

Abstract
We screened the NIH’s Molecular Libraries Small Molecule Repository for inhibitors of cytotoxic T lymphocyte (CTL) lytic granule exocytosis by measuring binding of an antibody in the extracellular solution to a lysosomal membrane protein (LAMP-1) that is transferred to the plasma membrane by exocytosis. We used TALL-104 human leukemic CTLs stimulated with soluble chemicals. Using high-throughput cluster cytometry to screen 364202 compounds in 1536-well plate format, identifying 2404 initial hits. 161 were confirmed on retesting, and dose-response measurements were performed. 75 of those compounds were obtained, and 48 were confirmed active. Experiments were conducted to determine the molecular mechanism of action (MMOA) of the active compounds. Fifteen blocked increases in intracellular calcium >50%. Seven blocked phosphorylation of ERK by upstream MAP kinase kinases >50%. One completely blocked the activity of the calcium-dependent phosphatase calcineurin. None blocked ERK catalytic activity. Eight blocked more than one pathway. For eight compounds, we were unable to determine an MMOA. The activity of one of these compounds was confirmed from powder resupply. We conclude that a screen based on antibody binding to CTLs is a good means of identifying novel candidate immunosuppressants with either known or unknown MMOA.
Keywords: Immunosuppressor, lymphocyte, high throughput screening (HTS), chemical biology, signal transduction
INTRODUCTION
Immune suppression is critical to organ transplantation and treatment of autoimmune diseases. Because of problems with current agents, there is a widely-recognized need for novel immune-suppressing compounds that could serve as leads for new drugs 1. We recently developed an assay for inhibitors of cytotoxic T lymphocyte (CTL) lytic granule exocytosis 2. CTLs kill cancerous and virus-infected cells and participate in immune processes that lead to graft rejection. One mechanism CTLs use to kill is the exocytosis of lytic granules which contain cytolytic peptides 3, 4. Our assay uses TALL-104 human leukemic CTLs 5 stimulated with soluble agents that bypass the T cell receptor, triggering maximal granule exocytosis. The screen detects transfer of lysosome-associated membrane protein 1 (LAMP-1) from lytic granules to the plasma membrane via exocytosis 6, 7. This results in exposure of an intraluminal epitope on LAMP-1 to antibodies in the external solution, enabling quantitative fluorescent detection of cells that have responded. High-throughput screening (HTS) is feasible because sampling flow cytometers make it possible to measure cell-associated fluorescence without washing away unbound antibody. The use of a cell line and a maximal- albeit unphysiological- stimulation was chosen to ensure consistent and large enough responses for HTS.
We reasoned that HTS conducted with our method on a large and diverse compound collection could be useful for a variety of reasons. First, since the signals that control lytic granule exocytosis overlap fairly extensively with the signals that control the function of helper T cells 8, 9 which synthesize and release cytokines when stimulated, screening for inhibitors of CTL function would be expected to reveal compounds that also inhibit helper T cell function. These would be broadly immunosuppressive and could help fulfill the need for novel immunosuppressants outlined above. Granule exocytosis occurs in minutes, whereas helper T cells take hours to synthesize and release cytokines, so assay is better suited to large-scale screening than cytokine production or reporter gene expression. Second, screening lytic granule exocytosis could reveal compounds that specifically inhibit CTLs, which would enable a more specific kind of immune modulation than is currently available. Third, we surmised that a screen based on the endpoint of a cellular function controlled by a number of important signaling pathways 8, 9 would offer a means of identifying compounds working on known targets which could be identified in follow-up experiments, acting as a novel type of high-content screening. Finally, we anticipated that assessing the molecular mechanism of action (MMOA) of hits following screening could lead to the identification of compounds that do not work via any of the known pathways interrogated. Such compounds might involve novel molecular targets. They could serve as leads for new classes of immunosuppressives, and could also be used to explore the pathways that control lymphocyte function.
Here we describe the results of our screening of the NIH’s Molecular Libraries Small Molecule Repository (MLSMR) of 364202 compounds with a 1536-well version of the assay, and follow-up experiments to explore the cellular action of confirmed active compounds, with an important goal being the identification of compounds with unknown MMOA. We identified 31 compounds that likely inhibit lytic granule exocytosis by blocking important pathways known to be involved in T cell function, as well as eight compounds, the actions of one of which has been confirmed from powder, for which we could not assign a MMOA. These eight compounds may be of potential utility in identifying novel pathways that control lymphocyte function, and may represent leads to new classes of immunosuppressants.
MATERIALS AND METHODS
Chemicals, cells and solutions
TALL-104 human leukemic CTLs were maintained as described previously. Experimental Saline (ES) was composed of 155mM NaCl, 4.5 mM KCl, 1mM MgCl2, 2mM CaCl2, 5mM HEPES and 10mM glucose. The pH was adjusted to 7.4 with NaOH, and 2% BSA was added. Powder resupply of SID 7977862 was from Chembridge (San Diego, CA).
HTS of the MLSMR
A BiotekMicroFlo Dispenser was used to sequentially transfer to individual wells of a 1536 well plate (Greiner #782101 HyBase): i) 4 μL of Ringers buffer (RB), ii) 4 μL of cells (5,000–6,000/well), and iii) 2 μL of a mixture of thapsigargin (1 μM final), PMA (50 nM final) and Alexa647-conjugated anti-LAMP antibodies (1:500 final dilution). Compounds (in 100% DMSO) or DMSO alone were transferred to wells after the RB addition step and prior to addition of cells using a Beckman Coulter FX Liquid Handler robot equipped with 100 nL pintools (V&B Scientific). Final compound and DMSO concentrations were 10 μM and 1%, respectively. Plates were then sealed with aluminum foil sealing film (AlumaSeal, Excel Scientific) and continuously rotated end-over-end at 8 RPM to maintain cells in suspension (surface tension retains fluid volumes at well bottom when plates are inverted) and to facilitate mixing of well contents. Plates were first rotated for 2 hours at 24°C, then transferred to a 4°C cold room for continuing rotation overnight. The following day plates were processed in 4-plate groups in which plates in each group were first allowed to warm to 24°C (10 min or more while being continuously rotated), then sequentially transferred to a custom HyperCyt HTS platform configured with 4 sampling probes linked to 4 Accuri C6 flow cytometers (BD Biosciences). Each probe sampled wells from a separate quadrant of the 1536-well plate at a combined rate for all 4 probes of 130–160 wells/min and 10–12 min/plate. This platform has been previously described and operationally validated elsewhere 10. Custom modified HyperView software (IntelliCyt, Albuquerque, NM) was used to analyze data. First, flow cytometer data files were parsed by software-based well identification algorithms to segregate data from individual wells. Next, viable cells were identified and gated based on forward and side light scatter profiles elicited by the 488 nm laser. These were then analyzed with respect to Alexa647 fluorescence intensity (FL4, 663–688 nm) excited by the 640 nm laser. A pooled fluorescence intensity histogram of unstimulated cells from all control wells containing DMSO alone (positive controls) was used to establish an intensity threshold above which we detected fewer than 5% of the unstimulated cells. Cells from compound-containing wells with fluorescence intensity exceeding this threshold were considered to be responsive to the thapsigargin/PMA stimulus mixture and sub-threshold cells were considered to be unresponsive. Typically, 80–100% of cells from control wells containing only DMSO and stimulus mixture (negative controls) were responsive.
The response range for the assay was defined as the difference between the averages of % responsive (%Resp) cells in the negative control (NCntrl, stimulated) wells and in the positive control (PCntrl, unstimulated) wells, respectively. Response inhibition mediated by compounds in sample wells was calculated as (Equation 1):
Compounds inhibiting 55% or more were considered to be active, a threshold chosen to generate ~2500 active compounds. Wells in which fewer than 10 cells were detected within the viability gate were not analyzed further due to insufficient sample size; 767 of 364202 tested compounds (0.2%) were labeled undetermined as a result.
LAMP/BLT-esterase assays
100 μl of ES-washed TALL-104 cells (2.5×106/ml) were added to each well, mixed and incubated at 37 deg. C for 30 minutes. 5 μl of stimulation solution (ES supplemented with 20 μM thapsigargin and 1 μM PMA) or control solution (ES + 4% DMSO) was added and mixed, and the plate was incubated for an additional 90 minutes in the dark at room temperature. The plate was centrifuged, and 50 μl of supernatant was transferred to a new plate for BLT-esterase assays, which measure the release of granzyme B by monitoring the cleavage of a synthetic substrate, benzyloxycarbonyl-L-lysine thiobenzyl ester 11. Meanwhile, 50 μl of ES containing 0.6 μg/ml anti-LAMP antibody was added to the wells containing the pellets, mixed and incubated for 30 minutes in the dark at room temperature with constant rotation prior to the addition of 100 μl/well 2% PFA. The geometric mean value of LAMP-1 fluorescence or the absorbance values at 410 nm were determined from unstimulated (U) or stimulated (S) DMSO-treated or stimulated compound-treated (C) cells and the percent inhibition of LAMP externalization or BLT esterase secretion by each compound was calculated as (Equation 2):
Fura-2 measurements
90 μl ES was added to each well of a black flat-bottomed plate laid out with DMSO and compound-containing wells, mixed, and the fluorescence excited at 340 and 380 nm measured at 510nm emission (F340 and F380) was acquired (reading 1, blank B). TALL-104 cells were loaded with 0.5 μM Fura-2 AM for 25 minutes in the dark at room temperature, then washed with ES and incubated for 20 minutes. 2.5×107 cells in 10 μl ES were added to each well of the test plate and mixed. The plate was incubated for 30 minutes at 37 deg. C and read again (reading 2). Finally, 5 μl of stimulation solution (ES supplemented with 40 μM thapsigargin) or control solution (ES + 4 % DMSO) was added and mixed. After 50 minutes, third fluorescence reading was taken. Unstimulated calcium levels (U) was reading 2 from DMSO-treated cells. Stimulated calcium levels (S) were reading 3 from DMSO or compound-treated cells. Blanks for each well were subtracted for F340 and F380 prior to computation of the F340/F380 ratio. Percent inhibition of calcium rise was calculated using equation 2.
Assays for PKC, and MAPKK activity
100 μl ES-washed TALL-104 cells (2.5×106/ml) were added to Eppendorf tubes containing compound or DMSO, and incubated at 37 deg. C for 30 minutes. 5 μl of stimulation solution or control solution (as above for “LAMP/BLT measurements”) was added and mixed, and tubes were incubated for an additional 50 minutes in the dark at room temperature with constant rotation. After incubation, cells were fixed with 100 μl of 2% PFA and permeabilized by addition of 1 ml of ice-cold methanol. Cells were washed with FACS buffer, incubated with primary antibodies (rabbit anti-active PKC substrate, Cell Signaling 2261, 1:100 and mouse anti-p44/p42 MAPK Cell Signaling 9106, 1:1000) in FACS buffer then incubated with secondary antibodies (6 μg/ml Alexa488-conjugated donkey anti-rabbit and 7 μg/ml Cy 5-conjugated donkey anti-mouse) and washed. The geometric mean value of Alexa488 and Cy5 fluorescence was determined for unstimulated (U) or stimulated (S) DMSO-treated or stimulated compound-treated (C) cells. The percent inhibition of PKC substrate phosphorylation or ERK phosphorylation was calculated using equation 2.
Immunoblotting to detect effects on calcineurin activity
TALL-104 cells were nucleofected as described previously with CaNAR1 cDNA. 5 hours later, 1 ×106 ES-washed transfected cells in 400 μl were added to each tube containing 4 μl test compound at initial concentration of 3 mM or 4 μl DMSO, mixed, and incubated for an additional 30 minutes at 37 deg. C. 600 μl of stimulation solution (ES supplemented with 1.67μM TG and 83.3nM PMA) or control solution was added and mixed, and the tube was incubated for another 50 minutes in the dark at room temperature with constant rotation. Cell pellets were lysed in standard RIPA buffer supplemented with 10mM EGTA, 10mM EDTA, protease and phosphatase inhibitor cocktails and processed for immunoblotting on nitrocellulose membranes.
Immunocytochemical determination of compound effects on ERK catalytic activity
TALL-104 cells were nucleofected with EKAR cDNA. 5 hours later, 100 μl ES-washed transfected cells (2.5 ×106/ml) were treated with test compound or DMSO and stimulated. Cells were fixed and permeabilized, then and stained with rabbit anti-phospho Cdc25c antibody (Cell Signaling 9527), followed by a Cy5-conjugated secondary. FL-1 signals (corresponding to YFP expression level) and FL-4 signals (corresponding to anti-phospho-Cdc25C staining intensity) were collected. Percent inhibition was calculated as described above.
Plate measurements and flow cytometry
All plate measurements for followup experiments were performed with a Biotek Synergy 2 plate reader (Biotek, Winooski VT). Flow cytometry for follow-up experiments was performed on a FACSCalibur (BD Biosciences, Franklin Lake NJ) at the University of Connecticut Storrs Flow Cytometry Facility. Flow cytometry data were analyzed with FlowJo Software (Treestar, Ashland OR).
RESULTS
High throughput screening of the MLSMR
We performed HTS of the MLSMR in 1536-well format using clustered high throughput flow cytometers (Figure 1). In this implementation 10, plates are divided into four 384-well quadrants that are analyzed by dedicated cytometers. Representative data from a validation run with half unstimulated and half TG+PMA-stimulated wells are shown in Figures 1A–C. Using sampling of 900 ms duration (to aspirate ~2 μl sample) with an inter-sampling interval of 400 ms (to aspirate an air bubble to separate adjacent samples), it was possible to acquire a plate in ~10.5 minutes. In validation experiments, this resulted in acquisition of 1150 +/− 120 cells per sample (mean +/− S.D.), with Z′ 12 comparing stimulated and unstimulated cells > 0.8.
Figure 1. Screening the NIH’s MLSMR in 1536-well format with high-throughput cluster cytometry.

A) Plot of forward scatter vs. side scatter from an experiment validating the 1536-well format of the assay. The circled region represents live cells, which were selected for analysis. B) Representative data from row A (top) and the entire first quadrant (bottom) of a test plate in which half the columns contained unstimulated cells, while the other half contained stimulated cells. C) Histogram of FL-4 fluorescence for all live-cell events in a validation plate showing bivariate nature of the distribution of fluorescence corresponding to unstimulated (to the left of the line) and stimulated (to the right of the line) cells. D) Representative data from one row of a library screening plate. E) Heat map for a representative plate from the primary screening campaign. Arrows in the upper left indicate columns that were not stimulated to serve as positive controls. Arrow in the upper right designates stimulated negative controls. The color scale used to display data (indicated to the right) ranges from 0% response (purple) to 85% response (red). Dashed lines demarcate quadrants. E) Histogram of Z values for the 1208 quadrants analyzed.
We screened 364,202 compounds in 302 plates comprised of 1208 quadrants on 21 days over 12 weeks. Figure 1D shows a representative row of data and Figure 1E shows a heat map for a representative screening plate. Responses were analyzed by determining the percentage of responding cells (see below). Horizontal lines indicate the quadrants interrogated by the four cytometer. In each plate, two columns of DMSO-treated control cells remained unstimulated (left) to serve as positive controls, while one column of control cells was stimulated (right) to serve as a negative control. Samples were gated on forward and side scatter prior to analysis to exclude dead or dying cells.
We explored two different ways to analyze primary data from the plates. The first was to compute the mean fluorescence intensity (MFI) for anti-LAMP fluorescence for cells in the live cell gate. The second involved determine the percentage of responding cells (see Figure 1C). Analyzing data by determining the percentage of responding cells was found to give better reproducibility than the MFI analysis, and was therefore used to analyze primary screening data. Using that analysis, 1195/1208 plate quadrants (98.9%) had Z values 12 > 0.5, consistent with the idea that the assay had appropriate statistical reliability for HTS. Hits from plates that had Z < 0.5 were not excluded. 767 wells had fewer than 10 cells in the live cell gate and were scored inconclusive. Low event number may not reflect compound toxicity; a number of issues such as problems dispensing cells or acquiring samples can reduce event number.
We wanted to select ~ 2500 compounds for follow-up. Using an arbitrary threshold of 55% inhibition yielded 2404 hits. 1460 of these substances that were available and did not have obvious chemical liabilities (reactive groups, dye moieties, unsuitable properties for chemical optimization) were obtained and retested. 161 substances were confirmed active, and were selected for dose-response testing. Dose-response measurements were performed by testing the effects of compounds at nine concentrations ranging in a 3-fold dilution series from 7 nM to 15 μM. Data were fit to Equation 3:
where top is the maximum inhibition, bottom is the response of stimulated cells in the absence of compound, logEC50 is the logarithm of the EC50 (in μM), and Hillslope is the Hill coefficient. 45 compounds exhibited acceptable dose-dependent inhibitory curves (monotonic dose-dependence with EC50 < 10 μM and least one point defining an intermediate region of the curve) when analyzed using the percent positive analysis strategy described above. However, additional compounds demonstrated acceptable dose-response behavior when curves were fit to the MFI measurement. Based on these considerations and the availability of compounds, a resupply of 75 substances was obtained for further analysis. Supplemental Figure 1 outlines the decision points leading from 364202 substances screened to the 75 that were selected for follow up.
Confirming the activity of selected substances
We first confirmed the activity of the substances using a protocol that combined a repeat of the LAMP assay with BLT esterase assays 11, a standard means for measuring granule exocytosis (Figure 2, see also Supplemental Figure 2). We combined the two measures to minimize compound use, and to reduce the chance for error. Compounds were tested at 30 μM so as to achieve maximal inhibition of exocytosis. Additionally, since a major goal was to identify compounds with unknown MMOA, we felt that using a relatively high concentration would likely reveal any effects on known MMOA. We did not observe striking effects on the fraction of cells in the live cell gate in these experiments, suggesting that toxicity in the short term was not a problem.
Figure 2. Confirming compound activity.

Plot of inhibition measured in the LAMP assay vs. inhibition measured in the BLT esterase assay from the experiment shown in (A). The dashed line has a slope of 1 and passes through the origin, indicating the behavior expected for a perfect correlation between the two measures. The solid line is a best-fit regression line for the data.
Cells were pretreated with compounds or DMSO, then, except for control wells, stimulated with TG+PMA. 50 minutes after stimulation, plates were centrifuged, and samples of the supernatant were collected for BLT esterase assays. The pelleted cells were stained with anti-LAMP antibodies for 15–20 minutes then fixed and analyzed via flow cytometry. We have shown previously that staining cells after stimulation yields essentially similar results to stimulating them in the presence of the antibody 13.
We found that 48 substances blocked granule exocytosis by >50% as measured by LAMP staining. BLT esterase measurements reported on average ~20% less inhibition of exocytosis than LAMP staining. Despite this, 41 substances also inhibited lytic granule exocytosis > 50% measured with BLT esterase assay. For seven compounds there was a sufficient discrepancy between the two measures of exocytosis that compounds scored as active on the basis of LAMP externalization were scored as inactive based on BLT esterase assays. A number of factors, including a modest degree of compound toxicity could be responsible for this. Those compounds were further investigated.
A strategy for identifying MMOA of active substances
Follow-up experiments were intended to determine the mechanism by which hit compounds block exocytosis (see Supplemental Figure 3). We envisioned seven testable known MMOAs that could block lytic granule exocytosis. Sustained calcium influx, which is required for exocytosis (reviewed in 9), could be inhibited by two MMOAs: 1) block of store operated calcium channels, which are known to mediate calcium signals in CTLs 14 or 2) block of K+ channels, which maintain a favorable driving force for calcium entry (see 15, 16. 3) Inhibition of PKC could block exocytosis 17, as could 4) inhibition of the activation of the MAP kinase ERK 18 by upstream MAP kinase kinases or 5) block of ERK catalytic activity. Finally, since calcineurin is known to be required for exocytosis (see 9 for discussion) 6) calcineurin activity could be inhibited, either directly or 7) as a result of inhibition of calmodulin or of calmodulin binding to calcineurin. We reasoned that it would be most efficient to put assays that could be conducted entirely in plate format early in our experimental design. When possible, we interrogated multiple processes simultaneously. Additionally, we reasoned that we might not need to test each known MMOAs individually, provided we could interrogate a common output. For example, inhibition of calcium influx can result from block of calcium channels or block of K+ channels; either will be revealed by assessing intracellular calcium increases. Similarly, block of either calmodulin or calcineurin’s catalytic activity will result in decreased calcineurin-dependent phosphorylation; both can be assessed by examining dephosphorylation of calcineurin substrates. In the end, four sets of experiments allowed us to test all of the MMOAs described above. All experiments to determine MMOA described below were performed at least twice, and average results are reported.
Testing for inhibition of [Ca2+]i increases
To asses effects on intracellular [Ca2+] (Figure 3), we measured fluorescence of substance-containing solutions at 340 and 380 nm excitation prior to adding fura-2 loaded cells. We then dispensed fura-2 loaded cells into the wells, and incubated them for 15 minutes with test substances. We measured fluorescence at 340 and 380 nm excitation to estimate resting [Ca2+]i levels, then stimulated cells with TG and measured fluorescence again after 50 minutes, a time point at which [Ca2+] elevations depend entirely on Ca2+ influx. We found that 8 of the substances that blocked lytic granule exocytosis had sufficiently high fluorescence at 340 and/or 380 nm excitation that we were not able to acquire meaningful fura-2 signals from cells treated with them. Of the substances with sufficiently low fluorescence that we could acquire Fura-2 signals, fifteen inhibited increases in the fura 340/380 ratio by 50% or more. We retested the highly fluorescent substances with the related dye fura-red in flow cytometry, since this dye has the same Kd for calcium as fura-2 but different spectral characteristics. We loaded cells with fura red, then treated them with substances for 15 minutes at 37 degrees C. Half of the substance-treated cells were stimulated with TG+PMA, and half were left unstimulated. Samples were analyzed 50 minutes later. We found that six out of the eight substances decreased the fura-red fluorescence of unstimulated cells in at least one of the trials, an effect that could be an artifact related to leakage of the dye from cells or that could reflect elevation of resting calcium levels. Since these possibilities cannot be distinguished with a single wavelength indicator, we were only able to measure with confidence the blocking effects of two substances, neither of which inhibited calcium signals > 50%. Figure 3 plots the percent inhibition of LAMP-1 responses vs. the percent inhibition of fura signals for substances that decreased exocytosis by >50% (filled squares). The six substances for which we could not reliably measure effects on [Ca2+]i with Fura-2 or Fura-red were not pursued further.
Figure 3. Measuring effects of compounds on calcium influx.
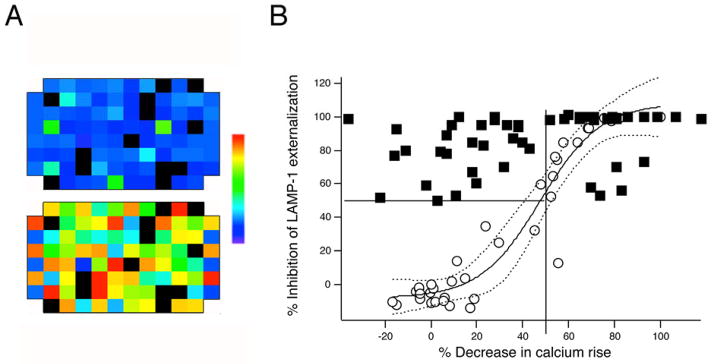
A) Pseudo color representations of Fura-2 ratios prior to (top) and after (bottom) stimulation. Black indicates wells for which fluorescence measured prior to addition of cells was sufficiently high that ratios could not be acquired. The leftmost and rightmost columns contain DMSO-treated cells. Half these wells were stimulated, while half were left unstimulated. The color scale used to display data (indicated to the right) ranges from ratio of 0.04 (purple) to 0.5 (red). B) Filled squares plot the inhibition of LAMP responses as a function of the inhibition of Fura-2 signal for all test substances that inhibited exocytosis >50% in the experiments shown in Figure 2. Open circles are plots of LAMP responses vs. inhibition of fura signals caused by varying the concentration of extracellular Ca2+, normalized to the responses at 2 mM Ca2+. The solid curve is a fit of a sigmoidal function to the data in the open circles. The dashed lines outside the solid curve represent 99% confidence bands for the fit. The straight lines denote 50% levels for Fura-2 and LAMP inhibition, respectively.
Overall, 15 of 48 exocytosis-inhibiting substances blocked fura signals by >50% (Figure 3 and Table 1), a level that we thought appropriate to use as a cut-off to discriminate compounds likely to work by blocking Ca2+ increases. (All hits are listed by their substance IDs (SIDS), and information about them can be found by searching PubChem Substance (http://www.ncbi.nlm.nih.gov/pcsubstance)). To confirm that this level of inhibition of Ca2+ mobilization is sufficient to block exocytosis, we measured Fura-2 ratios and lytic granule exocytosis while varying extracellular Ca2+ from 0 to 2 mM (Figure 3, open circles). We plotted the relative amount of exocytosis at each concentration of Ca2+ vs. the fura-2 ratio we measured, allowing us to estimate the dependence of granule exocytosis on fura-reported measures of intracellular [Ca2+]. Inhibiting the increase in fura ratios by 50% or more reduced lytic granule exocytosis by >50%. Thus, we considered compounds that block exocytosis by >50% but inhibit Ca2+ increases < 50% as likely to have an alternate MMOA.
Table I.
Summary of MMOA Testing
| Substance ID (Pubchem) | EC50 for LAMP inhibition (μM)1 | Inhibition of LAMP signal (%)2 | Inhibition of BLT esterase Release (%)2 | Inhibition of Fura-2 signal (%)3 | Inhibition of MAPKK phosphorylation (%)4 | Inhibition of calcineurin activity (%)5 | Inhibition of Cdc25C phosphorylation (%)6 |
|---|---|---|---|---|---|---|---|
| Calcium Inhibitors | |||||||
| 24817911 | 3.4% | 101 | 91 | 59 | |||
| 22417005 | 6.0% | 100 | 95 | 78 | |||
| 51090459 | 6.0/M | 100 | 91 | 69 | |||
| 17408817 | 1.1M | 100 | 90 | 63 | |||
| 92764822 | 3.2% | 100 | 81 | 67 | |||
| 49673940 | 4.5% | 100 | 96 | 91 | |||
| 92764425 | 0.5M | 99 | 88 | 57 | |||
| 49721823 | 0.3% | 99 | 97 | 76 | |||
| 4246714 | 2.3% | 99 | 94 | 116 | |||
| 49714966 | 4.0% | 99 | 88 | 79 | |||
| 99495188 | 2.1% | 98 | 86 | 70 | |||
| 51089860 | 0.6M | 98 | 67 | 51 | |||
| 26728166 | 1.8M | 73 | 63 | 91 | |||
| 26728904 | 3.2M | 70 | 68 | 80 | |||
| 99456627 | 5.0M | 56 | 68 | 81 | |||
| MAPKK Inhibitors | |||||||
| 3713060 | 2.5% | 100 | 99 | 84 | |||
| 248109231 | 1.4% | 99 | 80 | 79 | |||
| 124755744 | 4.0% | 95 | 76 | 51 | |||
| 26727153 | 3.7% | 95 | 92 | 66 | |||
| 56324549 | 1.1% | 95 | 55 | 87 | |||
| 17414945 | 0.3% | 89 | 72 | 67 | |||
| 99359838 | 1.4M | 78 | 61 | 53 | |||
| Calcineurin Inhibitors | |||||||
| 92764285 | 1.5% | 100 | 97 | 100 | |||
| Inhibitors of more than one pathway | |||||||
| 24840307 | 0.8M | 98 | 83 | 27 | 36 | ||
| 124755743 | 4.9M | 94 | 64 | 37 | 37 | ||
| 99359840 | 1.4M | 60 | 45 | 20 | 35 | 13 | |
| 124755736 | 5.0% | 95 | 74 | 38 | 28 | 6 | |
| 24815073 | 3.3M | 81 | 51 | 42 | 12 | 14 | |
| 14737268 | 1.6% | 85 | 73 | 18 | 40 | 16 | |
| 7977862 | 0.8% | 93 | 89 | 38 | 17 | ||
| 49665798 | 4.9% | 85 | 67 | 40 | 40 | ||
| Unknown MMOA | |||||||
| 7977862 | 0.9% | 93 | 89 | 0 | 0 | 17 | |
| 103159345 | 5.1M | 79 | 54 | 0 | 0 | 0 | |
| 103060008 | 3.5% | 83 | 77 | 23 | 0 | 0 | |
| 103073958 | 3.1% | 70 | 63 | 32 | 0 | 0 | |
| 03075418 | 3.9% | 53 | 41 | 11 | 0 | 0 | |
| 103074366 | 1.8M | 53 | 64 | 40 | 0 | 0 | |
| 49677461 | 2.1M | 87 | 52 | 36 | 0 | 0 | |
| 17387000 | 2.2M | 80 | 68 | 0 | 38 | 0 | |
All data are the average of at least two separate determinations
Obtained from fits to dose-response data. % indicates that the fit was to data analyzed by computing the % positive cells, while M indicates that mean fluorescence intensity was used.
Defined as in Figure 2.
From data presented in Figure 3.
From data in Figure 4.
From data in Figure 5.
From data in Figure 6.
Testing for effects on PKC and activation of ERK MAP kinases
We tested the substances whose effects we concluded were not likely due to effects on [Ca2+]i for inhibition activity of PKC and activation of the MAPK ERK by upstream MAP kinase kinases (Figure 4A). As a control, we treated cells with the compound Ro31-8220, which blocks PKC and thus PMA-stimulated activation of MAPKKs 19. Ro31-8220 inhibited PKC substrate phosphorylation by 82% and ERK phosphorylation by 95%. Seven of the exocytosis-blocking compounds tested inhibited ERK phosphorylation by >50%, but none significantly reduced the fluorescence intensity of anti-phospho PKC substrate staining (see Table 1). We conclude that seven compounds likely inhibit granule exocytosis by inhibiting MAPKK activation downstream of PKC.
Figure 4. Testing the effects of compounds on PKC and MAPKK activity, NFAT dephosphorylation and ERK catalytic activity.

A) Histograms of anti-phospho PKC substrate antibody staining (left) and anti-phospho ERK staining (right) are shown for unstimulated control cells, stimulated control cells, cells treated with Ro31-8220 and cells treated with SID 56324549, one of 7 substances (see Table 1) that we conclude inhibit exocytosis by blocking MAPKK activity. B) Western blotting to assess calcineurin activity was performed on control and compound-treated lysates from cells transfected with CANAR. The image displayed is a montage. Each row is from a single blot. Columns corresponding to molecular weight standards have been digitally removed, and lanes to their right have been moved horizontally to the left. In each row, samples in column 1 are from unstimulated DMSO-treated cells, and samples in column 2 are from DMSO-treated cells stimulated with TG+PMA. In blot 4, the sample in column 5 was prepared from cells treated with the calcineurin inhibitor cyclosporin A, which serves as a positive control, while the sample in column 4 was treated with Ro31-8220, serving as an additional negative control. Note the single experimental sample (blot 3, lane 7) with high molecular weight immunoreactivity corresponding to inhibition of calcineurin. C) Flow cytometry was performed on EKAR-transfected cells to assess ERK catalytic activity. i) Contour plots of anti-phospho Cdc25C staining intensity vs. YFP fluorescence for unstimulated control cells (top) and for cells stimulated with TG+PMA (bottom). The vertical line indicates the cutoff that was used to gate YFP positive cells. ii) Histograms of anti-phospho Cdc25C staining intensity for YFP positive cells from the samples shown in (i), and also for stimulated cells treated with the positive control Ro31-8220 and with a test substance, SID 16952891. Stimulation with TG+PMA causes a shift to higher anti-phospho Cdc25c fluorescence intensity that is inhibited by Ro31-8220.
Testing for effects on calcineurin activity
We tested the effects of substances whose effects on exocytosis were not due to effects on Ca2+, PKC or ERK for their ability to inhibit the activity of the Ca2+-dependent phosphatase calcineurin, which is required for granule exocytosis. To measure calcineurin activity, we used a genetically-encoded reporter construct 20 based on the transcription factor NFAT, the best-known calcineurin substrate (Figure 4B). Because NFAT is highly phosphorylated in resting cells, dephosphorylation resulting from calcineurin activity increases the reporter construct’s apparent mobility on SDS-PAGE gels from ~ 125 kD to ~110 kD. Four sets of gels/transfers were required to run all the samples; Figure 4B is a montage of the results (see legend for details of its assembly). In unstimulated cells (column 1), immunoreactivity was present as two bands. As expected, stimulation of cells caused a shift in the pattern of immunoreactivity to a single band of low molecular weight (column 2). Treatment of cells with the calcineurin inhibitor cyclosporin A prior to stimulation caused immunoreactivity to shift exclusively to the high molecular weight form (blot 4, column 5). One of the substances we tested, SID 92764285 (blot 3, column 7), completely blocked the shift of the sensor to lower molecular weight, resulting in a single band of high molecular weight, identical to the effect of cyclosporin A. Other substances were without apparent effect.
Testing effects on ERK catalytic activity
Last, we tested whether compounds for which an MMOA had not yet been determined blocked the catalytic activity of ERK (Figure 4C), as this possibility was not excluded by our experiments with the phospho-ERK antibody, which detects activation of ERK by upstream MAPKKs. We used cyto-EKAR 21, a genetically-encoded sensor of ERK activity that contains the phosphorylation site of Cdc25c and an ERK docking motif (Figure 4C). We used an anti-phospho Cdc25c antibody to detect activity. In unstimulated cells, increasing expression of the sensor (as detected by YFP fluorescence) was accompanied by higher levels of anti-pCdc25c staining. Stimulating cells with TG+PMA increased anti- pCdc25c staining specifically in the YFP-positive cells (Figure 4Ci). This increase in anti phospho-Cdc25c staining was inhibited when cells were treated with the PKC inhibitor Ro31-8220 (Fig 4Cii), a result consistent with the involvement of PKC activation in activating ERK. We found that none of the compounds inhibited the increase in anti-pCdc25c staining by >30%, while Ro31-8220 blocked by 83%. This suggests that no compound is likely to block lytic granule exocytosis solely by inhibiting ERK catalytic activity.
Identification of a compound with unknown MMOA
Sixteen compounds did not inhibit a single pathway tested by >50%. However, eight of those exerted effects on multiple pathways that, although separately of insufficient magnitude to account for the block of granule exocytosis, could be imagined to exert summed effects large enough to inhibit release. The compounds with significant multiple inhibitory effects are detailed in Table 1. Since we know relatively little about how varying the strength of the different signals affects exocytosis, it is difficult to determine whether the effects of these compounds on known MMOAs completely account for their ability to block exocytosis, or whether they have additional effects. Our choice to use a relatively high concentration of compounds for MMOA testing may play a role in generating cases of apparent multiple activities. However, the final eight compounds had summed inhibitory effects that seemed unlikely to be of sufficient magnitude to account for block. We were able to obtain powder resupply of one of them, SID 7977862. On retest, this compound blocked exocytosis with an EC50 of ~ 4 μM, had no effect on cell viability in the short term when tested at 100 μM, and was without inhibitory effect on calcium signals, ERK activation, ERK catalytic activity or calcineurin activation (Figure 5).
Figure 5. Confirming the effects of SID 7977862 from powder.

A) Structure of SID 7977862. B) Histograms of anti-LAMP fluorescence for unstimulated control (blue), stimulated control (red) and SID 7977862-treated cells. Experiment was conducted in NR. C) Dose-response testing using LAMP-1 externalization. Data are the average of 2 separate experiments conducted in NR+ 2% BSA. D) Assessing toxicity using Prestoblue reagent. Cells were pretreated with 100 μM SID 7877962 (triangle, 250,000 cells) or DMSO (circles, different numbers of cells), and Prestoblue fluorescence was measured. Different numbers of DMSO-treated cells treated with Triton X-100 (open circles) were used as an additional positive control. E) Testing for inhibition of calcium influx. F) Measuring effects on ERK phosphorylation. Experiments were conducted as in Figure 4A. G) Measuring effects on ERK catalytic activity. Experiments were conducted as in Figure 4C. H) Testing for inhibition of calcineurin. Experiments were conducted essentially as in Figure 4B.
DISCUSSION
Our initial HTS campaign worked essentially as anticipated. Using clustered cytometers, we were able to miniaturize the assay to 1536-well format, resulting in a substantial decrease in the amount of reagents- primarily cells and antibody- required. Overall, we had 161 confirmed hits, a number that seemed low to us considering the complexity of the cellular phenotype we were interrogating. However, this hit rate appears consistent with results from the most directly comparable screening campaign conducted on the MLSMR that we were able to identify. That screen looked for inhibitors of thrombin-stimulated platelet dense granule release using luciferase to detect ATP release accompanying granule exocytosis. Primary screening (AID 1663) of 302517 compounds identified 661 that were active. Subsequent confirmation (AID 1189) and counter-screening for compounds that inhibit luciferase activity (AID 1891) yielded ~250 active compounds that inhibited platelet granule release. This effort resulted in discovery of a probe that targeted protease-activated receptor 1 22. Surprisingly, given what we would anticipate would be many similarities between exocytosis in CTLs and platelets, including the involvement of signaling pathways like PKC and calcium influx, as well as the involvement of SNARE proteins, (see 23 for a recent review of signaling in platelets and 24 for a discussion of the exocytic machinery), there was only one compound active in that screen that we also found to be active. SID 17414945, which we found blocked lytic granule exocytosis and MAPKK activation, inhibited lytic granule exocytosis with an EC50 of ~0.3 μM, but apparently inhibited platelet granule release much less potently, with an EC50 of 19 μM. We cannot account for these results. It seems unlikely to result from our use of a cell line, as TALL-104 cells recapitulate key features of CTL function. A second potentially comparable phenotypic screening campaign used a luciferase reporter construct to look for inhibitors of NF-κB activation in Jurkat cells stimulated with PMA and ionomycin. That effort identified 735 confirmed active compounds (initial screen in AID 435003, confirmation in AID 435020). Although the hit rate in that screening campaign was ~5 times as high as ours, the phenotype interrogated includes the entire transcriptional and translational apparatus required to generate the reporter, together with the proteins required for ubiquitination 25, which may account for the larger number of hits.
One of our hypotheses was that a phenotypic cellular endpoint assay followed by appropriately-designed mechanism-testing could be used as a high-content screen for important targets. Consistent with this idea, our experiments revealed a number of substances that act on important pathways, including fifteen inhibitors of calcium influx, seven MAPKK inhibitors and one calcineurin inhibitor. Were any of these activities defined in previous screens of the MLSMR? Determining this is unfortunately not straightforward, and was not facilitated by the structure and query capabilities of the PubChem web interface. To the best of our knowledge, no screen has looked for inhibitors of calcium influx in nonexcitable cells. However, we were able to identify three screens of >100,000 MLSMR compounds conducted on calcium-permeable channels and five conducted on potassium channels. Blockers of these channels could be envisioned to block calcium or potassium channels in CTLs, although this is by no means certain. None of the substances we defined as inhibiting calcium signals were reported to block calcium-permeable CAV3 (confirmation in AID 489005), TRPC4 (confirmation in AID 2247) or TRPC6 (confirmation in AID 488961) channels. One substance that blocked Fura-2 signals, SID 49721823, was reported to inhibit KCNK9 potassium channels (confirmation in AID 492992). Interestingly, KCNK9 (also known as TASK-3) channels have been found in lymphocytes, and inhibitors reduce proliferation and cytokine production 26. SID 49677461 blocked lytic granule exocytosis and KIR2.1 potassium channels (confirmation in AID 2032), but gave inconclusive results in our Fura assays. Similarly, SID 4964129 blocked lytic granule exocytosis and KCNK3 potassium channels (confirmation in AID 651638), but also gave inconclusive results in our Fura assays.
There has been a screen of MLSMR compounds on ERK activation by MAP kinase kinases. That effort (AID 1454) used AlphaScreen technology to assess levels of phosphorylated ERK in lysates of cells treated with EGF. Of the eight MAPKK inhibitors we found, only two appear to have been tested in that screen, which tested an earlier version of the MLSMR containing fewer compounds. Of the two substances both active in our assay and tested in theirs, they found SID 24810923 to be inactive and SID 26727153 gave inconclusive results. We were unable to identify any screens of the MLSMR that might have been expected to reveal calmodulin or calcineurin inhibitors. The substance that we found to inhibit NFAT dephosphorylation (SID 92764285, 2-chloro-3-(3,5-dimethylmorpholin-4-yl) naphthalene-1,4-dione), was, as expected based on its chemistry, fairly broadly active in deposited assays, but had no confirmed effects that suggested to us possible effects on either calmodulin or calcineurin.
In addition to revealing important actions of compounds on known targets, we suspected that a screen of lytic granule exocytosis could reveal substances that might work via an unknown MMOA. This appears to be the case, as we identified SID 7977862 which blocks lytic granule exocytosis without inhibiting any of the pathways we tested. Inspecting the reported effects of this compound does not lead to well-supported hypotheses about its MMOA. An unconfirmed high content screen (AID 1381) found that it inhibited dynein motor protein activity, but, while dynein has been reported to be involved in reorientation of lytic granules to the site of contact with target cells 27, there is no evidence for dynein involvement in TCR-independent exocytosis. Furthermore, >1000 other compounds were active in AID 1381, but none of them inhibited lytic granule exocytosis in our screen. If dynein were a common target in both assays, we would have expected more extensive overlap. Examination of the confirmed activities reported in the PubChem database for each of the seven other substances for which we were unable to determine a MMOA also failed to suggest plausible mechanistic hypotheses for any of their actions. In addition to the MMOAs we were able to test, other possible targets include SNARE proteins and associated components of the exocytic machinery (see 28 for a recent review). Note that in preliminary experiments we tested the effects of a cherry-pick of DMSO stocks of all eight substances with undefined MMOA (including SID 7977862) on CD3-stimulated IL-2 secretion by Jurkat human leukemic T lymphocytes, and found that all inhibited, suggesting that each is likely to have broad immunosuppressive activity rather than being a specific inhibitor of lytic granule exocytosis (J. Doucette and A. Zweifach, unpublished observations).
Our assay was configured specifically to detect inhibitors. One way we achieved that was the use of very strong artificial stimulation with TG and PMA. We therefore did not- and did not expect to- detect enhancers of exocytosis. A recent study with human NK cells screened the Prestwick Compound Library (PCL) for agents modulating FCGRIII-stimulated lytic granule exocytosis, integrin activation and protein synthesis 29. This study found 56 inhibitors and 12 enhancers. Our screen of the PCL identified several of the inhibitors they found, including amoxapine, desloratidine, thimerosol and zotepine. Differences in assay conditions- particularly protein concentration in the buffer used- likely account for incomplete overlap of inhibitors.
Despite an emphasis in recent years on target-based approaches, a recent analysis demonstrates that a majority of first-in-class new drugs released between 1999 and 2008 were discovered by phenotypic screens 30. Our screen of lytic granule exocytosis identified a number of previously-undescribed actions of compounds on known targets that could serve as leads for immunosuppressants, and also identified several compounds that may work via an unknown MMOA. We determined that at least one of these has probe-like properties. This confirms that our phenotypic screen based on lytic granule exocytosis may be a good means of identifying immunosuppressive compounds. Furthermore, we suggest that our basic strategy of screening a complex cellular phenotype that incorporates multiple potential targets and then defining MMOAs for active compounds may be a useful means of identifying interesting biologically active small molecules in compound collections.
Acknowledgments
We would like to thank Amy E. Florian for conjugating the anti-LAMP antibody and for helping grow and ship cells from UConn to UNM. Christopher K. Lepensky also helped grow and ship cells. This work was supported by R21 NS066462 to AZ and U54 MH084690 to LAS.
References
- 1.Azimzadeh AM, Lees JR, Ding Y, et al. Immunobiology of transplantation: impact on targets for large and small molecules. Clin Pharmacol Ther. 2011;90:229–242. doi: 10.1038/clpt.2011.106. [DOI] [PubMed] [Google Scholar]
- 2.Florian AE, Lepensky CK, Kwon O, et al. Flow cytometry enables a high-throughput homogeneous fluorescent antibody-binding assay for cytotoxic T cell lytic granule exocytosis. J Biomol Screen. 2013;18:420–429. doi: 10.1177/1087057112466697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen MH, Schrama D, Thor Straten P, et al. Cytotoxic T cells. J Invest Dermatol. 2006;126:32–41. doi: 10.1038/sj.jid.5700001. [DOI] [PubMed] [Google Scholar]
- 4.Bossi G, Griffiths GM. CTL secretory lysosomes: biogenesis and secretion of a harmful organelle. Semin Immunol. 2005;17:87–94. doi: 10.1016/j.smim.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Cesano A, Santoli D. Two unique human leukemic T-cell lines endowed with a stable cytotoxic function and a different spectrum of target reactivity analysis and modulation of their lytic mechanisms. In Vitro Cell Dev Biol. 1992;28A:648–656. doi: 10.1007/BF02631041. [DOI] [PubMed] [Google Scholar]
- 6.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 8.Radoja S, Frey AB, Vukmanovic S. T-cell receptor signaling events triggering granule exocytosis. Crit Rev Immunol. 2006;26:265–290. doi: 10.1615/critrevimmunol.v26.i3.40. [DOI] [PubMed] [Google Scholar]
- 9.Pores-Fernando AT, Zweifach A. Calcium influx and signaling in cytotoxic T-lymphocyte lytic granule exocytosis. Immunol Rev. 2009;231:160–173. doi: 10.1111/j.1600-065X.2009.00809.x. [DOI] [PubMed] [Google Scholar]
- 10.Edwards BS, Zhu J, Chen J, et al. Cluster cytometry for high capacity bioanalysis. Cytometry Part A. 2012;81:419–429. doi: 10.1002/cyto.a.22039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takayama H, Trenn G, Humphrey W, Jr, et al. Antigen receptor-triggered secretion of a trypsin-type esterase from cytotoxic T lymphocytes. J Immunol. 1987;138:566–569. [PubMed] [Google Scholar]
- 12.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 13.Pores-Fernando AT, Bauer RA, Wurth GA, et al. Exocytic responses of single leukaemic human cytotoxic T lymphocytes stimulated by agents that bypass the T cell receptor. J Physiol. 2005;567:891–903. doi: 10.1113/jphysiol.2005.089565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maul-Pavicic A, Chiang SC, Rensing-Ehl A, et al. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A. 2011;108:3324–3329. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis RS. Calcium signaling mechanisms in T lymphocytes. Ann Rev Immunol. 2001;19:497–521. doi: 10.1146/annurev.immunol.19.1.497. [DOI] [PubMed] [Google Scholar]
- 16.Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev. 2009;231:59–87. doi: 10.1111/j.1600-065X.2009.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haverstick DM, Engelhard VH, Gray lS. Three intracellular signals for cytotoxic T lymphocyte-mediated killing: independent roles for protein kinase C, calcium influx, and calcium release from intracellular stores. J Immunol. 1991;146:3306–3313. [PubMed] [Google Scholar]
- 18.Puente LG, Stone JC, Ostergaard HL. Evidence for protein kinase C-dependent and -independent activation of mitogen-activated protein kinase in T cells: potential role of additional diacylglycerol binding proteins. J Immunol. 2000;165:6865–6871. doi: 10.4049/jimmunol.165.12.6865. [DOI] [PubMed] [Google Scholar]
- 19.Pores-Fernando AT, Gaur S, Grybko MJ, et al. ERK activation is only one role of PKC in TCR-independent cytotoxic T cell granule exocytosis. Biochem Biophys Res Commun. 2008;371:630–634. doi: 10.1016/j.bbrc.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newman RH, Zhang J. Visualization of phosphatase activity in living cells with a FRET-based calcineurin activity sensor. Mol Biosyst. 2008;4:496–501. doi: 10.1039/b720034j. [DOI] [PubMed] [Google Scholar]
- 21.Harvey CD, Ehrhardt AG, Cellurale C, et al. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci U S A. 2008;105:19264–19269. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dockendorff C, Aisiku O, Verplank L, et al. Discovery of 1,3-Diaminobenzenes as Selective Inhibitors of Platelet Activation at the PAR1 Receptor. ACS Med Chem Lett. 2012;3:232–237. doi: 10.1021/ml2002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Delaney MK, O’Brien KA, et al. Signaling during platelet adhesion and activation. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koseoglu S, Flaumenhaft R. Advances in platelet granule biology. Curr Opin Hematol. 2013;20:464–471. doi: 10.1097/MOH.0b013e3283632e6b. [DOI] [PubMed] [Google Scholar]
- 25.Chen ZJ. Ubiquitin signalling in the NF-κB pathway. Nature cell biology. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meuth SG, Bittner S, Meuth P, et al. TWIK-related acid-sensitive K+ channel 1 (TASK1) and TASK3 critically influence T lymphocyte effector functions. Journal of biological chemistry. 2008;283:14559–14570. doi: 10.1074/jbc.M800637200. [DOI] [PubMed] [Google Scholar]
- 27.Mentlik AN, Sanborn KB, Holzbaur EL, et al. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Molecular biology of the cell. 2010;21:2241–2256. doi: 10.1091/mbc.E09-11-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwarz EC, Qu B, Hoth M. Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2013;1833:1603–1611. doi: 10.1016/j.bbamcr.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 29.Theorell J, Gustavsson AL, Tesi B, et al. Immunomodulatory activity of commonly used drugs on Fc-receptor-mediated human natural killer cell activation. Cancer Immunology, Immunotherapy. 2014;63:627–641. doi: 10.1007/s00262-014-1539-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]