645,MDP0000181606, MDP0000200319,MDP0000296716 | 5 |
| MDP0000137561 | |||||
| 166,167,169,858 | regulation of flower development | GO:0009909 | 0.00012 | MDP0000050082,MDP0000319957,MDP0000164531,MDP0000143276 | 5 |
| MDP0000887107 | |||||
| 156,160,164,166, | reproductive structure development | GO:0048608 | 8.20E-17 | MDP0000146640,MDP0000155354,MDP0000297978,MDP0000131481, | 8 |
| MDP0000221322,MDP0000256621,MDP0000298182,MDP0000005879 | |||||
| 167,168,169,172 | reproductive process | GO:0022414 | 2.70E-13 | MDP0000153538,MDP0000137461, MDP0000232417,MDP0000069525, | 7 |
| MDP0000161046,MDP0000164531,MDP0000296716 | |||||
| 3627,393,398,858 | reproductive developmental process | GO:0003006 | 3.70E-17 | MDP0000133746,MDP0000258197,MDP0000125975,MDP0000203334 | 8 |
| MDP0000268652,MDP0000308890,MDP0000193167,MDP0000143276 | |||||
| 159,172 | sexual reproduction | GO:0019953 | 0.14 | MDP0000147309,MDP0000163645,MDP0000181606,MDP0000200319, | 6 |
| MDP0000296716,MDP0000137561 | |||||
| 172 | specification of organ identity | GO:0010092 | 6.90E-06 | MDP0000163645,MDP0000181606, MDP0000200319,MDP0000296716 | 5 |
| MDP0000137561 | |||||
| 160,162,164 | organ development | GO:0048513 | 3.10E-26 | MDP0000131481, MDP0000221322,MDP0000256621,MDP0000273491, | 7 |
| MDP0000187512,MDP0000298182, MDP0000528658 | |||||
| 156,159, 858 | leaf development | GO:0048366 | 5.90E-12 | MDP0000146640,MDP0000155354,MDP0000297978,MDP0000249364, | 6 |
| MDP0000147309, MDP0000143276 | |||||
| 156, 3627,393 | shoot development | GO:0048367 | 4.60E-13 | MDP0000155354,MDP0000297978,MDP0000133746,MDP0000258197, | 7 |
| MDP0000125975,MDP0000203334,MDP0000268652 | |||||
| 159,166, 858 | leaf morphogenesis | GO:0009965 | 4.60E-13 | MDP0000147309,MDP0000005879,MDP0000050082,MDP0000133817, | 5 |
| MDP0000143276 | |||||
| 166,167,168 | xylem and phloem pattern formation | GO:0010051 | 2.70E-14 | MDP0000005879,MDP0000319957,MDP0000153538,MDP0000137461, | 5 |
| MDP0000069525 | |||||
| 160, 393,828 | root development | GO:0048364 | 7.60E-22 | MDP0000131481, MDP0000221322,MDP0000256621,MDP0000125975 | 8 |
| MDP0000203334,MDP0000268652,MDP0000143276,MDP0000253904 | |||||
| 169,172,858 | fruit development | GO:0010154 | 6.50E-06 | MDP0000296077,MDP0000296716,MDP0000137561,MDP0000143276, | 6 |
| MDP0000140609,MDP0000887107 | |||||
| 156,159,160 | phyllome development | GO:0048827 | 1.80E-11 | MDP0000155354,MDP0000297978,MDP0000249364,MDP0000147309 | 5 |
| MDP0000778465 | |||||
| 156,162, 858 | tissue development | GO:0009888 | 5.30E-21 | MDP0000146640,MDP0000155354,MDP0000297978,MDP0000187512, | 5 |
| MDP0000143276 | |||||
| 164, 168,172 | meristem development | GO:0048507 | 6.30E-27 | MDP0000298182, MDP0000528658,MDP0000069525,MDP0000161046, | 6 |
| MDP0000296716,MDP0000137561 | |||||
| 156,162,166 | regulation of meristem development | GO:0048509 | 1.30E-08 | MDP0000146640,MDP0000155354,MDP0000297978,MDP0000187512 | 6 |
| MDP0000005879,MDP0000126553 | |||||
| 164, 167,168 | meristem structural organization | GO:0009933 | 8.40E-15 | MDP0000298182,MDP0000550049,MDP0000232417,MDP0000069525, | 5 |
| MDP0000305971 | |||||
| 162,166 | meristem growth | GO:0035266 | 3.40E-10 | MDP0000187512,MDP0000298182, MDP0000528658,MDP0000050082, | 5 |
| MDP0000943529 |
A total of 26 KEGG pathways were enriched for targets of known miRNAs (Additional file 10). The categories of plant hormone signal transduction and metabolic pathways contained the most targets, at 14.93%. The majority of the targets were involved in starch and sucrose metabolism, and plant hormone signal transduction, which play important roles in plant growth and development. A few of these targets were involved in the p53 signaling pathway, glycan degradation and purine metabolism, which may regulate metabolism and synthesis in plants. Additionally, we found that 13 of the 108 pathways containing targets of novel miRNAs were detected at significantly high abundance levels (p < 0.05) (Additional file 11). The majority of novel miRNA targets were associated with RNA polymerase or involved in plant-pathogen interactions and peptidoglycan biosynthesis.
Identification by qRT-PCR of differentially expressed miRNAs and their targets in A and J leaves
To examine the expression levels of miRNAs and their targets during different developmental leaf tissue stages (3, 4, 5, 6, 7 and 8 months) between the A and J samples, as well as to confirm the sequencing results, we examined expression levels of 11 miRNAs and 16 targets by qRT-PCR (Figure 7). The up-regulation in J compared with A was confirmed for mdm-miR156 during leaf development (Figure 7A), while its targets, SPL2 and SPL9, showed higher expression levels in A than in J from March to August, with their levels gradually increasing (Figure 7A). The qRT-PCR experiments also validated the deep-sequencing results of the down-regulation in J compared with A leaves for mdm-miR172, mdm-miR398a and mdm-miR398a in April, May and June (Figure 7B,H and I). The expression levels of mdm-miR172 targets AP2 and AP2-like were significantly higher in J than in A leaves during early leaf development (from March to May); however, they were relatively low later (from June to August) (Figure 7B). The DC19 (mdm-miR398a’s target) and ADH2 (mdm-miR398b’s target) gene patterns were similar to those of AP2 and AP2-like (Figure 7 H and I). mdm-miR160 and mdm-miR393 were up-regulated in J compared with A during the early leaf development stage (from March to June), while most of their targets showed significantly higher expression levels in A than in J. Additionally, we found that AFB2 and AFB3 (mdm-miR393’s targets) were detected in A but were almost undetectable in J leaves (May and June) (Figure 7C). The expression of TIR1 (mdm-miR393’s target) was higher in A than in J during April, May and August, but was lower in A than in J during July and August (Figure 7C).
Figure 7.
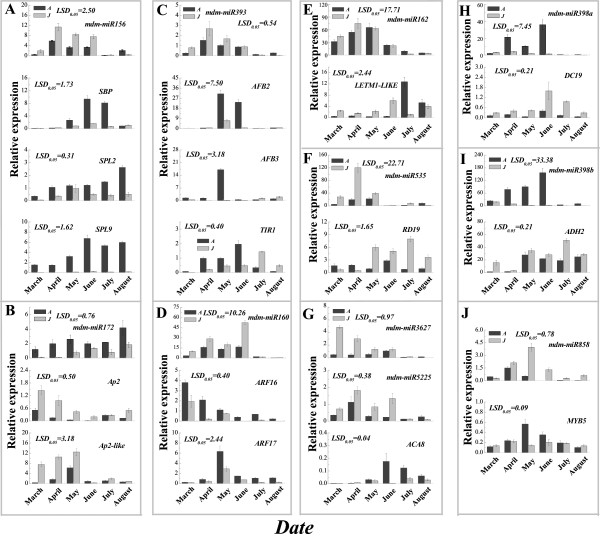
Identification by qRT-PCR of miRNA and target expression patterns in Malus hupehensis leaves. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. The expression of miRNAs and their targets in A and J leaves: mdm-miR156 (A); mdm-miR172 (B); mdm-miR393 (C); mdm-miR160 (D); mdm-miR162 (E); mdm-miR535 (F); mdm-miR3627and mdm-miR5225 (G); mdm-miR398a (H); mdm-miR398b (I); and mdm-miR858 (J).
Hierarchical clustering of known miRNAs and targets by expression levels in the A and J leaves of M. hupehensis resulted in six and five major clusters, respectively (Additional file 12). The miRNAs within cluster 1 (miR172 for A and J) typically displayed high expression levels during the later stages of leaf development (August), but miRNAs within cluster 6 (miR156, 169, 393 and 858 for A and miR156 and 5225 for J) displayed opposite results, with high expression levels during the early stages (April and May) (Figure 7 and Additional file 12A). Additionally, the targets of cluster 1 (AP2 and ARF16 for A and J, respectively) displayed high expression levels in the early stages of leaf development (March to April); however, the cluster 5 targets (such as SPL9, SBP and TIR1) showed the opposite results, with higher expression levels in later stages (July and August) (Figure 7 and Additional file 12B). These data suggest that the expression patterns of miRNAs and their targets, such as miR156 and SPL9, display opposite trends during leaf development.
Identification by qRT-PCR of miRNA and target expression patterns in leaves of different ages
We validated miRNA and target expression profiles in leaves from the tops of trees of different ages (1, 2, 3, 4, 5 and 6 years old) via qRT-PCR (Figure 8 and Additional file 15). mdm-miR156 had significantly higher expression levels in younger (except 1-year-olds) than in older tree leaves (4, 5, and 6 years old) (Figure 8A), whereas its targets (SBP, SPL2 and SPL9) had relatively higher expression levels in older tree leaves (5 and 6 years old) than in younger tree leaves (1 and 2 years old) (Figure 8B). Compared with the expression profile of mdm-miRNA156, mdm-miR172 showed a high expression level in older tree leaves (4-, 5- and 6-years-old) and this expression increased gradually in 1- to 6-year-olds (Figure 8A,B). A perfect inverse expression pattern was found for its targets, AP2 and AP2-like genes, which were expressed higher in younger than in older tree leaves (Figure 8B). Mdm-miR160 exhibited strong expression in 1-year-old leaves and the expression decreased gradually in 1- to 6-year-olds, whereas its targets’ (ARF16 and ARF17) expression increased to their highest levels in 4-year-olds but then decreased to relatively low levels in 5- and 6-year-olds (Figure 8D). The expression of mdm-miR393 could barely be detected in older tree leaves (4-, 5- and 6-year-olds) but was relatively high in young tree leaves (1, 2 and 3 years old) (Figure 8C). Interestingly, its targets (AFB2, AFB3 and TIR1) had an inverse expression pattern that was higher in older leaves (4 and 5 years old) (Figure 8C).
Figure 8.
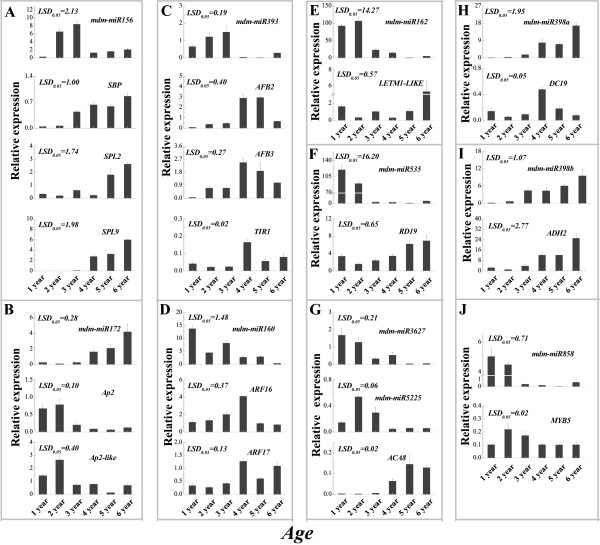
Identification by qRT-PCR of miRNA and target expression patterns in Malus hupehensis leaves of different ages. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. The expression of miRNAs and their targets in leaves of different ages: mdm-miR156 (A); mdm-miR172 (B); mdm-miR393 (C); mdm-miR160 (D); mdm-miR162 (E); mdm-miR535 (F); mdm-miR3627 and mdm-miR5225 (G); mdm-miR398a (H); mdm-miR398b (I); and mdm-miR858 (J).
Additionally, the hierarchical cluster analysis resulted in these known miRNAs and target genes forming three major clusters each, and it also showed that the expression levels of most known miRNAs were inversely correlated with those of the corresponding targets among leaves of different ages (Additional file 13).
Identification by qRT-PCR of miRNA and target expression patterns in different tissues
Tissue-specific expression and hierarchical cluster analyses, which allowed known miRNAs and their target genes into three major clusters each (Additional file 14),revealed that miRNAs and their targets in different tissues (roots, stem, flower, leaves and fruit) presented a variety of expression patterns (Figure 9). The expression patterns of these miRNAs and their targets could be divided into four types: (1) mdm-miR156, mdm-miR160, mdm-miR535 and their targets, the SBP, AP2, AP2-like, ARF16, AFB, DC19 and RD19 genes, had the highest expression levels in roots but relatively low expression levels in fruit (Figure 9A,B,D,F and H); (2) mdm-miR393, mdm-miR398a, mdm-miR398b and their targets, the SPL2, SPL9 and ACA8 genes, were found to be expressed most abundantly in flowers but had relatively low expression levels in fruit and roots (Figure 9A,C,E,F,G and H); (3) mdm-miR172, mdm-miR162, mdm-miR162, mdm-miR5225 and their targets, the ARF17, LETM1-LIKE and ADH2 genes, showed high expression levels in leaf tissue but relatively low levels were observed in stems (Figure 9B,D,E,G and I); (4) mdm-miR858 and mdm-miR3627 and their targets, the TIR1 and MYB5 genes, had high expression levels in fruit but low levels in stems (Figure 9C,G and J).
Figure 9.
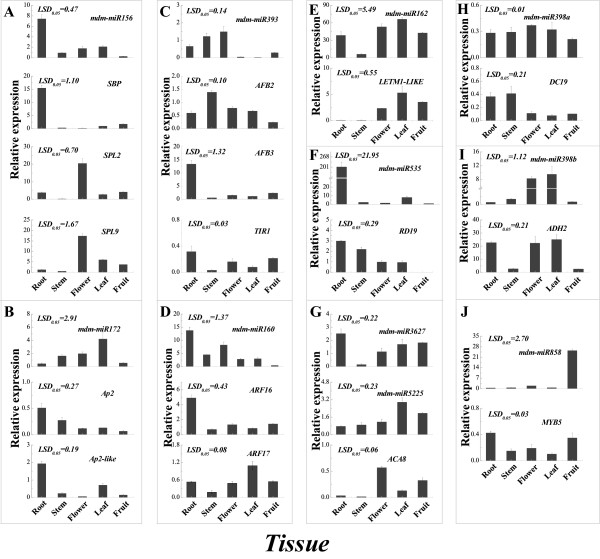
Identification by qRT-PCR of miRNA and target expression patterns in different Malus hupehensis ’ tissues. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. Expression of miRNAs and their targets in different tissue: mdm-miR156 (A); mdm-miR172 (B); mdm-miR393 (C); mdm-miR160 (D); mdm-miR162 (E); mdm-miR535 (F); mdm-miR3627 and mdm-miR5225 (G); mdm-miR398a (H); mdm-miR398b (I); and mdm-miR858 (J).
Identification of novel miRNA expression patterns in M. hupehensisby qRT-PCR
We have verified the expression of some novel miRNAs by qRT-PCR in A and J leaves of M. hupehensis, as well as leaves of different ages and different tissues (Additional file 15). Novel-miR486 and novel-miR492 were detected as up-regulated in J compared with A from April to June, while novel-miR207 and novel-miR329 showed significantly higher expression levels in J than in A from May to June. We also found that novel-miR207, 329, 486 and 492 had relatively higher expression levels in leaves from young trees than from older trees (Additional files 15 and 16). A tissue-specific expression analysis revealed that novel-miR329 and 492 expressed at a higher level in flower, while novel-miR486 had the highest expression in root (Additional files 15 and 16).
Leaf morphology characteristics and hormone content analysis
To determine the characteristics of A and J during leaf development, we measured leaf length, width and area, and dry weight from March to August, all of which were significantly higher in A compared with J (Figure 10). Additionally, the leaf AUX content increased from March to July and then decreased in August for A. The pattern of AUX expression for J had a similar trend to A, but reached a peak in May, and the AUX content was significantly higher in A than in J during later leaf growth stages (May to August) (Figure 11A). The leaf CK content decreased gradually from March to August for both A and J, and was significantly higher in A than in J from March to June. However, the CK content was almost undetectable during July and August in both A and J leaves (Figure 11C). The leaf GA content was significantly higher in J than in A from April to May, but there was no significant difference at any other time (Figure 11B). The leaf ABA content was significantly higher in A than in J from March to May, but this was reversed in July and August (Figure 11D). The different levels of hormones between A and J leaves indicated that hormones play a vital role in leaf growth and development.
Figure 10.
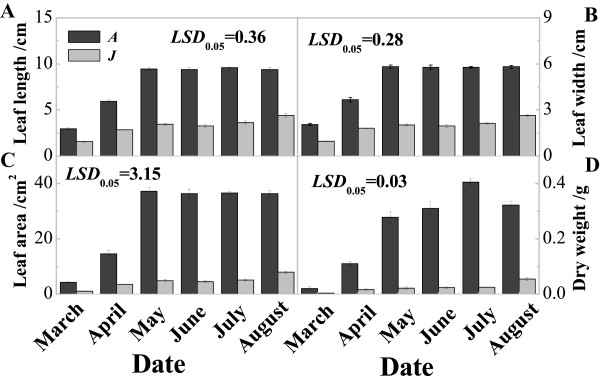
Malus hupehensis leaf characteristics in the adult and juvenile phases. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. Leaf length (A); Leaf width (B); Leaf area (C); and Dry weight (D).
Figure 11.
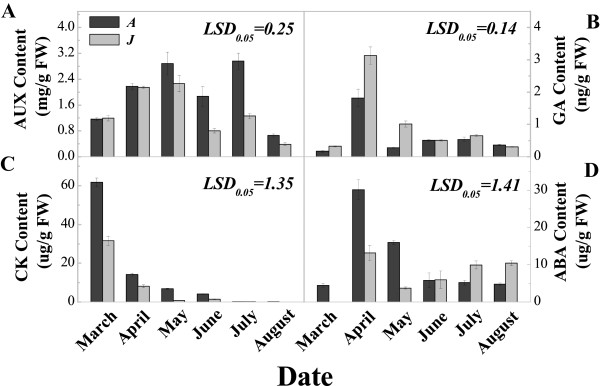
Auxin (AUX), cytokinin (CK), gibberellic acid (GA) and abscisic acid (ABA) contents in adult and juvenile Malus hupehensis leaves. A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. AUX content (A); GA content (B); CK content (C); and ABA content (D).
Discussion
Leaf morphology is very different between the juvenile phase at the base of the trees and the adult phase at the top of the trees. Our results showed that leaf traits, such as size and dry weight, were significantly higher in A than in J leaves (Figure 10), which was consistent with a previous study in A. thaliana[7]. Plant growth hormones, such as AUXs, CKs, GA and ABA, have important roles in leaf development during the transition from vegetative to reproductive growth [34]. Spraying exogenous GAs can cause a reversion from the adult to the juvenile vegetative phase, producing juvenile traits in newly formed leaves [29, 35]. The level of GA was higher in juvenile tissue than in adult tissue of trees [35], which was consistent with our results (Figure 11). In this study, AUX, CK and ABA contents were significantly higher in A than in J leaves, but the GA content showed the opposite result, indicating that hormone levels play an important role in the physiological processes surrounding the phase transition (Figure 11).
In this study, by constructing two sRNA libraries of M. hupehensis (Pamp.) Rehd. using high-throughput sequencing, we identified 207 known miRNAs that belonged to 42 miRNA families (Additional file 4). The majority were conserved in A. thaliana and peach [30, 32, 36]. Among them, 12 of 17 known miRNA families had higher expression levels in the J library than in the A library (Figure 5A and B), while others showed an opposite expression pattern (Figure 5C and D). Additionally, 172 novel miRNAs were identified based on the universal rules for novel miRNA prediction and annotation [37]. Of these, 42 were only expressed in the J library (Figure 6A) and 48 were expressed only in the A library (Figure 6C). The differential expression of known and novel miRNAs may involve various biological functions in the regulation of leaf development, phase transition and reproductive growth. Using degradome analysis, we also identified a total of 127 targets for 25 known miRNA families and 168 targets for 35 unique novel miRNAs (Additional files 6 and 7). The majority of these targets were relatively conserved in other species [1] and regulated a variety of biological processes involved in developmental growth, hormone-mediated signaling pathways, flower development and reproductive processes in plants.
Because we established only one miRNA library from juvenile leaves and one from adult leaf, our results are limited and may not include all of the differences present (Figures 3, 4, 5 and 6). A study on miRNA control of vegetative phase change in the gymnosperm Sequoia sempervirens and the identification of sRNAs also provide evidence supporting our results [1, 18].
The GO analyses revealed that miRNA targets were mainly associated with the juvenile to adult transition during plant development (Table 5). The majority of GO terms were associated with plant growth and development (GO:0040007, GO:0021700, GO:0048638 and GO:0048589), plant tissue growth and formation (GO:0010092, GO:0048366, GO:0048367, GO:0009965 and GO:0010051), meristem development (GO:0009888, GO:0048507 and GO:0009933) and hormone-mediated signaling pathways (GO:0009734, GO:0009755, GO:0009737 and GO:0009733), which are all involved in the juvenile to adult transition-associated biological processes (Table 5). The KEGG analysis also revealed that the genes targeted by known and novel miRNAs were largely involved in starch and sucrose metabolism, plant hormone signal transduction, p53 signaling and the glycan degradation pathway, which play important roles in the juvenile to adult transition and reproductive growth in plants (Additional files 10 and 11).
It was reported that miR156 regulated leaf development, showed juvenile characteristics when overexpressed in plants and had reduced expression levels in adult leaves [12, 38]. This was consistent with our results that the expression level of mdm-miR156 in the J library was significantly higher than in the A library (Figure 5). Additionally, miR156 regulated its targets, the SPL family, through translational inhibition and gene silencing in A. thaliana[39]. The overexpression of the targets SPL3, SPL9 and SPL15 resulted in increased cell numbers in leaves [7, 40]. This lead to an increased leaf size in plants overexpressing SPL3 and SPL9. Additionally, the overexpression of SPL9 and SPL15 controlled shoot maturation and leaf initiation [41]. In this study, we confirmed the up-regulation of mdm-miR156 in J leaves compared with A leaves during leaf development (Figure 7A). However, two of its targets, SPL2 and SPL9, showed higher expression levels in A than in J leaves (Figures 7A and 8A). Previous research also showed that SPL9 was expressed in the vegetative shoot apices, although the expression level of miRNA156 was almost undetectable [42]. SPL9, as well as SPL10, expression levels in the leaf primordia affect the initiation of new leaves at the shoot apical meristem [40], while expression levels of SPL2, along with SPL11, control the leaf lamina shape during shoot maturation in the reproductive phase [8]. Additionally, mdm-miR156 was highly expressed in roots but had almost no expression in flowers; however, its targets, SPL2 and SPL9, were more highly expressed in flowers than in other tissues (Figure 9A). Expression levels of miRNA156 and their targets were associated with the transition from vegetative to reproductive growth and the transition to flowering.
The increased expression of miRNA156 and decreased expression of its targets (SPLs) delayed flowering, whereas inhibiting miR156 expression accelerated flowering [43, 44]. Some genes involved in flowering were regulated by the expression levels of miRNA156 and their targets in plants. For example, miR156 regulates FLOWERING LOCUS T expression in apical meristem to control temperature-responsive flowering in A. thaliana[45]. LFY, FUL and AP1 genes were directly activated by the miRNA-targeted SPL3 to control the timing of flower formation in A. thaliana[13]. SPL9 and SPL3 also directly regulated and controlled MADS-box gene expression levels that promote flowering [40]. Our results showed that leaves in the juvenile phase were much smaller and had higher miRNA156 expression levels and lower SBP, SPL2 and SPL9 expression levels compared with adult leaves, implying that miRNA156 and its targets may play important roles in the juvenile to adult transition, leaf development and the transition to flowering.
Previous research showed that miR172 could promote flowering, but that its targets were floral repressors, such as AP2-like, AP2, EAT1 and EAT2, which play important roles in the regulation of leaf traits in A. thaliana[44, 46]. miR172 may be involved in regulating the juvenile to adult transition during developmental stages [47, 48]. We also found that the sit-miR172 family members’ expression levels were significantly higher in adult leaves and flower tissues in olives (Olea europaea L.) [47]. In our study, the expression of mdm-miR172 family members was significantly higher in the A leaf library than in the J leaf library, implying that they were active in adult stage maintenance (Figure 5C). The qRT-PCR experiments also validated the deep-sequencing results of a down-regulation of mdm-miR172 in the J library compared with the A library during leaf development (Figure 7B). However, the expression levels of the targets AP2 and AP2-like (mdm-miR172 targets) were significantly higher in the J than in the A leaves from March to May (Figure 7B). These expression trends were also detected in the leaves of trees of different ages (Figure 8B). Additionally, AP2 and AP2-like genes exhibited their highest expression levels in roots but had relatively low expression levels in flowers (Figure 9B). Our research showed that miRNA172 and its targets participated in the regulation of the juvenile to adult phase transition and the formation of floral organs.
Plant hormones, as major regulators, have roles in leaf growth, juvenile to adult phase transitioning and flowering [19, 49, 50]. AUX is a key hormone that is important in hormone-mediated responses during plant development [51–54]. It has been reported that AUX response factors, ARF16 and ARF17, are targets of miRNA160 and regulate various biological processes of plant development in Arabidopsis, maize and rice [55–57]. The expression level of miRNA160 was different between the on- and off-year, and its expression was higher in juvenile leaves than in mature leaves in olive trees [47]. In our study, we also found that mdm-miR160 was up-regulated in J leaves whereas its targets were up-regulated in A leaves, supporting the high-throughput data on miRNA160 (Figures 5B, 7D). mdm-miR160 exhibited high expression levels in young leaves, but ARF16 and ARF17 expressed higher in the relatively mature leaves (Figure 9D). Additionally, the AUX signal F-box genes TIR1, AFB2 and AFB3 were negatively regulated by miRNA393 [58, 59], confirming the high-throughput sequencing and qRT-PCR results. These showed that miRNA393 expressed highly in J leaves, while its targets showed almost no expression (Figures 5B, 7C). An exogenous AUX treatment could enhance miRNA393 transcription and induce miR393 accumulation, indicating miRNA393 regulated TIR1 through a feedback control during the plant development process [60]. Our results showed that the AUX content and the expression of the targets (ARF16, ARF17, TIR1, AFB2 and AFB3) of miRNA160 and miRNA393 were significantly higher in A leaves than in J leaves (Figure 7C and D), indicating an important contribution of hormone-mediated responses to leaf maturation, reproductive growth and the flowering transition.
We also found that other miRNA family members identified by high-throughput sequencing were differentially expressed between the A and J libraries (Figure 7). These results were corroborated by qRT-PCR (Figures 7, 8 and 9). The expression profiles of mdm-miR162, mdm-miR535, mdm-miR858, mdm-miR3727 and miR5225 were up-regulated in J leaves compared with A leaves during leaf development, while mdm-miR398a and mdm-miR398b were down-regulated in J leaves compared with A leaves, implying that these miRNAs may participate in the regulating leaf development and other biological processes (Figure 7F,G,H and I). Meanwhile, the expression profiles of their targets presented opposite results for A and J leaves, which was also supported by qRT-PCR results (Figure 7F,G,H and I). This was consistent with a previous study on cotton, which showed that the expression of miRNAs (398a and miR398b) and their targets (RD19 and ADH2, respectively) had opposite expression patterns [61, 62]. The expression patterns of miR858 and their targets (MYB family) were also similar to those found in apple [22] and peach trees (Figures 8 and 9) [36].
Conclusions
A comprehensive study on M. hupehensis miRNAs related to the juvenile to adult phase transition was performed. In this study, we identified 42 known miRNA families and 172 novel miRNAs from two sRNA libraries. Additionally, using a degradome analysis, we identified 127 targets of the 25 known miRNA families and 168 targets of the 35 unique novel miRNAs. A GO analysis showed that these miRNAs and their targets participated in regulating phase transition and reproductive growth during plant development. Our results showed that the juvenile to adult phase transition and flowering were controlled by mdm-miRNA156 and mdm-miRNA172. mdm-miR156 is highly abundant in J leaves and decreases in A leaves, while mdm-miR172 has the opposite expression pattern in the two leave types. The miRNA-mediated regulation of multiple plant hormone pathways, such as the GA, AUX, CK and ABA, also plays key roles in phase transition and flowering during the plant life cycle. The identification of the mdm-miR160–target (ARF16 and ARF17) and mdm-miR393–target (AFB2, AFB3 and TIR1) hormone-mediated expression patterns significantly improves our understanding of the roles miRNAs play in the regulation of plant growth, development, reproductive phase transition and flowering. In general, the combination of sRNA and degradome sequencing can better illustrate the profiles of hormone-regulated miRNAs and miRNA targets involved in complex regulatory networks, thus contributing to the understanding of miRNA functions during growth, phase transition and reproductive growth in perennial woody fruit trees.
Electronic supplementary material
Additional file 1: Targets of known miRNAs in Malus hupehensis identified by degradome analysis. (DOCX 21 KB)
Additional file 2: Primer sequences of selected known and novel miRNAs and target genes for qRT-PCR validation experiments in Malus hupehensis . (XLSX 12 KB)
Additional file 3: The length distribution of small RNAs in the Malus hupehensis juvenile leaf degradome library. (DOCX 46 KB)
Additional file 4: Identified known miRNAs in Malus hupehensis and their read counts. (DOCX 42 KB)
Additional file 5: Identified novel miRNAs in Malus hupehensis and their read counts. (DOCX 46 KB)
Additional file 6: The total number of distinct transcripts targeted by unique known miRNAs detected in the Malus hupehensis . (XLSX 55 KB)
Additional file 7: The total number of distinct transcripts targeted by unique novel miRNAs detected in the Malus hupehensis . (XLSX 34 KB)
Additional file 8: GO functional analysis of identified targets of known miRNAs identified in the Malus hupehensis . (XLSX 21 KB)
Additional file 9: GO functional analysis of identified targets of novel miRNAs identified in the Malus hupehensis . (XLSX 16 KB)
Additional file 10: KEGG functional analysis of identified targets of known miRNAs identified in the Malus hupehensis . (XLSX 12 KB)
Additional file 11: KEGG functional analysis of identified targets of novel miRNAs identified in the Malus hupehensis . (XLSX 19 KB)
Additional file 12: Hierarchical clustering of known miRNAs (A) and targets (B) by expression levels in adult and juvenile leaves of Malus hupehensis . Samples are reported on the top side of the heat map with the following codes: Date (from March to August). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 131 KB)
Additional file 13: Hierarchical clustering of known miRNAs (A) and targets (B) by expression levels in Malus hupehensis leaves of different ages. Samples are reported on the top side of the heat map with the following codes: Age (from 1 to 6 years). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 75 KB)
Additional file 14: Hierarchical clustering of known miRNAs (A) and targets (B) by expression levels in different Malus hupehensis ’ tissues. Samples are reported on the top side of the heat map with the following codes: Tissues (root, stem, flower, leaf and fruit). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 78 KB)
Additional file 15: Hierarchical clustering of novel miRNAs with expression levels in adult and juvenile leaves of Malus hupehensis (A) in leaves of different ages (B) and in different tissues (C). Samples are reported on the top side of the heat map with the following codes: Date (From March to August) (A); Age (from 1 to 6 years) (B); Tissues (root, stem, flower, leaf and fruit) (c). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 83 KB)
Additional file 16: Identification by qRT-PCR of novel miRNA expression patterns in adult and juvenile leaves of Malus hupehensis (A) in leaves of different ages (B) and in different tissues (C). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 75 KB)
Acknowledgements
This work was supported by National Apple Industry Technology System of Agriculture Ministry of China (CARS-28), National Science and Technology Supporting Project (2013BAD20B03), The Major Innovation of Science and Technology Project of Shaanxi Province (2011KTZB02-02-05) and The China Postdoctoral Science Foundation (No. 2014 M56806).
Abbreviations
- AUX
Auxin
- CK
Cytokinin
- ABA
Abscisic acid
- GA
Gibberellic acid
- A
Adult
- J
Juvenile.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MH, DZ and LX designed the study. LX and YL performed data analysis. SZ, YS and YL performed the hormone measurements. LX,NA and DZ did the qRT-PCR analysis. MH and CZ provided guidance on the whole study. LX and DZ wrote and revised the manuscript. All authors approved the final manuscript.
Contributor Information
Libo Xing, Email: xinglibo1986@live.com.
Dong Zhang, Email: afant@nwsuaf.edu.cn.
Youmei Li, Email: lym052589@163.com.
Caiping Zhao, Email: cpzhao403@163.com.
Songwen Zhang, Email: zhangsongwen686@163.com.
Yawen Shen, Email: 15229037807@163.com.
Na An, Email: 312586091@qq.com.
Mingyu Han, Email: hanmy@nwsuaf.edu.cn.
References
- 1.Wang J-W, Park MY, Wang L-J, Koo Y, Chen X-Y, Weigel D. Poethig RS: miRNA control of vegetative phase change in trees. PLoS Genet. 2011;7(2):e1002012. doi: 10.1371/journal.pgen.1002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Airoldi CA, Bergonzi S, Davies B. Single amino acid change alters the ability to specify male or female organ identity. Proc Natl Acad Sci. 2010;107(44):18898–18902. doi: 10.1073/pnas.1009050107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cacau FV, Reis GG, Reis MGF, Leite HG, Alves FF, Souza FC. Juvenile eucalypt plant coppicing and sprout management in agroforestry system. Pesquisa Agropecuária Brasileira. 2008;43(11):1457–1465. doi: 10.1590/S0100-204X2008001100003. [DOI] [Google Scholar]
- 4.Li YN. Research of germplasm resources in Malus Mill. Beijing: China Agriculture Press; 2001. [Google Scholar]
- 5.Chen XS, Han MY, Su GL, Liu FZ, Guo GN, Jiang YM, Mao ZQ, Peng FT, Su HR. Discussion on today’s world apple industry trends and the suggestions on sustainable and efficient development of apple industry. China J Fruit Sci. 2010;27(04):598–604. [Google Scholar]
- 6.Kang G-Z, Xu W, Liu G-Q, Peng X-Q, Guo T-C, Bell J. Comprehensive analysis of the transcription of starch synthesis genes and the transcription factor RSR1 in wheat (Triticum aestivum) endosperm. Genome. 2012;56(2):115–122. doi: 10.1139/gen-2012-0146. [DOI] [PubMed] [Google Scholar]
- 7.Usami T, Horiguchi G, Yano S, Tsukaya H. The more and smaller cells mutants of Arabidopsis thaliana identify novel roles for SQUAMOSA PROMOTER BINDING PROTEIN-LIKE genes in the control of heteroblasty. Development. 2009;136(6):955–964. doi: 10.1242/dev.028613. [DOI] [PubMed] [Google Scholar]
- 8.Shikata M, Koyama T, Mitsuda N, Ohme-Takagi M. Arabidopsis SBP-box genes SPL10, SPL11 and SPL2 control morphological change in association with shoot maturation in the reproductive phase. Physiol. 2009;50(12):2133–2145. doi: 10.1093/pcp/pcp148. [DOI] [PubMed] [Google Scholar]
- 9.Mohamed R, Wang CT, Ma C, Shevchenko O, Dye SJ, Puzey JR, Etherington E, Sheng X, Meilan R, Strauss SH. Populus CEN/TFL1 regulates first onset of flowering, axillary meristem identity and dormancy release in Populus. Plant J. 2010;62(4):674–688. doi: 10.1111/j.1365-313X.2010.04185.x. [DOI] [PubMed] [Google Scholar]
- 10.Kotoda N, Wada M. MdTFL1, a TFL1-like gene of apple retards the transition from the vegetative to reproductive phase in transgenic Arabidopsis. Plant Sci. 2005;168(1):95–104. doi: 10.1016/j.plantsci.2004.07.024. [DOI] [Google Scholar]
- 11.Castillo M-C, Forment J, Gadea J, Carrasco JL, Juarez J, Navarro L, Ancillo G. Identification of transcription factors potentially involved in the juvenile to adult phase transition in Citrus. Ann Bot. 2013;112(7):1371–1381. doi: 10.1093/aob/mct211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu G, Park MY, Conway SR, Wang J-W, Weigel D, Poethig RS. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell. 2009;138(4):750–759. doi: 10.1016/j.cell.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi A, Wu M-F, Yang L, Wu G, Poethig RS, Wagner D. The MicroRNA-Regulated SBP-Box Transcription Factor SPL3 Is a Direct Upstream Activator of LEAFY FRUITFULL and APETALA1. Dev Cell. 2009;17(2):268–278. doi: 10.1016/j.devcel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fornara F, Coupland G. Plant phase transitions make a SPLash. Cell. 2009;138(4):625–627. doi: 10.1016/j.cell.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Lauter N, Kampani A, Carlson S, Goebel M. Moose SP: microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc Natl Acad Sci U S A. 2005;102(26):9412–9417. doi: 10.1073/pnas.0503927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bond BJ. Age-related changes in photosynthesis of woody plants. Trends Plant Sci. 2000;5(8):349–353. doi: 10.1016/S1360-1385(00)01691-5. [DOI] [PubMed] [Google Scholar]
- 17.Day M, Greenwood M, Diaz-Sala C. Age-and size-related trends in woody plant shoot development: regulatory pathways and evidence for genetic control. Tree Physiol. 2002;22(8):507–513. doi: 10.1093/treephys/22.8.507. [DOI] [PubMed] [Google Scholar]
- 18.Chen YT, Shen CH, Lin WD, Chu HA, Huang BL, Kuo CI, Yeh KW, Huang LC, Chang IF. Small RNAs of Sequoia sempervirens during rejuvenation and phase change. Plant Biol. 2013;15(1):27–36. doi: 10.1111/j.1438-8677.2012.00622.x. [DOI] [PubMed] [Google Scholar]
- 19.Scarpella E, Barkoulas M, Tsiantis M. Control of leaf and vein development by auxin. Cold Spring Harb Perspect Biol. 2010;2(1):a001511. doi: 10.1101/cshperspect.a001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curaba J, Singh MB. Bhalla PL: miRNAs in the crosstalk between phytohormone signalling pathways. J Exp Bot. 2014;65(6):1425–1438. doi: 10.1093/jxb/eru002. [DOI] [PubMed] [Google Scholar]
- 21.Yu S, Galvão VC, Zhang Y-C, Horrer D, Zhang T-Q, Hao Y-H, Feng Y-Q, Wang S, Schmid M, Wang J-W. Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA PROMOTER BINDING–LIKE transcription factors. Plant Cell Online. 2012;24(8):3320–3332. doi: 10.1105/tpc.112.101014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia R, Zhu H. An Y-q, Beers EP, Liu Z: Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 2012;13(6):R47. doi: 10.1186/gb-2012-13-6-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hafner M, Landgraf P, Ludwig J, Rice A, Ojo T, Lin C, Holoch D, Lim C, Tuschl T. Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods. 2008;44(1):3–12. doi: 10.1016/j.ymeth.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25(1):130–131. doi: 10.1093/bioinformatics/btn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffiths-Jones S, Saini HK, van Dongen S. Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(suppl 1):D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.German MA, Pillay M, Jeong D-H, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R. Global identification of microRNA–target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26(8):941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- 27.Du Z, Zhou X, Ling Y, Zhang Z. Su Z: agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010;38(suppl 2):W64–W70. doi: 10.1093/nar/gkq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gómez-Cadenas A, Mehouachi J, Tadeo FR, Primo-Millo E, Talon M. Hormonal regulation of fruitlet abscission induced by carbohydrate shortage in citrus. Planta. 2000;210(4):636–643. doi: 10.1007/s004250050054. [DOI] [PubMed] [Google Scholar]
- 29.Zimmerman RH, Hackett WP, Pharis RP. Hormonal aspects of phase change and precocious flowering. In: Pharis RP, editor. Hormonal Regulation of Development III. 1. Berlin Heidelberg: Springer; 1985. pp. 79–115. [Google Scholar]
- 30.Fahlgren N, Jogdeo S, Kasschau KD, Sullivan CM, Chapman EJ, Laubinger S, Smith LM, Dasenko M, Givan SA, Weigel D. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell Online. 2010;22(4):1074–1089. doi: 10.1105/tpc.110.073999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava S, Srivastava AK, Suprasanna P, D’Souza S. Identification and profiling of arsenic stress-induced microRNAs in Brassica juncea. J Exp Bot. 2013;64(1):303–315. doi: 10.1093/jxb/ers333. [DOI] [PubMed] [Google Scholar]
- 32.Wang K, Zhu X, Zhong L, Chen R. Small RNA in rice genome. Sci China C Life Sci. 2002;45(5):497–503. doi: 10.1360/02yc9054. [DOI] [PubMed] [Google Scholar]
- 33.Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 34.Ulger S, Sonmez S, Karkacier M, Ertoy N, Akdesir O, Aksu M. Determination of endogenous hormones, sugars and mineral nutrition levels during the induction, initiation and differentiation stage and their effects on flower formation in olive. Plant Growth Regul. 2004;42(1):89–95. doi: 10.1023/B:GROW.0000014897.22172.7d. [DOI] [Google Scholar]
- 35.Hackett WP. Juvenility, maturation, and rejuvenation in woody plants. Horticultural Reviews. 1985;7:109–155. [Google Scholar]
- 36.Zhu H, Xia R, Zhao B. An Y-q, Dardick CD, Callahan AM, Liu Z: Unique expression, processing regulation, and regulatory network of peach (Prunus persica) miRNAs. BMC Plant Biol. 2012;12(1):149. doi: 10.1186/1471-2229-12-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ. Criteria for annotation of plant MicroRNAs. Plant Cell Online. 2008;20(12):3186–3190. doi: 10.1105/tpc.108.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu G, Poethig RS. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development. 2006;133(18):3539–3547. doi: 10.1242/dev.02521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gandikota M, Birkenbihl RP, Höhmann S, Cardon GH, Saedler H, Huijser P. The miRNA156/157 recognition element in the 3′ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007;49(4):683–693. doi: 10.1111/j.1365-313X.2006.02983.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang J-W, Schwab R, Czech B, Mica E, Weigel D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell Online. 2008;20(5):1231–1243. doi: 10.1105/tpc.108.058180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwarz S, Grande AV, Bujdoso N, Saedler H, Huijser P. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant Mol Biol. 2008;67(1–2):183–195. doi: 10.1007/s11103-008-9310-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann JU. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005;37(5):501–506. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- 43.Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, García JA, Paz-Ares J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007;39(8):1033–1037. doi: 10.1038/ng2079. [DOI] [PubMed] [Google Scholar]
- 44.Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D. Specific effects of microRNAs on the plant transcriptome. Dev Cell. 2005;8(4):517–527. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 45.Gou J-Y, Felippes FF, Liu C-J, Weigel D, Wang J-W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell Online. 2011;23(4):1512–1522. doi: 10.1105/tpc.111.084525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yant L, Mathieu J, Dinh TT, Ott F, Lanz C, Wollmann H, Chen X, Schmid M. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell Online. 2010;22(7):2156–2170. doi: 10.1105/tpc.110.075606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yanik H, Turktas M, Dundar E, Hernandez P, Dorado G, Unver T. Genome-wide identification of alternate bearing-associated microRNAs (miRNAs) in olive (Olea europaea L.) BMC Plant Biol. 2013;13(1):10. doi: 10.1186/1471-2229-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Q-H, Upadhyaya NM, Gubler F, Helliwell CA. Over-expression of miR172 causes loss of spikelet determinacy and floral organ abnormalities in rice (Oryza sativa) BMC Plant Biol. 2009;9(1):149. doi: 10.1186/1471-2229-9-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen E, Xie Z, Gustafson AM. Carrington JC: microRNA-Directed Phasing during Trans−Acting siRNA Biogenesis in Plants. Cell. 2005;121(2):207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 50.Scarpella E, Marcos D, Friml J, Berleth T. Control of leaf vascular patterning by polar auxin transport. Genes Dev. 2006;20(8):1015–1027. doi: 10.1101/gad.1402406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friml J, Vieten A, Sauer M, Weijers D, Schwarz H, Hamann T, Offringa R, Jürgens G. Efflux-dependent auxin gradients establish the apical–basal axis of Arabidopsis. Nature. 2003;426(6963):147–153. doi: 10.1038/nature02085. [DOI] [PubMed] [Google Scholar]
- 52.Okushima Y, Fukaki H, Onoda M, Theologis A, Tasaka M. ARF7 and ARF19 regulate lateral root formation via direct activation of LBD/ASL genes in Arabidopsis. Plant Cell Online. 2007;19(1):118–130. doi: 10.1105/tpc.106.047761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okushima Y, Overvoorde PJ, Arima K, Alonso JM, Chan A, Chang C, Ecker JR, Hughes B, Lui A, Nguyen D. Functional genomic analysis of the AUXIN RESPONSE FACTOR gene family members in Arabidopsis thaliana: unique and overlapping functions of ARF7 and ARF19. Plant Cell Online. 2005;17(2):444–463. doi: 10.1105/tpc.104.028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Overvoorde PJ, Okushima Y, Alonso JM, Chan A, Chang C, Ecker JR, Hughes B, Liu A, Onodera C, Quach H. Functional genomic analysis of the AUXIN/INDOLE-3-ACETIC ACID gene family members in Arabidopsis thaliana. Plant Cell Online. 2005;17(12):3282–3300. doi: 10.1105/tpc.105.036723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mallory AC, Bartel DP, Bartel B. MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell Online. 2005;17(5):1360–1375. doi: 10.1105/tpc.105.031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xing H, Pudake RN, Guo G, Xing G, Hu Z, Zhang Y, Sun Q, Ni Z. Genome-wide identification and expression profiling of auxin response factor (ARF) gene family in maize. BMC Genomics. 2011;12(1):178. doi: 10.1186/1471-2164-12-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang L, Chia J-M, Kumari S, Stein JC, Liu Z, Narechania A, Maher CA, Guill K, McMullen MD, Ware D. A genome-wide characterization of microRNA genes in maize. PLoS Genet. 2009;5(11):e1000716. doi: 10.1371/journal.pgen.1000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Si-Ammour A, Windels D, Arn-Bouldoires E, Kutter C, Ailhas J, Meins F. Vazquez F: miR393 and secondary siRNAs regulate expression of the TIR1/AFB2 auxin receptor clade and auxin-related development of Arabidopsis leaves. Plant Physiol. 2011;157(2):683–691. doi: 10.1104/pp.111.180083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanneste S, Friml J. Auxin: a trigger for change in plant development. Cell. 2009;136(6):1005–1016. doi: 10.1016/j.cell.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Chen Z-H, Bao M-L, Sun Y-Z, Yang Y-J, Xu X-H, Wang J-H, Han N, Bian H-W, Zhu M-Y. Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol Biol. 2011;77(6):619–629. doi: 10.1007/s11103-011-9838-1. [DOI] [PubMed] [Google Scholar]
- 61.Dugas DV, Bartel B. Sucrose induction of Arabidopsis miR398 represses two Cu/Zn superoxide dismutases. Plant Mol Biol. 2008;67(4):403–417. doi: 10.1007/s11103-008-9329-1. [DOI] [PubMed] [Google Scholar]
- 62.Yang X, Wang L, Yuan D, Lindsey K, Zhang X. Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J Exp Bot. 2013;64(6):1521–1536. doi: 10.1093/jxb/ert013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Targets of known miRNAs in Malus hupehensis identified by degradome analysis. (DOCX 21 KB)
Additional file 2: Primer sequences of selected known and novel miRNAs and target genes for qRT-PCR validation experiments in Malus hupehensis . (XLSX 12 KB)
Additional file 3: The length distribution of small RNAs in the Malus hupehensis juvenile leaf degradome library. (DOCX 46 KB)
Additional file 4: Identified known miRNAs in Malus hupehensis and their read counts. (DOCX 42 KB)
Additional file 5: Identified novel miRNAs in Malus hupehensis and their read counts. (DOCX 46 KB)
Additional file 6: The total number of distinct transcripts targeted by unique known miRNAs detected in the Malus hupehensis . (XLSX 55 KB)
Additional file 7: The total number of distinct transcripts targeted by unique novel miRNAs detected in the Malus hupehensis . (XLSX 34 KB)
Additional file 8: GO functional analysis of identified targets of known miRNAs identified in the Malus hupehensis . (XLSX 21 KB)
Additional file 9: GO functional analysis of identified targets of novel miRNAs identified in the Malus hupehensis . (XLSX 16 KB)
Additional file 10: KEGG functional analysis of identified targets of known miRNAs identified in the Malus hupehensis . (XLSX 12 KB)
Additional file 11: KEGG functional analysis of identified targets of novel miRNAs identified in the Malus hupehensis . (XLSX 19 KB)
Additional file 12: Hierarchical clustering of known miRNAs (A) and targets (B) by expression levels in adult and juvenile leaves of Malus hupehensis . Samples are reported on the top side of the heat map with the following codes: Date (from March to August). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 131 KB)
Additional file 13: Hierarchical clustering of known miRNAs (A) and targets (B) by expression levels in Malus hupehensis leaves of different ages. Samples are reported on the top side of the heat map with the following codes: Age (from 1 to 6 years). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 75 KB)
Additional file 14: Hierarchical clustering of known miRNAs (A) and targets (B) by expression levels in different Malus hupehensis ’ tissues. Samples are reported on the top side of the heat map with the following codes: Tissues (root, stem, flower, leaf and fruit). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 78 KB)
Additional file 15: Hierarchical clustering of novel miRNAs with expression levels in adult and juvenile leaves of Malus hupehensis (A) in leaves of different ages (B) and in different tissues (C). Samples are reported on the top side of the heat map with the following codes: Date (From March to August) (A); Age (from 1 to 6 years) (B); Tissues (root, stem, flower, leaf and fruit) (c). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 83 KB)
Additional file 16: Identification by qRT-PCR of novel miRNA expression patterns in adult and juvenile leaves of Malus hupehensis (A) in leaves of different ages (B) and in different tissues (C). A: Adult phase leaves from the tree top; J: Juvenile phase leaves from the tree base. (DOCX 75 KB)