

Abstract
This review concerns how the primary inflammation preceding the generation of certain key damage-associated molecular patterns (DAMPs) arises in Alzheimer’s disease (AD). In doing so, it places soluble amyloid β (Aβ), a protein hitherto considered as a primary initiator of AD, in a novel perspective. We note here that increased soluble Aβ is one of the proinflammatory cytokine-induced DAMPs recognized by at least one of the toll-like receptors on and in various cell types. Moreover, Aβ is best regarded as belonging to a class of DAMPs, as do the S100 proteins and HMBG1, that further exacerbate production of these same proinflammatory cytokines, which are already enhanced, and induces them further. Moreover, variation in levels of other DAMPs of this same class in AD may explain why normal elderly patients can exhibit high Aβ plaque levels, and why removing Aβ or its plaque does not retard disease progression. It may also explain why mouse transgenic models, having been designed to generate high Aβ, can be treated successfully by this approach.
Tables of Links
| TARGETS | |
|---|---|
| Catalytic receptorsa | Enzymesb |
| TLR4 | Dnmt1, DNA methyltransferase 1 |
| TLR7 | α secretase (ADAM10) |
| TLR9 | BACE1, β secretase |
| LIGANDS | |
|---|---|
| Amyloid β | Lead |
| Cadmium | LPS |
| Exendin-4 | Mercury |
| GLP-1, glucagon-like peptide-1 | Thalidomide |
| IL-1 | TNF |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Despite its dominance of the publications on the pathogenesis of Alzheimer’s disease (AD), the amyloid theory is yet to provide any positive clinical outcome, and still contains uncertainties. Others (Castellani and Smith, 2011; Mullane and Williams, 2013; Castello et al., 2014) have extensively summarized the amyloid theory and the difficulties it has encountered. These include the presence of abundant amyloid in sections from many cognitively normal older brains, and the failure, to date, of being able to replicate in humans, the anti-amyloid immunotherapy that performed well in mice. Recently, we (Morris et al., 2014) extensively reviewed the complexities, inconsistencies and controversies that have now surrounded the amyloid theory, and discussed the bias of preclinical AD models towards the amyloid hypothesis. We also illustrated how extensive data cited in support of the amyloid hypothesis, including genetic links to disease, can be interpreted independently of a role for amyloid β (Aβ), and summarized the case for the validity of the argument for proinflammatory cytokines having a central role, and therefore being a valid pharmacological target. Here we expand this section of our recent review (Morris et al., 2014) by going back to the roots of our understanding of innate immunity while still providing a role for Aβ. For this role of Aβ to become clear, we first consider the cytokine output of the innate immune system, and the pathogen-associated molecular pattern (PAMP) and damage-associated molecular pattern (DAMP) terminology that allows a workable framework for describing how this output is triggered through this primitive, but ever present, immune system recognizing its surroundings.
The immune system, for decades concerned with adaptive immunity against pathogens, is now, through innate immunity, recognized as being allied to the inflammatory response. This has brought together the basis of the pathogenesis of infectious disease, sterile inflammatory states such as AD and Parkinson’s disease (PD), and also stroke and traumatic brain injury (TBI) (Arvin et al., 1996; Tarkowski et al., 2003; Esiri, 2007; Clark et al., 2010; Eikelenboom et al., 2011; Howcroft et al., 2013). The general perception of inflammation as a complex interaction of cellular responses orchestrated by chemokines and cytokines rightly includes TNF and IL-1. But being termed proinflammatory cytokines often leads this closely linked pair to be regarded simply as biomarkers for the presence of inflammation, whereas their pleiotropy includes many roles in all tissues, including such diverse roles as physiological cerebral transmitters, particularly in brain homeostasis (Stellwagen and Malenka, 2006; McAfoose and Baune, 2009), which is otherwise unrelated to inflammation. As recently reviewed (Clark and Vissel, 2014), TNF and IL-1 closely mimic each other, and occur together, but for various reasons, including that anti-TNF antibody also reduces IL-1 (Brennan et al., 1989), TNF dominates the literature.
The ubiquity and importance of TNF in biology, innate immunity and disease
The polypeptide TNF is arguably the centrepiece of the mammalian innate immune system. Yet it is extremely well preserved in phylogeny, huTNF recognizing and being very widely recognized, even by corals (Quistad et al., 2014). The ubiquity of TNF in biology is demonstrated by the presence of many more entries in PubMed than any other proinflammatory cytokine, let alone Alzheimer’s or Aβ. It is one of the pillars of normal physiology, including metabolism. The fundamental roles of lower concentrations of TNF and related cytokines in normal physiology, involving all organs but not least in the brain (Vitkovic et al., 2000a,b), nowadays outnumber references to their proinflammatory and immunological roles. For instance, TNF and IL-1β are released during physiological neuronal activity and, as reviewed (Marin and Kipnis, 2013), play a crucial role in regulating the strength of normal synaptic transmission. TNF, of itself rather than through the inflammatory cascade it can trigger, is also involved in normal transmission via modulating excitatory neurotransmission (Pickering et al., 2005), trafficking of AMPA receptors (Ferguson et al., 2008), homeostatic synaptic scaling (Stellwagen and Malenka, 2006), long-term potentiation (Cumiskey et al., 2007) and maintaining normal background levels of neurogenesis (Bernardino et al., 2008). Mitochondrial function depends on TNF (Sanchez-Alcazar et al., 2000), as does regulation of the neurotransmitter, orexin (Zhan et al., 2011), which, as recently reviewed in a brain disease context (Clark and Vissel, 2014), controls sleep, motor control, focused effort, appetite and water intake. TNF also regulates neuronal type 1 inositol trisphosphate receptors, which are central to neuronal Ca++ homeostasis, and thus the ionic signalling cascades on which normal function of these cells depends (Park et al., 2008). Likewise, glycine receptors, which are structurally related to GABA receptors and have a similar inhibitory role, are influenced by proinflammatory cytokines (Chirila et al., 2014). Clearly, all these functions are susceptible to TNF and/or IL-1 being outside their homeostatic range.
Yet TNF is much more than normal physiology. An awareness of TNF began with its detection in the serum of mice receiving Gram-negative bacterial endotoxin, that is, LPS, several weeks after they were infected with Bacillus Calmette–Guérin (BCG), an attenuated strain of Mycobacterium bovis. On transfer to mice bearing transplanted sarcomas, this novel protein caused necrosis of these tumours as effectively as did LPS, but contained no LPS (Carswell et al., 1975). The argument that excessive TNF and IL-1 both controlled pathogens and generated disease was first put forward, in collaboration with Carswell, with respect to malaria (Clark et al., 1981) and sepsis (Clark, 1982). Excessive production of TNF and related cytokines was soon recognized as mediating the rapid response of non-specific, or innate, immunity against malaria parasites, and subsequently many other pathogens, as well as the pathogenesis of the diseases these organisms induce (Clark et al., 1981; Rook et al., 1987; Clark and Cowden, 1989; Raziuddin et al., 1994; Peper and Vancampen, 1995; Arsenijevic et al., 1997; Bhutta et al., 1997; Nakane et al., 1999). TNF has also key roles in physiological functions (see later). Its control over insulin signalling, reviewed in an AD context (Talbot and Wang, 2014), will extend greatly its known influence in the brain and elsewhere, in both normal and disease states (Chiu et al., 2008; Chiu and Cline, 2010).
Cloning of TNF (Aggarwal et al., 1985) and LPS protection experiments based on this technology (Beutler et al., 1985) produced data consistent with the above predictions. Thus, the groundwork on these cytokines mediating disease was in place before the first proposal that TNF and IL-1 were associated with inflammation (Nawroth et al., 1986). Soon rTNF, when trialled against tumours in patients (Sherman et al., 1988; Spriggs et al., 1988), caused side effects that mimicked not only the disease seen in influenza and malaria but also the aphasia seen in stroke and AD. As we have discussed previously (Clark et al., 2010), proinflammatory cytokines are enhanced very early in AD. For example, using a novel high-sensitivity proteomic neuroimaging technique, increased plasma levels of clusterin (apolipoprotein J), proved to be intimately associated with onset, progression and severity of AD (Thambisetty et al., 2010). Increased clusterin follows even slightly enhanced levels of pro-inflammatory cytokines such as TNF and IL-1 (Hardardottir et al., 1994). For all these reasons, it is essential to appreciate what generates TNF in AD. The recognized steps of TNF generation in innate immunity, and thence disease, are discussed next.
What controls the TNF response in innate immunity and disease?
The earlier observations tie together much diverse physiology and disease pathogenesis, so pose many important questions. For example, why should the same array of functionally related primitive cytokines, dominated by TNF, be generated in strikingly different circumstances? Our interest in this question arose from trying to understand spectacular protective outcomes of systemic exposure of mice to a then inexplicably wide array of agents, infectious and otherwise, weeks prior to infection with haemoprotozoan parasites (Clark et al., 1976; 1977,; Clark, 1979a,b). Intriguingly, such protection was functionally related to the onset of the non-specific systemic disease, akin to that seen in bacterial and viral infections, caused by these parasites (Clark et al., 1981). At a major symposium in 1989, within the topic of the evolution of immune recognition, Janeway (1989) offered the argument of a primitive ability, retained in humans, of effector cells to recognize what he termed a range of ‘pathogen-associated molecular patterns’ on or secreted by infectious agents. The name stuck, and the now-familiar acronym PAMP, came into use. Five years later this concept was incorporated into a proposal that the immune system may have evolved to distinguish between danger and non-danger, as distinct from self and non-self (Matzinger, 1994; 2002,; Gallucci and Matzinger, 2001). These authors saw PAMPs as a type of DAMP, and part of an overall damage-associated scheme (Seong and Matzinger, 2004). Hence, a disparate collection of signals triggering the same functional outcome fits within a framework that encompasses their ability to trigger the release of proinflammatory cytokines, with the capacity to kill pathogens through innate immunity, but also, in excess, to cause disease.
Thus, infectious agents provide triggers, collectively termed PAMPs, for release of TNF, and the rest of the proinflammatory cytokine cascade. Other triggers, either of host origin or exogenous, and usually termed damage-associated molecular patterns, or DAMPs, ultimately function in the same way as PAMPs. In effect, host function is inadvertently harmed in lieu of often non-existent pathogens. Others have elected, with commendable simplicity, to use the term alarmins to encompass both PAMPs and DAMPs (Oppenheim et al., 2007; Chan et al., 2012). Activation occurs when they are seen by the pattern recognition receptors (PRRs) (Janeway, 1989), the toll-like receptors (TLRs) (Poltorak et al., 1998) being one of the best described PRRs families. Many PAMPs and DAMPs important in instigating disease onset are seen by TLR4, on the cell surface, and others, typically those arising from modified RNA and DNA, are recognized intracellularly by TLR9, on the endoplasmic reticulum. In either case the outcome is very similar from a disease pathogenesis perspective. TLRs were well summarized recently in a myocardial context (de Haan et al., 2013), a text that also notes that DAMPs can be usefully divided into the constitutive and inducible, or secondary, groupings used here.
PAMPs implicated in chronic neuroinflammatory diseases
A key precursor of the present concept of PAMPs was the insight gained by early experience with the functional subtleties of BCG, which led to the original awareness that TNF exists, as outlined earlier. BCG is a pathogen, albeit attenuated, so by definition a source of PAMPs, and LPS is a PAMP derived from Gram-negative bacterial cell walls. Patients convalescing from typhoid (Neva and Morgan, 1950) and malaria (Rubenstein et al., 1965) are tolerant to LPS, whereas chronic, non-resolving infections, such as caused by BCG in mice, cause a LPS-sensitive state (Suter et al., 1958). A now historic set of experiments on this post-BCG LPS-sensitive state in Lloyd Old’s laboratory, as recently recounted (Carswell-Richards and Williamson, 2012), led to the isolation of a peptide they termed TNF (Carswell et al., 1975). This proved to be an invaluable tool in understanding details of a wide range of physiology, as well as innate immunity.
Acute severe infectious diseases, as well as sterile conditions such as stroke and TBI, can be forerunners to delirium, a transient AD-like condition associated with acute proinflammatory signals, and aptly described as the extreme end of the sickness behaviour spectrum (Cunningham and Maclullich, 2013). From an exceptionally large dataset, dementia, a related longer lasting state, proved to be more common in patients with more systemic infections, that is, exposure to PAMPs (Dunn et al., 2005). Another comprehensive study found the rate of cognitive decline in AD to be higher in patients who experienced more systemic inflammatory events associated with increased serum levels of TNF (Holmes et al., 2009). Likewise, others have recently compiled an intriguing overview of the influence of pathogenic microbes and the largely gut-located microbiome on the pathogenesis of AD and other chronic CNS disease (Hill et al., 2014). As they note, new technologies now allow the balance between pathogens and homeostatic commensals to be monitored. The work on Helicobacter pylori (Alvarez-Arellano and Maldonado-Bernal, 2014), including the reported beneficial effects of its eradication on 5 year survival in AD (Kountouras et al., 2010), is a specific example. Clearly, these groups’ studies are readily expressed in PAMP terminology. Consistent with the accepted multifactorial origins of AD, any of these PAMPs, or the DAMPs discussed below, can also be expected to increase the rate of cognitive decline through influencing TLR-dependent production of TNF and similar CNS-active cytokines (Figure 1).
Figure 1.
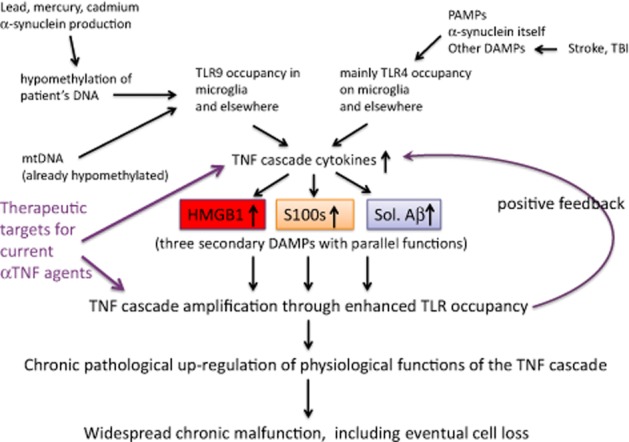
Late onset AD (LOAD). A representation of the array of DAMPs and PAMPs that, through triggering TLRs, can initiate release of proinflammatory cytokines. These cause changes that include enhancement of HMGB1, S100 proteins and soluble forms of Aβ in late onset AD, three secondary DAMPs that independently further enhance levels of the cytokines that induced them. Thus, chronic functional change and damage occurs within the brain.
MicroRNAs (miRNA) and mitochondrial DNA (mtDNA)as DAMPs in AD
Many miRNAs, small non-coding RNAs, are increased in the CSF and plasma in AD (Lukiw, 2007; Cogswell et al., 2008; Lukiw et al., 2012b; Alexandrov et al., 2014). This down-regulates complement factor H, a repressor of the innate immune response (Lukiw and Alexandrov, 2012a), thus enhancing this response, a key contributor to AD pathogenesis. As reviewed (Alexandrov et al., 2014), miRNA146a is up-regulated in the anatomical regions of the brain affected by AD, but not in the thalamus and brain stem of the same brain, and is induced by IL-1 and TNF, as well as by Aβ42 peptides, which act as DAMPs to induce TNF (Rowan et al., 2007). In addition, let-7, one of the most abundant of the hundreds of the miRNAs expressed in the human brain, and increased in the CSF in AD, has been reported to act, through activating TLR7, a reliable TNF trigger (Lehmann et al., 2012). These authors also observed that introducing let-7b into the CSF of mice resulted in neurodegeneration in intact, but not TLR7-deficient, mice. Intrauterine transfection with TLR7 restored activity.
Circulating mtDNA increases with age, which is associated with AD and PD, and the degree of increase is a familiar trait (Pinti et al., 2014). mtRNA is increased in human plasma soon after trauma (Lam et al., 2004), and has a bacterial DNA-like capacity to act as a danger signal, being similarly hypomethylated, and therefore sensed by TLR9 (Zhang et al., 2009). This is in keeping with its pathogen ancestry (Margulis and Chapman, 1998; Emelyanov, 2001) identifying it as a PAMP that has evolved into a DAMP, but normally harmless provided that it remains in the mitochondrion, without access to TLR9. It is considerably more sensitive to oxidative stress than is mammalian nuclear DNA (Strand et al., 2014). Oxidatively degraded mtDNA is a particularly aggressive DAMPs, proposed to participate in neurodegenerative processes (Mathew et al., 2012). Others have reported that occupancy of several TLRs simultaneously enhances oxidative stress (Lavieri et al., 2014), consistent with this increased DAMP potency of mtDNA.
Increased CSF levels of mtDNA have recently been correlated with severity in paediatric TBI cases (Walko et al., 2014). It is yet to be investigated whether mtDNA variants associated with AD and PD (Coskun et al., 2012) differ in their degradation rates. Likewise, uncertainty still surrounds CSF levels of mtRNA in AD. They have been argued to be reduced (Podlesniy et al., 2013), but other have proposed that technical error has left the question unresolved (Sondheimer et al., 2014).
Toxic metals and excess α-synuclein production generating DAMPs in AD
In brief, the evidence is consistent with lead (Pb) turning mammalian nuclear DNA into a DAMP. As discussed elsewhere (Clark and Vissel, 2013), Pb hypomethylates DNA that then recognizes TLR9, and generates TNF (Guo et al., 1996; Cheng et al., 2006). Fetal exposure to Pb also leads, via chronic TNF generation, to amyloid deposition later in life (Basha et al., 2005; Bihaqi et al., 2011). As reviewed (Wang et al., 2008), the case for epigenetic involvement in the pathogenesis of AD is well known: any discernible inheritance of late onset AD is non-Mendelian, concordance rates in monozygotic twins are low and levels of folate and homocysteine in the AD brain fit abnormal methylation homeostasis. Others have independently expanded these concepts in AD (Mastroeni et al., 2010; 2011,; Bakulski et al., 2012; Bihaqi et al., 2012) and PD (Iraola-Guzman et al., 2011; Kaut et al., 2012).
We (Clark and Vissel, 2013) have also discussed the publications on mercury and cadmium which show that lead is not the only contaminant metal associated with DNA hypomethylation (Hanna et al., 2012; Goodrich et al., 2013), an inflammatory response (Gardner et al., 2009; Olszowski et al., 2012), and Aβ accumulation (Song and Choi, 2013; Notarachille et al., 2014). We also summarized the implications of increased intraneural levels of soluble α-synuclein in human AD brains being a much better correlate with cognitive impairment than are levsl of the soluble forms of Aβ or phosphorylated tau (Larson et al., 2012). The actual process of generating excessive α-synuclein hypomethylates the DNA of the cell producing it (Desplats et al., 2011). These authors examined the intracellular location of α-synuclein as well as of Dnmt1, the major DNA methylation enzyme, in neurons from PD and dementia with Lewy bodies brains, and reported a cytoplasmic, rather than nuclear, location of this protein in neurons that overexpress it. Crucially, this cytoplasmic α-synuclein sequestered Dnmt1, reducing its levels by almost 50% in the nucleus, where it normally keeps DNA highly methylated. Consequently, a 30% decrease in local global DNA methylation occurred. Hence, the events leading up to increased soluble α-synuclein (Larson et al., 2012) give DAMP activity to this DNA, leading to up-regulation of proinflammatory cytokines when sensed by TLR9.
High-mobility group box 1 (HMGB1), S100 proteins and Aβ: three potent secondary DAMPs
Certain DAMPs incriminated in generating neuroinflammatory disease can themselves be induced by proinflammatory cytokines of infectious or sterile origin. They may therefore be termed secondary DAMPs (de Haan et al., 2013), and can also be regarded as positive feedback DAMPs, being further generated by the proinflammatory cytokines they themselves induce. This would thereby perpetuate and worsen disease, as happens in AD. Some mediators such as HMGB1 are constitutive in cells and, before they encounter the TLRs or other PRRs that enable them to display their proinflammatory potential, require relocating from their usual physiological niche by proinflammatory cytokines (Wang et al., 1999b) or by tissue damage. HMGB1 is a non-histone nuclear protein that, when extracellular, functions as a proinflammatory cytokine generator (Andersson et al., 2000), exacerbating inflammation. It is released in sepsis (Wang et al., 1999a; Andersson and Tracey, 2003), malaria (Alleva et al., 2005) and influenza (Alleva et al., 2008), and on recognition by TLR4 and the receptor for advanced glycation end products (RAGE) enhances inflammation through inducing cytokines such as TNF (van Zoelen et al., 2009). HMGB1 is essential to the chain of events that mediates cognitive impairment in sepsis survivors (Chavan et al., 2012) and memory impairment (Mazarati et al., 2011). It is released during trauma (Cohen et al., 2009), and involved in post-operative cognitive dysfunction (POCD) (He et al., 2012). When injected i.c.v. HMGB1 worsens, and anti-HMGB1 monoclonal antibody ameliorates, infarction in experimental cerebral ischaemia in rats (Liu et al., 2007). Recently, HMGB1 has proved to be a long-lasting component of the inflammatory response of stroke (Schulze et al., 2013). Increased extracellular HMGB1 has a well-documented involvement in a range of chronic inflammatory CNS states, including AD (Fang et al., 2012).
The S100 proteins are constitutive calcium-binding molecules present in cytoplasm, where they have homeostatic roles, but when released to the extracellular compartment they operate as proinflammatory danger signals, that is, as DAMPs. They are induced (Yen et al., 1997) and released extracellularly by proinflammatory signals, for instance from astrocytes by TNF (Edwards and Robinson, 2006), and therefore are also pro-inflammatory (Ryckman et al., 2003; Simard et al., 2013). S100B is increased in the CSF in the early stages of AD (Peskind et al., 2001), and S100A9 and S100A12 are enhanced in autopsy brains of both familial and sporadic AD (Shepherd et al., 2006). S100 proteins are well represented in the publications on TBI, stroke and PD. For example, S100B is increased in CSF of paediatric TBI cases (Berger et al., 2002), as are mtDNA and HMGB1 (Walko et al., 2014), as discussed earlier. It is regarded as a DAMP in PD (Sathe et al., 2012). Indeed, as discussed (Foell et al., 2007), the S100 proteins are standard DAMPs, by the same criteria as are HMGB1 and mtDNA.
The soluble Aβ proteins, a term encompassing a range of oligomers, are normally present in cells (Selkoe et al., 1996; Ghiso et al., 1997). They have various physiological functions including synapse elimination in brain development (Wasling et al., 2009) and in the normal hippocampus (Puzzo et al., 2011). Although when in excess soluble Aβ is often regarded as the initiator of AD, it is not specific to this condition, being documented in lead exposure (Basha et al., 2005; Bihaqi et al., 2011) and in post-stroke patients (Lee et al., 2005). As reviewed recently (Knowles et al., 2014), many more proteins than previously suspected are inherently unstable, and can therefore misfold. Such prefibrillar states, analogous to Aβ oligomers, can be expected to allow PRRs to sense chemical groupings not normally accessible to the cellular environment, and therefore merit investigation as DAMPs in disease pathogenesis (Stefani and Dobson, 2003). To date some 50 conditions, including AD and the spongiform encephalopathies, have been associated with such aggregations (Chiti and Dobson, 2006; Knowles et al., 2014). Indeed, the finding that this phenomenon was common to these two diseases apparently inspired the idea of Aβ plaques causing AD. As recorded (Schnabel, 2011), this recognition of the histological similarities of scrapie prions and plaque in AD (Prusiner et al., 1983; Prusiner, 1984; Masters, 1985) arose from the meeting of like minds who saw similarities between histological features as implying similar function. The idea received encouragement from the ability of products of the amyloid cascade to kill neurons directly (Yankner et al., 1989), with its scope eventually widening to encompass a direct capacity to impair synapse function (Beyreuther et al., 1993). Coming at a time when AD research needed direction, these ideas quickly dominated the field, and still have formidable momentum, despite increasing criticism and repeated trial failure. Once it became evident that the plaque formed from aggregated Aβ was inert in terms of disease pathogenesis (Holmes et al., 2008), the focus of amyloid research transferred to the soluble oligomers of this peptide.
Nevertheless, as the progenitor of amyloid plaque, soluble Aβ has a front-row seat in the experimental world of AD pathogenesis, with HMGB1 and the S100s well to the rear. The built-in bias towards Aβ in the transgenic APP-based models (below) has also muddied the waters. Soluble Aβ has been referred to as a constitutive DAMP (Shichita et al., 2012), because when enhanced it exacerbates levels of proinflammatory cytokines, mainly through activating TLR4 (Reed-Geaghan et al., 2009; Stewart et al., 2010; Vollmar et al., 2010). It is, without doubt, also generated to excess in the various infectious diseases in which amyloid plaque is histologically evident, such as neuroborreliosis (Miklossy et al., 2006), cerebral Chlamydia infections (Little et al., 2004) and HIV dementia (Soontornniyomkij et al., 2012). Important cerebral functional consequences of Aβ-induced inflammation have been documented for some time (Wang et al., 2005; Rowan et al., 2007), and new data continue to emerge (Lourenco et al., 2013).
Clearly, Aβ production is controlled by proinflammatory cytokines, as well as generating them. Studies on the secretases have, as reviewed (Gandy, 2005; Zhang and Song, 2013), demonstrated this. For example, genetically inhibiting TNF signalling (He et al., 2007), or administering thalidomide, an inhibitor of TNF (He et al., 2013), reduces both β secretase (BACE1) and Aβ load. TNF also up-regulates BACE1 (Yamamoto et al., 2007; Zhao et al., 2011) and γ secretase (Liao et al., 2004), another secretase variant involved in Aβ enhancement. Moreover, a 3,6 dithio variant of thalidomide, which inhibits TNF production, prevents (Gabbita et al., 2012) and reverses (Tweedie et al., 2012) disease in mouse models of AD. Likewise, glucagon-like peptide-1 (GLP-1), which has several mimetics in routine clinical use against type 2 diabetes mellitus, enhances α secretase (ADAM10) (Ohtake et al., 2014). This shifts the cleavage of the amyloid precursor protein away from the Aβ producing β-secretase pathway and towards the growth-signalling pathway, reducing the brain levels of Aβ. Data generated 10 years ago with exendin-4, a GLP-1 mimetic (Perry et al., 2003), are consistent with this. The GLP-1 mimetics have been well reviewed as plausible AD treatments (Greig et al., 2004; Holscher and Li, 2010) and have complex functions that can broadly be described as anti-inflammatory, including, as recently reviewed (Clark et al., 2012; Clark and Vissel, 2013), countering the insulin resistance generated by an inflammatory milieu. These mimetics protect against (McClean et al., 2011) and reverse (McClean and Holscher, 2014) experimental AD, and are in clinical trials (NCT01255163, NCT01843075).
POCD as an illustrative microcosm
As discussed, the inflammation-induced, inflammation-generating nature of these three secondary DAMPs provides parallel positive feedback mechanisms operating to enhance the original inflammatory cascade in AD (Figure 1). Post-surgery patients provide a convenient example of how the big picture has been missed. Transient delirium is common in intensive care units, and is, as noted earlier, an extreme manifestation of the sickness behaviour caused by systemic inflammation (Cunningham and Maclullich, 2013). A characteristic of post-surgery patients, particularly the more elderly, is the persistent self-propagating inflammatory syndrome, in which case it is referred to as POCD, with changes analogous to those seen in AD (Newman et al., 2007; Steinmetz et al., 2009). Indeed, in some studies the conversion rates to dementia are up to 70% in patients who are 65 years or older (Vanderweyde et al., 2010).
The publications on POCD show how a field can be obscured by focusing on individual jigsaw pieces rather than constructing the wider picture. For example, at least three groups have explored both inflammatory cytokines and HMGB1 in POCD (Terrando et al., 2010; He et al., 2012; Lin et al., 2014). Notably, all three groups considered HMGB1 in isolation from S100s or Aβ. Likewise, while others (Linstedt et al., 2002; Rohan et al., 2005; Leiendecker et al., 2010; Li et al., 2012; Lili et al., 2013) showed increased S100s in POCD, two of these co-assaying for an inflammatory cytokine (Li et al., 2012; Lili et al., 2013), and none for HMGB1 or Aβ. In the same vein, others have published on Aβ in POCD (Xie and Tanzi, 2006; Ji et al., 2013; Reinsfelt et al., 2013; Xu et al., 2014), but few discuss inflammatory cytokines (Ji et al., 2013; Reinsfelt et al., 2013), and none, so far as we are aware, co-investigated HMGB1or S100s. All this is consistent with the concept, based on mouse studies (Terrando et al., 2010), of preventing POCD by pre-emptively treating at-risk surgical patients with anti-TNF antibody.
The bias built into transgenic AD models and caused by injecting soluble Aβ
Could mouse transgenic models, which encouraged the argument that anti-amyloid immunotherapy approaches were ready for human trials (Janus et al., 2000; Morgan et al., 2000), have led researchers astray? The same question mark may hang over the impressive outcome in which ultrasound scanning, rather than passive or active antibody, was recently used to remove Aβ and restore normal function in another mouse strain commonly used as an AD model (Leinenga and Gotz, 2015). Because these genetically modified mouse strains overexpress human AβPP and therefore Aβ, any other secondary DAMP, such as HMGB1 or S100s, would become relatively insignificant (Figure 2), allowing Aβ removal, by whatever method, to be sufficient to block the secondary DAMP step in the pathogenesis pathway. Whereas these mouse models are an argument in favour of anti-amyloid immunotherapy for early-onset human AD (EOAD), which is characterized by mutations that lead to high Aβ expression (Kowalska, 2003), the same does not hold for the much more common, sporadic, late onset form of the disease, in which there is no reason to presume, as in mouse models and EOAD, that secondary DAMP function is dominated by Aβ rather than shared with HMGB1 and S100 proteins.
Figure 2.
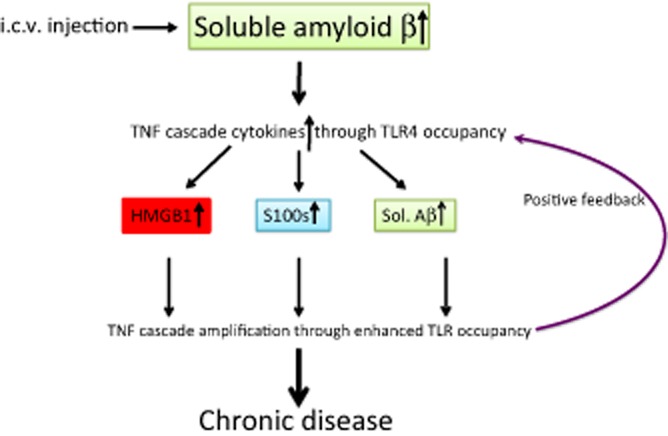
Model of AD induced by i.c.v. injection of Aβ in mice. Partial mimicry of LOAD, but the pathway is artificially biased towards of an end result that is Aβ dependent, and therefore responds to therapy that reduces a TNF cascade that was initially induced by the injected Aβ.
It has become common practice (Maurice et al., 1996; Kim et al., 2014) to strengthen the amyloid case by transiently reproducing aspects of AD by injecting soluble Aβ into experimental animals. As with transgenic mice, such experiments have limited relevance to the clinical disease without HMBG1 and S100 proteins, the other two secondary DAMPs we have discussed, being brought into the equation.
Total PAMP plus DAMP determines outcome
We have made the case that PAMPs and DAMPs may start the chain of proinflammatory events leading to the pathogenesis of the chronic neurodegenerative diseases, including being incorporated into the AD pathogenesis pathway. A most prescient publication has proposed a damage signal hypothesis of AD pathogenesis in which long-term activation of the innate immune system was central (Fernandez et al., 2008). In essence, the authors reasoned that what matters is not whether a particular danger signal is present, but whether the total sum of their activity and persistence, and thus the chronic level of the proinflammatory cytokine they induce, are sufficient to initiate and drive neurodegenerative disease. Although much more is known nowadays about the range of possible DAMPs, including the presence of Aβ in their ranks, the idea that the danger signals discussed earlier all converge to provide harmful levels of the same proinflammatory cytokines (Fernandez et al., 2008) still rings true. Our awareness of the details of outcomes when a number of TLRs are activated simultaneously is, however, still in its infancy (Rosenberger et al., 2014).
Aβ in perspective
One consequence of the prolonged enthusiasm for Aβ has been a relative ignorance in this field of the other secondary DAMPs, such as HMGB1 and S100s, which are increased in AD but remain little tested in this context. Until all three are given equal consideration, there seems little rationale for implying that Aβ is more potent than the other two. Nevertheless, plaque is certainly an instructive histological footprint from which to infer long-term DAMP activity by soluble Aβ. For example, the presence of excessive plaque in the absence of cognitive loss (Schmitt et al., 2000) may indicate that few if any other PAMPs or DAMPs were up-regulated in that individual. Hence increased Aβ alone may, in this circumstance, have been insufficient to raise the net load of proinflammatory cytokines above threshold required for disease onset. If, on the other hand, HMGB1 and the S100s – as well as other inflammation-enhancing DAMPs of which we are as yet unaware – are plentiful, immunotherapeutically removing soluble Aβ, no matter how diligently or on how grand a scale, as in recent random trials (Doody et al., 2014; Salloway et al., 2014), is unlikely to be helpful to AD patients because the contributions from other secondary DAMPs ensure that the total proinflammatory cytokine load remains high enough to maintain illness.
We propose that, as one of the secondary DAMPs able to further enhance inflammatory cytokine levels, soluble Aβ has a middle-ranking role in AD pathogenesis, no more or less essential than those of HMGB1 or the S100 proteins. This questions the continuing stream of literature assuming oligomer versions of Aβ are the primary initiators of AD pathogenesis, either implying direct harmfulness or acting via the proinflammatory cytokines it induces. As noted earlier, even the post-Aβ proinflammatory cytokine step is still often omitted from the AD pathogenesis narrative, even though the capacity of Aβ to act as a DAMP, a link first recognized in 2005 (Wang et al., 2005), is now clear. As noted earlier, Aβ is recognized by various TLRs (Reed-Geaghan et al., 2009; Stewart et al., 2010; Vollmar et al., 2010) and the field is continuing to expand (Lourenco et al., 2013). Particularly telling recent evidence is that etanercept, the anti-TNF agent used off-label via an apparent i.c.v. equivalent route for treating AD and stroke (Tobinick and Vega, 2006a; Tobinick et al., 2006b; 2012,), has been reported to prevent memory deficits caused by administering Aβ to mice i.c.v. (Detrait et al., 2014). Notably, publications ignoring the effects of post-Aβ TNF includes new evidence on GABA from reactive astrocytes impairing memory in mouse models of AD (Jo et al., 2014). TLR4s, which sense Aβ, are on astrocytes (Gorina et al., 2011) and oligomeric Ab induces high levels of TNF in these cells (White et al., 2005).
Even so, understanding the secondary DAMP character of Aβ, in line with that of HMGB1 and the S100 proteins, requires an awareness that the proinflammatory cytokines that mediate the harm caused by Aβ had also been instrumental in inducing Aβ (Liao et al., 2004; He et al., 2007; 2013,; Yamamoto et al., 2007; Zhao et al., 2011). Given these shared positive feedback functions of HMGB1, S100 and Aβ for proinflammatory cytokines, it is intriguing to consider the history of AD research priorities, and the number and influence of consequent publications, if either or both of these other two DAMPs, as well as Aβ, had left histologically spectacular plaques as a persistent footprint of their past formation.
Parallel circumstances in related conditions
This review is not complete without noting the parallel world within the publications on the AD-related conditions, stroke and TBI (Hua et al., 2007; Cohen et al., 2009; Hyakkoku et al., 2010; Su et al., 2011; Tsai et al., 2011; Shichita et al., 2012). Indeed, a narrative largely parallel to ours could be constructed, focusing on either stroke or TBI, with a similar degree of reference to the other two neurodegenerative states as all three conditions are now described in terms of the innate immunity cytokines and have an appreciable body of publications on HMBG1, S100 proteins and Aβ. Thus, the best way to advance rational treatment of this close knit trio of neurodegenerative conditions seems to be to focus on what they have in common, despite their disparate clinical origins. As reviewed (Clark and Vissel, 2013), a range of studies point to efficacy of anti-TNF agents and GLP-1 mimetics, which, as TNF induces insulin resistance, ameliorate consecutive harmful steps in those brain disease states with excess TNF, whatever their traditional, clinically based, disparate nomenclatures.
In summary, a clear perspective on the role of soluble Aβ in AD is most rationally gained by visualizing it in the company of other secondary DAMPs, such as HMBG1 and S100 proteins, rather than in isolation. When this is performed, the presence of high amyloid levels in many cognitively normal older brains, and the failure to replicate in humans the anti-amyloid immunotherapy, successful in transgenic mice, can be better understood.
Acknowledgments
No funding was sought or received for this study.
Glossary
- AD
Alzheimer’s disease
- BACE1
β secretase
- DAMP
damage-associated molecular pattern
- EOAD
early onset human AD
- HMGB1
high-mobility group box 1
- LOAD
late onset human AD
- PAMP
pathogen-associated molecular pattern
- PD
Parkinson’s disease
- POCD
post-operative cognitive dysfunction
- TLR
toll-like receptor
Conflict of interest
The authors declare that they have not any conflict of interest.
References
- Aggarwal BB, Kohr WJ, Hass PE, Moffat B, Spencer SA, Henzel WJ, et al. Human tumor necrosis factor: production, purification, and characterization. J Biol Chem. 1985;260:2345–2354. [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov PN, Dua P, Lukiw WJ. Up-regulation of miRNA-146a in progressive, age-related inflammatory neurodegenerative disorders of the human CNS. Front Neurol. 2014;5:181. doi: 10.3389/fneur.2014.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alleva LM, Yang H, Tracey KJ, Clark IA. High mobility group box 1 (HMGB1) protein: possible amplification signal in the pathogenesis of falciparum malaria. Trans R Soc Trop Med Hyg. 2005;99:171–175. doi: 10.1016/j.trstmh.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Alleva LM, Budd AC, Clark IA. Systemic release of high mobility group box 1 protein during severe murine influenza. J Immunol. 2008;181:1454–1459. doi: 10.4049/jimmunol.181.2.1454. [DOI] [PubMed] [Google Scholar]
- Alvarez-Arellano L, Maldonado-Bernal C. Helicobacter pylori and neurological diseases: married by the laws of inflammation. World J Gastrointest Pathophysiol. 2014;5:400–404. doi: 10.4291/wjgp.v5.i4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Tracey KJ. HMGB1 in sepsis. Scand J Infect Dis. 2003;35:577–584. doi: 10.1080/00365540310016286. [DOI] [PubMed] [Google Scholar]
- Andersson U, Wang HC, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenijevic D, Girardier L, Seydoux J, Chang HR, Dulloo AG. Altered energy balance and cytokine gene expression in a murine model of chronic infection with Toxoplasma gondii. Am J Physiol. 1997;272:E908–E917. doi: 10.1152/ajpendo.1997.272.5.E908. [DOI] [PubMed] [Google Scholar]
- Arvin B, Neville LF, Barone FC, Feuerstein GZ. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev. 1996;20:445–452. doi: 10.1016/0149-7634(95)00026-7. [DOI] [PubMed] [Google Scholar]
- Bakulski KM, Dolinoy DC, Sartor MA, Paulson HL, Konen JR, Lieberman AP, et al. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J Alzheimers Dis. 2012;29:571–588. doi: 10.3233/JAD-2012-111223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, et al. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25:823–829. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger RP, Pierce MC, Wisniewski SR, Adelson PD, Clark RS, Ruppel RA, et al. Neuron-specific enolase and S100B in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatrics. 2002;109:E31. doi: 10.1542/peds.109.2.e31. [DOI] [PubMed] [Google Scholar]
- Bernardino L, Agasse F, Silva B, Ferreira R, Grade S, Malva JO. Tumor necrosis factor-alpha modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells. 2008;26:2361–2371. doi: 10.1634/stemcells.2007-0914. [DOI] [PubMed] [Google Scholar]
- Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effects of endotoxin. Science. 1985;229:869–871. doi: 10.1126/science.3895437. [DOI] [PubMed] [Google Scholar]
- Beyreuther K, Pollwein P, Multhaup G, Monning U, Konig G, Dyrks T, et al. Regulation and expression of the Alzheimer’s beta/A4 amyloid protein precursor in health, disease, and Down’s syndrome. Ann N Y Acad Sci. 1993;695:91–102. doi: 10.1111/j.1749-6632.1993.tb23035.x. [DOI] [PubMed] [Google Scholar]
- Bhutta ZA, Mansoorali N, Hussain R. Plasma cytokines in paediatric typhoidal salmonellosis: correlation with clinical course and outcome. J Infect. 1997;35:253–256. doi: 10.1016/s0163-4453(97)93004-8. [DOI] [PubMed] [Google Scholar]
- Bihaqi SW, Huang H, Wu J, Zawia NH. Infant exposure to lead (Pb) and epigenetic modifications in the aging primate brain: implications for Alzheimer’s disease. J Alzheimers Dis. 2011;27:819–833. doi: 10.3233/JAD-2011-111013. [DOI] [PubMed] [Google Scholar]
- Bihaqi SW, Schumacher A, Maloney B, Lahiri DK, Zawia NH. Do epigenetic pathways initiate late onset Alzheimer disease (LOAD): towards a new paradigm. Curr Alzheimer Res. 2012;9:574–588. doi: 10.2174/156720512800617982. [DOI] [PubMed] [Google Scholar]
- Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–247. doi: 10.1016/s0140-6736(89)90430-3. [DOI] [PubMed] [Google Scholar]
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carswell-Richards EA, Williamson BD. A man of vision and the discovery of tumor necrosis factor. Cancer Immun. 2012;12:1–4. [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Smith MA. Compounding artefacts with uncertainty, and an amyloid cascade hypothesis that is ‘too big to fail’. J Pathol. 2011;224:147–152. doi: 10.1002/path.2885. [DOI] [PubMed] [Google Scholar]
- Castello MA, Jeppson JD, Soriano S. Moving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s disease. BMC Neurol. 2014;14:169. doi: 10.1186/s12883-014-0169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest. 2012;122:2711–2719. doi: 10.1172/JCI62423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, et al. HMGB1 mediates cognitive impairment in sepsis survivors. Mol Med. 2012;18:930–937. doi: 10.2119/molmed.2012.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YJ, Yang BC, Liu MY. Lead increases lipopolysaccharide-induced liver-injury through tumor necrosis factor-alpha overexpression by monocytes/macrophages: role of protein kinase C and P42/44 mitogen-activated protein kinase. Environ Health Perspect. 2006;114:507–513. doi: 10.1289/ehp.8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirila AM, Brown TE, Bishop RA, Bellono NW, Pucci FG, Kauer JA. Long-term potentiation of glycinergic synapses triggered by interleukin 1beta. Proc Natl Acad Sci U S A. 2014;111:8263–8268. doi: 10.1073/pnas.1401013111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Chiu SL, Cline HT. Insulin receptor signaling in the development of neuronal structure and function. Neural Dev. 2010;5:7. doi: 10.1186/1749-8104-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SL, Chen CM, Cline HT. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron. 2008;58:708–719. doi: 10.1016/j.neuron.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark I, Atwood C, Bowen R, Paz-Filho G, Vissel B. Tumor necrosis factor-induced cerebral insulin resistance in Alzheimer’s disease links numerous treatment rationales. Pharmacol Rev. 2012;64:1004–1026. doi: 10.1124/pr.112.005850. [DOI] [PubMed] [Google Scholar]
- Clark IA. Protection of mice against Babesia microti with cord factor, COAM, zymosan, glucan, Salmonella and Listeria. Parasite Immunol. 1979a;1:179–196. doi: 10.1111/j.1365-3024.1979.tb00705.x. [DOI] [PubMed] [Google Scholar]
- Clark IA. Resistance to Babesia spp. and Plasmodium sp. in mice pretreated with an extract of Coxiella burnetii. Infect Immun. 1979b;24:319–325. doi: 10.1128/iai.24.2.319-325.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA. Suggested importance of monokines in pathophysiology of endotoxin shock and malaria. Klin Wochenschr. 1982;60:756–758. doi: 10.1007/BF01716573. [DOI] [PubMed] [Google Scholar]
- Clark IA, Cowden WB. Is TNF a key to acute infectious illness? Today’s Life Sci. 1989;1:26–29. [Google Scholar]
- Clark IA, Vissel B. Treatment implications of the altered cytokine-insulin axis in neurodegenerative disease. Biochem Pharmacol. 2013;86:862–871. doi: 10.1016/j.bcp.2013.07.030. [DOI] [PubMed] [Google Scholar]
- Clark IA, Vissel B. Inflammation-sleep interface in brain disease: TNF, insulin, orexin. J Neuroinflammation. 2014;11:51. doi: 10.1186/1742-2094-11-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA, Allison AC, Cox FE. Protection of mice against Babesia and Plasmodium with BCG. Nature. 1976;259:309–311. doi: 10.1038/259309a0. [DOI] [PubMed] [Google Scholar]
- Clark IA, Cox FE, Allison AC. Protection of mice against Babesia spp. and Plasmodium spp. with killed Corynebacterium parvum. Parasitology. 1977;74:9–18. doi: 10.1017/s003118200004748x. [DOI] [PubMed] [Google Scholar]
- Clark IA, Virelizier J-L, Carswell EA, Wood PR. Possible importance of macrophage-derived mediators in acute malaria. Infect Immun. 1981;32:1058–1066. doi: 10.1128/iai.32.3.1058-1066.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA, Alleva LM, Vissel B. The roles of TNF in brain dysfunction and disease. Pharmacol Ther. 2010;128:519–548. doi: 10.1016/j.pharmthera.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]
- Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care. 2009;13:R174. doi: 10.1186/cc8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumiskey D, Butler MP, Moynagh PN, O’Connor JJ. Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor-alpha on long-term potentiation. Brain Res. 2007;1136:13–19. doi: 10.1016/j.brainres.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Maclullich AM. At the extreme end of the psychoneuroimmunological spectrum: delirium as a maladaptive sickness behaviour response. Brain Behav Immun. 2013;28:1–13. doi: 10.1016/j.bbi.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, et al. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrait ER, Danis B, Lamberty Y, Foerch P. Peripheral administration of an anti-TNF-alpha receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-alpha levels and memory deficits in mice. Neurochem Int. 2014;72:10–13. doi: 10.1016/j.neuint.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- Dunn N, Mullee M, Perry VH, Holmes C. Association between dementia and infectious disease: evidence from a case-control study. Alzheimer Dis Assoc Disord. 2005;19:91–94. doi: 10.1097/01.wad.0000165511.52746.1f. [DOI] [PubMed] [Google Scholar]
- Edwards MM, Robinson SR. TNF alpha affects the expression of GFAP and S100B: implications for Alzheimer’s disease. J Neural Transm. 2006;113:1709–1715. doi: 10.1007/s00702-006-0479-5. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Veerhuis R, van Exel E, Hoozemans JJ, Rozemuller AJ, van Gool WA. The early involvement of the innate immunity in the pathogenesis of late-onset Alzheimer’s disease: neuropathological, epidemiological and genetic evidence. Curr Alzheimer Res. 2011;8:142–150. doi: 10.2174/156720511795256080. [DOI] [PubMed] [Google Scholar]
- Emelyanov VV. Evolutionary relationship of Rickettsiae and mitochondria. FEBS Lett. 2001;501:11–18. doi: 10.1016/s0014-5793(01)02618-7. [DOI] [PubMed] [Google Scholar]
- Esiri MM. The interplay between inflammation and neurodegeneration in CNS disease. J Neuroimmunol. 2007;184:4–16. doi: 10.1016/j.jneuroim.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Fang P, Schachner M, Shen YQ. HMGB1 in development and diseases of the central nervous system. Mol Neurobiol. 2012;45:499–506. doi: 10.1007/s12035-012-8264-y. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, et al. Cell death after spinal cord injury is exacerbated by rapid TNFalpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez JA, Rojo L, Kuljis RO, Maccioni RB. The damage signals hypothesis of Alzheimer’s disease pathogenesis. J Alzheimers Dis. 2008;14:329–333. doi: 10.3233/jad-2008-14307. [DOI] [PubMed] [Google Scholar]
- Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Srivastava MK, Eslami P, Johnson MF, Kobritz NK, Tweedie D, et al. Early intervention with a small molecule inhibitor for tumor necrosis factor-alpha prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2012;9:99. doi: 10.1186/1742-2094-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RM, Nyland JF, Evans SL, Wang SB, Doyle KM, Crainiceanu CM, et al. Mercury induces an unopposed inflammatory response in human peripheral blood mononuclear cells in vitro. Environ Health Perspect. 2009;117:1932–1938. doi: 10.1289/ehp.0900855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiso J, Calero M, Matsubara E, Governale S, Chuba J, Beavis R, et al. Alzheimer’s soluble amyloid beta is a normal component of human urine. FEBS Lett. 1997;408:105–108. doi: 10.1016/s0014-5793(97)00400-6. [DOI] [PubMed] [Google Scholar]
- Goodrich JM, Basu N, Franzblau A, Dolinoy DC. Mercury biomarkers and DNA methylation among Michigan dental professionals. Environ Mol Mutagen. 2013;54:195–203. doi: 10.1002/em.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- Greig NH, Mattson MP, Perry T, Chan SL, Giordano T, Sambamurti K, et al. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann N Y Acad Sci. 2004;1033:290–315. doi: 10.1196/annals.1332.018. [DOI] [PubMed] [Google Scholar]
- Guo TL, Mudzinski SP, Lawrence DA. The heavy metal lead modulates the expression of both TNF-alpha and TNF-alpha receptors in lipopolysaccharide-activated human peripheral blood mononuclear cells. J Leukoc Biol. 1996;59:932–939. doi: 10.1002/jlb.59.6.932. [DOI] [PubMed] [Google Scholar]
- de Haan JJ, Smeets MB, Pasterkamp G, Arslan F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediators Inflamm. 2013;2013:206039. doi: 10.1155/2013/206039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna CW, Bloom MS, Robinson WP, Kim D, Parsons PJ, vom Saal FS, et al. DNA methylation changes in whole blood is associated with exposure to the environmental contaminants, mercury, lead, cadmium and bisphenol A, in women undergoing ovarian stimulation for IVF. Hum Reprod. 2012;27:1401–1410. doi: 10.1093/humrep/des038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardardottir I, Kunitake ST, Moser AH, Doerrler WT, Rapp JH, Grunfeld C, et al. Endotoxin and cytokines increase hepatic messenger RNA levels and serum concentrations of apolipoprotein J (clusterin) in Syrian hamsters. J Clin Invest. 1994;94:1304–1309. doi: 10.1172/JCI117449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HJ, Wang Y, Le Y, Duan KM, Yan XB, Liao Q, et al. Surgery upregulates high mobility group box-1 and disrupts the blood-brain barrier causing cognitive dysfunction in aged rats. CNS Neurosci Ther. 2012;18:994–1002. doi: 10.1111/cns.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, et al. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Cheng X, Staufenbiel M, Li R, Shen Y. Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of beta-secretase in a mouse model of Alzheimer’s disease. PLoS ONE. 2013;8:e55091. doi: 10.1371/journal.pone.0055091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Clement C, Pogue AI, Bhattacharjee S, Zhao Y, Lukiw WJ. Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD) Front Aging Neurosci. 2014;6:127. doi: 10.3389/fnagi.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C, Li L. New roles for insulin-like hormones in neuronal signalling and protection: new hopes for novel treatments of Alzheimer’s disease? Neurobiol Aging. 2010;31:1495–1502. doi: 10.1016/j.neurobiolaging.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Howcroft TK, Campisi J, Louis GB, Smith MT, Wise B, Wyss-Coray T, et al. The role of inflammation in age-related disease. Aging (Albany NY) 2013;5:84–93. doi: 10.18632/aging.100531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Xia Y, Kelley J, Williams DL, et al. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience. 2010;171:258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- Iraola-Guzman S, Estivill X, Rabionet R. DNA methylation in neurodegenerative disorders: a missing link between genome and environment? Clin Genet. 2011;80:1–14. doi: 10.1111/j.1399-0004.2011.01673.x. [DOI] [PubMed] [Google Scholar]
- Janeway CA., Jr Pillars article: approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Ji MH, Yuan HM, Zhang GF, Li XM, Dong L, Li WY, et al. Changes in plasma and cerebrospinal fluid biomarkers in aged patients with early postoperative cognitive dysfunction following total hip-replacement surgery. J Anesth. 2013;27:236–242. doi: 10.1007/s00540-012-1506-3. [DOI] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaut O, Schmitt I, Wullner U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics. 2012;13:87–91. doi: 10.1007/s10048-011-0308-3. [DOI] [PubMed] [Google Scholar]
- Kim E, Jung YS, Kim H, Kim JS, Park M, Jeong J, et al. Metabolomic signatures in peripheral blood associated with Alzheimer’s disease amyloid-beta-induced neuroinflammation. J Alzheimers Dis. 2014;42:421–433. doi: 10.3233/JAD-132165. [DOI] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Kountouras J, Boziki M, Gavalas E, Zavos C, Deretzi G, Chatzigeorgiou S, et al. Five-year survival after Helicobacter pylori eradication in Alzheimer disease patients. Cogn Behav Neurol. 2010;23:199–204. doi: 10.1097/WNN.0b013e3181df3034. [DOI] [PubMed] [Google Scholar]
- Kowalska A. Amyloid precursor protein gene mutations responsible for early-onset autosomal dominant Alzheimer’s disease. Folia Neuropathol. 2003;41:35–40. [PubMed] [Google Scholar]
- Lam NY, Rainer TH, Chiu RW, Joynt GM, Lo YM. Plasma mitochondrial DNA concentrations after trauma. Clin Chem. 2004;50:213–216. doi: 10.1373/clinchem.2003.025783. [DOI] [PubMed] [Google Scholar]
- Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, et al. Soluble alpha-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J Neurosci. 2012;32:10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieri R, Piccioli P, Carta S, Delfino L, Castellani P, Rubartelli A. TLR costimulation causes oxidative stress with unbalance of proinflammatory and anti-inflammatory cytokine production. J Immunol. 2014;192:5373–5381. doi: 10.4049/jimmunol.1303480. [DOI] [PubMed] [Google Scholar]
- Lee PH, Bang OY, Hwang EM, Lee JS, Joo US, Mook-Jung I, et al. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J Neural Transm. 2005;112:1371–1379. doi: 10.1007/s00702-004-0274-0. [DOI] [PubMed] [Google Scholar]
- Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, et al. An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 2012;15:827–835. doi: 10.1038/nn.3113. [DOI] [PubMed] [Google Scholar]
- Leiendecker J, Hocker J, Meybohm P, Fudickar A, Bein B. Postoperative neurocognitive function and microembolus detection in patients undergoing neck dissection: a pilot study. Eur J Anaesthesiol. 2010;27:417–424. doi: 10.1097/eja.0b013e328336c633. [DOI] [PubMed] [Google Scholar]
- Leinenga G, Gotz J. Scanning ultrasound removes amyloid-beta and restores memory in an Alzheimer’s disease mouse model. Sci Transl Med. 2015;7:278ra33. doi: 10.1126/scitranslmed.aaa2512. [DOI] [PubMed] [Google Scholar]
- Li YC, Xi CH, An YF, Dong WH, Zhou M. Perioperative inflammatory response and protein S-100beta concentrations – relationship with post-operative cognitive dysfunction in elderly patients. Acta Anaesthesiol Scand. 2012;56:595–600. doi: 10.1111/j.1399-6576.2011.02616.x. [DOI] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- Lili X, Zhiyong H, Jianjun S. A preliminary study of the effects of ulinastatin on early postoperative cognition function in patients undergoing abdominal surgery. Neurosci Lett. 2013;541:15–19. doi: 10.1016/j.neulet.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Lin GX, Wang T, Chen MH, Hu ZH, Ouyang W. Serum high-mobility group box 1 protein correlates with cognitive decline after gastrointestinal surgery. Acta Anaesthesiol Scand. 2014;58:668–674. doi: 10.1111/aas.12320. [DOI] [PubMed] [Google Scholar]
- Linstedt U, Meyer O, Kropp P, Berkau A, Tapp E, Zenz M. Serum concentration of S-100 protein in assessment of cognitive dysfunction after general anesthesia in different types of surgery. Acta Anaesthesiol Scand. 2002;46:384–389. doi: 10.1034/j.1399-6576.2002.460409.x. [DOI] [PubMed] [Google Scholar]
- Little CS, Hammond CJ, MacIntyre A, Balin BJ, Appelt DM. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004;25:419–429. doi: 10.1016/S0197-4580(03)00127-1. [DOI] [PubMed] [Google Scholar]
- Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18:831–843. doi: 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport. 2007;18:297–300. doi: 10.1097/WNR.0b013e3280148e8b. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Alexandrov PN. Regulation of complement factor H (CFH) by multiple miRNAs in Alzheimer’s disease (AD) brain. Mol Neurobiol. 2012a;46:11–19. doi: 10.1007/s12035-012-8234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukiw WJ, Andreeva TV, Grigorenko AP, Rogaev EI. Studying micro RNA function and dysfunction in Alzheimer’s disease. Front Genet. 2012b;3:327. doi: 10.3389/fgene.2012.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulis L, Chapman MJ. Endosymbioses: cyclical and permanent in evolution. Trends Microbiol. 1998;6:342–345. doi: 10.1016/s0966-842x(98)01325-0. [DOI] [PubMed] [Google Scholar]
- Marin I, Kipnis J. Learning and memory … and the immune system. Learn Mem. 2013;20:601–606. doi: 10.1101/lm.028357.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters C. Perspectives on prions. Nature. 1985;314:15–16. doi: 10.1038/314015a0. [DOI] [PubMed] [Google Scholar]
- Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging. 2010;31:2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging. 2011;32:1161–1180. doi: 10.1016/j.neurobiolaging.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, et al. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimers Dis. 2012;30:617–627. doi: 10.3233/JAD-2012-120145. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- Maurice T, Lockhart BP, Privat A. Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996;706:181–193. doi: 10.1016/0006-8993(95)01032-7. [DOI] [PubMed] [Google Scholar]
- Mazarati A, Maroso M, Iori V, Vezzani A, Carli M. High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and Receptor for Advanced Glycation End Products. Exp Neurol. 2011;232:143–148. doi: 10.1016/j.expneurol.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfoose J, Baune BT. Evidence for a cytokine model of cognitive function. Neurosci Behav Rev. 2009;34:615–619. doi: 10.1016/j.neubiorev.2008.10.005. [DOI] [PubMed] [Google Scholar]
- McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacol. 2014;76(Pt A):57–67. doi: 10.1016/j.neuropharm.2013.08.005. [DOI] [PubMed] [Google Scholar]
- McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklossy J, Kis A, Radenovic A, Miller L, Forro L, Martins R, et al. Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol Aging. 2006;27:228–236. doi: 10.1016/j.neurobiolaging.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Vissel B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun. 2014;2:135. doi: 10.1186/s40478-014-0135-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullane K, Williams M. Alzheimer’s therapeutics: continued clinical failures question the validity of the amyloid hypothesis-but what lies beyond? Biochem Pharmacol. 2013;85:289–305. doi: 10.1016/j.bcp.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Nakane A, Yamada K, Hasegawa S, Mizuki D, Mizuki M, Sasaki S, et al. Endogenous cytokines during a lethal infection with Listeria monocytogenes in mice. FEMS Microbiol Lett. 1999;175:133–142. doi: 10.1111/j.1574-6968.1999.tb13612.x. [DOI] [PubMed] [Google Scholar]
- Nawroth PP, Bank I, Handley D, Cassimeris J, Chess L, Stern D. Tumor necrosis factor/cachectin interacts with endothelial cell receptors to induce release of interleukin 1. J Exp Med. 1986;163:1363–1375. doi: 10.1084/jem.163.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neva FA, Morgan HR. Tolerance to the actions of endotoxins of enteric bacilli in patients convalescent from typhoid and paratyphoid fevers. J Lab Clin Med. 1950;35:911–921. [PubMed] [Google Scholar]
- Newman S, Stygall J, Hirani S, Shaefi S, Maze M. Postoperative cognitive dysfunction after noncardiac surgery: a systematic review. Anesthesiology. 2007;106:572–590. doi: 10.1097/00000542-200703000-00023. [DOI] [PubMed] [Google Scholar]
- Notarachille G, Arnesano F, Calo V, Meleleo D. Heavy metals toxicity: effect of cadmium ions on amyloid beta protein 1–42. Possible implications for Alzheimer’s disease. Biometals. 2014;27:371–388. doi: 10.1007/s10534-014-9719-6. [DOI] [PubMed] [Google Scholar]
- Ohtake N, Saito M, Eto M, Seki K. Exendin-4 promotes the membrane trafficking of the AMPA receptor GluR1 subunit and ADAM10 in the mouse neocortex. Regul Pept. 2014;190–191:1–11. doi: 10.1016/j.regpep.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Olszowski T, Baranowska-Bosiacka I, Gutowska I, Chlubek D. Pro-inflammatory properties of cadmium. Acta Biochim Pol. 2012;59:475–482. [PubMed] [Google Scholar]
- Oppenheim JJ, Tewary P, de la Rosa G, Yang D. Alarmins initiate host defense. Adv Exp Med Biol. 2007;601:185–194. doi: 10.1007/978-0-387-72005-0_19. [DOI] [PubMed] [Google Scholar]
- Park KM, Yule DI, Bowers WJ. Tumor necrosis factor-alpha potentiates intraneuronal CA2+ signaling via regulation of the inositol 1,4,5-trisphosphate receptor. J Biol Chem. 2008;283:33069–33079. doi: 10.1074/jbc.M802209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peper RL, Vancampen H. Tumor necrosis factor as a mediator of inflammation in influenza A viral pneumonia. Microb Pathog. 1995;19:175–183. doi: 10.1006/mpat.1995.0056. [DOI] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Sambamurti K, Chen D, Mattson MP, Egan JM, et al. Glucagon-like peptide-1 decreases endogenous amyloid-beta peptide (Abeta) levels and protects hippocampal neurons from death induced by Abeta and iron. J Neurosci Res. 2003;72:603–612. doi: 10.1002/jnr.10611. [DOI] [PubMed] [Google Scholar]
- Peskind ER, Griffin WS, Akama KT, Raskind MA, Van Eldik LJ. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer’s disease. Neurochem Int. 2001;39:409–413. doi: 10.1016/s0197-0186(01)00048-1. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for ‘inflamm-aging’. Eur J Immunol. 2014;44:1552–1562. doi: 10.1002/eji.201343921. [DOI] [PubMed] [Google Scholar]
- Podlesniy P, Figueiro-Silva J, Llado A, Antonell A, Sanchez-Valle R, Alcolea D, et al. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol. 2013;74:655–668. doi: 10.1002/ana.23955. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Some speculations about prions, amyloid, and Alzheimer’s disease. N Engl J Med. 1984;310:661–663. doi: 10.1056/NEJM198403083101021. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, McKinley MP, Bowman KA, Bolton DC, Bendheim PE, Groth DF, et al. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 1983;35(2 Pt 1):349–358. doi: 10.1016/0092-8674(83)90168-x. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F, et al. Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann Neurol. 2011;69:819–830. doi: 10.1002/ana.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistad SD, Stotland A, Barott KL, Smurthwaite CA, Hilton BJ, Grasis JA, et al. Evolution of TNF-induced apoptosis reveals 550 My of functional conservation. Proc Natl Acad Sci U S A. 2014;111:9567–9572. doi: 10.1073/pnas.1405912111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raziuddin S, Abdalla RE, el Awad EH, al Janadi M. Immunoregulatory and proinflammatory cytokine production in visceral and cutaneous leishmaniasis. J Infect Dis. 1994;170:1037–1040. doi: 10.1093/infdis/170.4.1037. [DOI] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–11992. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinsfelt B, Westerlind A, Blennow K, Zetterberg H, Ricksten SE. Open-heart surgery increases cerebrospinal fluid levels of Alzheimer-associated amyloid beta. Acta Anaesthesiol Scand. 2013;57:82–88. doi: 10.1111/j.1399-6576.2012.02769.x. [DOI] [PubMed] [Google Scholar]
- Rohan D, Buggy DJ, Crowley S, Ling FK, Gallagher H, Regan C, et al. Increased incidence of postoperative cognitive dysfunction 24 h after minor surgery in the elderly. Can J Anaesth. 2005;52:137–142. doi: 10.1007/BF03027718. [DOI] [PubMed] [Google Scholar]
- Rook GAW, Taverne J, Leveton C, Steele J. The role of gamma-interferon, vitamin D3 metabolites and tumour necrosis factor in the pathogenesis of tuberculosis. Immunology. 1987;62:229–234. [PMC free article] [PubMed] [Google Scholar]
- Rosenberger K, Derkow K, Dembny P, Krüger C, Schott E, Lehnardt S. The impact of single and pairwise Toll-like receptor activation on neuroinflammation and neurodegeneration. J Neuroinflammation. 2014;11:166. doi: 10.1186/s12974-014-0166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R. Synaptic memory mechanisms: Alzheimer’s disease amyloid beta-peptide-induced dysfunction. Biochem Soc Trans. 2007;35(Pt 5):1219–1223. doi: 10.1042/BST0351219. [DOI] [PubMed] [Google Scholar]
- Rubenstein M, Mulholland JH, Jeffery GM, Wolff SM. Malaria-induced endotoxin tolerance. Proc Soc Exp Biol Med. 1965;118:283–287. doi: 10.3181/00379727-118-29820. [DOI] [PubMed] [Google Scholar]
- Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170:3233–3242. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Alcazar JA, Schneider E, Martinez MA, Carmona P, Hernandez-Munoz I, Siles E, et al. Tumor necrosis factor-alpha increases the steady-state reduction of cytochrome b of the mitochondrial respiratory chain in metabolically inhibited L929 cells. J Biol Chem. 2000;275:13353–13361. doi: 10.1074/jbc.275.18.13353. [DOI] [PubMed] [Google Scholar]
- Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, et al. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain. 2012;135(Pt 11):3336–3347. doi: 10.1093/brain/aws250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. Preclinical’ AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55:370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- Schnabel J. Amyloid: little proteins, big clues. Nature. 2011;475:S12–S14. doi: 10.1038/475S12a. [DOI] [PubMed] [Google Scholar]
- Schulze J, Zierath D, Tanzi P, Cain K, Shibata D, Dressel A, et al. Severe stroke induces long-lasting alterations of high-mobility group box 1. Stroke. 2013;44:246–248. doi: 10.1161/STROKEAHA.112.676072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, et al. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, et al. Inflammatory S100A9 and S100A12 proteins in Alzheimer’s disease. Neurobiol Aging. 2006;27:1554–1563. doi: 10.1016/j.neurobiolaging.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Sherman ML, Spriggs DR, Arthur KA, Imamura K, Frei EF, Kufe DW. Recombinant human tumor necrosis factor administered as a five-day continuous infusion in cancer patients: phase 1 toxicity and effects on lipid metabolism. J Clin Oncol. 1988;6:344–350. doi: 10.1200/JCO.1988.6.2.344. [DOI] [PubMed] [Google Scholar]
- Shichita T, Sakaguchi R, Suzuki M, Yoshimura A. Post-ischemic inflammation in the brain. Front Immunol. 2012;3:132. doi: 10.3389/fimmu.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JC, Cesaro A, Chapeton-Montes J, Tardif M, Antoine F, Girard D, et al. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-kappaB(1) PLoS ONE. 2013;8:e72138. doi: 10.1371/journal.pone.0072138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer N, Zollo O, Van Deerlin V, Trojanowski JQ. Analysis of cerebrospinal fluid mitochondrial DNA levels in Alzheimer disease. Ann Neurol. 2014;75:458–460. doi: 10.1002/ana.24107. [DOI] [PubMed] [Google Scholar]
- Song JW, Choi BS. Mercury induced the accumulation of amyloid beta (abeta) in PC12 Cells: the role of production and degradation of abeta. Toxicol Res. 2013;29:235–240. doi: 10.5487/TR.2013.29.4.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soontornniyomkij V, Moore DJ, Gouaux B, Soontornniyomkij B, Tatro ET, Umlauf A, et al. Cerebral beta-amyloid deposition predicts HIV-associated neurocognitive disorders in APOE epsilon4 carriers. AIDS. 2012;26:2327–2335. doi: 10.1097/QAD.0b013e32835a117c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs DR, Sherman ML, Michie H, Arthur KA, Imamura K, Wilmore D, et al. Recombinant human tumor necrosis factor administered as a 24-hour intravenous infusion. A phase 1 and pharmacologic study. J Natl Cancer Inst. 1988;80:1039–1044. doi: 10.1093/jnci/80.13.1039. [DOI] [PubMed] [Google Scholar]
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl) 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- Steinmetz J, Christensen KB, Lund T, Lohse N, Rasmussen LS. Long-term consequences of postoperative cognitive dysfunction. Anesthesiology. 2009;110:548–555. doi: 10.1097/ALN.0b013e318195b569. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand JM, Scheffler K, Bjoras M, Eide L. The distribution of DNA damage is defined by region-specific susceptibility to DNA damage formation rather than repair differences. DNA Repair (Amst) 2014;18:44–51. doi: 10.1016/j.dnarep.2014.03.003. [DOI] [PubMed] [Google Scholar]
- Su X, Wang H, Zhao J, Pan H, Mao L. Beneficial effects of ethyl pyruvate through inhibiting high-mobility group box 1 expression and TLR4/NF-kappaB pathway after traumatic brain injury in the rat. Mediators Inflamm. 2011;2011:807142. doi: 10.1155/2011/807142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter E, Ullman GE, Hoffman RG. Sensitivity of mice to endotoxin after vaccination with BCG (Bacillus Calmette-Guérin) Proc Soc Exp Biol Med. 1958;99:167–169. doi: 10.3181/00379727-99-24282. [DOI] [PubMed] [Google Scholar]
- Talbot K, Wang HY. The nature, significance, and glucagon-like peptide-1 analog treatment of brain insulin resistance in Alzheimer’s disease. Alzheimers Dement. 2014;10(1 Suppl):S12–S25. doi: 10.1016/j.jalz.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Liljeroth AM, Minthon L, Tarkowski A, Wallin A, Blennow K. Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Res Bull. 2003;61:255–260. doi: 10.1016/s0361-9230(03)00088-1. [DOI] [PubMed] [Google Scholar]
- Terrando N, Monaco C, Ma D, Foxwell BM, Feldmann M, Maze M. Tumor necrosis factor-alpha triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci U S A. 2010;107:20518–20522. doi: 10.1073/pnas.1014557107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer’s disease. Arch Gen Psychiatry. 2010;67:739–748. doi: 10.1001/archgenpsychiatry.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobinick E, Vega CP. The cerebrospinal venous system: anatomy, physiology, and clinical implications. Medgenmed. 2006a;8:53. [PubMed] [Google Scholar]
- Tobinick E, Kim NM, Reyzin G, Rodriguez-Romanacce H, DePuy V. Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs. 2012;26:1051–1070. doi: 10.1007/s40263-012-0013-2. [DOI] [PubMed] [Google Scholar]
- Tobinick EL, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer’s disease: a 6-month pilot study. MedGenMed. 2006b;8:25. [PMC free article] [PubMed] [Google Scholar]
- Tsai NW, Lin TK, Chen SD, Chang WN, Wang HC, Yang TM, et al. The value of serial plasma nuclear and mitochondrial DNA levels in patients with acute ischemic stroke. Clin Chim Acta. 2011;412:476–479. doi: 10.1016/j.cca.2010.11.036. [DOI] [PubMed] [Google Scholar]
- Tweedie D, Ferguson RA, Fishman K, Frankola KA, Van Praag H, Holloway HW, et al. Tumor necrosis factor-alpha synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J Neuroinflammation. 2012;9:106. doi: 10.1186/1742-2094-9-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Bednar MM, Forman SA, Wolozin B. Iatrogenic risk factors for Alzheimer’s disease: surgery and anesthesia. J Alzheimers Dis. 2010;22(Suppl. 3):91–104. doi: 10.3233/JAD-2010-100843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkovic L, Bockaert J, Jacque C. Inflammatory’ cytokines: neuromodulators in normal brain? J Neurochem. 2000a;74:457–471. doi: 10.1046/j.1471-4159.2000.740457.x. [DOI] [PubMed] [Google Scholar]
- Vitkovic L, Konsman JP, Bockaert J, Dantzer R, Homburger V, Jacque C. Cytokine signals propagate through the brain. Mol Psychiatry. 2000b;5:604–615. doi: 10.1038/sj.mp.4000813. [DOI] [PubMed] [Google Scholar]
- Vollmar P, Kullmann JS, Thilo B, Claussen MC, Rothhammer V, Jacobi H, et al. Active immunization with amyloid-beta 1–42 impairs memory performance through TLR2/4-dependent activation of the innate immune system. J Immunol. 2010;185:6338–6347. doi: 10.4049/jimmunol.1001765. [DOI] [PubMed] [Google Scholar]
- Walko TD, 3rd, Bola RA, Hong JD, Au AK, Bell MJ, Kochanek PM, et al. Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock. 2014;41:499–503. doi: 10.1097/SHK.0000000000000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999a;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Wang H, Vishnubhakat JM, Bloom O, Zhang M, Ombrellino M, Sama A, et al. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery. 1999b;126:389–392. [PubMed] [Google Scholar]
- Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS One. 2008;3:e2698. doi: 10.1371/journal.pone.0002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QW, Wu JQ, Rowan MJ, Anwyl R. beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci. 2005;22:2827–2832. doi: 10.1111/j.1460-9568.2005.04457.x. [DOI] [PubMed] [Google Scholar]
- Wasling P, Daborg J, Riebe I, Andersson M, Portelius E, Blennow K, et al. Synaptic retrogenesis and amyloid-beta in Alzheimer’s disease. J Alzheimers Dis. 2009;16:1–14. doi: 10.3233/JAD-2009-0918. [DOI] [PubMed] [Google Scholar]
- White JA, Manelli AM, Holmberg KH, Van Eldik LJ, Ladu MJ. Differential effects of oligomeric and fibrillar amyloid-beta 1–42 on astrocyte-mediated inflammation. Neurobiol Dis. 2005;18:459–465. doi: 10.1016/j.nbd.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Xie Z, Tanzi RE. Alzheimer’s disease and post-operative cognitive dysfunction. Exp Gerontol. 2006;41:346–359. doi: 10.1016/j.exger.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Xu Z, Dong Y, Wang H, Culley DJ, Marcantonio ER, Crosby G, et al. Age-dependent postoperative cognitive impairment and Alzheimer-related neuropathology in mice. Sci Rep. 2014;4:3766. doi: 10.1038/srep03766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, et al. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- Yen T, Harrison CA, Devery JM, Leong S, Iismaa SE, Yoshimura T, et al. Induction of the S100 chemotactic protein, CP-10, in murine microvascular endothelial cells by proinflammatory stimuli. Blood. 1997;90:4812–4821. [PubMed] [Google Scholar]
- Zhan S, Cai GQ, Zheng A, Wang Y, Jia J, Fang H, et al. Tumor necrosis factor-alpha regulates the hypocretin system via mRNA degradation and ubiquitination. Biochim Biophys Acta. 2011;1812:565–571. doi: 10.1016/j.bbadis.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 MAP-kinase. Shock. 2009;34:55–59. doi: 10.1097/SHK.0b013e3181cd8c08. [DOI] [PubMed] [Google Scholar]
- Zhang X, Song W. The role of APP and BACE1 trafficking in APP processing and amyloid-beta generation. Alzheimers Res Ther. 2013;5:46. doi: 10.1186/alzrt211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Abeta production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2011;8:150. doi: 10.1186/1742-2094-8-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zoelen MA, Yang H, Florquin S, Meijers JC, Akira S, Arnold B, et al. Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock. 2009;31:280–284. doi: 10.1097/SHK.0b013e318186262d. [DOI] [PMC free article] [PubMed] [Google Scholar]