

Abstract
Background and Purpose
Dimethyl fumarate (DMF) is a newly approved drug for the treatment of relapsing forms of multiple sclerosis and relapsing-remitting multiple sclerosis. Here, we investigated the effects of DMF and its metabolites mono-methylfumarate (MMF and methanol) on different gastrointestinal cancer cell lines and the underlying molecular mechanisms involved.
Experimental Approach
Cell viability was measured by the MTT or CCK8 assay. Protein expressions were measured by Western blot analysis. LDH release, live- and dead-cell staining, intracellular GSH levels, and mitochondrial membrane potential were examined by using commercial kits.
Key Results
DMF but not MMF induced cell necroptosis, as demonstrated by the pharmacological tool necrostatin-1, transmission electron microscopy, LDH and HMGB1 release in CT26 cells. The DMF-induced decrease in cellular GSH levels as well as cell viability and increase in reactive oxygen species (ROS) were inhibited by co-treatment with GSH and N-acetylcysteine (NAC) in CT26 cells. DMF activated JNK, p38 and ERK MAPKs in CT26 cells and JNK, p38 and ERK inhibitors partially reversed the DMF-induced decrease in cell viability. GSH or NAC treatment inhibited DMF-induced JNK, p38, and ERK activation in CT26 cells. DMF but not MMF increased autophagy responses in SGC-7901, HCT116, HT29 and CT26 cancer cells, but autophagy inhibition did not prevent the DMF-induced decrease in cell viability.
Conclusion and Implications
DMF but not its metabolite MMF induced necroptosis in colon cancer cells through a mechanism involving the depletion of GSH, an increase in ROS and activation of MAPKs.
Tables of Links
| TARGETS | |
|---|---|
| Caspase 3 | JNK |
| ERK | p38 |
| LIGANDS | |
|---|---|
| Cyclosporine A (CsA) | PD98059 |
| Dimethyl fumarate (DMF) | SB203580 |
| GSH | SP600125 |
These tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Dimethyl fumarate (DMF), the methyl ester of fumaric acid, was initially recognized as an effective hypoxic cell radiosensitizer (Held et al., 1988). Recently, DMF (BG-12, Tecfidera®, BIOGEN IDEC Inc., Weston, MA, USA) was approved by the FDA and European Medicines Agency as a new oral drug for the treatment of relapsing forms of multiple sclerosis and relapsing-remitting multiple sclerosis (Burness and Deeks, 2014; Fox et al., 2014). DMF suppresses the expression of inflammatory cytokines such as inducible NOS, IL-1β, IL-6 in glial cells (Wierinckx et al., 2005; Wilms et al., 2010), and exerts neuroprotective effects via the antioxidant pathway in the CNS (Linker et al., 2011; Scannevin et al., 2012). All of these effects can account for the efficacy of DMF at treating multiple sclerosis.
Furthermore, in the field of multiple sclerosis, DMF has also been found to exhibit anti-tumour effects. Loewe et al. reported that DMF inhibited melanoma growth and metastasis both in vitro and vivo (Loewe et al., 2006); Kirlin et al. reported that DMF induced apoptosis in HT29 colon carcinoma cells (Kirlin et al., 1999), and others have shown that DMF enhances the anti-tumour effects of some chemotherapy drugs (Gu and DeAngelis, 2005; Valero et al., 2010). However, it should be noted that, recent studies revealed that DMF could not be detected in plasma because it was immediately and completely hydrolyzed by esterases to mono-methylfumarate (MMF) (Nibbering et al., 1993; Werdenberg et al., 2003), and during this process, methanol was also generated (Rostami-Yazdi et al., 2010). Therefore, it is essential to identify the effects of DMF and its metabolites (MMF and methanol) on cancer cells and the underlying molecular mechanisms.
Necroptosis is a necrotic-like, caspase-independent cell death that is characterized by loss of plasma membrane integrity, excessive autophagy, loss of mitochondria membrane potential and the generation of reactive oxygen species (ROS) is enhanced in necrotic cells (Degterev et al., 2005). Necroptosis is emerging as an important target for anticancer treatment (Fulda, 2014); targeting necropotosis has been shown to overcome drug resistance in cancer chemotherapy (Han et al., 2007; Steinhart et al., 2013). Many necroptotic stimuli, such as shiconin, smac mimetic and granulysin, have been shown to have antitumour effects or potentiate the effect of chemotherapeutic drugs (Han et al., 2007; Zhang et al., 2009; Steinhart et al., 2013). Here, we used different gastrointestinal cancer cell lines to investigate the effects of DMF and its metabolites (MMF and methanol), and the underlying molecular mechanisms involved. We found that DMF but not MMF induced necroptosis in colon cancer cells by depleting cellular GSH and subsequently activating MAPKs. We propose that DMF has the potential to be an anticancer drug provided an appropriate drug delivery system can be devised for it.
Methods
Cell culture
CT26, HT29, HCT116 and SGC-7901 cells were maintained in DMEM containing 25 mmoL·L−1 glucose supplemented with 10% FBS, 100 u·mL−1 penicillin and 100 μg·mL−1 streptomycin at 37°C, 5%CO2. The time of treatment and concentration of agents were shown in the figures and/or corresponding figure legends.
Western blot analysis
Western blot analysis was performed as described in our previous work (Sheng et al., 2014). Cells were lysed with RIPA buffer containing 1% protease inhibitor and centrifuged at 16 500× g for 15 min at 4°C. Then the supernatants were collected and the protein concentrations were determined by BCA assay kit (Beyotime). The protein was applied to 10% to 12% SDS-PAGE gels, transferred to nitrocellulose membranes. After incubation with the appropriate primary and secondary antibodies, Western blot bands were quantified by using Odyssey infrared imaging system (Li-Cor Inc., Lincoln, NE, USA) and Odyssey v3.0 software.
Cell viability measurement
Cell viability was measured by using either the MTT or CCK8 assay, as indicated in Figures and/or corresponding Figure legends.
LDH assay
Cell culture medium was collected for LDH determination according to the manufacturer’s instructions (LDH cytotoxicity assay kit; Beyotime Biotech). LDH can catalyze the synthesis of pyruvic acid from lactic acid and then pyruvic acid reacts to form 2,4-dinitrophenyl-hydrazine, which shows up as a brownish red colour in basic solution. After the reaction, the absorbance was read at a wavelength 490 nm. LDH release reflected the cell death. The cell death ratio was calculated by the following formula according to the manufacturer’s instructions:
Transmission electron microscopy (TEM)
TEM was performed to identify the cells undergoing necroptosis. The cells were fixed with ice-cold 2.5% glutaraldehyde in PBS (pH 7.3) at 4°C for 4 h. Fixed cells were post-fixed in 2% OsO4, dehydrated in graded alcohol, embedded in Epon 812 (Electron Microscopy Sciences, Fort Washington, PA, USA), sectioned with ultramicrotome, and stained with uranyl acetate and lead citrate. The sections were examined with a TEM (Technai 10; Philips, Eindhoven, The Netherlands).
Live- and dead-cell staining
The LIVE/DEAD® Viability/Cytotoxicity Assay Kit (Invitrogen) was used to detect the live and dead cells. Briefly, cells were grown on coverslips at a density of 3.75 × 104 cells mL−1 and incubated overnight at 37°C in a humidified 5% CO2 incubator. The cells were washed with PBS and dyed according to the manufacturer’s instructions. The labelled cells were photographed under a fluorescence microscope. The live cells fluoresce green and dead cells fluoresce red.
Measurement of intracellular GSH
Intracellular GSH contents were measured using a total GSH assay kit (Beyotime Biotech) according to the manufacturer’s instructions. Cells were harvested and lysed in protein removal solution S provided in the kit, then repeatedly (twice) frozen and thawed in liquid nitrogen and 37°C water respectively. After incubation for 5 min at 4°C, the samples were centrifuged at 10000× g for 10 min at 4°C. The supernatant was treated with Ellman’s reagent (DTNB) in combination with GSH reductase enzyme and NADPH. Finally the absorbance values were measured at a wavelength of 412 nm by a microplate reader. The intracellular GSH content was quantified against the corresponding standard curves.
Measurement of mitochondrial membrane potential (MMP)
The MMP was measured by using a MMP assay kit (Beyotime Biotech) according to the manufacturer’s instructions. Cells were washed with PBS and then incubated in the culture medium containing 1 μg·mL−1 JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide) for 20 min at 37°C. Then, the supernatant was removed and the cells were washed with JC-1 buffer solution twice. The fluorescence intensity was detected by a fluorescence microscope. JC-1 monomer fluorescence (green), which indicates a low MMP, was observed at the wavelength of excitation/emission = 485/530 nm. JC-1 aggregate fluorescence (red), which indicates a high MMP, was detected at the wavelength of excitation/emission = 485/590 nm.
Measurement of intracellular ROS accumulation
Intracellular ROS accumulation was determined by an ROS assay kit (Beyotime Biotech) that utilizes 2′,7′-dichlorofluorescin diacetate (DCFH-DA) as a fluorescent probe. In principle, DCFH-DA penetrates the cell membrane freely and is hydrolysed by esterase into DCFH, which cannot escape from the cell, then ROS further oxidizes DCFH to DCF, which fluoresces bright green. After drug treatments, cells were incubated with 10 μM DCFH-DA for 20 min at 37°C and then were washed with DMEM three times. Intracellular ROS were determined by a fluorescence microscope at an excitation wavelength of 488 nm and an emission wavelength of 525 nm. The fluorescence intensity reflected the amount of ROS generated, an increase in fluorescence intensity represents an enhanced generation of ROS.
Detection for HMGB1 release
CT26 cells were seeded in 6-well plates, and then incubated at 37°C, 5%CO2 overnight. The culture medium was collected after different durations of DMF treatment. The samples were centrifuged at 1000× g for 20 min, then the HMGB1 protein in supernatants was detected by using an HMGB1 ELISA kit (Cloud-Clone Corporation, Houston, TX, USA) according to the manufacturer’s instructions.
PARP-1 activity assay
PARP-1 activity in HCT116 cells was examined by using a universal colorimetric PARP-1 assay kit (Trevigen, Gaithersburg, MD, USA). After drug treatment, cells were collected and suspended in PARP lysis buffer, which contained protease inhibitor. Then, the samples were centrifuged at 16 500× g for 15 min at 4°C, and the protein concentration was assayed using a BCA protein assay kit (Beyotime); 50 μg of protein from each sample were loaded into a 96-well plate coated with histones and biotinylated poly ADP-ribose, and PARP cocktail was also added to each well and then incubated for 60 min at room temperature. Next, the supernatant was aspirated and then the diluted Strep-HRP was added and the samples were incubated for another 60 min at room temperature followed by TACS-Sapphire treatment for 15 min in the dark. To stop the reaction, 0.2 M HCl was added to each well and the plate was read at 450 nm.
Data analysis
Data are presented as mean ± SEM. Significance was determined by using anova, followed by Holm–Sidak. P < 0.05 was considered significant.
Cell lines and agents
The murine colon adenocarcinoma cell line (CT26), human colon adenocarcinoma cell line (HT29), human colon cancer cell line (HCT116) and human gastric cancer cell line (SGC-7901) were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). DMEM was purchased from Hyclone (Logan, UT, USA). FBS was purchased from Biological Industries (Beit Haemek, Israel). The LIVE/DEAD® Viability/Cytotoxicity Assay Kit was obtained from Invitrogen (Eugene, OR, USA). Cyclosporine A (CsA), Ac-DEVD-CHO and LDH assay kits were purchased from Beyotime Institute of Biotechnology (Shanghai, China). Methanol was purchased from Tianjin Yongda Chemical Regent Company (Tianjin, China), DMF, mono-methyl fumarate, DMH1(4-[6-(4-isopropoxyphenyl)-pyrazolo [1,5-a]pyrimidin-3-yl] quinoline) and 3-methyladenine (3-MA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Necrostatin-1 (Nec) and PARP-1 antibody were purchased from Santa Cruz (Eugene, OR, USA). Anti-LC3B antibody was purchased from Sigma-Aldrich. Anti-bax, Anti-bcl-2, Anti-gapdh and Anti-actin antibodies were purchased from Sangon Biotech (Shanghai, China). Anti-cleaved caspase-3 antibody was obtained from Cell Signaling Technology (Beverly, MA, USA). MTT and DMSO were obtained from Amresco (Solon, OH, USA). Cell counting kit-8 (CCK-8) was obtained from Dojindo (Kumamoto, Japan). DMSO was used as a solvent for stock solutions and its final concentration was limited to 4%, which showed no effect on cell viability. The positive inducers of apoptosis, apopida and apobid, were purchased from the Beyotime Institute of Biotechnology.
Results
DMF but not MMF or methanol reduces cell viability in SGC-7901, HT29, HCT116 and CT26 cancer cells
DMF was reported to inhibit melanoma growth and metastasis in vivo and in vitro (Loewe et al., 2006; Yamazoe et al., 2009). The concentration of DMF commonly used in these in vitro experiments is in the range 50 to 200 μM (Loewe et al., 2006; Ghods et al., 2013). Hence, we initially examined the effects of DMF 20–200 μM on cell viability in four types of gastrointestinal cancer cell lines, SGC-7901, HT29, HCT116 and CT26. Because DMF is metabolized into MMF and methanol in the body (Werdenberg et al., 2003; Rostami-Yazdi et al., 2010), we further tested the effects of MMF and methanol on the cell viability in these cell lines. As shown in Figure 1A, DMF dose-dependently reduced the viability of SGC-7901, HT29, HCT116 and CT26 cancer cells in the range 20–200 μM. However, MMF and methanol at the same concentrations showed no significant effects on cell viability, with the exception that MMF at 100 and 200 μM slightly reduced the viability of CT26 cells (Figure 2A and B).
Figure 1.

DMF, but not MMF or methanol, reduces the viability of SGC-7901, HT29, HCT116 and CT26 cancer cells after a 24 h treatment. (A) DMF dose-dependently reduced the viability of SGC-7901, HCT116, HT29, and CT26 cancer cells. **P < 0.01 versus CTL. CTL, control. (B–C) MMF and methanol showed no significant effect on the viability of SGC-7901, HCT116, HT29 and CT26 cancer cells, except that MMF at 100 and 200 μM slightly reduced CT26 cell viability. **P < 0.01 versus CTL. CTL, control.
Figure 2.

DMF does not affect apoptotic parameters in HCT116 and CT26 cancer cells. (A–B) DMF in the range from 20 μM to 200 μM showed no effect on bcl-2, bax, cleaved caspase-3 protein expressions in HCT116 and CT26 cells after 24 h treatment. (C–D) DMF (100 μM) showed no effect on bax, cleaved caspase-3 protein expressions in HCT116 and CT26 cells after different treatment durations (3, 6, 12 and 24 h). (E) DMF-induced reduction in cell viability of CT26 cells was not affected by pretreatment with the caspase-3 inhibitor Ac-DEVD-HO (100 μM) for 30 min. The treatment time of DMF was 24 h. **P < 0.01 versus CTL. CTL, control. (F) The positive apoptosis inducer increased bax protein expression in HCT116 and CT26 cells; n = 6, **P < 0.01 versus control. (G) The positive apoptosis inducer increased the cleaved caspase-3 protein expression in HCT116 cells; n = 5, **P < 0.01 versus control. The positive apoptosis inducer was apopida and apobid at a concentration of 1:1000 in the medium.
DMF does not induce cell apoptosis in HCT116 and CT26 cancer cells
DMF was reported to induce cell apoptosis in human mast cells (Forster et al., 2013). Since DMF reduced the viability of SGC-7901, HT29, HCT116 and CT26 cells, we further investigated whether the reduced cell viability was due to the induction of apoptosis in HCT116 and CT26 cells by measuring different apoptosis-related factors, bcl-2, bax, caspase-3, in these cells after DMF treatment. As shown in Figure 2A and B, DMF in the range 20 to 200 μM showed no effect on the proptein expressions of bcl-2, bax, cleaved caspase-3 in HCT116 and CT26 cells after a 24 h treatment. DMF at 100 μM also showed no effect on bax, cleaved caspase-3 in HCT116 and CT26 cells after treatment from 3 to 24 h (Figure 2C and D). Furthermore, DMF-induced decrease in cell viability in CT26 cells was not affected by the caspase-3 inhibitor Ac-DEVD-HO (Figure 2E). The above data indicated that DMF-induced decrease in cell viability was not due to the induction of apoptosis. In order to confirm our experimental conditions, we treated these cells with a positive apoptosis inducer and measured the protein expressions of bax and cleaved caspase-3 protein; this positive apoptosis inducer increased the expression of bax and cleaved caspase-3 (Figure 2F and G).
DMF induces cell necroptosis
LDH release is a typical property of cell necrosis (Huang et al., 2013). The LDH release from CT26 cells treated with 100 μM DMF for different time was measured to determine whether necrosis was involved in DMF-induced decrease in cell viability. As shown in Figure 3A, DMF (100 μM) time-dependently increased the release of LDH, which reflected necrotic cell death. We further examined the release of HMGB1 and the activation of PARP-1, which reflect cell necroptosis. As shown in Figure 3B, DMF induced HMGB1 release in CT26 cells. It had no significant effect on the protein expression of PARP-1 (Figure 3C), but DMF treatment significantly increased PARP-1 activity (Figure 3D). Nec, an inhibitor of receptor interacting protein-1 kinase, is a pharmacological tool used to identify cell necroptosis (Degterev et al., 2008); the dose of Nec needed to have this effect has been shown to range from 30 to 100 μM (Degterev et al., 2008; El-Mesery et al., 2015). We found that Nec at 20 μM showed no significant effect on DMF-induced decrease in cell viability, but it significantly attenuated DMF-induced decrease of cell viability when the dose was increased to 60 and 100 μM (Figure 3E). Optical photography also showed that DMF (100 μM) reduced the cell density and Nec treatment partially reversed this decrease in cell density (Figure 3F). We further tested the effect of Nec using the LIVE/DEAD® Viability/Cytotoxicity Assay. As shown in Figure 3G, DMF (100 μM) significantly reduced the number of green cells and increased the number of red cells, and these changes were partially reversed by Nec treatment. In principal, the red staining reflects the cell membrane integrity. Therefore, we speculated that DMF disrupted the cell membrane integrity. We further used TEM to observe the DMF-induced cell death in CT26 cells. As shown in Figure 3H, DMF (100 μM) increased cell death, which was characterized by mitochondrial swelling, reticulum expansion and autolysosome formation. Pretreatment with Nec (100 μM) for 1 h significantly attenuated DMF-induced morphological changes in CT26 cells. Taken together, the above results indicate that DMF induced cell necroptosis.
Figure 3.

Nec inhibits DMF-induced cell necroptosis. (A) DMF (100 μM) time-dependently increased the cell death ratio in CT26 cells. The treatment time of DMF was set as 3, 6, 12 and 24 h. (B) DMF induced HMGB1 release in CT26 cells. The treatment time of DMF was set as 3, 6, 12 and 24 h. *P < 0.05, **P < 0.01 versus control; n = 4. (C) DMF treatment showed no significant effect on PARP-1 protein expression in HCT116 cells. The treatment time of DMF was set as 3, 6, 12 and 24 h. (D) DMF treatment significantly increased PARP1 activity in HCT116 cells. **P < 0.01 versus control; n = 6. (E) CCK8 cell viability assay showed that Nec inhibited DMF-induced decrease in CT26 cell viability. Nec alone had no effect on CT26 cell viability. Cells were pretreated with Nec for 1 h, then treated with both DMF and Nec for 24 h. **P < 0.01 versus control. ##P < 0.01 versus DMF. (F) Representative photos of CT26 cells treated with DMF (100 μM) and DMF (100 μM) plus Nec (100 μM). Cells were pretreated with Nec for 1 h, then treated with both DMF and Nec for 24 h. (G) The LIVE/DEAD® Viability/Cytotoxicity assay showed that DMF(100 μM) induced CT26 cell death, which was partially reversed by Nec (100 μM) treatment. The live cells appear as green and dead cells as red. The number of cells counted: 4136, 5874, 2276 and 2122 in control, DMF, DMF plus Nec (60 μM), and DMF plus Nec (100 μM) respectively. Cells were pretreated with Nec for 1 h, then treated with both DMF and Nec for 24 h. χ2 test was used, **P < 0.01 versus control; ##P < 0.01 versus DMF. (H) Representative electron microscope photographs showed that Nec attenuated DMF-induced cell necroptosis. (a) CT26 cells treated with DMSO were considered as control. (b, c) DMF (100 μM) treatment led to cell necrosis, which is characterized by a disrupted cell membrane, mitochondrial swelling, reticulum expansion and autolysosome formation in CT26 cells as indicated by arrows. (d) Pretreatment with Nec(100 μM) for 1 h attenuated DMF-induced necroptosis in CT26 cells with intact cell membrane and reduced autolysosome formation.
Necroptosis involves the active disintegration of mitochondrial membranes (Han et al., 2007; Vandenabeele et al., 2010). CsA inhibits mitochondrial permeability transition pore (MPTP) opening and attenuates necrotic damage (Petrosillo et al., 2009; Vaseva et al., 2012; Zhang et al., 2014). Hence, we investigated the effect of CsA on DMF-induced cell death in CT26 and HT29 cells using the LIVE/DEAD® Viability/Cytotoxicity Assay. As shown in Figure 4A and B, CsA treatment significantly inhibited DMF-induced cell death in CT26 and HT29 cells. MMF and methanol treatments were investigated as parallel controls and showed no effects. Indeed, CsA treatment also inhibited DMF-induced mitochondrial membrane depolarization (Figure 4C). These data indicate that MPTP opening was involved in DMF-induced cell necroptosis.
Figure 4.

CsA inhibited DMF-induced cell death. (A, B) CsA inhibited DMF-induced cell death in CT26 and HT29 cells as indicated by using live/dead assay. MMF and methanol did not induce cell death. The live cells appear as green and dead cells as red. (A) The number of cells counted: 7833, 1921, 6826, 1123, 14381 and 12381 in control, DMF (100 μM), DMF (200 μM), DMF (100 μM) + CsA (1 μM), MMF (200 μM) and methanol (200 μM) respectively. (B) The number of cells counted: 4592, 4038, 5429, 1189, 4756 and 3568 in control, DMF (100 μM), DMF (100 μM) + CsA (1 μM), DMF (100 μM) + CsA (5 μM), MMF (200 μM) and methanol (200 μM) respectively. Cells were pretreated with CsA for 30 min, then treated with both DMF and CsA for 24 h. χ2 test was used, **P < 0.01 versus control; ##P < 0.01 versus DMF. (C) CsA treatment attenuated DMF-induced mitochondrial membrane depolarization in CT26 cells, evaluated by JC-1 staining. In normal conditions, JC-1 accumulated in the mitochondrial matrix to form J-aggregates, which fluoresced red and indicated a high MMP. When cell MMP was low, JC-1 would not accumulate in the mitochondrial matrix and exists in its monomer form, which fluoresced green.
DMF induces cell necroptosis by depleting GSH
GSH is the major redox buffer in cells (Mailloux et al., 2013). Supplements of exogenous GSH or the GSH precursor N-acetylcysteine (NAC) can attenuate hemin-induced astrocyte and oridonin-induced hepatic stellate cell death (Laird et al., 2008; Kuo et al., 2014). DMF has been shown to deplete cellular GSH (Lehmann et al., 2007; Schmidt et al., 2007), indicating that DMF-induced GSH depletion might contribute to DMF-induced cell necroptosis. As shown in Figure 5A and B, the DMF-induced decrease in cellular GSH level and cell viability was inhibited by co-treatment with GSH and NAC in CT26 cells, but Nec and CsA treatments showed no effects on DMF-induced decrease of cellular GSH (Figure 5C). The protective effects of GSH and NAC against DMF-induced cell death were further confirmed by using the LIVE/DEAD® Viability/Cytotoxicity Assay (Figure 5D). The DMF-induced increase in ROS generation was also inhibited by the co-treatment with GSH and NAC (Figure 5F), and DMF-induced mitochondrial depolarization was also inhibited by co-treatment with GSH, NAC and Nec in CT26 cells (Figure 5F).
Figure 5.

GSH and NAC attenuate DMF-induced CT26 cell necroptosis. (A) DMF induced reduction in cellular GSH levels in CT26 cells and the reversal of this reduction by GSH and NAC treatment. Cells were pretreated with DMF(100 μM) for 1 h, then GSH(1 mM) and NAC(1 mM) were added for 2 h. **P < 0.01 versus control. (B) GSH and NAC treatment alleviated DMF-induced the decrease of CT26 cell viability. Cell were pretreated with DMF(100 μM) for 1 h, then treated with both DMF(100 μM) and GSH(0.5 mM) or NAC(0.5 mM) for 24 h. **P < 0.01 versus control; ##P < 0.01 versus DMF(100 μM). (C) Nec and CsA treatment did not affect DMF-induced reduction of cellular GSH levels in CT26 cells. Cells were pretreated with Nec (100 μM) for 1 h and CsA(5 μM) for 30 min, then DMF(100 μM) was added until 3 h. **P < 0.01 versus control. (D) GSH and NAC treatment attenuated DMF-induced cell death in CT26 cells, shown using the LIVE/DEAD staining assay. Cells were pretreated with DMF(100 μM) for 1 h, then GSH(1 mM) and NAC(1 mM) were added until 24 h. The live cells appear as green and dead cells as red. The total number of cells counted cells: 4136, 1857, 3988 and 2811 in control, DMF (100 μM), DMF (100 μM) plus GSH (1 mM) and DMF (100 μM) plus NAC (1 mM) respectively. χ2 test was used, **P < 0.01 versus control; ##P < 0.01 versus DMF. (E) GSH and NAC treatment attenuated the DMF-induced ROS increase in CT26 cells as shown using DCFH-DA staining. Cells were pretreated with DMF(100 μM) for 1 h, then GSH(1 mM) and NAC(1 mM) were added until 3 h. DMF treatment increased the cell fluorescence intensity. (F) DMF-induced mitochondrial depolarization was inhibited by GSH, NAC and Nec treatment in CT26 cells. Cell were pretreated with DMF(100 μM) for 1 h, then GSH (1 mM) and NAC (1 mM) were added for 5 h. For Nec treatment, cells were pretreated with Nec (100 μM) for 1 h, then DMF (100 μM) was added for 6 h. The MMP was evaluated by JC-1 staining. Red fluorescence indicates a high MMP and green fluorescence indicates a low MMP concentration.
MAPKs activation is involved in DMF-induced cell necroptosis
MAPKs are the important downstream signals of ROS. Since DMF induced ROS generation, MAPKs would be subsequently activated. As shown in Figure 6A, DMF (100 μM) significantly activated JNK, p38 and ERK in CT26 cells after treatment from 3 to 24 h, and the maximal activation appeared at the 6 h time point. Treatment with the JNK inhibitor SP6001257, p38 inhibitor SB202190 or ERK inhibitor PD98059 partially reversed DMF-induced decrease in cell viability (Figure 6B).
Figure 6.

DMF activates JNK, p38 and ERK MAPKs in CT26 cells. (A) The time course of DMF-induced JNK, p38 and ERK activation in CT26 cells.*P < 0.05, **P < 0.01 versus control. (B) JNK, p38 and ERK inhibitors attenuated DMF-induced cell death in CT26 cells as shown by the CCK8 assay. SP, JNK inhibitor SP600125; SB, p38 inhibitor SB203580; PD, ERK inhibitor PD98059. **P < 0.01 versus control; ##P < 0.01 versus DMF. Cells were pretreated with the JNK inhibitor SP600125, p38 inhibitor SB203580, or ERK inhibitor PD98059 for 1 h, then DMF(100 μM) was added until 24 h.
Previous data showed that co-treatment with GSH or NAC inhibited DMF-induced necroptosis (Figure 5B). Similarly, co-treatment with GSH or NAC inhibited DMF-induced JNK, p38 and ERK activation in CT26 cells (Figure 7). Although Nec restored DMF-induced necroptosis, Nec did not inhibit DMF-induced JNK, p38 and ERK activation (Figure 7), indicating that the target of Nec was downstream of the MAPKs signals.
Figure 7.
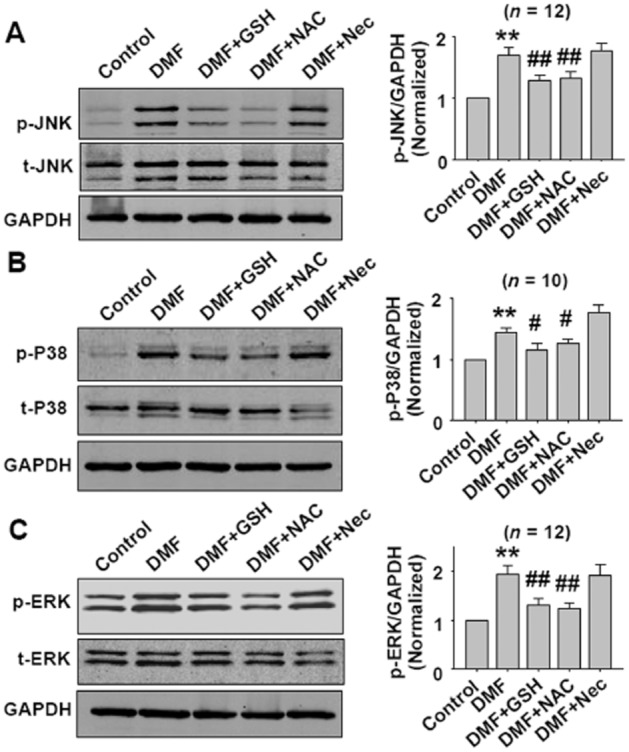
GSH and NAC treatment inhibits DMF-induced JNK, p38 and ERK activation in CT26 cells. Cells were pretreated with DMF (100 μM) for 1 h, then treated with DMF plus GSH or DMF plus NAC for 5 h. For Nec treatment, cells were pretreated with Nec (100 μM) for 1 h, then DMF (100 μM) was added for 6 h. The concentrations of DMF, GSH, NAC and Nec were 100 μM, 1 mM, 1 mM and 100 μM respectively. **P < 0.01 versus control. #P < 0.05, ##P < 0.01 versus DMF.
DMF induces cell autophagy
Autophagy is a natural cellular response to many stimuli, and is usually represented by LC3-II protein expression. Since DMF induced cell necroptosis, the effects of DMF on the levels of LC3-II in CT26, HT29, HCT116 and SGC-7901 cells were examined. As shown in Figure 8A–8D, DMF, but not MMF, increased autophagy responses in SGC-7901, HCT116, HT29 and CT26 cells. DMH1, an inhibitor of autophagy (Sheng et al., 2014), inhibited the DMF-induced increased autophagy response in CT26 cells (Figure 8E). Although DMH1 inhibited the DMF-induced increase in autophagy in CT26 cells, it did not prevent the DMF-induced decrease in cell viability (Figure 8F).
Figure 8.

DMF induces autophagy responses in SGC-7901, HCT116, HT29 and CT26 cancer cells. (A–D) Western blot results showing that DMF but not MMF increased LC3-II levels in SGC-7901, HCT116, HT29 and CT26 cancer cells. (E) DMH1 inhibited DMF-induced increase in LC3-II in CT26 cells. *P < 0.05 versus control. #P < 0.05 versus DMF (100 μM). CTL, control. (F) DMH1 did not affect DMF-induced decease in CT26 cell viability. **P < 0.01 versus control.
In order to identify the role of autophagy in DMF-induced cell necroptosis, we further studied the effects of Nec and CsA on basal and DMF-induced autophagy responses. As shown in Figure 9, Nec or CsA treatment attenuated DMF-induced autophagy responses, but Nec or CsA alone did not affect the basal or starvation-induced autophagy responses in CT26 cells. These data indicate that DMF-induced autophagy was a secondary response in these cells.
Figure 9.
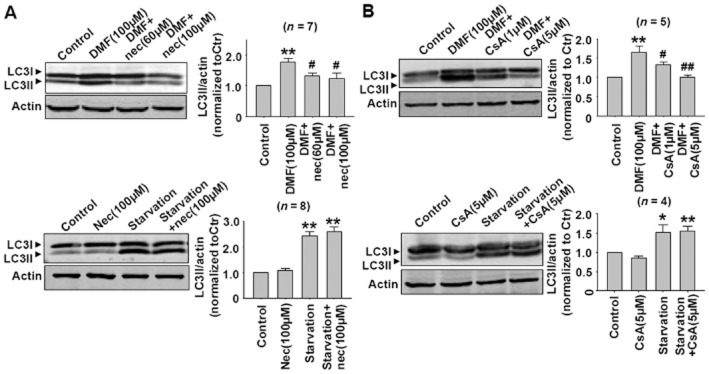
(A) Nec treatment attenuated DMF-induced autophagy responses, but Nec alone did not affect the basal and starvation-induced autophagy in CT26 cells. **P < 0.01 versus control. #P < 0.05 versus DMF (100 μM). (B) CsA treatment attenuated DMF-induced autophagy responses, but CsA alone did not affect the basal and starvation-induced autophagy in CT26 cells. *P < 0.05, **P < 0.01 versus control. #P < 0.05, ##P < 0.01 versus DMF (100 μM). The starvation condition was serum-free DMEM (5.5 mM glucose).
Discussion
DMF, the methyl ester of fumaric acid, has been approved for the treatment of relapsing forms of multiple sclerosis and relapsing-remitting multiple sclerosis clinically (Burness and Deeks, 2014; Fox et al., 2014). In the present study, we found that DMF but not its metabolite MMF induces necroptosis in colon cancer cells and the mechanism involves GSH depletion, an increase in ROS and activation of MAPKs-mediated signalling. The mechanism of DMF-induced cell necroptosis is summarized in Figure 10. In the present study, we examined the effects of DMF on cell viability in four types of cancer cell lines, SGC-7901, HT29, HCT116 and CT26, and then mainly used CT26 cells to elucidate the mechanism of DMF’s action.
Figure 10.
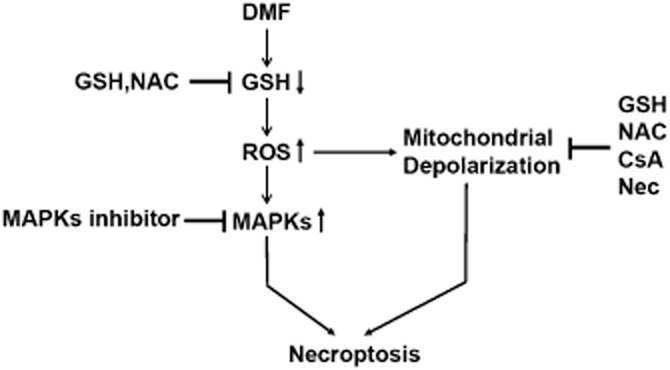
The mechanism of DMF-induced cell necroptosis.
DMF and its in vivo metabolite MMF are fumaric acid esters and an important property of this class of compound is its ability to induce antioxidant effects by up-regulating the expression of nuclear factor (erythroid-derived 2)-like 2 (Nrf2)-driven antioxidant response genes (Gill and Kolson, 2013). In both the nervous and cardiovascular systems, oxidative stress is associated with numerous diseases; thus, fumaric acid esters show protective effects on a wide variety of disorders. DMF and MMF protect neurons and astrocytes against oxidative stress-induced cellular injury and loss (Scannevin et al., 2012), and protect neurons and glia from oxidative stress-induced cell death (Linker et al., 2011). DMF treatment attenuates axonal loss and reduces astrocyte activation in wild-type mice, but not in Nrf2−/− knockout mice, suggesting a role for Nrf2-dependent antioxidant response element activation in the neuroprotection mediated by fumaric acid esters (Linker et al., 2011). In the cardiovascular system, augmentation of myocardial fumarate, whether through fumarate hydratase depletion or by DMF treatment, was found to be cardioprotective against ischaemia-reperfusion injury (Ashrafian et al., 2012). In spontaneously hypertensive rats that transgenically express human C-reactive protein, fumaric acid esters ameliorated inflammation, oxidative stress and metabolic disturbances (Silhavy et al., 2014). However, from the present data, we speculated that DMF can both induce and inhibit oxidative stress, its effect depending on the dose used.
Previous studies showed that DMF or MMF at 10 μM had protective effects produced through the induction of antioxidant response genes (Ashrafian et al., 2012; Scannevin et al., 2012). We found that DMF at 10 μM showed no cytotoxic effects on colon cancer cells. However, when the concentration of DMF was increased (50 to 100 μM), significant cytotoxic effects occurred. Furthermore, the rapid cytotoxic effects of DMF (100 μM) appeared 3 h after DMF treatment and the GSH level decreased simultaneously. Thus, even if DMF at this concentration could induce an up-regulation of antioxidant response genes, the cytotoxic effects were so rapid that there is not enough time for the DMF to up-regulate the expression of antioxidant genes, which would protect the cells. GSH scavenges ROS with the aid of GSH peroxidase (Mailloux et al., 2013), and GSH depletion can result in cell death due to increased levels of ROS (You and Park, 2010; Franco and Cidlowski, 2012). As DMF is an α,β-unsaturated carboxylic acid ester, it can react spontaneously with thiols in GSH via a Michael-type reaction, whereas MMF reacts at a much lower rate (Schmidt et al., 2007). Consistent with this, we found that MMF showed no significant effects on cell viability or on the cellular level of GSH.
Apoptosis is defined as a process driven by a set of molecular mechanisms that programmes the cells to die. Apoptotic cell death involves the engagement of pathways that result in the activation of caspase proteases. Necroptosis is defined as a receptor-interacting protein kinase-3 dependent cell death, and is implicated in a variety of disease states, such as stroke, atherosclerosis, ischaemia-reperfusion injury and pancreatitis (Linkermann and Green, 2014). Cell necroptosis has different characteristics compared with cell apoptosis, both biochemically and morphologically, and has been well reviewed (Christofferson and Yuan, 2010; Galluzzi et al., 2012; Linkermann and Green, 2014). It has been proposed that targeting cancer cells by induction of necroptosis may have significant potential in cancer chemotherapy, especially in the apoptotic/drug resistance cancers (Hu et al., 2007). Indeed, recently this hypothesis has been substantiated by the results of several studies (Grassilli et al., 2013; Steinhart et al., 2013; Yan et al., 2013; Chromik et al., 2014). In the present study, we found that DMF did not significantly affect apoptotic parameters, such as the expression of bcl-2, bax, cleaved caspase-3 protein, but it did induce cell necroptosis as demonstrated by use of the pharmacological tool Nec, TEM and LDH release. We did not exclude the possibility that DMF induces cell apoptosis, but necroptosis was found to be the main method by which DMF induced cell death. Our results showed that DMF increased cellular ROS and induced cell necroptosis, indeed, ROS also induces cell apoptosis. We speculate that DMF depleted GSH and triggered cell necroptosis so rapidly that there was not enough time for the initiation of apoptosis. The ability of DMF to induce necroptosis would be an advantage for DMF as a potential anticancer drug. GSH levels in cancer cells are much higher than that in normal tissues (Perry et al., 1993; Berger et al., 1994) and elevated intracellular GSH is associated with drug resistance in cancer chemotherapy (Berger et al., 1994); therefore, this effect of DMF would be relatively selective for cancer cells because of its ability to deplete GSH.
Autophagy is a critical lysosomal process that maintains cellular homeostasis through the degradation of cellular components such as organelles and proteins, but when extensive, the autophagy response leads to autophagic cell death (Yang and Klionsky, 2010). Autophagy plays a complicated role in tumourigenesis and treatment responsiveness. On the one hand, induction of autophagy seems to be beneficial for cancer prevention and induction of autophagic cell death is considered as a therapeutic strategy; on the other hand, inhibiting autophagy can enhance the efficacy of anticancer therapies (Choi, 2012). Since DMF induced cell necroptosis, we further studied the effects of DMF on autophagy responses and the role of autophagy responses in DMF-induced cell death. We found that DMF increased cell autophagy responses, but inhibiting DMF-induced autophagy by DMH1 did not prevent the DMF-induced decrease in cell viability. Nec or CsA treatment, which showed cytoprotective effects, attenuated DMF-induced autophagy responses, but Nec or CsA alone did not affect the basal and starvation-induced cell autophagy responses. These results indicate that the changes in cell autophagy were secondary responses to DMF stimulation. In the present study, we also used another classical autophagy inhibitor 3-MA. Our previous work and other studies have shown that 3-MA is not a stable autophagy inhibitor and has some non-specific effects (Wu et al., 2010; Sheng et al., 2013). Here, we found that 3-MA did not inhibit the DMF-induced increased autophagy.
We also measured the effects of DMF on the viability of normal cultured cardiomyocytes and cardiofibroblasts and found that DMF reduced the viability of these cells in a similar manner to the cancer cells (data not shown), indicating that these cytotoxic effects of DMF are non-specific. Currently, no necroptosis responses have been reported as a side effect of the clinical use of DMF. MMF, but not DMF, can be detected in serum after oral DMF ingestion (Werdenberg et al., 2003; Rostami-Yazdi et al., 2010). MMF is biologically active but not cytotoxic, so, the metabolism of DMF in the intestine might be a process by which the systemic toxicity of DMF is reduced. These properties of DMF indicate that local administration of high concentrations of DMF into the intestine would be needed to directly kill cancer cells, but this route of administration would reduce its systemic toxicity by increasing its degradation to MMF. Therefore, DMF might have potential as a drug for localized colorectal cancer therapy. Colorectal cancer is a common and lethal disease, and tends to have drug resistance (Panczyk, 2014). At present, targeted drug delivery systems are being investigated for the direct administration of chemotherapeutic and chemopreventive agents to the colon and rectum (Krishnaiah and Khan, 2012). DMF is a promising drug candidate for colorectal cancer therapy provided an appropriate local drug delivery system can be devised.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 81373406, 81421063).
Glossary
- 3-MA
3-methyladenine
- CsA
cyclosporine A
- DMF
dimethyl fumarate
- DMH1
4-[6-(4-isopropoxyphenyl)pyrazolo [1,5-a]pyrimidin-3-yl] quinolone
- EthD-1
ethidium-homodimer-1
- HMGB1
high-mobility group box 1
- MMF
mono-methylfumarate
- NAC
N-acetylcysteine
- Nec
necrostatin-1
- ROS
reactive oxygen species
- TEM
transmission electron microscopy
Author contributions
X X, Y Z, C-Y M, X-M X, Y-Q Z, C-G W, J J, X S, J-L G, N L performed the cell culture, Western blot and parameter detection by commercial kits. D-L D and Z-J S designed the project. D-L D wrote the paper.
Conflict of interest
The authors declare no conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafian H, Czibik G, Bellahcene M, Aksentijevic D, Smith AC, Mitchell SJ, et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012;15:361–371. doi: 10.1016/j.cmet.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SJ, Gosky D, Zborowska E, Willson JK, Berger NA. Sensitive enzymatic cycling assay for glutathione: measurements of glutathione content and its modulation by buthionine sulfoximine in vivo and in vitro in human colon cancer. Cancer Res. 1994;54:4077–4083. [PubMed] [Google Scholar]
- Burness CB, Deeks ED. Dimethyl fumarate: a review of its use in patients with relapsing-remitting multiple sclerosis. CNS Drugs. 2014;28:373–387. doi: 10.1007/s40263-014-0155-5. [DOI] [PubMed] [Google Scholar]
- Choi KS. Autophagy and cancer. Exp Mol Med. 2012;44:109–120. doi: 10.3858/emm.2012.44.2.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chromik J, Safferthal C, Serve H, Fulda S. Smac mimetic primes apoptosis-resistant acute myeloid leukaemia cells for cytarabine-induced cell death by triggering necroptosis. Cancer Lett. 2014;344:101–109. doi: 10.1016/j.canlet.2013.10.018. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mesery M, Seher A, Stuhmer T, Siegmund D, Wajant H. MLN4924 sensitizes monocytes and maturing dendritic cells for TNF-dependent and -independent necroptosis. Br J Pharmacol. 2015;172:1222–1236. doi: 10.1111/bph.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster A, Preussner LM, Seeger JM, Rabenhorst A, Kashkar H, Mrowietz U, et al. Dimethylfumarate induces apoptosis in human mast cells. Exp Dermatol. 2013;22:719–724. doi: 10.1111/exd.12247. [DOI] [PubMed] [Google Scholar]
- Fox RJ, Kita M, Cohan SL, Henson LJ, Zambrano J, Scannevin RH, et al. BG-12 (dimethyl fumarate): a review of mechanism of action, efficacy, and safety. Curr Med Res Opin. 2014;30:251–262. doi: 10.1185/03007995.2013.849236. [DOI] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. Glutathione efflux and cell death. Antioxid Redox Signal. 2012;17:1694–1713. doi: 10.1089/ars.2012.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S. Therapeutic exploitation of necroptosis for cancer therapy. Semin Cell Dev Biol. 2014;35:51–56. doi: 10.1016/j.semcdb.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghods AJ, Glick R, Braun D, Feinstein D. Beneficial actions of the anti-inflammatory dimethyl fumarate in glioblastomas. Surg Neurol Int. 2013;4:160–165. doi: 10.4103/2152-7806.123656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill AJ, Kolson DL. Dimethyl fumarate modulation of immune and antioxidant responses: application to HIV therapy. Crit Rev Immunol. 2013;33:307–359. doi: 10.1615/critrevimmunol.2013007247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassilli E, Narloch R, Federzoni E, Ianzano L, Pisano F, Giovannoni R, et al. Inhibition of GSK3B bypass drug resistance of p53-null colon carcinomas by enabling necroptosis in response to chemotherapy. Clin Cancer Res. 2013;19:3820–3831. doi: 10.1158/1078-0432.CCR-12-3289. [DOI] [PubMed] [Google Scholar]
- Gu B, DeAngelis LM. Enhanced cytotoxicity of bioreductive antitumor agents with dimethyl fumarate in human glioblastoma cells. Anticancer Drugs. 2005;16:167–174. doi: 10.1097/00001813-200502000-00008. [DOI] [PubMed] [Google Scholar]
- Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–1649. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- Held KD, Epp ER, Clark EP, Biaglow JE. Effect of dimethyl fumarate on the radiation sensitivity of mammalian cells in vitro. Radiat Res. 1988;115:495–502. [PubMed] [Google Scholar]
- Hu X, Han W, Li L. Targeting the weak point of cancer by induction of necroptosis. Autophagy. 2007;3:490–492. doi: 10.4161/auto.4592. [DOI] [PubMed] [Google Scholar]
- Huang CY, Kuo WT, Huang YC, Lee TC, Yu LC. Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis. 2013;4:e622. doi: 10.1038/cddis.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirlin WG, Cai J, DeLong MJ, Patten EJ, Jones DP. Dietary compounds that induce cancer preventive phase 2 enzymes activate apoptosis at comparable doses in HT29 colon carcinoma cells. J Nutr. 1999;129:1827–1835. doi: 10.1093/jn/129.10.1827. [DOI] [PubMed] [Google Scholar]
- Krishnaiah YS, Khan MA. Strategies of targeting oral drug delivery systems to the colon and their potential use for the treatment of colorectal cancer. Pharm Dev Technol. 2012;17:521–540. doi: 10.3109/10837450.2012.696268. [DOI] [PubMed] [Google Scholar]
- Kuo LM, Kuo CY, Lin CY, Hung MF, Shen JJ, Hwang TL. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules. 2014;19:3327–3344. doi: 10.3390/molecules19033327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Wakade C, Alleyne CH, Jr, Dhandapani KM. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic Biol Med. 2008;45:1103–1114. doi: 10.1016/j.freeradbiomed.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Lehmann JC, Listopad JJ, Rentzsch CU, Igney FH, von Bonin A, Hennekes HH, et al. Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J Invest Dermatol. 2007;127:835–845. doi: 10.1038/sj.jid.5700686. [DOI] [PubMed] [Google Scholar]
- Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewe R, Valero T, Kremling S, Pratscher B, Kunstfeld R, Pehamberger H, et al. Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res. 2006;66:11888–11896. doi: 10.1158/0008-5472.CAN-06-2397. [DOI] [PubMed] [Google Scholar]
- Mailloux RJ, McBride SL, Harper ME. Unearthing the secrets of mitochondrial ROS and glutathione in bioenergetics. Trends Biochem Sci. 2013;38:592–602. doi: 10.1016/j.tibs.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Nibbering PH, Thio B, Zomerdijk TP, Bezemer AC, Beijersbergen RL, van Furth R. Effects of monomethylfumarate on human granulocytes. J Invest Dermatol. 1993;101:37–42. doi: 10.1111/1523-1747.ep12358715. [DOI] [PubMed] [Google Scholar]
- Panczyk M. Pharmacogenetics research on chemotherapy resistance in colorectal cancer over the last 20 years. World J Gastroenterol. 2014;20:9775–9827. doi: 10.3748/wjg.v20.i29.9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RR, Mazetta JA, Levin M, Barranco SC. Glutathione levels and variability in breast tumors and normal tissue. Cancer. 1993;72:783–787. doi: 10.1002/1097-0142(19930801)72:3<783::aid-cncr2820720325>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Colantuono G, Moro N, Ruggiero FM, Tiravanti E, Di Venosa N, et al. Melatonin protects against heart ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Am J Physiol Heart Circ Physiol. 2009;297:H1487–H1493. doi: 10.1152/ajpheart.00163.2009. [DOI] [PubMed] [Google Scholar]
- Rostami-Yazdi M, Clement B, Mrowietz U. Pharmacokinetics of anti-psoriatic fumaric acid esters in psoriasis patients. Arch Dermatol Res. 2010;302:531–538. doi: 10.1007/s00403-010-1061-4. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- Schmidt TJ, Ak M, Mrowietz U. Reactivity of dimethyl fumarate and methylhydrogen fumarate towards glutathione and N-acetyl-L-cysteine–preparation of S-substituted thiosuccinic acid esters. Bioorg Med Chem. 2007;15:333–342. doi: 10.1016/j.bmc.2006.09.053. [DOI] [PubMed] [Google Scholar]
- Sheng Y, Sun B, Guo WT, Zhang YH, Liu X, Xing Y, et al. 3-Methyladenine induces cell death and its interaction with chemotherapeutic drugs is independent of autophagy. Biochem Biophys Res Commun. 2013;432:5–9. doi: 10.1016/j.bbrc.2013.01.106. [DOI] [PubMed] [Google Scholar]
- Sheng Y, Sun B, Guo WT, Liu X, Wang YC, Xie X, et al. DMH1 (4-[6-(4-Isopropoxyphenyl)pyrazolo [1,5-a]pyrimidin-3-yl] quinoline) is a novel autophagy inhibitor. Br J Pharmacol. 2014;171:4970–4980. doi: 10.1111/bph.12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy J, Zidek V, Mlejnek P, Landa V, Simakova M, Strnad H, et al. Fumaric acid esters can block pro-inflammatory actions of human CRP and ameliorate metabolic disturbances in transgenic spontaneously hypertensive rats. PLoS ONE. 2014;9:e101906. doi: 10.1371/journal.pone.0101906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhart L, Belz K, Fulda S. Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis. 2013;4:e802. doi: 10.1038/cddis.2013.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero T, Steele S, Neumuller K, Bracher A, Niederleithner H, Pehamberger H, et al. Combination of dacarbazine and dimethylfumarate efficiently reduces melanoma lymph node metastasis. J Invest Dermatol. 2010;130:1087–1094. doi: 10.1038/jid.2009.368. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werdenberg D, Joshi R, Wolffram S, Merkle HP, Langguth P. Presystemic metabolism and intestinal absorption of antipsoriatic fumaric acid esters. Biopharm Drug Dispos. 2003;24:259–273. doi: 10.1002/bdd.364. [DOI] [PubMed] [Google Scholar]
- Wierinckx A, Breve J, Mercier D, Schultzberg M, Drukarch B, Van Dam AM. Detoxication enzyme inducers modify cytokine production in rat mixed glial cells. J Neuroimmunol. 2005;166:132–143. doi: 10.1016/j.jneuroim.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation. 2010;7:30–37. doi: 10.1186/1742-2094-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazoe Y, Tsubaki M, Matsuoka H, Satou T, Itoh T, Kusunoki T, et al. Dimethylfumarate inhibits tumor cell invasion and metastasis by suppressing the expression and activities of matrix metalloproteinases in melanoma cells. Cell Biol Int. 2009;33:1087–1094. doi: 10.1016/j.cellbi.2009.06.027. [DOI] [PubMed] [Google Scholar]
- Yan C, Oh JS, Yoo SH, Lee JS, Yoon YG, Oh YJ, et al. The targeted inhibition of mitochondrial Hsp90 overcomes the apoptosis resistance conferred by Bcl-2 in Hep3B cells via necroptosis. Toxicol Appl Pharmacol. 2013;266:9–18. doi: 10.1016/j.taap.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You BR, Park WH. Gallic acid-induced lung cancer cell death is related to glutathione depletion as well as reactive oxygen species increase. Toxicol in Vitro. 2010;24:1356–1362. doi: 10.1016/j.tiv.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhong C, Shi L, Guo Y, Fan Z. Granulysin induces cathepsin B release from lysosomes of target tumor cells to attack mitochondria through processing of bid leading to necroptosis. J Immunol. 2009;182:6993–7000. doi: 10.4049/jimmunol.0802502. [DOI] [PubMed] [Google Scholar]
- Zhang LY, Wu YL, Gao XH, Guo F. Mitochondrial protein cyclophilin-D-mediated programmed necrosis attributes to berberine-induced cytotoxicity in cultured prostate cancer cells. Biochem Biophys Res Commun. 2014;450:697–703. doi: 10.1016/j.bbrc.2014.06.039. [DOI] [PubMed] [Google Scholar]