

Abstract
A small series of norbornane bisether diguanidines have been synthesized and evaluated as antibacterial agents. The key transformation—bisalkylation of norbornane diol 6—was not successful using Williamson methodology but has been accomplished using Ag2O mediated alkylation. Further functionalization to incorporate two guanidinium groups gave rise to a series of structurally rigid cationic amphiphiles; several of which (16d, 16g and 16h) exhibited antibiotic activity. For example, compound 16d was active against a broad range of bacteria including Pseudomonas aeruginosa (MIC = 8 µg/mL), Escherichia coli (MIC = 8 µg/mL) and methicillin-resistant Staphylococcus aureus (MIC = 8 µg/mL).
Introduction
Antibacterial resistance is now a global medical concern.1–4 A decline in the number of pharmaceutical companies pursuing new therapeutics, and the continued misuse of antibiotics has only served to exacerbate the problem.1–5 Many antibacterial agents that target the lipopolysaccharide (LPS) layer of Gram-negative bacteria either possess or adopt an amphiphilic structure.6–10 It has also been shown that appropriate functionalization of a rigidified core (such as a calixarene) enables facially amphiphilic compounds to be constructed.7–11 The bicyclo[2.2.1]heptane (norbornane) scaffold represents one of the most accessible, preorganized frameworks available and has a history of use in many fields of chemistry. Biologically active examples include: naxifylline 1 (an A1-adenosine receptor antagonist),12 N6-(5,6-epoxynorbornyl)adenonsines (A1-adenosine receptor agonists)13–15 and diguanidine 3, (active against Gram-negative bacteria).16 In supramolecular chemistry norbornanes have found use in organogel formation,17 whilst fused polynorbornanes have been successfully employed as scaffolds for both anion recognition,18–21 and as ligands for the construction of metal-organic cages.22 In the field of asymmetric synthesis, chiral auxiliaries (such as 2) based on the norbornane framework have also been successfully employed.23, 24
It is known that ethers typically demonstrate better in vivo stability than acetals,25 therefore, analogs of antibacterial 316 (Figure 1) that followed the general structure 4 (Scheme 1) were considered attractive targets. In order to construct this class of compounds it was envisaged that bisether diacids (5) would be useful building blocks and these could in turn be synthesized from norbornane diol 6 (Scheme 1).
Figure 1.

Examples of functionalized norbornanes.
Scheme 1.

Access to bisether diguanidine 4 from diol 6.
The direct alkylation of norbornane diol 6 is as yet unreported in the literature, which is presumably because the reaction is troublesome. Indeed, it has been shown that in some cases alkylation of alcohols using typical Williamson protocols gives lower than expected yields, or no desired product.26, 27 Indeed, in circumstances where a stabilized leaving group can be formed the basic conditions employed can lead to an elimination event.28, 29 An example of this competitive elimination was described by Hergueta et al. (Scheme 2); alkoxide 7, derived from a norbornane fused to a quinoxaline, undergoes elimination to give a carbanion (8) which is stabilized by the aromatic heterocyclic.30
Scheme 2.
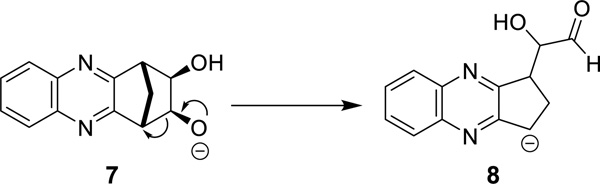
Ring opening to give stabilized ‘benzylic’ carbanion as observed by Hergueta et al30
The use of silver(I) oxide for the transformation of an alcohol to a methyl ether has been known for well over a century.31 The Irvine-Purdie method has been used to achieve i) mono-alkylations;32–36 ii) bisalkylation of unhindered diols such as cycohexanediol;37 and iii) per-methylation of carbohydrates such as glucose, galactose and fructose.31, 38 Inefficient reaction conditions (e.g. multiple additions of the alkylating agent34 or carrying the reaction out in neat alkylating agent) means the Irvine-Purdie alkylation is often overlooked in favor of Williamson methodology. To the best of our knowledge, the use of silver(I) oxide for the bisalkylation of eclipsed syn-1,2-diols has not been previously described.
Herein, we report a protocol for the bisalkylation of the sterically hindered 1,2-diol 6 using silver(I) oxide and a small excess (4.6 equiv) of suitable alkyl halides. Hydrolysis of the esters gave access to compounds of the general structure 5, which were shown to exist in a sterically congested environment around the two alkyl groups by X-ray crystallography. Subsequent functionalization afforded a series of diguanidines (4, Scheme 1), that were tested against a range of Gram-negative and Gram-positive bacteria (Table 3 and 4).
Table 3.
Zone of inhibition (ZOI) as measured (in mm) using disk diffusion.a
| Compound |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 16a | 16b | 16c | 16d | 16e | 16f | 16g | 16h | 16i | COLc | |
|
A. baumannii ATCC 19606 |
NTb | NT | - | - | - | - | 15 | - | - | |
|
P. aeruginosa ATCC 27853 |
- | 11 | 16 | 15 | 16 | 15 | 16 | 15 | - | 19 |
|
K. pneumoniae ATCC13883 |
- | - | 14 | 16 | 14 | 14 | 15 | 8 | - | 20 |
|
S. aureus MRSA ATCC 43300 |
- | - | 15 | 17 | 11 | 11 | 17 | 18 | - | - |
|
E. faecium VREATCC 700221 |
- | - | - | 18 | - | - | 7 | 16 | - | - |
Measured after incubation of disk (6 mm diameter, 50 µg/disk) at 37 °C for 20 hours.
NT = not tested.
Tested at 10 µg/disk.
Table 4.
MIC values (µg/mL)
| Compound |
||||||||
|---|---|---|---|---|---|---|---|---|
| 16c | 16d | 16e | 16f | 16g | 16h | COLb | VANc | |
|
A. baumannii ATCC 19606 |
>32 | 32 | >32 | >32 | >32 | >32 | 0.06 | NT |
|
P. aeruginosa ATCC 27853 |
>32 | 8 | >32 | >32 | 32 | >32 | 0.25 | NT |
|
K. pneumoniae ATCC 700603 |
>32 | >32 | >32 | >32 | >32 | >32 | 0.03 | NT |
|
E. coli ATCC 25922 |
>32 | 8 | >32 | >32 | >32 | >32 | 0.06 | NT |
|
S. aureus MRSA ATCC 43300 |
>32 | 8 | >32 | >32 | 16 | 32 | NT | 1 |
|
S. aureus mMRSA |
NTa | 16 | NT | NT | 32 | NT | NT | 1 |
|
S. aureus GISA, NRS 17 |
NT | 8 | NT | NT | 16 | NT | NT | 4 |
|
S. aureus VISANRS 1 |
NT | 16 | NT | NT | 32 | NT | NT | 8 |
|
S. aureus MRSA |
NT | 16 | NT | NT | 32 | NT | NT | 2 |
|
S. aureus NARSA VRS 10 |
NT | 8 | NT | NT | 16 | NT | NT | 2 |
|
S. pneumoniae MDR ATCC 700677 |
NT | 8 | NT | NT | 8 | NT | NT | 2 |
| VanAE. faecalis | NT | 8 | NT | NT | 32 | NT | NT | >32 |
NT = not tested due to lack of activity against the primary gram-positive strain (MRSA ATCC 43300).
COL = Colistin sulphate.
VAN = Vancomycin.
Results & Discussion
Chemistry
The starting diester diol 6 was synthesized in two steps from dimethyl fumarate.16, 39 The norbornane framework was constructed using either standard Diels–Alder conditions (99%) or by a solvent-free, microwave-assisted approach (2 hours at 150 °C, 98%) similar to that reported by Nencka and co-workers.39 The syn-1,2-diol 6 was synthesized in excellent yields (94%) on multigram scales (up to 4 g), using OsO4 (0.1 mol%) mediated dihydroxylation of the norbornene using 4-methylmorpholine N-oxide (NMO) as a co-oxidant.16, 40 Dihydroxylation using KMnO4, as detailed by Donohoe,41 was also successful, albeit in lower yield (58%) (see Supporting Information for full reaction conditions).
Williamson methodology was initially trialled using 6, NaH, MeI, at 55 °C (Scheme 2) and after 16 hours the diol had been completely consumed (as determined by TLC analysis). However, examination of the crude product using 1H NMR spectroscopy showed no signs of the desired bisether 9a. Instead, a complex mixture (inseparable by column chromatography) was produced. Deprotonation with n-BuLi in THF at −78–0 °C was also trialled but again the reaction was unsuccessful. Given that similar norbornane diols undergo base-induced ring opening to give stabilized anions (Scheme 2),30 we propose that following deprotonation, a Grob-type fragmentation occurs to give a stabilized enolate (11, Scheme 3).42
Scheme 3.

Failed attempt to access bisether 9a and the possible pathway leading to enolate 11.
The Sakai group have described the reduction of esters to the corresponding ethers using Et3SiH in the presence of catalytic InBr3.43 Unfortunately, in the current work, after acetylation of diol 6 to form diacetate 20 (63%, see experimental section),41 reaction with InBr3 failed to produce the bisether product; instead 1H NMR analysis indicated that a complex mixture of products had formed.
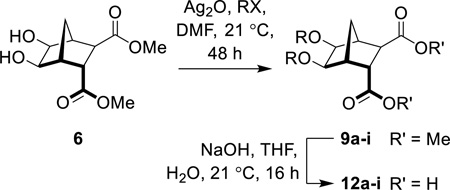
In light of these failures, attention turned towards the use of Ag2O.32–37, 44, 45 Indeed, when diol 6 was reacted with Ag2O (1.6 equiv) and MeI (4.6 equiv) in DMF at ambient temperature for 48 hours (Scheme 4), bisether 9a was isolated in good yield (67%).33 Saponification of diester 9a gave the required bisether diacid (12a) in 73% yield (Scheme 4). Extended reaction times, adding further portions of MeI throughout the reaction, and heating the reaction (both conventional and microwave irradiation), did not lead to increased yields. Using Ag2CO3, AgNO3, AgBF4 or AgPF6 gave none of the desired product with only starting material recovered.
Scheme 4.

Synthesis of diacid 12a
To further test the scope of these reaction conditions a range of benzyl halides were used. Using benzyl bromide (Table 1, entry 2), bisether diacid 12b was attained over two steps (18% using 300 mg of 6). Pleasingly, when the reaction was performed on a larger scale (900 mg of 6), the yield increased to 37%. A Finkelstein approach46 using NaI did not increase the yield of bisether diacid 12b. The low yields can be rationalized somewhat by the steric bulk introduced as a result of the first benzylation—the reaction with the second equivalent benzyl halide is considerably inhibited. Indeed, an appreciable amounts of the monoalkylated regioisomer (37%) were isolated along with the desired bisether product 12b.
Table 1.
Synthesis of bisether diacids 12a–i
 | |||
|---|---|---|---|
| Entry | RX | Diacid | Yield (%)c |
| 1 | MeI | 12a | 48 |
| 2 | 12b | 37 | |
| 3 | 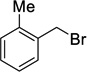 |
12c | 19 |
| 4 | 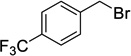 |
12d | 51 |
| 5 | 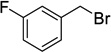 |
12e | 55 |
| 6 | 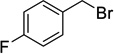 |
12f | 25 |
| 7 | 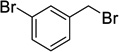 |
12g | 24 |
| 8 | 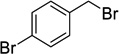 |
12h | 28 |
| 9a | 12i | 25 | |
| 10 | 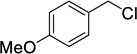 |
N.Rb | N.R |
| 11 | 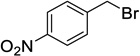 |
N.R | N.R |
| 12 | N.R | N.R | |
Reaction was stirred for 4 days.
N.R = no reaction.
Yield calculated over two steps.
Similarly, using 2-methylbenzyl bromide, 12c was accessed in a 19% yield (Table 1, entry 3), and a reasonable quantity of a monoalkylated intermediate was isolated (39 %). However, despite the modest yields, this protocol provided access to the desired norbornane bisethers—a previously inaccessible family of compounds.
When a range of fluorinated benzyl bromides were used good yields of the desired bisether products were obtained. Following the aforementioned two-step process, 12d (51%) and 12e (55%) were accessed in good yields using 4-(trifluoromethyl)benzyl bromide and 3-fluorobenzyl bromide respectively (Table 1, entries 4 and 5). Furthermore, when 4-fluorobenzyl bromide was employed to alkylate diol 6, and following hydrolysis of the ester groups using NaOH/THF (Table 1, entry 6) diacid 12f was obtained in moderate yield (25%) over the two steps.
Using 3-bromobenzyl bromide and 4-bromobenzyl bromide the synthesis of bisether diacids 12g and 12h (24 and 28% over two steps respectively, Table 1, entries 7 and 8) were carried out in the same fashion. Crystals suitable for X-ray diffraction were obtained for bisether diacid 12h after recrystallization from EtOH/pet. spirits. The resulting structure contained a unit cell comprised of two conformational isomers (as shown in Figure 2). The O···O distance (ca. 2.6 Å), clearly illustrates the proximity of the two benzyl groups and their non-symmetric orientation (presumably due to steric constraints).
Figure 2.

Thermal ellipsoid plot of the two independent molecular confirmations (Ellipsoids at 20% probability level) of compound 12h.
When allyl bromide was used stirring for 4 days was required to consume all the starting material; subsequent ester hydrolysis gave diacid 12i in a 25% yield over the two steps (Table 1, entry 9). Unfortunately, reactions with 4-methoxybenzyl chloride and 4-nitrobenzyl bromide (Table 1, entries 10 and 11) failed to give any of the desired product with only minimal consumption of starting material taking place (as evidenced by 1H NMR analysis of the crude reaction mixture). Also, when 1-iodooctane (Table 1, entry 12) was employed alkylation was unsuccessful.
In six of the alkylation reactions (12b, 12d–12h) transesterification occurred to a small extent (6–12%, as determined using 1H NMR spectroscopy). This mixed-ester side-product (example 13 from synthesis of 9b, Figure 3) was difficult to separate from the desired dimethyl ester product using column chromatography. The presumption that the more sterically accessible exo methyl ester was replaced is based on a previous report by Niwayama and co-workers that illustrated how the exo face is less hindered than the endo face of related norbornane diesters.40 Despite the presence of small amounts of mixed-ester by-products, in each case hydrolysis proceeded smoothly and pure diacids were isolated in every instance.
Figure 3.

Representative structure of proposed mixed-ester 13 formed during reaction with BnBr.
With bisethers diacids (12a–12i) in hand, functionalization to the required amphiphiles was pursued. Attachment of 2-[2,3-bis(tert-butoxycarbonyl)guanidino]ethylamine (14)47, 48 to the norbornane scaffold was carried out using EDCI and HOBt in either DMF or CHCl3 using microwave irradiation at 50 °C for 30 minutes to give compounds 15a–15i in moderate to good yields (26–72%, Table 2). Subsequent deprotection was accomplished using HCl (generated in situ from AcCl in MeOH) to give the desired guanidines as the guanidinium chloride salts (16a–16i, Table 2).
Table 2.
Formation of HCl salts 16a–i and associated cLogP values
 | ||||
|---|---|---|---|---|
| Diacid | R | Diamide (yield, %) |
Diguanidineb (yield, %) |
cLogPc |
| 12a | Me | 15a (72) | 16a (96) | −4.80 |
| 12ba | 15b (57) | 16b (96) | −2.04 | |
| 12c | 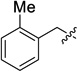 |
15c (54) | 16c (24) | −1.23 |
| 12d | 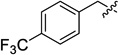 |
15d (56) | 16d (66) | −0.24 |
| 12e | 15e (26) | 16e (99) | −1.76 | |
| 12fa | 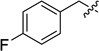 |
15f (45) | 16f (77) | −1.71 |
| 12ga | 15g (53) | 16g (97) | −0.47 | |
| 12h | 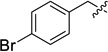 |
15h (26) | 16h (99) | −0.42 |
| 12i | 15i (63) | 16i (31) | −3.94 | |
Reaction was performed in CHCl3.
Product was isolated as an HCl salt.
Calculated using www.molinspiration.com software.
A balance of both hydrophobicity and hydrophilicity is essential for the antibacterial activity of structural amphiphiles.49 In light of this, the calculated LogP (cLogP) values (the log of the octanol/water partition coefficient) were determined for bisether diguanidines (16a–16i) using www.molinspiration.com software (Table 2) and these will be discussed in relation to activity in the following section.
Biological evaluation
The antibacterial activity of these compounds was evaluated against a range of Gram-negative and Gram-positive bacteria, including members of the ESKAPE pathogens;1 first using the disk diffusion assay to identify active compounds then micro-broth dilution assays to determine minimum inhibitory concentrations (MICs).
Bis-methyl ether 16a and allyl ether 16i, did not show any inhibition in disk diffusion studies at 50 µg/disk (Table 3). For the bis-benzyl ether 16b, a noticeable zone of inhibition (ZOI) was observed (11 mm) against Pseudomonas aeruginosa. The inclusion of small substituents to the phenyl rings, such as 2-methyl (16c), 3-fluoro (16e) and 4-fluoro (16f), led to improved activity as shown by ZOI’s of 14–16 mm against P. aeruginosa and Klebsiella pneumoniae (Table 3). Furthermore, appreciable ZOI’s (11–15 mm) were also observed against Gram-positive methicillin-resistant Staphylococcus aureus (MRSA). When larger substituents occupied the 4-position of the phenyl ring, inhibition of vancomycin-resistant Enterococcus faecium (VRE) was also seen in addition to the aforementioned strains. The 4-(trifluoromethyl)benzyl (16d) and 4-bromobenzyl (16h) analogs showed ZOI of 18 and 16 mm respectively for VRE (Table 3). The 3-bromobenzyl derivative (16g) was active against all pathogens assessed in this study, and was the sole compound to exhibit activity against Acinetobacter baumannii (ZOI = 15 mm). Comparison of ZOI between 16g and 16h indicates that activity against A. baumannii, K. pneumoniae and E. faecium can be influenced by subtle changes to substituent locations and may have implications in the design of subsequent compounds.
Given the promising results in the disk diffusion screen the substituted benzyl ethers were subjected to a micro-broth dilution assay to quantify MICs (Table 4). When smaller substituents; 2-methyl (16c), 3-fluoro (16e) and 4-fluoro (16f) displaying no MIC ≤ 32 µg/mL against any bacterial strain tested (Table 4). When larger substituents were included on the benzyl rings such as 4-(trifluoromethyl)benzyl (16d) MIC values of 32 µg/mL against A. baumannii, and 8 µg/mL against each of P. aeruginosa, Escherichia coli and MRSA (Table 4) were observed. The 3-bromo benzyl-substituted analog (16g) showed a reasonable MIC against both Gram-negative P. aeruginosa (32 µg/mL) and Gram-positive MRSA (16 µg/mL) bacterial strains. Furthermore, an MIC of 32 µg/mL was observed for the 4-bromo benzyl-substituted analog (16h) against MRSA (Table 4). A correlation between antibacterial activity and cLogP was apparent with compounds with higher cLogP values (Table 2) showing stronger antibacterial activity.
Given the encouraging MIC values for compounds 16d and 16g against MRSA (Table 4), both compounds were evaluated against seven additional Gram-positive bacterial strains including such S. aureus bacterial isolates as multi-resistant methicillin resistant (mMRSA), glycopeptide-intermediate (GISA) and vancomycin-intermediate (VISA), as well as Streptococcus pneumoniae and E. faecalis (Table 5). The 4-(trifluoromethyl)benzyl bisether (16d) was again the most active compound; exhibiting antibacterial activity (MIC = 8–16 µg/mL) against all eight bacterial strains tested. Furthermore, activity was observed by 3-bromobenzyl bisether (16g) against all Gram-positive bacterial strains tested which was highlighted by an MIC of 8 µg/mL against S. pneumoniae (Table 4).
Table 5.
Cytotoxicity values (% of cell survival at 24 h)
| Compound |
||
|---|---|---|
| 16d | 16g | |
|
ATCC CRL-1573 HEK293 |
43 | 90 |
|
ATCC HB-8065 HepG2 |
56 | 96 |
The cell viability (% survival) against human embryonic kidney cells (HEK293) and hepatocellular carcinoma (HepG2) was determined after exposure to compounds 16d or 16g at 100 µM for 24 hours (Table 6). The 4-(trifluoromethyl)benzyl bisether (16d) exhibited moderate cytotoxicity with 43 and 56% cell survival observed against HEK293 and HepG2 respectively (Table 5). In the case of bis-3-bromobenzyl ether (16g) the cell viability was determined to be 90% and 96% against HEK293 and HepG2 respectively (IC50 < 100 µM). The cytotoxicity profile for these compounds is acceptable when compared to previously reported antibacterial agents.50
Conclusions
The previously inaccessible norbornane bisether diacids 12a–i, were successfully prepared using Ag2O and a suitable alkyl or benzyl halide as the key step. An X-ray crystal structure of bisether diacid 12h highlighted the sterically crowded environment of the ethers; which presumably hindered the second etherification step and resulted in lower yields. Nevertheless, the protocol presented here provides a viable alternative for the alkylation of congested syn-diols or base-sensitive alcohols where typical Williamson ether synthesis conditions fail. Further functionalization of bisether diacids (12a–i) gave a series of bisguanidines as hydrochloride salts (16a–i).
Several of the compounds (16d, 16g and 16h) displayed antibacterial activity, with MIC values as low as 8 µg/mL, against a range of problematic bacterial species including P. aeruginosa, E. coli, S. pneumonia, E. faecalis and several strains of S. aureus. The results presented here reinforce the notion that the activity of cationic antimicrobial peptides can be mimicked by relatively small, structurally rigid amphiphiles. Indeed, when compared to other synthetic scaffolds (such as calixarenes) which are used to generate antibacterial amphiphiles,9 the low molecular weight of these compounds and their reasonable antibacterial activity make them an attractive class of compounds worthy of further investigation.
Experimental Section
The following compounds were prepared using literature methods and full reaction details can be found in the supplementary information; Dimethyl bicyclo[2.2.1]hept-5-ene-3-endo-2-exo-dicarboxylate (17),39, 51 616, 41 and 14.47
General Information
All microwave reactions were conducted using a CEM Discover S-Class Explorer 48 Microwave Reactor, operating on a frequency of 50/60 Hz and continuous irradiation power from 0–300 W. All reactions were performed in sealed reaction vessels. All melting points are uncorrected. All 1H, 13C and 19F NMR spectra were collected on either a 270 MHz FT-NMR spectrometer, a 400 MHz FT-NMR spectrometer, or a 500 MHz FT-NMR spectrometer where indicated. All 2D NMR experiments were performed on a 500 MHz FT-NMR spectrometer. Variable temperature (VT) NMR experiments were performed on a 270 MHz FT-NMR spectrometer. Samples were dissolved in CDCl3, DMSO-d6 or CD3OD where specified with the residual solvent peak used as the internal reference – CDCl3; 7.26 (1H) and 77.0 (13C), DMSO-d6; 2.50 (1H) and 39.52 (13C), CD3OD; 3.31 (1H) and 49.0 (13C).52 Proton spectra are reported as chemical shift δ (ppm) (integral, multiplicity (s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, quin = quintet and m = multiplet), coupling constant (Hz), assignment). Carbon spectra are reported as chemical shift δ (ppm) (integral, multiplicity (d = doublet and q = quartet), coupling constant (Hz)) were appropriate. Fluorine spectra are reported as chemical shift δ (ppm) and were externally referenced using 0.05% α,α,α-trifluorotoluene in CDCl3; −63.72 (19F).
High resolution mass spectral data was collected on using a QTOF mass spectrometer (LC-1200 series) under the following conditions: gas temperature (300 °C), nitrogen drying gas (10.0 L min−1), capillary voltage (3500 V), fragmentor (140 V), and nebuliser (45 psi) in a 80% MeCN in H2O solvent system. Analyte solutions were prepared in HPLC grade methanol (conc. ~ 1 mg mL−1).
All chemicals and solvents were used as received without further purification unless otherwise stated. Column chromatography was performed on silica gel (230–400 mesh).
Dimethyl 5,6-diacetate bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (20)
The stirring solution of diol 6 (130 mg, 0.532 mmol), DMAP (13 mg, 0.11 mmol), pyridine (171 µL, 2.13 mmol) and CH2Cl2 (1.1 mL) was treated with AcCl (130 µL, 1.81 mmol) slowly and the reaction was stirred at ambient temperature for 16 h. The resulting bright orange solution was quenched with sat. NaHCO3 (6 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was washed with 1M HCl (10 mL), dried (MgSO4), filtered and concentrated in vacuo to give a clear oil that was purified by column chromatography (35–50% EtOAc in pet. spirits). The title compound (110 mg, 63%) was isolated as a clear oil; Rf = 0.68 (50% EtOAc in pet. spirits). 1H NMR (270 MHz, CDCl3) δ 1.53 (1H, dt, J = 11.0, 1.4 Hz, H7s), 1.94 (1H, dd, J = 11.0, 1.6 Hz, H7a), 2.01 (6H, s, 2 × Me), 2.61 (1H, br s, H1), 2.64–2.66 (1H, m, H4), 2.87 (1H, dd, J = 5.6, 1.2 Hz, H2), 3.23 (1H, app. t, J = 5.6 Hz, H3), 3.69 (3H, s, Me), 3.73 (3H, s, Me), 4.75 (1H, dd, J = 6.0, 1.6 Hz, H6), 4.87 (1H, dd, J = 6.0, 1.4 Hz, H5). 13C NMR (67.5 MHz, CDCl3) δ 20.6, 20.7, 33.7, 44.6, 44.7, 46.1, 46.2, 52.4, 52.5, 72.4, 74.8, 169.7, 169.9, 172.2, 173.7. HRMS (ESI, m/z) for C15H20O8 [M + H]+ calc. 329.1231 found 329.1233.
General procedure A for the bis-alkylation of diol 6
Anhydrous DMF (2.4 mL) was added to a pre-dried round-bottom flask protected from light, containing diol 6 (3.7 mmol) and Ag2O (1.6 equiv) at ambient temperature. To the stirring solution was added the appropriate alkylating agent (4.6 equiv) and the reaction was stirred for 48 h before the reaction vessel was cooled to 4 °C (refrigerator) for a further 16 h without agitation. The resulting precipitate was removed by vacuum filtration and washed with EtOAc (15 mL). The filtrate was washed with H2O (3 × 10 mL), dried (MgSO4), filtered and concentrated in vacuo to afford the crude product which was purified by column chromatography to give the desired bisether.
Dimethyl 5,6-bis(methoxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9a)
Compound 9a was prepared from diol 6 (915 mg, 3.74 mmol) and iodomethane (1.1 mL, 17.7 mmol) according to general procedure A and was purified by column chromatography (20% EtOAc in pet. spirits) to give the title compound (682 mg, 67%) as a clear viscous oil; Rf = 0.17 (20% EtOAc in pet. spirits). 1H NMR (270 MHz, CDCl3) δ 1.39 (1H, dquin, J = 10.7, 1.6 Hz, H7s), 1.89 (1H, ddd, J = 10.7, 1.6, 1.5 Hz, H7a), 2.64–2.69 (3H, m, H1, H2, H4), 3.20 (1H, dd, J = 5.5, 0.8 Hz, H3), 3.29 (1H, dd, J = 6.0, 1.7 Hz, H5), 3.37 (3H, s, Me), 3.42 (3H, s, Me), 3.46 (1H, dd, J = 6.1, 1.8 Hz, H6), 3.70 (3H, s, Me), 3.72 (3H, s, Me). 13C NMR (67.5 MHz, CDCl3) δ 32.9, 43.3, 44.8, 45.0, 46.4, 52.2, 52.4, 58.7, 58.8, 81.0, 84.1, 173.2, 174.2. HRMS (ESI, m/z) for C13H20O6 [M + H]+ calc. 273.1333; found 273.1328.
Dimethyl 5,6-bis(benzyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9b)
Compound 9b was prepared from diol 6 (917 mg, 3.75 mmol) and benzyl bromide (2.1 mL, 17.3 mmol) according to general procedure A and was purified by column chromatography (10% EtOAc in pet. spirits) to give the title compound (743 mg, 47%) as a clear oil; Rf = 0.43 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.45 (1H, dt, J = 10.7, 1.5 Hz, H7s), 2.10 (1H, dd, J = 10.7, 1.6 Hz, H7a), 2.64–2.65 (2H, m, H1, H2), 2.70–2.71 (1H, m, H4), 3.19 (1H, app. t, J = 5.3 Hz, H3), 3.46 (1H, dd, J = 5.8, 1.6 Hz, H5), 3.60 (3H, s, Me), 3.62 (1H, dd, J = 5.9, 1.4 Hz, H6), 3.69 (3H, s, Me), 4.53–4.62 (4H, m, 2 × ArCH2), 7.25–7.38 (10H, m, ArH). 13C NMR (125 MHz, CDCl3) δ 33.4, 44.2, 45.1, 45.9, 46.4, 52.2, 52.4, 72.5, 72.7, 77.9, 81.7, 127.7 (2 × C), 127.9 (2 × C), 128.0 (2 × C), 128.4 (2 × C), 128.5 (2 × C), 138.4, 138.5, 173.1, 174.3. HRMS (ESI, m/z) for C25H28O6 [M + Na]+ calc. 447.1778; found 447.1754.
Dimethyl 5,6-bis[(2-methylbenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9c)
Compound 9c was prepared from diol 6 (321 mg, 1.31 mmol) and 2-methylbenzyl bromide (810 µL, 6.03 mmol) according to general procedure A and was purified by column chromatography (10–20% EtOAc in pet. spirits) to give the title compound (115 mg, 19%) as a clear oil; Rf = 0.50 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.45 (1H, d, J = 10.6 Hz, H7s), 2.10 (1H, dd, J = 10.6, 1.4 Hz, H7a), 2.27 (3H, s, ArMe), 2.30 (3H, s, ArMe), 2.67–2.68 (2H, m, H1, H2), 2.70–2.71 (1H, m, H4), 3.19 (1H, app. t, J = 5.3 Hz, H3), 3.49 (1H, dd, J = 5.8, 1.4 Hz, H5), 3.62 (3H, s, Me), 3.64 (1H, dd, J = 5.7, 1.2 Hz, H6), 3.70 (3H, s, Me), 4.49–4.61 (4H, m, 2 × ArCH2), 7.10–7.20 (6H, m, ArH), 7.24–7.25 (1H, m, ArH), 7.31–7.33 (1H, m, ArH). 13C NMR (125 MHz, CDCl3) δ 18.9, 19.0, 33.4, 44.4, 45.1, 45.7, 46.5, 52.2, 52.4, 70.9, 71.0, 78.3, 82.0, 125.8, 125.9, 127.8 (2 × C), 128.8, 128.9, 130.2, 130.3, 136.3, 136.4, 136.7, 136.9, 173.2, 174.3. HRMS (ESI, m/z) for C27H32O6 [M + Na]+ calc. 475.2091; found 475.2073.
Dimethyl 5,6-bis[(4-trifluoromethyl)benzyloxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9d)
Compound 9d was prepared from diol 6 (315 mg, 1.29 mmol) and 4-(trifluoromethyl)benzyl bromide (910 µL, 5.93 mmol) according to general A procedure and was purified by column chromatography (10–20% EtOAc in pet. spirits) to give the title compound (461 mg, 64%) as a clear oil; Rf = 0.26 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.49 (1H, dt, J = 10.7, 1.4 Hz, H7s), 2.10 (1H, dd, J = 10.7, 1.6 Hz, H7a), 2.66–2.68 (2H, m, H1, H2), 2.72–2.75 (1H, m, H4), 3.22 (1H, app. t, J = 5.2 Hz, H3), 3.52 (1H, dd, J = 5.8, 1.6 Hz, H5), 3.63 (3H, s, Me), 3.67 (1H, dd, J = 5.8, 1.4 Hz, H6), 3.71 (3H, s, Me), 4.57–4.68 (4H, m, 2 × ArCH2), 7.38–7.42 (4H, m, ArH), 7.54–7.57 (4H, m, ArH). 13C NMR (125 MHz, CDCl3) δ 33.3, 44.2, 45.0, 45.8, 46.3, 52.3, 52.5, 71.8, 71.9, 78.6, 82.2, 124.2 (q, JCF = 270.6 Hz, 2 × CF3), 125.3 (q, 3JCF = 3.6 Hz, 2 × CH), 125.4 (q, 3JCF = 3.6 Hz, 2 × CH), 127.7 (2 × C), 127.8 (2 × C), 129.9 (q, 2JCF = 32.3 Hz), 130.0 (q, 2JCF = 32.3 Hz), 142.4 (2 × C), 173.1, 174.0. 19F NMR (470 MHz, CDCl3) δ −63.02, −63.00. HRMS (ESI, m/z) for C27H26O6F6 [M + H]+ calc. 561.1706; found 561.1718.
Dimethyl 5,6-bis[(3-fluorobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9e)
Compound 9e was prepared from diol 6 (333 mg, 1.36 mmol) and 3-fluorobenzyl bromide (770 µL, 6.26 mmol) according to general procedure A and was purified by column chromatography (10–20% EtOAc in pet. spirits) to give the title compound (326 mg, 52%) as a clear oil; Rf = 0.31 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.47 (1H, dt, J = 10.7, 1.5 Hz, H7s), 2.09 (1H, dd, J = 10.7, 1.6 Hz, H7a), 2.65–2.67 (2H, m, H1, H2), 2.73 (1H, m, H4), 3.21 (1H, app. t, J = 5.3 Hz, H3), 3.47 (1H, dd, J = 5.9, 1.6 Hz, H5), 3.62–3.64 (4H, m, Me, H6), 3.70 (3H, s, Me), 4.52–4.62 (4H, m, 2 × ArCH2), 6.94–7.13 (6H, m, ArH), 7.25–7.30 (2H, m, ArH). 13C NMR (125 MHz, CDCl3) δ 33.3, 44.1, 45.0, 45.8, 46.3, 52.2, 52.5, 71.7, 71.9, 78.2, 81.9, 114.52 (d, 2JCF = 21.6 Hz, 2 × CH), 114.59 (d, 2JCF = 21.1 Hz), 114.62 (d, 2JCF = 21.6 Hz), 123.07 (d, 3JCF = 2.8 Hz), 123.23 (d, 3JCF = 2.8 Hz), 129.94 (d, 4JCF = 1.6 Hz), 130.00 (d, 4JCF = 1.7 Hz), 140.98 (d, 3JCF = 2.4 Hz), 141.04 (d, 3JCF = 2.3 Hz), 163.00 (d, 1JCF = 244.6 Hz), 163.05 (d, JCF = 244.4 Hz), 173.1, 174.1. 19F NMR (470 MHz, CDCl3) δ −113.77. HRMS (ESI, m/z) for C25H26O6F2 [M + H]+ calc. 461.1770; found 461.1784.
Dimethyl 5,6-bis[(4-fluorobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9f)
Compound 9f was prepared from diol 6 (308 mg, 1.26 mmol) and 4-fluorobenzyl bromide (720 µL, 5.80 mmol) according to general procedure A and was purified by column chromatography (10–20% EtOAc in pet. spirits) to give the title compound (157 mg, 27%) as a clear oil; Rf = 0.30 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.45 (1H, dt, J = 10.7, 1.5 Hz, H7s), 2.07 (1H, dd, J = 10.7, 1.6 Hz, H7a), 2.64–2.65 (1H, m, H2), 2.66 (1H, m, H1), 2.69 (1H, dd, J = 4.6, 1.4 Hz, H4), 3.20 (1H, app. t, J = 5.1 Hz, H3), 3.47 (1H, dd, J = 5.8, 1.6 Hz, H5), 3.62 (1H, dd, J = 5.8, 1.5 Hz, H6), 3.64 (3H, s, Me), 3.70 (3H, s, Me), 4.47–4.53 (2H, m, ArCH2), 4.54 (2H, m, ArCH2), 6.96–7.01 (4H, m, ArH), 7.23–7.30 (4H, m, ArH). 13C NMR (125 MHz, CDCl3) δ 33.3, 44.2, 45.1, 45.9, 46.3, 52.2, 52.4, 71.8, 72.0, 78.1, 81.8, 115.2 (d, 2JCF = 21.2 Hz, 2 × CH), 115.3 (d, 2JCF = 21.1 Hz, 2 × CH), 129.6 (d, 3JCF = 9.2 Hz, 2 × CH), 129.7 (d, 3JCF = 8.4 Hz, 2 × CH), 134.1 (d, 4JCF = 3.4 Hz), 134.2 (d, 4JCF = 2.9 Hz), 162.4 (d, 1JCF = 244.0 Hz), 162.5 (d, 1JCF = 244.3 Hz), 173.1, 174.1. 19F NMR (470 MHz, CDCl3) δ −115.36, −115.30. HRMS (ESI, m/z) for C25H26O6F2 [M + Na]+ calc. 483.1590; found 483.1594.
Dimethyl 5,6-bis[(3-bromobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9g)
Compound 9g was prepared from diol 6 (315 mg, 1.29 mmol) and 3-bromobenzyl bromide (1.48 g, 5.93 mmol) according to general procedure A and was purified by column chromatography (10–20% EtOAc in pet. spirits) to give the title compound (266 mg, 35%) as a clear oil; Rf = 0.27 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.47 (1H, dt, J = 10.7, 1.4 Hz, H7s), 2.09 (1H, dd, J = 10.7, 1.5 Hz, H7a), 2.66–2.67 (2H, m, H1, H2), 2.72 (1H, dd, J = 4.6, 1.3 Hz, H4), 3.21 (1H, app. t, J = 4.9 Hz, H3), 3.48 (1H, dd, J = 6.0, 1.7 Hz, H5), 3.63 (1H, dd, J = 5.7, 1.4 Hz, H6), 3.66 (3H, s, Me), 3.71 (3H, s, Me), 4.49–4.59 (4H, m, 2 × ArCH2), 7.15–7.29 (4H, m, ArH), 7.39–7.53 (4H, m, ArH). 13C NMR (125 MHz, CDCl3) δ 33.3, 44.2, 45.1, 45.9, 46.3, 52.3, 52.5, 71.7, 71.9, 78.3, 82.0, 122.6, 122.7, 126.2, 126.3, 130.1 (2 × C), 130.7, 130.8 (3 × C), 140.7, 140.8, 173.1, 174.1. HRMS (ESI, m/z) for C25H26O6Br2 [M + H]+ calc. 581.0169; found 581.0181.
Dimethyl 5,6-bis[(4-bromobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9h)
Compound 9h was prepared from diol 6 (333 mg, 1.36 mmol) and 4-bromobenzyl bromide (1.57 g, 6.26 mmol) according to general procedure A and was purified by column chromatography (10–20% EtOAc in pet. spirits) to give the title compound (279 mg, 35%) as a clear oil. Rf = 0.36 (20% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.45 (1H, d, J = 10.7 Hz, H7s), 2.06 (1H, d, J = 10.6 Hz, H7a), 2.64–2.65 (2H, m, H1, H2), 2.68–2.69 (1H, m, H4), 3.20 (1H, app. t, J = 5.0 Hz, H3), 3.46 (1H, dd, J = 5.8, 1.6 Hz, H5), 3.61 (1H, dd, J = 5.8, 1.3 Hz, H6), 3.64 (3H, s, Me), 3.70 (3H, s, Me), 4.45–4.53 (4H, m, 2 × ArCH2), 7.14–7.19 (4H, m, ArH), 7.42–7.45 (4H, m, ArH). 13C NMR (125 MHz, CDCl3) δ 33.3, 41.2, 45.1, 45.9, 46.3, 52.3, 52.5, 71.8, 72.0, 78.3, 81.9, 121.6 (2 × C), 129.4 (2 × C), 129.6 (2 × C), 131.5 (2 × C), 131.6 (2 × C), 137.4, 137.5, 173.1, 174.1. HRMS (ESI, m/z) for C25H26O6Br2 [M + H]+ calc. 581.0169; found 581.0182.
Dimethyl 5,6-bis(allyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylate (9i)
Compound 9i was prepared from diol 6 (302 mg, 1.24 mmol) and allyl bromide (530 µL, 5.70 mmol) according to general procedure A (but was stirred for 4 days) and was purified by column chromatography (20% EtOAc in pet. spirits) to give the title compound (107 mg, 29%) as a clear oil; Rf = 0.44 (20% EtOAc in pet. spirits). 1H NMR (270 MHz, CDCl3) δ 1.41 (1H, dt, J = 10.6, 1.5 Hz, H7s), 1.99 (1H, dd, J = 10.7, 1.6 Hz, H7a), 2.58–2.59 (1H, m, H1), 2.64–2.68 (2H, m, H2, H4), 3.18 (1H, dd, J = 5.3, 4.8 Hz, H3), 3.43 (1H, dd, J = 5.9, 1.7 Hz, H5), 3.56 (1H, dd, J = 5.9, 1.6 Hz, H6), 3.69 (3H, s, Me), 3.70 (3H, s, Me), 4.01 (2H, dt, J = 5.6, 1.4 Hz, OCH2), 4.06 (2H, dt, J = 5.6, 1.4 Hz, OCH2), 5.13–5.31 (4H, m, 2 × CH2CH), 5.81–5.98 (2H, m, 2 × CH2CH). 13C NMR (67.5 MHz, CDCl3) δ 33.3, 44.2, 45.1, 46.0, 46.4, 52.2, 52.4, 71.8, 71.9, 78.3, 81.8, 117.1 (2 × C), 135.0 (2 × C), 173.2, 174.3. HRMS (ESI, m/z) for C17H24O6 [M + Na]+ calc. 347.1465; found 347.1479.
General procedure B for the hydrolysis of diesters 9a–i
A biphasic solution of the appropriate diester (0.71 mmol), 2 M NaOH (1.5 mL) and THF (3.0 mL) was stirred at ambient temperature for 16 h. All organic impurities were extracted with CH2Cl2 (2 × 5 mL) and the remaining aqueous phase was acidified using 4 M HCl (pH = 1) and extracted with EtOAc (3 × 10 mL). The combined organic phase was washed with brine (10 mL), dried (MgSO4), filtered and concentrated in vacuo to afford the required diacid.
5,6-Bis(methoxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12a)
The title compound 12a was prepared from diester 9a (194 mg, 0.71 mmol) according to general procedure B and isolated as a white waxy solid (127 mg, 73%). 1H NMR (270 MHz, CDCl3) δ 1.43 (1H, d, J = 10.7 Hz, H7s), 1.96 (1H, d, J = 10.0 Hz, H7a), 2.67–2.76 (3H, m, H1, H2, H4), 3.25 (1H, app. t, J = 5.4 Hz, H3), 3.41 (3H, s, Me), 3.44 (3H, s, Me), 3.45–3.50 (2H, m, H5, H6). 13C NMR (67.5 MHz, CDCl3) δ 33.1, 43.1, 44.8, 44.9, 46.3, 58.8, 58.9, 80.8, 84.1, 178.0, 179.2. HRMS (ESI, m/z) for C11H16O6 [M + Cl]− calc. 279.0641; found 279.0652.
5,6-Bis(benzyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12b)
The title compound 12b was prepared from diester 9b (717 mg, 1.69 mmol) according to general procedure B and isolated as a white powder (525 mg, 78%). 1H NMR (270 MHz, CDCl3) δ 1.48 (1H, d, J = 10.9 Hz, H7s), 2.17 (1H, d, J = 10.5 Hz, H7a), 2.63 (1H, d, J = 5.6 Hz, H1), 2.73 (1H, br s, H2), 2.79 (1H, d, J = 4.0 Hz, H4), 3.23 (1H, app. t, J = 5.4 Hz, H3), 3.63 (2H, t, J = 6.4 Hz, H5, H6), 4.57–4.67 (4H, m, 2 × ArCH2), 7.23–7.35 (10H, m, ArH). 13C NMR (67.5 MHz, DMSO-d6) δ 32.9, 43.6, 44.7, 45.3, 46.1, 71.6, 71.7, 78.1, 81.5, 127.4 (2 × C), 127.7 (4 × C), 128.1 (2 × C), 128.1 (2 × C), 138.5, 138.7, 173.6, 174.8. HRMS (ESI, m/z) for C23H24O6 [M – H]− calc. 395.1501; found 395.1504.
5,6-Bis(2-methylbenzyloxy) bicyclo[2.2.1]heptane-2-endo-2-exo-dicarboxylic acid (12c)
The title compound 12c was prepared from diester 9c (121 mg, 0.27 mmol) according to general procedure B and isolated as a clear oil (113 mg, 99%). 1H NMR (500 MHz, DMSO-d6) δ 1.31 (1H, d, J = 10.1 Hz, H7s), 1.85 (1H, d, J = 9.4 Hz, H7a), 2.19 (3H, s, Me), 2.21 (3H, s, Me), 2.47 (1H, d, J = 5.5 Hz, H4), 2.55 (1H, br s, H1), 2.61 (1H, d, J = 3.3 Hz, H2), 2.99 (1H, app. t, J = 5.0 Hz, H3), 3.59 (1H, d, J = 5.7 Hz, H5), 3.64 (1H, d, J = 5.6 Hz, H6), 4.44–4.49 (2H, m, ArCH2), 4.53 (2H, br s, ArCH2), 7.08–7.18 (6H, m, ArH), 7.22–7.27 (2H, m, ArH), 12.54 (2H, br s, 2 × COOH). 13C NMR (125 MHz, DMSO-d6) δ 18.3 (2 × C), 33.0, 43.6, 44.7, 45.2, 46.1, 70.0, 70.1, 78.5, 81.8, 125.5 (2 × C), 127.5 (2 × C), 128.3 (2 × C), 129.8 (2 × C), 136.1 (2 × C), 136.5, 136.6, 173.6, 174.8. HRMS (ESI, m/z) for C25H28O6 [M + Na]+ calc. 447.1778; found 447.1787.
5,6-Bis[(4-trifluoromethyl)benzyloxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12d)
The title compound 12d was prepared from diester 9d (423 mg, 0.755 mmol) according to general procedure B and isolated as a white powder (316 mg, 79%); m.p: 114.8–143.7 °C. 1H NMR (500 MHz, DMSO-d6) δ 1.34 (1H, d, J = 10.1 Hz, H7s), 1.90 (1H, d, J = 9.7 Hz, H7a), 2.48 (1H, m, H2), 2.58 (1H, br s, H1), 2.62–2.63 (1H, m, H4), 3.01 (1H, dd, J = 5.4, 4.8 Hz, H3), 3.62 (1H, d, J = 5.6 Hz, H6), 3.72 (1H, d, J = 5.5 Hz, H5), 4.57–4.69 (4H, m, 2 × ArCH2), 7.46–7.50 (4H, m, ArH), 7.60–7.63 (4H, m, ArH), 12.58 (2H, br s, 2 × COOH).13C NMR (125 MHz, DMSO-d6) δ 33.0, 43.6, 44.6, 45.3, 46.0, 70.7, 70.8, 78.4, 81.7, 124.4 (q, 1JCF = 270 Hz, 2 × CF3), 124.9 (q, 3JCF = 3.3 Hz, 2 × CH), 125.0 (q, 3JCF = 3.3 Hz, 2 × CH), 127.83, (2 × C), 127.84 (q, 2JCF = 31.6 Hz, 2 × C), 127.9 (2 × C), 143.5, 143.6, 173.6, 174.8. 19F NMR (470 MHz, DMSO-d6) δ −61.43. HRMS (ESI, m/z) for C25H22O6F6 [M + Na]+ calc. 555.1213; found 555.1211.
5,6-Bis(3-fluorobenzyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12e)
The title compound 12e was prepared from diester 9e (292 mg, 0.635 mmol) according to general procedure B and isolated as a clear waxy solid (206 mg, 75%). 1H NMR (500 MHz, CD3OD) δ 1.45 (1H, d, J = 10.5 Hz, H7s), 2.04 (1H, dd, J = 10.5, 1.5 Hz, H7a), 2.61 (1H, dd, J = 5.6, 0.9 Hz, H2), 2.65 (1H, br s, H1), 2.70 (1H, dd, J = 4.5, 1.4 Hz, H4), 3.16 (1H, dd, J = 5.5, 4.8 Hz, H3), 3.67–3.71 (2H, m, H5, H6), 4.56 (2H, s, ArCH2), 4.61 (2H, s, ArCH2), 6.96–7.00 (2H, m, ArH), 7.04–7.14 (4H, m, ArH), 7.27–7.33 (2H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 33.9, 45.2, 46.2, 47.2, 47.6, 73.7 (d, 4JCF = 1.3 Hz), 72.8 (d, 4JCF = 1.5 Hz), 80.0, 83.4, 115.2 (d, 2JCF = 21.1 Hz), 115.3 (d, 2JCF = 21.6 Hz), 115.4 (d, 2JCF = 22.3 Hz), 115.5 (d, 2JCF = 21.7 Hz), 124.4 (d, 3JCF = 2.7 Hz), 124.5 (d, 3JCF = 2.7 Hz), 130.98, 131.04, 142.6 (d, 3JCF = 7.2 Hz), 142.7 (d, 3JCF = 7.2 Hz), 164.26 (d, 1JCF = 242.9 Hz), 164.28 (d, 1JCF = 242.8 Hz), 175.7, 176.9. 19F NMR (470 MHz, CD3OD) δ −116.06, −116.03. HRMS (ESI, m/z) for C23H22O6F2 [M + Na]+ calc. 455.1277; found 455.1264.
5,6-Bis(4-fluorobenzyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12f)
The title compound 12f was prepared from diester 9f (115 mg, 0.25 mmol) according to general procedure B and isolated as a white powder (103 mg, 94%); m.p: 198.1–199.5 °C. 1H NMR (270 MHz, CDCl3) δ 1.48 (1H, d, J = 10.6 Hz, H7s), 2.13 (1H, d, J = 10.8 Hz, H7a), 2.64 (1H, d, J = 5.8 Hz, H2), 2.72 (1H, br s, H1), 2.76–2.78 (1H, m, H4), 3.24 (1H, dd, J = 5.7, 4.8 Hz, H3), 3.56–3.64 (2H, m, H5, H6), 4.53 (2H, br s, ArCH2), 4.56 (2H, br s, ArCH2), 6.93–7.02 (4H, m, ArH), 7.23–7.31 (4H, m, ArH). 13C NMR (67.5 MHz, DMSO-d6) δ 32.9, 43.6, 44.6, 45.3, 46.0, 70.8, 70.9, 78.0, 81.4, 114.8 (d, 2JCF = 21.1 Hz, 2 × CH), 114.9 (d, 2JCF = 21.0 Hz, 2 × CH), 129.6 (d, 3JCF = 8.0 Hz, 2 × CH), 129.7 (d, 3JCF = 8.1 Hz, 2 × CH), 134.7 (d, 4JCF = 3.0 Hz), 134.9 (d, 4JCF = 3.0 Hz), 161.5 (d, 1JCF = 241.3 Hz, 2 × CF), 173.5, 174.7. 19F NMR (470 MHz, DMSO-d6) δ −116.68. HRMS (ESI, m/z) for C23H22O6F2 [M + Na]+ calc. 455.1277; found 455.1270.
5,6-Bis(3-bromobenzyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12g)
The title compound 12g was prepared from diester 9g (265 mg, 0.455 mmol) according to general procedure B and isolated as a white powder (171 mg, 68%); m.p: 76.0–78.1 °C. 1H NMR (270 MHz, CDCl3) δ 1.50 (1H, d, J = 11.3 Hz, H7s), 2.16 (1H, d, J = 11.1 Hz, H7a), 2.67 (1H, d, J = 4.9 Hz, H2), 2.75 (1H, br s, H1), 2.79 (1H, d, J = 4.3 Hz, H4), 3.27 (1H, app. t, J = 5.6 Hz, H3), 3.62–3.67 (2H, m, H5, H6), 4.55 (2H, s, ArCH2), 4.58 (2H, s, ArCH2), 7.14–7.24 (4H, m, ArH), 7.36–7.49 (4H, m, ArH). 13C NMR (67.5 MHz, CDCl3) δ 33.5, 43.9, 44.9, 45.9, 46.2, 71.9, 72.0, 78.5, 81.9, 122.6, 122.7, 126.2, 126.4, 130.1, 130.2, 130.7, 130.8, 130.9 (2 × C), 140.5, 140.6, 177.5, 178.6. HRMS (ESI, m/z) for C23H22O6Br2 [M – H]− calc. 550.9710; found 550.9729.
5,6-Bis(4-bromobenzyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12h)
The title compound 12h was prepared from diester 9h (159 mg, 0.273 mmol) according to general procedure B and isolated as a white powder (119 mg, 79%); m.p: 119.7–124.9 °C. 1H NMR (500 MHz, CD3OD) δ 1.43 (1H, d, J = 10.4 Hz, H7s), 2.02 (1H, dd, J = 10.4, 1.1 Hz, H7a), 2.60 (1H, d, J = 5.4 Hz, H2), 2.63 (1H, br s, H1), 2.68 (1H, dd, J = 4.5, 1.3 Hz, H4), 3.15 (1H, app. t, J = 5.4 Hz, H3), 3.65–3.68 (2H, m, H5, H6), 4.50 (2H, m, ArCH2), 4.54–4.56 (2H, m, ArCH2), 7.19–7.24 (4H, m, ArH), 7.42–7.44 (4H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 33.9, 45.2, 46.2, 47.2, 47.6, 72.7, 72.8, 79.8, 83.3, 122.3, 122.4, 130.8 (2 × C), 130.9 (2 × C), 132.4 (4 × C), 138.9, 139.1, 173.6, 174.8. HRMS (ESI, m/z) for C23H22O6Br2 [M – H]− calc. 550.9710; found 550.9723.
5,6-Bis(allyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxylic acid (12i)
The title compound 12i was prepared from diester 9i (41 mg, 0.127 mmol) according to general procedure B and isolated as a viscous yellow oil (127 mg, 73%). 1H NMR (270 MHz, CDCl3) δ 1.41–1.45 (1H, m, H7s), 2.04 (1H, d, J = 10.4 Hz, H7a), 2.64–2.72 (3H, m, H1, H2, H4), 3.22 (1H, dd, J = 5.5, 4.7 Hz, H3), 3.56–3.61 (2H, m, H5, H6), 4.03–4.10 (4H, m, 2 × OCH2), 5.12–5.32 (4H, m, 2 × CH2CH), 5.82–5.99 (2H, m, CH2CH). 13C NMR (125 MHz, CDCl3) δ 33.2, 43.9, 44.9, 45.8, 46.2, 71.7, 71.8, 78.2, 81.6, 117.2, 117.3, 134.6, 134.7, 178.1, 179.2. HRMS (ESI, m/z) for C15H20O6 [M + Na]+ calc. 319.1152; found 319.1152.
General procedure C for the amidation of diacids 12a–i
A microwave vial was charged with the appropriate carboxylic acid, EDCI (3.0 equiv), HOBt (0.1 equiv) and dry DMF and was stirred at ambient temperature for 30 min. Aminoethylguanidine 14 (3.0 equiv) was then added and the reaction was irradiated to 50 °C for 30 min. The resulting homogenous clear mixture was diluted with EtOAc (15 mL), washed with H2O (3 × 8 mL), brine (8 mL), dried (MgSO4), filtered, and concentrated in vacuo to afford a solid that was purified by column chromatography (as specified below) to give the title compound.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(methoxy)bicyclo[2.2.1]heptane (15a)
Compound 15a was prepared from diacid 12a (96 mg, 0.39 mmol) and amine 14 (357 mg, 1.18 mmol) according to general procedure C and after purification by column chromatography (EtOAc) was isolated as a clear oil (230 mg, 72%); Rf = 0.25 (EtOAc). 1H NMR (500 MHz, CDCl3) δ 1.45–1.47 (36H, m, 4 × t-Bu), 1.56 (1H, d, J = 10.2 Hz, H7a), 1.89 (1H, d, J = 10.2 Hz, H7s), 2.45 (1H, d, J = 6.3 Hz, H2), 2.56 (1H, br s, H1), 2.68 (1H, br s, H4), 2.82 (1H, app. t, J = 5.3 Hz, H3), 3.34 (3H, s, OMe), 3.36 (3H, s, OMe), 3.38–3.60 (10H, m, 4 × CH2, H5, H6), 6.69 (1H, t, J = 5.4 Hz, NH), 8.02 (1H, t, J = 3.4 Hz, NH), 8.45 (1H, t, J = 5.5 Hz, NH), 8.65 (1H, t, J = 5.6 Hz, NH), 11.44 (1H, s, NH), 11.48 (1H, s, NH). 13C NMR (125 MHz, CDCl3) δ 28.1, 28.2, 28.3, 28.4, 33.8, 39.4, 39.8, 40.1, 40.3, 42.4, 42.9, 43.7, 45.0, 49.3, 58.4, 58.6, 79.5, 80.0, 81.1, 83.3, 83.8, 84.2, 153.1, 153.2, 156.8, 157.9, 163.0, 163.5, 172.3, 174.4. HRMS (ESI, m/z) for C37H64O12N8 [M + H]+ calc. 813.4717; found 813.4733.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(2-methylbenzyloxy)bicyclo[2.2.1]heptane (15c)
Compound 15c was prepared from diacid 12c (62 mg, 0.15 mmol) and amine 14 (140 mg, 0.45 mmol) according to general procedure C and after purification by column chromatography (50–70% EtOAc in pet. spirits) was isolated as a white solid (36 mg, 54%); Rf = 0.71 (70% EtOAc in pet. spirits); m.p: 117.7–120.7 °C. 1H NMR (500 MHz, CDCl3) δ 1.46–1.48 (36H, m, 4 × t-Bu), 1.63 (1H, d, J = 10.2 Hz, H7s), 2.12 (1H, d, J = 9.3, Hz, H7a), 2.25 (3H, s, ArMe), 2.27 (3H, s, ArMe), 2.48 (1H, d, J = 6.2 Hz, H2), 2.60 (1H, br s, H1), 2.73 (1H, d, J = 3.1 Hz, H4), 2.82 (1H, app. t, J = 4.7 Hz, H3), 3.23–3.55 (8H, m, 4 × CH2), 3.58 (1H, d, J = 5.5 Hz, H5), 3.61 (1H, d, J = 5.8 Hz, H9), 4.50–4.58 (4H, m, 2 × CH2Ar), 6.52 (1H, br s, NH), 7.08–7.19 (6H, m, ArH), 7.27–7.31 (2H, m, ArH), 8.00 (1H, t, J = 4.1 Hz, NH), 8.48 (1H, br s, NH), 8.67 (1H, t, J = 5.6 Hz, NH), 11.45 (1H, s, NH), 11.50 (1H, s, NH). 13C NMR (125 MHz, CDCl3) δ 18.9, 19.0, 28.1, 28.2, 28.40, 28.42, 34.4, 39.2, 40.3, 40.5, 42.3, 44.1, 44.7, 45.2, 49.5, 70.8, 70.9, 78.7, 79.6, 80.2, 82.1, 83.4, 83.8, 125.7, 125.8, 127.6, 127.8, 128.7, 128.9, 130.1, 130.2, 136.6, 136.67, 136.69, 136.8, 153.19, 153.23, 156.7, 157.9, 172.3, 174.6. HRMS (ESI, m/z) for C51H76O12N8 [M + H]+ calc. 993.5655; found 993.5654.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis[(4-trifluoromethyl)benzyloxy]bicyclo[2.2.1]heptane (15d)
Compound 15d was prepared from diacid 12d (151 mg, 0.28 mmol) and amine 14 (260 mg, 0.85 mmol) according to general procedure C and after purification by flash column chromatography (EtOAc) was isolated as a clear oil (174 mg, 56%); Rf = 0.45 (EtOAc). 1H NMR (500 MHz, CDCl3) δ 1.44–1.52 (36H, m, 4 × t-Bu), 1.68 (1H, d, J = 10.3 Hz, H7s), 2.13 (1H, d, J = 9.3 Hz, H7a), 2.48 (1H, d, J = 6.0 Hz, H2), 2.65 (1H, br s, H1), 2.79–2.80 (1H, m, H4), 2.85 (1H, dd, J = 5.7, 4.5 Hz, H3), 3.23–3.56 (8H, m, 4 × CH2), 3.58 (1H, d, J = 6.1 Hz, H5), 3.62 (1H, dd, J = 5.8, 1.2 Hz, H9), 4.55–4.67 (4H, m, 2 × CH2Ar), 6.68 (1H, m, NH), 7.38–7.40 (4H, m, ArH), 7.51–7.54 (4H, m, ArH), 8.16 (1H, t, J = 4.0 Hz, NH), 8.49 (1H, m, NH), 8.72 (1H, t, J = 6.0 Hz, NH), 11.48 (1H, s, NH), 11.53 (1H, s, NH). 13C NMR (125 MHz, CDCl3) δ 28.1, 28.2, 28.4, 34.3, 39.4, 40.1, 40.4, 42.5, 43.8, 44.6, 45.1, 49.4, 71.5, 71.6, 78.9, 79.7, 80.3, 82.1, 83.5, 84.0, 125.3 (q, 3JCF = 3.5 Hz, 4 × CH), 126.4 (q, 1JCF = 270 Hz, 2 × CF3), 127.66 (2 × C), 127.72 (2 × C), 129.7 (q, 2JCF = 32.6 Hz), 129.8 (q, 2JCF = 32.6 Hz), 142.7, 142.8, 153.2 (2 × C), 156.7, 158.1, 163.0, 163.4, 172.2, 174.3. 19F NMR (470 MHz, CDCl3) δ −62.97. HRMS (ESI, m/z) for C51H70O12N8F6 [M + H]+ calc. 1101.5090; found 1101.5110.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(3-fluorobenzyloxy)bicyclo[2.2.1]heptane (15e)
Compound 15e was prepared from diacid 12e (184 mg, 0.43 mmol) and amine 14 (290 mg, 1.28 mmol) according to general procedure C and after purification by column chromatography (50–70% EtOAc in pet. spirits) was isolated as a white solid (111 mg, 26%); Rf = 0.58 (70% EtOAc in pet. spirits); m.p: 214.5–258.2 °C (slow decomposition). 1H NMR (500 MHz, CDCl3) δ 1.46–1.49 (36H, m, 4 × t-Bu), 1.66 (1H, d, J = 10.3 Hz, H7s), 2.12 (1H, d, J = 9.3 Hz, H7a), 2.47 (1H, d, J = 6.1 Hz, H1), 2.63 (1H, br s, H4), 2.78–2.79 (1H, m, H1), 2.84 (1H, app. t, J = 6.0 Hz, H3), 3.25–3.60 (10H, m, 4 × CH2, H5, H6), 4.51–4.61 (4H, m, 2 × CH2Ar), 6.62 (1H, br s, NH), 6.91–7.07 (6H, m, ArH), 7.22–7.28 (2H, m, ArH), 8.15 (1H, t, J = 4.0 Hz, NH), 8.50 (1H, br s, NH), 8.72 (1H, br s, NH), 11.47 (1H, s, NH), 11.53 (1H, s, NH). 13C NMR (125 MHz, CDCl3) δ 28.1, 28.2, 28.4, 34.3, 39.3, 40.2, 40.6, 42.4, 43.7, 44.7, 45.1, 49.4, 71.5, 71.6, 78.5, 80.0, 80.4, 81.9, 83.6, 84.0, 114.3 (d, 2JCF = 21.4 Hz), 114.4 (d, 2JCF = 21.1 Hz), 114.5 (d, 2JCF = 21.2 Hz), 114.6 (d, 2JCF = 21.4 Hz), 123.07 (d, 3JCF = 2.8 Hz), 123.16 (d, 3JCF = 2.9 Hz), 129.8, 129.9, 141.3 (d, 3JCF = 7.0 Hz), 141.4 (d, 3JCF = 7.1 Hz), 153.1, 153.2, 156.6, 158.0, 162.71, 162.73, 162.96 (d, 1JCF = 244.0 Hz), 163.02 (d, 1JCF = 244.0 Hz), 172.3, 174.5. 19F NMR (470 MHz, CDCl3) δ −113.92, −113.86. HRMS (ESI, m/z) for C49H70O12N8F2 [M + H]+ calc. 1001.5154; found 1001.5165.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(4-bromobenzyloxy)bicyclo[2.2.1]heptane (15h)
Compound 15h was prepared from diacid 12h (171 mg, 0.31 mmol) and amine 14 (281 mg, 0.93 mmol) according to general procedure C and after purification by column chromatography (50–70% EtOAc in pet. spirits) was isolated as a clear oil (91 mg, 26%); Rf = 0.48 (70% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.46–1.48 (36H, m, 4 × t-Bu), 1.64 (1H, d, J = 10.3 Hz, H7s), 2.09 (1H, d, J = 9.4 Hz, H7a), 2.46 (1H, d, J = 6.1 Hz, H1), 2.60 (1H, br s, H4), 2.74 (1H, m, H2), 2.83 (1H, app. t, J = 4.6 Hz, H3), 3.22–3.46 (4H, m, 2 × CH2), 3.52–3.58 (6H, m, 2 × CH2, H5, H6), 4.46–4.55 (4H, m, 2 × CH2Ar), 6.69 (1H, m, NH), 7.14–7.17 (4H, m, ArH), 7.39–7.42 (4H, m, ArH), 8.09 (1H, t, J = 4.0 Hz, NH), 8.54 (1H, m, NH), 8.73 (1H, t, J = 5.1 Hz, NH), 11.47 (1H, s, NH), 11.51 (1H, s, NH). 13C NMR (125 MHz, CDCl3) δ 28.1, 28.2, 28.4, 34.3, 39.3, 40.2, 40.5, 42.4, 43.8, 44.6, 45.1, 49.4, 71.56, 71.61, 78.6, 80.0, 80.4, 81.8, 83.6, 84.0, 121.4, 121.5, 129.5 (2 × C), 129.6 (2 × C), 131.4 (2 × C), 131.5 (2 × C), 137.6, 137.7, 153.1, 153.2, 156.7, 158.0, 162.8, 163.2, 172.2, 174.4. HRMS (ESI, m/z) for C49H70O12N8Br2 [M + H]+ calc. 1121.3553; found 1121.3550.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(allyloxy)bicyclo[2.2.1]heptane (15i)
Compound 15i was prepared from diacid 12i (70 mg, 0.24 mmol) and amine 14 (214 mg, 0.71 mmol) according to general procedure C and after purification by column chromatography (50–70% EtOAc in pet. spirits) was isolated as a clear oil (128 mg, 63%); Rf = 0.34 (70% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.49–1.50 (36H, m, 4 × t-Bu), 1.60 (1H, d, J = 10.3 Hz, H7s), 2.02 (1H, d, J = 10.0 Hz, H7a), 2.48 (1H, d, J = 6.2 Hz, H4), 2.54 (1H, br s, H1), 2.66 (1H, d, J = 2.9 Hz, H2), 2.82 (1H, dd, J = 6.0, 4.6 Hz, H3), 3.33–3.60 (10H, m, 4 × CH2, H5, H6), 3.98–4.04 (4H, m, 2 × OCH2), 5.10–5.14 (2H, m, CH2CH), 5.20–5.27 (2H, m, CH2CH), 5.84–5.94 (2H, m, 2 × CH2CH), 6.58 (1H, t, J = 5.1 Hz, NH), 8.03 (1H, t, J = 3.9 Hz, NH), 8.48 (1H, br s, NH), 8.68 (1H, t, J = 5.8 Hz, NH), 11.46 (1H, s, NH), 11.50 (1H, s, NH). 13C NMR (125 MHz, CDCl3) δ 28.17, 28.21, 28.41, 28.44, 34.2, 39.3, 40.2, 40.5, 42.5, 43.9, 44.9, 45.2, 49.4, 71.6, 71.7, 78.6, 80.2, 81.9, 83.4, 83.9 (2 × C), 116.6, 116.9, 135.2, 135.3, 153.23, 153.34, 156.7, 158.0, 172.4, 174.6. HRMS (ESI, m/z) for C41H68N8O12 [M + H]+ calc. 865.5030; found 865.5027.
General procedure D for the amidation of diacids 12a–i
A microwave vial was charged with the appropriate carboxylic acid, EDCI (3.0 equiv), HOBt (0.1 equiv) and dry CHCl3 and was stirred at ambient temperature for 30 min. Aminoethylguanidine 14 (3.0 equiv) was then added and the reaction was irradiated to 50 °C for 30 min. The resulting homogenous clear mixture was diluted with CHCl3 (20 mL), washed with H2O (2 × 10 mL), brine (8 mL), dried (MgSO4), filtered, and concentrated in vacuo to afford the crude material that was purified by column chromatography (as specified below) to give the title compound.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(benzyloxy)bicyclo[2.2.1]heptane (15b)
Compound 15b was prepared from diacid 12b (343 mg, 0.87 mmol) and amine 14 (786 mg, 2.60 mmol) according to general procedure D and after purification by column chromatography (70% EtOAc in pet. spirits) was isolated as a clear oil (563 mg, 57%); Rf = 0.43 (70% EtOAc in pet. spirits). 1H NMR (270 MHz, CDCl3) δ 1.45–1.49 (36H, m, 4 × t-Bu), 1.63 (1H, d, J = 10.8 Hz, H7s), 2.12 (1H, d, J = 10.4 Hz, H7a), 2.43 (1H, d, J = 6.2 Hz, H2), 2.60 (1H, s, H1), 2.74–2.76 (1H, m, H4), 2.79–2.83 (1H, m, H3), 3.16–3.57 (10H, m, 4 × CH2, H5, H6), 4.55 (2H, s, CH2Ar), 4.59 (2H, s, CH2Ar), 6.56 (1H, t, J = 5.8 Hz, NH), 7.22–7.33 (10H, m, ArH), 7.99 (1H, t, J = 4.0 Hz, NH), 8.45 (1H, t, J = 5.6 Hz, NH), 8.69 (1H, t, J = 4.0 Hz, NH), 11.45 (1H, s, NH), 11.53 (1H, s, NH). 13C NMR (67.5 MHz, CDCl3) δ 28.1, 28.2, 28.4, 34.4, 39.2, 40.1, 40.4, 42.5, 43.8, 44.6, 45.1, 49.5, 72.2, 72.3, 78.3, 79.5, 80.1, 81.6, 83.3, 83.8, 127.5, 127.6, 127.8, 128.1, 128.3, 128.4, 138.6, 138.7, 153.2, 156.7, 158.0, 163.1, 163.6, 172.3, 174.5. HRMS (ESI, m/z) for C49H72O12N8 [M + H]+ calc. 965.5343; found 965.5371.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(4-fluorobenzyloxy)bicyclo[2.2.1]heptane (15f)
Compound 15f was prepared from diacid 12f (86 mg, 0.20 mmol) and amine 14 (181 mg, 0.60 mmol) according to general procedure D and after purification by column chromatography (50–70% EtOAc in pet. spirits) was isolated as a clear oil (90 mg, 45%); Rf = 0.13 (50% EtOAc in pet. spirits). 1H NMR (270 MHz, CDCl3) δ 1.46–1.48 (36H, m, 4 × t-Bu), 1.62–1.66 (1H, m, H7s), 2.12 (1H, d, J = 10.7 Hz, H7a), 2.46 (1H, d, J = 7.3 Hz, H2), 2.60 (1H, br s, H1), 2.72–2.75 (1H, m, H4), 2.83 (1H, app. t, J = 5.2 Hz, H3), 3.28–3.60 (10H, m, 4 × CH2, H5, H6), 4.48–4.53 (4H, m, 2 × CH2Ar), 6.58 (1H, t, J = 5.3 Hz, NH), 6.92–7.00 (4H, m, ArH), 7.22–7.29 (4H, m, ArH), 8.07 (1H, t, J = 4.1 Hz, NH), 8.47 (1H, t, J = 5.7 Hz, NH), 8.70 (1H, t, J = 5.9 Hz, NH), 11.48 (1H, s, NH), 11.52 (1H, s, NH). 13C NMR (67.5 MHz, CDCl3) δ 28.1, 28.2, 28.4, 34.3, 39.4, 40.2, 40.4, 42.6, 43.8, 44.7, 45.2, 49.5, 71.6, 71.7, 78.6, 79.6, 80.2, 81.8, 83.4, 83.9, 115.0 (d, 2JCF = 18.4 Hz, 2 × CH), 115.3 (d, 2JCF = 18.5 Hz, 2 × CH), 129.5 (d, 3JCF = 9.8 Hz, 2 × CH), 129.7 (d, 3JCF = 9.9 Hz, 2 × CH), 134.4 (d, 4JCF = 3.2 Hz), 134.5 (d, 4JCF = 3.2 Hz), 162.3 (d, 1JCF = 243.7 Hz), 162.4 (d, 1JCF = 243.7 Hz), 153.2 (2 × C), 156.8, 158.1, 163.1, 163.6, 172.3, 174.4. 19F NMR (470 MHz, CDCl3) δ −115.17, −115.10. HRMS (ESI, m/z) for C49H70O12N8F2 [M + H]+ calc. 1001.5154; found 1001.5157.
3-endo-2-exo-Di[2′-(2′,3′-di-tert-butoxycarbonylguanidino)ethylcarbamoyl]-5,6-bis(3-bromobenzyloxy)bicyclo[2.2.1]heptane (15g)
Compound 15g was prepared from diacid 12g (170 mg, 0.31 mmol) and amine 14 (280 mg, 0.93 mmol) according to general procedure D and after purification by column chromatography (70% EtOAc in pet. spirits) was isolated as a clear oil (185 mg, 53%); Rf = 0.32 (70% EtOAc in pet. spirits). 1H NMR (500 MHz, CDCl3) δ 1.46–1.49 (36H, m, 4 × t-Bu), 1.66 (1H, d, J = 10.4 Hz, H7s), 2.12 (1H, dd, J = 10.4, 1.2 Hz, H7a), 2.47 (1H, d, J = 5.9 Hz, H2), 2.61 (1H, br s, H1), 2.78–2.79 (1H, m, H4), 2.85 (1H, dd, J = 6.0, 4.5 Hz, H3), 3.30–3.61 (10H, m, 4 × CH2, H5, H6), 4.47–4.57 (4H, m, 2 × CH2Ar), 6.59 (1H, t, J = 5.6 Hz, NH), 7.15–7.18 (2H, m, ArH), 7.22–7.24 (2H, m, ArH), 7.37–7.39 (2H, m, ArH), 7.44–7.47 (2H, m, ArH), 8.11 (1H, t, J = 4.0 Hz, NH), 8.47 (1H, t, J = 5.5 Hz, NH), 8.71 (1H, t, J = 6.1 Hz, NH), 11.47 (1H, s, NH), 11.53 (1H, s, NH). 13C NMR (125 MHz, CDCl3) δ 28.1, 28.2, 28.4, 34.3, 39.4, 40.3, 40.4, 42.5, 43.6, 44.8, 45.2, 49.4, 71.5, 71.7, 78.7, 79.6, 80.2, 82.1, 83.4, 83.9, 122.5, 122.6, 126.2, 126.3, 130.0, 130.1, 130.66, 130.68, 130.72, 130.75, 141.0, 141.1, 153.2 (2 × C), 156.9, 158.1, 163.1, 163.6, 172.2, 174.4. HRMS (ESI, m/z) for C49H70O12N8Br2 [M + H]+ calc. 1121.3553; found 1121.3541.
General procedure E for boc-removal of Boc-protected guanidines 15a–i
To the stirring solution of Boc-protected guanidine (0.05 mmol) and MeOH (520 µL), was added dropwise AcCl (20 equiv) and the reaction was stirred for 24 h at ambient temperature. The reaction mixture was concentrated under vacuum and the product was co-evaporated with MeOH (2 × 0.5 mL) to give the desired guandinium HCl salt.
5,6-Bis(methoxy)bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16a)
Compound 16a was prepared from Boc-protected diguanidine 15a (42 mg, 0.05 mmol) according to general procedure E as a white solid (24 mg, 96%); m.p: 161.5–195.0 °C (slow decomposition). 1H NMR (270 MHz, CD3OD) δ 1.50 (1H, d, J = 10.8 Hz, H7a), 1.80 (1H, d, J = 11.0 Hz, H7s), 2.46 (1H, br s, H2), 2.58–2.59 (1H, m, H1), 2.65 (1H, d, J = 5.3 Hz, H4), 3.17 (1H, app. t, J = 4.5 Hz, H3), 3.33 (3H, s, OMe), 3.41 (3H, s, OMe), 3.35–3.43 (9H, m, 4 × CH2, H5), 3.51 (1H, d, J = 6.3 Hz, H6), 8.22 (1H, t, J = 4.5 Hz, NH), 8.32 (1H, t, J = 4.6, NH). 13C NMR (67.5 MHz, CD3OD) δ 33.9, 39.5, 39.7, 42.0, 42.2, 45.4, 46.1, 47.6, 47.8, 58.8, 58.9, 81.8, 85.4, 158.9, 175.0, 176.7. HRMS (ESI, m/z) for C17H32O4N8 [M + 2H]2+ calc. 207.1346; found 207.1341.
5,6-Bis(benzyloxy)bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16)
Compound 16b was prepared from Boc-protected diguanidine 15b (76 mg, 0.08 mmol) according to general procedure E as a white residue (47 mg, 96%). 1H NMR (500 MHz, CD3OD) δ 1.56 (1H, d, J = 10.0 Hz, H7a), 2.02 (1H, d, J = 10.0 Hz, H7s), 2.53 (1H, br s, H2), 2.66–2.67 (2H, m, H1, H4), 3.18 (1H, app. t, J = 5.7 Hz, H3), 3.21–3.39 (8H, m, 4 × CH2), 3.62 (1H, d, J = 5.3 Hz, H5), 3.73 (1H, d, J = 5.5 Hz, H6), 4.50–4.65 (4H, m, 2 × ArCH2), 7.26–7.39 (10H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 34.3, 39.6, 39.7, 41.97, 42.01, 46.4 (2 × C), 47.8, 48.4, 73.4, 73.7, 78.9, 83.2, 128.70, 128.72, 129.2 (2 × C), 129.32 (3 × C), 129.33 (3 × C), 139.6, 139.7, 158.8, 158.9, 175.0, 176.7. HRMS (ESI, m/z) for C29H40O4N8 [M + 2H]2+ calc. 283.1659; found 283.1667.
5,6-Bis[(2-methylbenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16c)
Compound 16c was prepared from Boc-protected diguanidine 15c (54 mg, 0.05 mmol) according to general procedure E as a clear oil (8 mg, 24%). 1H NMR (500 MHz, CD3OD) δ 1.56 (1H, d, J = 10.3 Hz, H7a), 2.01 (1H, d, J = 9.5 Hz, H7s), 2.26 (3H, s, ArMe), 2.30 (3H, s, ArMe), 2.53 (1H, br s, H2), 2.63–2.69 (2H, m, H1, H4), 3.14–3.38 (9H, m, 4 × CH2, H3), 3.64 (1H, d, J = 5.1 Hz, H5), 3.76 (1H, d, J = 5.5 Hz, H6), 4.49–4.67 (4H, m, 2 × ArCH2), 7.08–7.20 (7H, m, ArH), 7.29 (1H, d, J = 7.3 Hz, ArH). 13C NMR (125 MHz, CD3OD) δ 19.07, 19.09, 34.4, 39.6, 39.7, 41.98, 42.04, 46.2, 46.3, 47.8, 48.4, 72.0, 72.1, 79.2, 83.6, 126.68, 126.73, 128.96, 128.99, 130.2, 130.3, 131.1, 131.2, 137.4, 137.6, 138.1, 138.2, 158.86, 158.89, 175.1, 176.8. HRMS (ESI, m/z) for C31H44N8O4 calc. [M + 2H]2+ 297.1816; found 297.1820.
5,6-Bis[(4-trifluoromethylbenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16d)
Compound 16d was prepared from Boc-protected diguanidine 15d (87 mg, 0.08 mmol) according to general procedure E as a colourless sticky residue (40 mg, 66%). 1H NMR (500 MHz, CD3OD) δ 1.60 (1H, d, J = 10.2 Hz, H7s), 2.06 (1H, d, J = 9.9 Hz, H7a), 2.59 (1H, br s, H2), 2.70–2.72 (2H, m, H1, H4), 3.21–3.38 (9H, m, 4 × CH2, H3), 3.69 (1H, d, J = 5.2 Hz, H5), 3.80 (1H, d, J = 5.5 Hz, H6), 4.63 (2H, s, ArCH2), 4.69–4.76 (2H, m, ArCH2), 7.46–7.48 (2H, m, ArH), 7.51–7.52 (2H, m, ArH), 7.55–7.58 (4H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 34.3, 39.6, 39.7, 41.9, 42.0, 46.1, 46.2, 47.8, 48.4, 72.6, 72.7, 79.6, 83.6, 125.69 (q, 1JCF = 269.7 Hz), 125.71 (q, 1JCF = 269.7 Hz), 126.1 (q, 3JCF = 3.4 Hz, 4 × CH), 129.2 (4 × C), 130.58 (q, 2JCF = 31.8 Hz), 130.60 (q, 2JCF = 31.7 Hz), 144.46, 144.54, 158.83, 158.85, 174.9, 176.6. 19F NMR (470 MHz, CD3OD) δ −65.41, −65.38. HRMS (ESI, m/z) for C31H38O4N8F6 [M + 2H]2+ calc. 351.1533; found 351.1532.
5,6-Bis[(3-fluorobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16e)
Compound 16e was prepared from Boc-protected diguanidine 15e (72 mg, 0.07 mmol) according to general procedure E as a yellow solid (46 mg, 99%); m.p: 77.4–82.4 °C (slow decomposition). 1H NMR (500 MHz, CD3OD) δ 1.58 (1H, d, J = 10.2 Hz, H7s), 2.02 (1H, d, J = 10.0 Hz, H7a), 2.57 (1H, br s, H2), 2.68–2.69 (2H, m, H1, H4), 3.19–3.38 (9H, m, 4 × CH2, H3), 3.65 (1H, d, J = 5.2 Hz, H5), 3.76 (1H, d, J = 5.6 Hz, H6), 4.53–4.58 (2H, m, ArCH2), 4.62–4.68 (2H, m, ArCH2), 6.97–7.00 (2H, m, ArH), 7.05 (1H, d, J = 9.8 Hz, ArH), 7.10–7.11 (2H, m, ArH), 7.16 (1H, d, J = 7.6 Hz, ArH), 7.28–7.34 (2H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 34.3, 39.6, 39.7, 41.9, 42.0, 46.1 (2 × C), 47.8, 48.3, 72.6, 72.7, 79.2, 83.4, 115.33 (d, 2JCF = 33.7 Hz), 115.26 (d, 2JCF = 26.6 Hz), 115.40 (d, 2JCF = 34.0 Hz), 115.43 (d, 2JCF = 27.0 Hz), 124.50 (d, 4JCF = 2.7 Hz), 124.52 (d, 4JCF = 2.6 Hz), 131.02 (d, 3JCF = 7.8 Hz), 131.08 (d, 3JCF = 6.6 Hz), 142.6 (d, 3JCF = 7.2 Hz), 142.8 (d, 3JCF = 7.1 Hz), 158.79, 158.81, 164.22 (d, 1JCF = 244.2 Hz), 164.23 (d, 1JCF = 242.8 Hz), 174.9, 176.6. 19F NMR (470 MHz, CDCl3) δ −116.03, −115.86. HRMS (ESI, m/z) for C29H38O4N8F2 [M + 2H]2+ calc. 301.1565; found 301.1576.
5,6-Bis[(4-fluorobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16f)
Compound (16f) was prepared from Boc-protected diguanidine 15f (90 mg, 0.09 mmol) according to general procedure E as a white solid (46 mg, 77%); m.p: 94.0–110.0 °C (slow decomposition). 1H NMR (500 MHz, CD3OD) δ 1.55 (1H, d, J = 10.1 Hz, H7a), 2.01 (1H, d, J = 9.0 Hz, H7s), 2.51 (1H, br s, H2), 2.62 (1H, m, H1), 2.67 (1H, d, J = 5.3 Hz, H4), 3.18–3.41 (9H, m, 4 × CH2, H3), 3.63 (1H, d, J = 5.8 Hz, H5), 3.73 (1H, d, J = 5.5 Hz, H6), 4.47–4.53 (2H, m, ArCH2), 4.59 (2H, s, ArCH2), 7.00–7.05 (4H, m, ArH), 7.29–7.31 (2H, m, ArH), 7.34–7.37 (2H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 34.3, 39.6, 39.7, 41.98, 42.04, 46.16, 46.20, 47.7, 48.6, 72.7, 72.9, 79.0, 83.2, 115.96 (d, 2JCF = 21.0 Hz), 115.97 (d, 2JCF = 21.6 Hz), 131.1 (d, 3JCF = 8.1 Hz, 4 × CH), 135.76 (d, 2JCF = 15.4 Hz), 135.78 (d, 2JCF = 15.6 Hz), 163.79 (d, 1JCF = 243.2 Hz), 163.81 (d, 1JCF = 242.9 Hz), 158.9 (2 × C), 175.0, 176.7. 19F NMR (470 MHz, CDCl3) δ −118.04, −117.89. HRMS (ESI, m/z) for C29H38O4N8F2 [M + 2H]2+ calc. 301.1565; found 301.1567.
5,6-Bis[(3-bromobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16g)
Compound 16g was prepared from Boc-protected diguanidine 15g (169 mg, 0.15 mmol) according to general procedure E as a white solid (115 mg, 97%); m.p: 99.9–132.8 °C (slow decomposition). 1H NMR (500 MHz, CD3OD) δ 1.57 (1H, d, J = 10.5 Hz, H7a), 2.01–2.04 (1H, m, H7s), 2.55 (1H, br s, H2), 2.66–2.69 (2H, m, H1, H4), 3.19–3.40 (9H, m, 4 × CH2, H3), 3.64 (1H, d, J = 5.6 Hz, H5), 3.75 (1H, d, J = 5.3 Hz, H6), 4.50–4.56 (2H, m, ArCH2), 4.59–4.65 (2H, m, ArCH2), 7.21–7.27 (3H, m, ArH), 7.32 (1H, d, J = 7.7 Hz, ArH), 7.40–7.43 (3H, m, ArH), 7.48 (1H, m, ArH), 7.53 (1H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 34.3, 39.6, 39.7, 41.96, 42.0, 46.13, 46.15, 47.7, 48.4, 72.6, 72.7, 79.3, 83.5, 123.32, 123.35, 127.59, 127.62, 131.16, 131.21, 131.66, 131.70, 131.8 (2 × C), 142.4, 142.5, 158.86, 158.87, 175.0, 176.7. HRMS (ESI, m/z) for C29H38O4N8Br2 [M + 2H]2+ calc. 361.0764; found 361.0774.
5,6-Bis[(4-bromobenzyl)oxy] bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16h)
Compound 16h was prepared from Boc-protected diguanidine 15h (57 mg, 0.05 mmol) according to general procedure E as a clear oil (40 mg, 99%). 1H NMR (500 MHz, CD3OD) δ 1.56 (1H, d, J = 10.2 Hz, H7s), 2.00 (1H, d, J = 9.9 Hz, H7a), 2.53 (1H, br s, H2), 2.64–2.67 (2H, m, H1, H4), 3.17–3.40 (9H, m, 4 × CH2, H3), 3.61 (1H, d, J = 5.6 Hz, H5), 3.73 (1H, d, J = 5.6 Hz, H6), 4.46–4.52 (2H, m, ArCH2), 4.56–4.62 (2H, m, ArCH2), 7.20–7.22 (2H, m, ArH), 7.26–7.28 (2H, m, ArH), 7.43–7.46 (4H, m, ArH). 13C NMR (125 MHz, CD3OD) δ 34.3, 39.6, 39.7, 41.9, 42.0, 46.1, 46.2, 47.7, 48.4, 72.6, 72.8, 79.1, 83.3, 122.36, 122.40, 131.0 (4 × C), 132.40 (2 × C), 132.41 (2 × C), 139.0, 139.1, 158.81, 158.84, 174.9, 176.6. HRMS (ESI, m/z) for C29H38O4N8Br2 [M + 2H]2+ calc. 361.0764; found 361.0769.
5,6-Bis(allyloxy) bicyclo[2.2.1]heptane-3-endo-2-exo-dicarboxamidoethylguanidine hydrogen chloride (16i)
Compound 16i was prepared from Boc-protected diguanidine 15i (126 mg, 0.15 mmol) according to general procedure E as a yellow solid (24 mg, 31%); m.p: 79.9–85.9 °C. 1H NMR (500 MHz, CD3OD) δ 1.52 (1H, d, J = 10.3 Hz, H7a), 1.92 (1H, d, J = 9.3 Hz, H7s), 2.44 (1H, br s, H2), 2.59 (1H, d, J = 3.5 Hz, H1), 2.66 (1H, d, J = 5.2 Hz, H4), 3.16 (1H, app. t, J = 5.1 Hz, H3), 3.30–3.46 (8H, m, 4 × CH2), 3.57 (1H, d, J = 6.0 Hz, H5), 3.63 (1H, d, J = 5.4 Hz, H6), 4.00–4.09 (4H, m, 2 × CH2O), 5.13–5.32 (4H, m, 2 × CH2CH), 5.86–5.98 (2H, m, 2 × CH2CH), 7.45–7.48 (2H, m, 2 × NH). 13C NMR (125 MHz, CD3OD) δ 34.2, 39.6, 39.7, 42.0, 42.2, 46.1, 46.2, 47.9, 48.4, 72.7, 72.8, 79.4, 83.3, 117.1, 117.2, 136.2 (2 × C), 158.87, 158.92, 175.0, 176.7. HRMS (ESI, m/z) for C21H36O4N8 [M + 2H]2+ calc. 233.1503; found 233.1502.
Crystallography
Intensity data were collected with an CCD diffractometer using Cu- Kα radiation, the temperature during data collection was maintained at 130.0(1) using an Oxford Cryosystems cooling device. The structure was solved by direct methods and difference Fourier synthesis.53 Thermal ellipsoid plots were generated using the program ORTEP-354 integrated within the WINGX55 suite of programs. Disordered solvent, assumed to be ethanol was removed using the Squeeze procedure.56
Disk Diffusion – Zone of Inhibition assay
A stock solution of 10 mg/mL was made for each compound under observation using DMSO as a solvent. Each of these stock solutions was then diluted by a factor of 1:2 to bring the concentration to 5 mg/mL. The diluted solutions were then filter-sterilized using a 0.2-µm nylon filter, and 10 µL of the 5 mg/mL stock was pipetted onto a blank disk (i.e. 50 µg/disk; Oxoid Limited, Hampshire, UK). All bacterial isolates were matched to a 0.5 McFarland standard (in 0.9% NaCl) before they were swabbed onto nutrient agar. The controls used were a 10 µg colistin disk (Oxoid), 10 µL of DMSO and a plate swabbed with saline from the dispenser used.
Minimum Inhibitory Concentration (MIC) determination
Bacteria were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) as listed in Table S2 (see ESI). Bacteria were cultured in Nutrient broth (NB; Bacto Laboratories, catalog No. 234000) or Muller-Hinton broth (MHB; Bacto Laboratories, catalog No. 211443) at 37 °C overnight with shaking (~180 RPM). A sample of each culture was diluted 50-fold in fresh MHB and incubated at 37 °C for 1.5–3 h with shaking (~180 RPM). Compound stock solutions were prepared as 10 mg/mL in DMSO and colistin was dissolved in milli-Q water at 5.12 mg/mL. The compounds, at twice the final desired concentration, were serially diluted 2-fold across the wells of 96–well plates (Non-Binding Surface, Corning, catalog No. 3641). Mid-log phase bacterial cultures (after 1.5–3 h incubation) were diluted to a final concentration of 5 × 105 colony forming units (CFU)/mL, and 50 µL was added to each well giving a final compound concentration range of 32 µg/mL to 0.015 µg/mL (DMSO ≤ 1%). MICs were determined visually after 20 h of incubation at 37 °C, with the MIC defined as the lowest compound concentration at which no bacterial growth was visible.
Cytotoxicity evaluation
HEK293 (ATCC CRL-1573) and HepG2 (ATCC HB-8065) cells were seeded as 3000 cells per well in a 384-well plate in in DMEM medium (GIBCO-Invitrogen #11995-073), in which 10% of FBS was added. Cells were incubated for 24 h at 37 °C, 5% CO2 to allow cells to attach to the plates. A concentration series of compounds was then added into each well. The cells were incubated with the compounds for 24 h at 37 °C, 5% CO2. After the incubation, 10 µM resazurin (dissolved in PBS) was added to each well. The plates were then incubated for 2 h at 37 °C, 5% CO2. The fluorescence intensity was read using Polarstar Omega with excitation/emission 560/590. The data was analysed by Prism software. Results are presented as the average percentage of control ± SD for each set of duplicate wells using the following equation: Percentage Viability = (FITEST – FINegative/FIUNTREATED –FINegative) × 100.
Supplementary Material
Figure 4.

Antibacterial activity of compound 16g (50 µg per disk) against P. aeruginosa (LHS) and MRSA (RHS) using a disk diffusion assay.
Acknowledgements
F.M.P., S.M.H. & T.D.A. thank the ARC (DP140100227) and the Strategic Research Centre for Chemistry and Biotechnology (Deakin University) for financial support and a top-up scholarship for S.M.H. The authors would also like to thank Dr Damien Callahan for his assistance with the collection of HRMS and the Australian Research Council for funding Deakin University’s Magnetic Resonance Facility through LIEF grant LE110100141. J.L. and R.L.N. are supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI098771). J.L. is an Australian NHMRC Senior Research Fellow. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. The MIC screening was done in collaboration with CO-ADD (The Community for Open Access Drug Discovery) and we thank David L. Paterson (UQCCR) for his kind donation of clinical Gram-positive strains used for testing.
Footnotes
Supporting Information Available: Crystal structure of compound 12h (CIF), synthetic procedures for all previously reported compounds and copies of NMR spectra (1H, 13C, 19F) for all new compounds. This material is available free of charge via the Internet at http://pubs.rsc.org
References
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Clin. Infec. Dis. 2009;48:1. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Talbot GH, Bradley J, Edwards JE, Gilbert D, Scheld M, Bartlett JG. Clin. Infec. Dis. 2006;42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 3.Cooper MA, Shlaes D. Nature. 2011;472:32–32. doi: 10.1038/472032a. [DOI] [PubMed] [Google Scholar]
- 4.Butler MS, Blaskovich MA, Cooper MA. J. Antibiot. 2013;66:571–591. doi: 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- 5.Yau W, Owen RJ, Poudyal A, Bell JM, Turnidge JD, Yu HH, Nation RL, Li J. J. Infect. 2009;58:138–144. doi: 10.1016/j.jinf.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Aumelas A, Mangoni M, Roumestand C, Chiche L, Despaux E, Grassy G, Calas B, Chavanieu A. Eur. J. Biochem. 1996;237:575–583. doi: 10.1111/j.1432-1033.1996.0575p.x. [DOI] [PubMed] [Google Scholar]
- 7.Thaker HD, Sgolastra F, Clements D, Scott RW, Tew GN. J. Med. Chem. 2011;7:2241–2254. doi: 10.1021/jm101410t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding B, Yin N, Liu Y, Cardenas-Garcia J, Evanson R, Orsak T, Fan M, Turin G, Savage PB. J. Am. Chem. Soc. 2004;126:13642–13648. doi: 10.1021/ja046909p. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Dings RPM, Nesmelova I, Debbert S, Haseman JR, Maxwell J, Hoye TR, Mayo KH. J. Med. Chem. 2006;49:7754–7765. doi: 10.1021/jm0610447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson M, Bremner JB, Coates J, Deadman J, Keller PA, Pyne SG, Somphol K, Rhodes DI. Eur. J. Med. Chem. 2011:4201–4211. doi: 10.1016/j.ejmech.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 11.Dings RP, Mayo KH. Acc. Chem. Res. 2007;40:1057–1065. doi: 10.1021/ar700086k. [DOI] [PubMed] [Google Scholar]
- 12.Pfister JR, Belardinelli L, Lee G, Lum RT, Milner P, Stanley WC, Linden J, Baker SP, Schreiner G. J. Med. Chem. 1997;40:1773–1778. doi: 10.1021/jm970013w. [DOI] [PubMed] [Google Scholar]
- 13.Scammells PJ, Baker SP, Bellardinelli L, Olsson RA, Russell RA, Knevitt SA. Bioorg. Med. Chem. Lett. 1996;6:811–814. [Google Scholar]
- 14.Wright DMJ, Baker SP, Stewart SG, Scammells PJ. Bioorg. Med. Chem. Lett. 1998;8:3647–3648. doi: 10.1016/s0960-894x(98)00668-4. [DOI] [PubMed] [Google Scholar]
- 15.Hutchinson SA, Baker SP, Scammells PJ. Biorg. Med. Chem. 2002;10:1115–1122. doi: 10.1016/s0968-0896(01)00384-4. [DOI] [PubMed] [Google Scholar]
- 16.Henderson LC, Li J, Nation RL, Velkov T, Pfeffer FM. Chem. Commun. 2010;46:3197–3199. doi: 10.1039/b925135a. [DOI] [PubMed] [Google Scholar]
- 17.Hickey SM, Tripcony SK, Li R, Williams RJ, Pfeffer FM. Supramol. Chem. 2014 [Google Scholar]
- 18.Lowe AJ, Pfeffer FM. Chem. Commun. 2008:1871–1873. doi: 10.1039/b801798k. [DOI] [PubMed] [Google Scholar]
- 19.Klärner F-G, Schrader T. Acc. Chem. Res. 2012;46:967–978. doi: 10.1021/ar300061c. [DOI] [PubMed] [Google Scholar]
- 20.Lowe AJ, Long BM, Pfeffer FM. J. Org. Chem. 2012;77:8507–8517. doi: 10.1021/jo301450b. [DOI] [PubMed] [Google Scholar]
- 21.Lowe AJ, Long BM, Pfeffer FM. Chem. Commun. 2013;49:3376–3388. doi: 10.1039/c3cc40702k. [DOI] [PubMed] [Google Scholar]
- 22.Johnstone MD, Frank M, Clever GH, Pfeffer FM. Eur. J. Org. Chem. 2013;2013:5848–5853. [Google Scholar]
- 23.Martínez AG, Vilar ET, Fraile AG, de la Moya Cerero S, Martínez Ruiz P, Díaz Morillo C. Tetrahedron: Asymmetry. 2007;18:742–749. [Google Scholar]
- 24.Lattanzi A, Iannece P, Vicinanza A, Scettri A. Chem. Commun. 2003:1440–1441. [PubMed] [Google Scholar]
- 25.Derksen JT, Baldeschwieler JD, Scherphof GL. Proc. Natl. Acad. Sci. U.S.A. 1988;85:9768–9772. doi: 10.1073/pnas.85.24.9768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho H, Choi Y, Yhei J. Derivatives of cyclic compound and the use thereof. 2008 WO2008/004788. [Google Scholar]
- 27.Bradshaw JS, Krakowiak KE, Huszthy P, Izatt RM. J. Heterocycl. Chem. 1991;28:773–775. [Google Scholar]
- 28.Mehta G, Mohal N. Tetrahedron Lett. 1999;40:5791–5794. [Google Scholar]
- 29.Mehta G, Mohal N, Lakshminath S. Tetrahedron Lett. 2000;41:3505–3508. [Google Scholar]
- 30.Hergueta AR, López C, García-Mera X, Fernández F. Tetrahedron. 2004;60:10343–10352. [Google Scholar]
- 31.Purdie T, Irvine JC. J. Chem. Soc. Trans. 1903;83:1021–1037. [Google Scholar]
- 32.Gould TJ, Balestra M, Wittman MD, Gary JA, Rossano LT, Kallmerten J. J. Org. Chem. 1987;52:3889–3901. [Google Scholar]
- 33.Van Hijfte L, Little RD, Petersen JL, Moeller KD. J. Org. Chem. 1987;52:4647–4661. [Google Scholar]
- 34.Ireland RE, Thaisrivongs S, Dussault PH. J. Am. Chem. Soc. 1988;110:5768–5779. [Google Scholar]
- 35.Mislow K. J. Am. Chem. Soc. 1951;73:4043–4044. [Google Scholar]
- 36.Bouzide A, Sauvé G. Tetrahedron Lett. 1997;38:5945–5948. [Google Scholar]
- 37.Mousseron M, Granger R, Merle A. Bull. Soc. Chim. Fr. 1947:459–461. [Google Scholar]
- 38.Walker HG, Gee M, McCready RM. J. Org. Chem. 1962;27:2100–2102. [Google Scholar]
- 39.Dejmek M, Hrebabecky H, Sala M, Dracinsky M, Nencka R. Synthesis. 2011:4077–4083. [Google Scholar]
- 40.Niwayama S, Cho H, Zabet-Moghaddam M, Whittlesey BR. J. Org. Chem. 2010;75:3775–3780. doi: 10.1021/jo100564e. [DOI] [PubMed] [Google Scholar]
- 41.Donohoe TJ, Jahanshahi A, Tucker MJ, Bhatti FL, Roslan IA, Kabeshov M, Wrigley G. Chem. Commun. 2011;47:5849–5851. doi: 10.1039/c1cc11654a. [DOI] [PubMed] [Google Scholar]
- 42.Grob CA. Angew. Chem. Int. Ed. 1969;8:535–546. [Google Scholar]
- 43.Sakai N, Moriya T, Konakahara T. J. Org. Chem. 2007;72:5920–5922. doi: 10.1021/jo070814z. [DOI] [PubMed] [Google Scholar]
- 44.Marshall JA, Seletsky BM, Luke GP. J. Org. Chem. 1994;59:3413–3420. [Google Scholar]
- 45.Lander GD. J. Chem. Soc., Trans. 1903;83:414–423. [Google Scholar]
- 46.Finkelstein H. Ber. Dtsch. Chem. Res. 1910;43:1528–1532. [Google Scholar]
- 47.Hickey SM, Ashton TD, Khosa SK, Pfeffer FM. Synlett. 2012;23:1779–1782. [Google Scholar]
- 48.Hickey SM, Ashton TD, Pfeffer FM. Asian J. Org. Chem. 2014 [Google Scholar]
- 49.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Deliver. Rev. 2012;64:4–17. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 50.Khalil ZG, Salim AA, Lacey E, Blumenthal A, Capon RJ. Org. Lett. 2014;16:5120–5123. doi: 10.1021/ol502472c. [DOI] [PubMed] [Google Scholar]
- 51.Flook MM, Börner J, Kilyanek SM, Gerber LCH, Schrock RR. Organometallics. 2012;31:6231–6243. [Google Scholar]
- 52.Gottlieb HE, Kotlyar V, Nudelman A. J. Org. Chem. 1997;62:7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 53.Sheldrick GM. Acta Cryst. 2007;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 54.Farrugia LJ. J. Appl. Cryst. 1997;30:565–565. [Google Scholar]
- 55.Farrugia LJ. J. Appl. Cryst. 1999;32:837–838. [Google Scholar]
- 56.Van der Sluis P, Spek A. Acta Cryst. 1990;46:194–201. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.