

Abstract
We present a simple method called ‘ClickSeq’ for Next-Generation Sequencing (NGS) library synthesis that uses click-chemistry rather than enzymatic reactions for the ligation of Illumina sequencing adaptors. In ClickSeq, randomly-primed reverse transcription reactions are supplemented with azido-2’,3’-dideoxynucleotides that randomly terminate DNA synthesis and release 3’-azido blocked cDNA fragments in a process akin to dideoxy-sanger sequencing. Purified fragments are ‘click-ligated’ via copper-catalyzed alkyne-azide cycloaddition to DNA oligos modified with a 5’-alkyne group. This generates ssDNA molecules containing an unnatural triazole-linked DNA backbone that is sufficiently biocompatible for PCR amplification to generate a cDNA library for RNAseq. Here, we analyze viral RNAs and mRNA to demonstrate that ClickSeq produces unbiased NGS libraries with low error-rates comparable to standard methods. Importantly, ClickSeq is robust against common artifacts of NGS such as chimera formation and artifactual recombination with fewer than 3 aberrant events detected per million reads.

Click-chemistry has emerged as a popular and widely applied technique for the conjugation of organic compounds onto biomolecules such as proteins and nucleic acids for a wide array of applications. Recent studies have demonstrated that click-chemistry can be used to synthesize large DNA molecules through the ‘click-ligation’ of smaller functionalized oligonucleotides1. DNA molecules containing unnatural triazole linked backbones can form stable double stranded DNA helices2 and remarkably have been demonstrated to be bio-compatible3. DNA templates generated in this manner can be transcribed in vitro4; 5, and in vivo in E. coli6; 7 and in eukaryotic cells8.
Next-Generation Sequencing has become a powerful tool in diverse fields of biological research with a corresponding variety of available strategies for synthesizing sequencing libraries9. However, a common feature is that DNA or RNA samples must be fragmented and enzymatically ligated to specific DNA adaptors required for PCR amplification, barcode or index insertion and annealing of the DNA fragment to the solid support of the sequencing platform. Fragmentation can be achieved via a variety of methods including mechanical sheering, ultrasonication, heat-treatment in the presence of divalent cations or through the use of enzymes such as exonucleases, restriction enzymes and transposases10. Here we provide an alternative to fragmentation and enzymatic ligation strategies by exploiting click-chemistry to make unnatural cDNA fragments as templates for PCR amplification and RNAseq.
To begin, RT-PCR is initiated from a random position in the un-fragmented sample RNA using a semi-random primer containing 6 random nucleotides and a constant region corresponding to the Illumina 3’ adaptor (Primer #1, Table S1). During nucleic acid synthesis, a free 3’-hydroxyl group is required in the nascent nucleic acid to react with the incoming deoxynucleotide (dNTP). Nucleotide analogues with modified 3’ groups such as dideoxynucleotides or 3’-azido-2’,3’-dideoxynucleotides (AzNTPs) (Figure 1A) can also be incorporated into nascent DNA chains, but induce chain termination. Here, in a process akin to dideoxy (Sanger) sequencing, we supplement the RT-PCR reaction containing dNTPs with small amounts of AzNTPs to randomly terminate chain elongation and release 3’-azido blocked cDNA fragments.
Figure 1.

ClickSeq uses bio-orthogonal click-chemistry to generate RNAseq libraries from stochastically terminated 3’-azido blocked cDNA fragments. A) Chemical structure of a 3’-azido-2’,3’-dideoxynucleotide (AzNTP). B) ‘Click-ligation’: Copper-catalyzed cycloaddition of a 5’-hexynyl modified adaptor DNA to a 3’-azido blocked cDNA fragment. C) Schematic illustration of ClickSeq. Template RNA is in red, and cDNA fragments are in black. The color of the sequence adaptors corresponds to those in Table S1.
After RT-PCR, unincorporated AzNTPs are removed using standard DNA cleaning/desalting columns. The purified 3’-azido blocked cDNA fragments are then mixed with excess Illumina 5’-adaptor that has been modified with a 5’-hexynyl (alkyne) group (the ‘click-adaptor’: Oligo #2, Table S1). Upon the addition of ascorbic acid (Vitamin C) and tris(benzyltriazolylmethyl)amine-conjugated copper(I) ions (Cu-TBTA) as catalysts, the 3’-azido-blocked cDNA fragments are ‘click-ligated’ to the 5’-hexynyl adaptor via copper(I)-catalysed azide-alkyne cycloaddition11 (Figure 1B). This reaction can be completed in a matter of minutes at room temperature.
This process generates random single-stranded cDNA fragments from the original RNA that are flanked by the 5’ and 3’ Illumina adaptors, but that contain an unnatural DNA backbone. The bulky triazole linkage (Figure 1B) can be read through by common DNA polymerases such as OneTaq polymerase (NEB) and Klenow DNA polymerase, albeit inefficiently. This is both remarkable and highly fortuitous, as the 5’-hexynyl modification was selected simply as it can be purchased from commercial vendors. Consequently, the click-ligated cDNA can be used directly in a PCR reaction to generate a cDNA library using Primers #3 and #4 (Table S1). cDNA fragment sizes appropriate for RNAseq are finally selected using AMPure beads and gel extraction. A schematic of the ClickSeq process is shown in Figure 1C.
We first set out to demonstrate that 3’-azido-terminated cDNA could be generated during RT-PCR and subsequently click-ligated efficiently to a 5’-alkyne-labelled oligo. An analysis was designed so that individual ssDNA components could be visualized and their relative amounts quantified via denaturing urea PAGE, as illustrated in Figure 2A. A template-specific primer to Flock House Virus (FHV) genomic RNA 1 (Primer #5, Table S1, Figure 2A-B, step 1) was designed that immediately precedes a 23 nt stretch of FHV RNA 1 devoid of adenine. Consequently, an RT-PCR reaction containing 100 ng purified FHV RNA performed in the presence of dATP, dGTP, dCTP and AzTTP (no dTTP) would extend this primer until termination at the first cognate adenine exactly 24 nts upstream, yielding a 3’-azido-terminated cDNA fragment 44nts in length (Figure 2A-B, step 2). This product was then mixed with the 62 nt 5’-hexynyl-modified click-adaptor (Oligo #2, Table S1, Figure 2A-B, steps 3a and 3b). The click-ligation was initiated via addition of Cu-TBTA and ascorbic acid catalysts (Figure 2A-B, step 4) yielding a ssDNA fragment of the expected length of 106 nts for a ligation product between the 3’-azido terminated cDNA fragment and the 5’-hexynyl oligo. However, some 3’-azido cDNA still remains indicating the reaction did not go to completion. Densitometry analysis of the urea PAGE gel indicates that approximately 10% of the 3’-azido-cDNA was ligated to the click-adaptor under the conditions used here.
Figure 2.

Electrophoretic analysis of click-reaction components, products and PCR amplification. A) Schematic illustration of primer extension by RT-PCR using FHV RNA and the click-ligation of the products to form a triazole-linked ssDNA fragment. B) The components and products were analysed by denaturing urea PAGE and quantified by densitometry. C) cDNA libraries with an increasing distribution of insert lengths were generated from Flock House Virus RNA using a range of AzNTP:dNTPs. The PCR amplified libraries were visualized by electrophoresis on a 1.5% agarose gel stained with Ethidium Bromide.
We next demonstrated that the triazole-linked ssDNA product could be used as a template in a conventional PCR reaction and that a range of AzNTP:dNTP ratios could be used in the RT-PCR reaction to generate longer cDNA fragments. We made cDNA libraries from 100 ng purified FHV RNA using the same template-specific primer as above (Primer #5, Table S1), but supplementing the reaction with a range of AzNTP:dNTP ratios: 1:0, 1:1, 1:5, 1:20, 1:100, 1:200, 1:500 and 0:1. After purification and click-ligation, the cDNA fragments were PCR amplified using Primers #3 and #5 and the products were analyzed by native gel electrophoresis. As shown in Figure 2C, the distribution of cDNA fragment lengths is determined by the AzNTP:dNTP ratio with shorter cDNA fragments being obtained in the presence of greater amounts of AzNTPs. The smear indicates a random distribution of cDNA fragments, as is desirable to provide distributed sequence coverage in RNAseq. No library amplification was observed when RT-PCR was performed in the absence of AzNTPs, demonstrating the requirement for 3'-azido-blocked cDNA.
We next generated cDNA libraries from 100 ng FHV RNA and 100 ng Cricket Paralysis Virus (CrPV) RNA using a 1:20 AzNTP:dNTP ratio and a semi-random primer (Primer # 1, Table S1) during RT-PCR (Figure S1). Final cDNA library fragment sizes of 300-500 nts were selected and sequenced on an Illumina HiSeq for single-end 100 nt reads. This sequencing data (hereafter termed the ClickSeq data) was compared to our previous analyses12; 13 that used the exact same RNA samples, but were prepared using the NEBNext RNAseq Library Prep Kit (hereafter termed the NEBNext data).
We first aligned the reads to the reference genomes for FHV (two RNAs, 3107 and 1400 nts in length), CrPV (one RNA, 9185 nts) and Drosophila melanogaster using the Bowtie short read aligner14. The alignment frequencies and the read coverage over the viral genomes are similar between the ClickSeq and NEBNext data (Table S2 and Figures 3A-C). Approximately 4% of the reads from each of the FHV datasets aligned to the host genome (as previously observed12; 15), allowing us to compare read coverage over individual mRNAs. These data are represented in Figure 3D, where the x-axis and y-axis gives the read coverage for each individual host mRNA using NEBNext and ClickSeq respectively. The data clusters along the x=y axis indicating good correlation between the two methods (R = 0.77). The average nucleotide mismatch rates were also similar, with 26 and 17 mismatches per 10,000 nucleotides in the FHV and CrPV ClickSeq datasets respectively and 13 and 15 mismatches per 10,000 nucleotides in NEBNext datasets (Figure S2). These values are within the range for base-calling errors for the Illumina HiSeq platform16. This demonstrates that ClickSeq produces even coverage with similar error rates to standard enzyme-based ligation NGS strategies, such as NEBNext.
Figure 3.
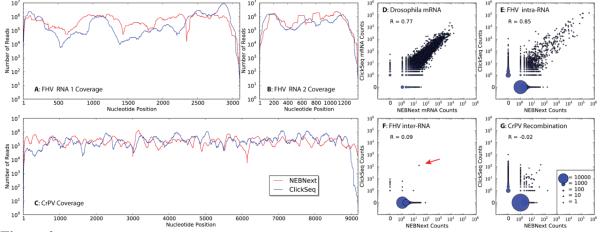
Read mapping to Flock House Virus (FHV), Cricket Paralysis Virus (CrPV), Drosophila melanogaster mRNAs and recombinations events using ClickSeq and NEBNext are compared. Coverage of sequences reads over A) FHV RNA 1, B) FHV RNA 2 and C) CrPV by NEBNext (red) and ClickSeq (blue). Scatter plots show the correlation between the frequencies of mapping events detected by NEBNext and ClickSeq: D) host mRNA in the FHV RNA datasets; E) Intra-molecular recombination in FHV; F) Inter-molecular recombination in FHV; and G) recombination in CrPV. Multiple overlapping points are merged to form a single larger point, as illustrated in the key.
We next searched for recombination events using ViReMa (Viral Recombination Mapper)17, a computational pipeline that reports recombination events both within and between viral and host genomes. As we previously reported13, we found numerous intra-RNA recombination events in FHV (Table S2). These correlate to defective-RNAs that accumulate during viral passaging in cell culture18. Importantly, these events are found in both FHV datasets with good correlation (Figure 3E, R = 0.85). Similarly, profiling the deletions across the FHV genome recapitulates our previous findings of genome conservation17 (Figure S3).
However, with ClickSeq we observed a dramatic reduction in recombination between the two FHV RNAs (Figure 3F). We observed 36,139 inter-RNA recombination events in the NEBNext dataset, but only 213 events in the ClickSeq dataset (Table S2) – a reduction of over 99%. This indicates that most of the recombination events in the NEBNext data were artifactual. Moreover, many of the events detected in the NEBNext datasets are not cross-validated by the ClickSeq data. These are illustrated by the voluminous data points along the y=0 axis for the scatter plots in Figures 3E-G.
On inspection of the inter-RNA recombination events, one event is predominant in the FHV ClickSeq dataset (122 reads) that fuses the 3’ terminus of RNA2 to nt 2720 of RNA1. This recombination event was also present in the NEBNext dataset, but only in 23 reads out of 36,139, thus obscuring its presence. This event is indicated in the scatter plot in Figure 3F with the red arrow. Interestingly, nt 2720 on RNA 1 corresponds to the exact 5’ terminus of the subgenomic RNA 3 generated during FHV replication (illustrated in Figure S4). Heterodimers of RNA2 and RNA3 have previously been characterized and observed to arise early during infections of FHV and other nodaviruses19 and may have an important role in the initiation and regulation of RNA2 replication20. The coverage of wild-type reads at nt 2720 of FHV RNA 1 is 767,479 in the FHV ClickSeq dataset, which indicates that the hetero-dimer is present less than once per 6000 virus genomes.
To directly measure the rate of artifactual recombination using ClickSeq, we made a third cDNA library from a mixture of 50 ng FHV RNA and 50 ng CrPV RNA. Any recombination between FHV and CrPV can only arise as an artifact during cDNA library generation. From 23.0 million reads, 13.0 million reads aligned to the CrPV genome (56%), while the rest aligned to FHV, the D. melanogaster genome and to recombinations within FHV as expected (Table S2). In total, we only found 25 CrPV to FHV recombination events. Similarly, only 8 recombination events were found between CrPV and host mRNAs (which are only present in the FHV RNA sample). This returns an artifactual recombination rate of only 33 reads per 13.0 million reads, or 2.5 events per million reads.
Artifactual recombination in NGS generates sequence chimeras that can be highly problematic in diverse areas of research including viral evolution and replication13; 21, bacterial recombination22, eukaryotic transcriptomics23; 24, mRNA splicing and detection of rare chromosomal rearrangements in oncogenic cells25. Artifactual chimeric DNA sequences also disrupt computational pipelines for the estimation of nucleotide polymorphisms frequency or species population diversity21; 26; 27; 28; 29 and de novo genome assembly by generating incorrect edges in de Bruijn graphs28; 30; 31; 32. Consequently, filtering of the data is required27; 28 and putative recombination events must be experimentally validated.
Artifactual recombination is largely attributed to template-switching between homologous DNA/RNA fragments during the PCR steps of cDNA library synthesis13; 33; 34; 35 and can account for over 10% of the output sequence reads26; 29; 33; 36; 37. This can be improved through careful optimization of reaction conditions38 or emulsion PCR to physically segregate the individual DNA/RNA molecules34; 39. In addition, common cDNA library preparations also fragment the original DNA/RNA sample, generating multiple new 5’ and 3’ ends to which sequencing adaptors are enzymatically ligated. A byproduct of this process is the ligation of the DNA/RNA fragments to one another, generating sequence chimeras.
ClickSeq overcomes these limitations by removing the fragmentation steps via the stochastic termination of cDNA synthesis and by replacing the enzymatic ligation steps with robust bio-orthogonal click-chemistry. This approach has three distinct strengths that impact artifactual recombination rates: 1) removal of the fragmentation step prevents the generation of RNA fragments that might be unintentionally ligated to one another; 2) bio-orthogonality ensures the ligation of 3’azido-cDNA only to alkyne-modified oligos and not to other cDNA fragments; and 3), 3’azido-blocked cDNAs cannot form a priming substrate as required for template switching. ClickSeq thus reduces artifactual recombination to 2.5 events per million reads even without careful optimization of PCR conditions or the use of emulsion-based PCRs. This low error-rate gives us confidence in our detection of rare recombination events, such as FHV RNA2-RNA3 heterodimers, and will allow for the reliable detection of other biologically important but rare recombination events.
One current limitation of ClickSeq is the efficiency with which 3’-azido-blocked cDNA fragments are captured during library generation. While approximately 10 % of the 3’-azido-blocked cDNA fragments in Figure 2B could be ligated to an alkyne-modified oligo, the conversion of this triazole-linked ssDNA into a double stranded DNA via the read-through of the triazole-linkage seems to be far less efficient. While the read-through is clearly sufficient to allow generation of unbiased cDNA libraries in a PCR reaction, we were unable to measure the rate of second-strand synthesis via single-primer extension of the triazole-linked cDNA via standard quantitative fluorometric approaches which would be sensitive to at least a 4% read-through. This implies that the read-through of the unnatural and bulky triazole linkage (Figure 1B) is highly inefficient. For standard RNAseq experiments such as the one described here, this is not an issue as we already under-sample the original RNA by multiple orders of magnitude (100 ng FHV RNA would correspond to 2 Trillion 100 nt reads at 1 X coverage). For applications requiring a thorough capture of the original RNA such as single-cell or single-molecule studies40; 41, the efficiency of the click-reaction and the subsequent read-though must be improved, for example, by using evolved or rationally-designed polymerases in optimized buffer conditions42.
Nonetheless, due to the suitability of laboratory-standard polymerases such as Taq, ClickSeq confers considerable practical advantages including its simplicity, cost-effectiveness and quick turnaround. There is no required specialized equipment such as sonicators or chemical synthesis. Consequently, ClickSeq is a highly accessible ‘home-brew’ method for making NGS libraries and illustrates how simple and robust chemistry can be harnessed in vitro to replace complex biological process using recombinant enzymes.
Supplementary Material
RESEARCH HIGHLIGHTS.
ClickSeq produces RNAseq libraries without sample fragmentation or adaptor ligation
cDNA fragments are made via stochastic RT-PCR termination with azido-nucleotides
Alkyne-modified adaptors are ligated using click-chemistry in place of enzymes
High-quality unbiased RNAseq libraries are made with low error-rates
Artifactual recombination is also reduced, resolving rare recombinant viral species
ACKNOWLEDGMENTS
We would like to thank Bruce Torbett and Tatiana Domitrovic for advice, discussions and proof reading. We would like to thank Anette Schneemann for providing CrPV inoculum and John Shimashita for technical support. This work was funded by The National Institutes of Health [5R01-GM54076 to J.E.J. and U19-A1063603 to S.R.H.].
GLOSSARY
- NGS
Next-Generation Sequencing
- FHV
Flock House Virus
- CrPV
Cricket Paralysis Virus
- AzNTP
3’-Azido-2’,3’-dideoxynucleotide
- dNTP
deoxynucleotide
- ViReMa
Viral Recombination Mapper
- TBTA
tris(benzyltriazolylmethyl)amine
- cDNA
complementary DNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
FastQ datasets used in this study are available from the Small Read Archive (NCBI) under accession codes SRP042304 and SRP042306.
REFERENCES
- 1.Qiu J, El-Sagheer AH, Brown T. Solid phase click ligation for the synthesis of very long oligonucleotides. Chem Commun (Camb) 2013;49:6959–61. doi: 10.1039/c3cc42451k. [DOI] [PubMed] [Google Scholar]
- 2.Isobe H, Fujino T, Yamazaki N, Guillot-Nieckowski M, Nakamura E. Triazole-linked analogue of deoxyribonucleic acid ((TL)DNA): design, synthesis, and double-strand formation with natural DNA. Org Lett. 2008;10:3729–32. doi: 10.1021/ol801230k. [DOI] [PubMed] [Google Scholar]
- 3.Dallmann A, El-Sagheer AH, Dehmel L, Mugge C, Griesinger C, Ernsting NP, Brown T. Structure and dynamics of triazole-linked DNA: biocompatibility explained. Chemistry. 2011;17:14714–7. doi: 10.1002/chem.201102979. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, El-Sagheer AH, Brown T. Reverse transcription through a bulky triazole linkage in RNA: implications for RNA sequencing. Chem Commun (Camb) 2014;50:7597–600. doi: 10.1039/c4cc03027c. [DOI] [PubMed] [Google Scholar]
- 5.el-Sagheer AH, Brown T. Efficient RNA synthesis by in vitro transcription of a triazole-modified DNA template. Chem Commun (Camb) 2011;47:12057–8. doi: 10.1039/c1cc14316f. [DOI] [PubMed] [Google Scholar]
- 6.El-Sagheer AH, Sanzone AP, Gao R, Tavassoli A, Brown T. Biocompatible artificial DNA linker that is read through by DNA polymerases and is functional in Escherichia coli. Proc Natl Acad Sci U S A. 2011;108:11338–43. doi: 10.1073/pnas.1101519108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Savolainen-Kopra C, Blomqvist S. Mechanisms of genetic variation in polioviruses. Rev Med Virol. 2010;20:358–71. doi: 10.1002/rmv.663. [DOI] [PubMed] [Google Scholar]
- 8.Birts CN, Sanzone AP, El-Sagheer AH, Blaydes JP, Brown T, Tavassoli A. Transcription of click-linked DNA in human cells. Angew Chem Int Ed Engl. 2014;53:2362–5. doi: 10.1002/anie.201308691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Head SR, Komori HK, LaMere SA, Whisenant T, Van Nieuwerburgh F, Salomon DR, Ordoukhanian P. Library construction for next-generation sequencing: overviews and challenges. Biotechniques. 2014;56:61–4. doi: 10.2144/000114133. 66, 68, passim. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knierim E, Lucke B, Schwarz JM, Schuelke M, Seelow D. Systematic comparison of three methods for fragmentation of long-range PCR products for next generation sequencing. PLoS One. 2011;6:e28240. doi: 10.1371/journal.pone.0028240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Routh A, Domitrovic T, Johnson JE. Host RNAs, including transposons, are encapsidated by a eukaryotic single-stranded RNA virus. Proc Natl Acad Sci U S A. 2012;109:1907–12. doi: 10.1073/pnas.1116168109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Routh A, Ordoukhanian P, Johnson JE. Nucleotide-resolution profiling of RNA recombination in the encapsidated genome of a eukaryotic RNA virus by next-generation sequencing. J Mol Biol. 2012;424:257–69. doi: 10.1016/j.jmb.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Routh A, Domitrovic T, Johnson JE. Packaging host RNAs in small RNA viruses: An inevitable consequence of an error-prone polymerase? Cell Cycle. 2012;11 doi: 10.4161/cc.22112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Minoche AE, Dohm JC, Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Routh A, Johnson JE. Discovery of functional genomic motifs in viruses with ViReMa-a Virus Recombination Mapper-for analysis of next-generation sequencing data. Nucleic Acids Res. 2014;42:e11. doi: 10.1093/nar/gkt916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jovel J, Schneemann A. Molecular characterization of Drosophila cells persistently infected with Flock House virus. Virology. 2011;419:43–53. doi: 10.1016/j.virol.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albarino CG, Price BD, Eckerle LD, Ball LA. Characterization and template properties of RNA dimers generated during flock house virus RNA replication. Virology. 2001;289:269–82. doi: 10.1006/viro.2001.1125. [DOI] [PubMed] [Google Scholar]
- 20.Eckerle LD, Ball LA. Replication of the RNA segments of a bipartite viral genome is coordinated by a transactivating subgenomic RNA. Virology. 2002;296:165–76. doi: 10.1006/viro.2002.1377. [DOI] [PubMed] [Google Scholar]
- 21.Shao W, Boltz VF, Spindler JE, Kearney MF, Maldarelli F, Mellors JW, Stewart C, Volfovsky N, Levitsky A, Stephens RM, Coffin JM. Analysis of 454 sequencing error rate, error sources, and artifact recombination for detection of Low-frequency drug resistance mutations in HIV-1 DNA. Retrovirology. 2013;10:18. doi: 10.1186/1742-4690-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marttinen P, Hanage WP, Croucher NJ, Connor TR, Harris SR, Bentley SD, Corander J. Detection of recombination events in bacterial genomes from large population samples. Nucleic Acids Res. 2012;40:e6. doi: 10.1093/nar/gkr928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kannan K, Wang L, Wang J, Ittmann MM, Li W, Yen L. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc Natl Acad Sci U S A. 2011;108:9172–7. doi: 10.1073/pnas.1100489108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, Luo S, Khrebtukova I, Barrette TR, Grasso C, Yu J, Lonigro RJ, Schroth G, Kumar-Sinha C, Chinnaiyan AM. Chimeric transcript discovery by paired- end transcriptome sequencing. Proc Natl Acad Sci U S A. 2009;106:12353–8. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6:e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wright ES, Yilmaz LS, Noguera DR. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol. 2012;78:717–25. doi: 10.1128/AEM.06516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li M, Stoneking M. A new approach for detecting low-level mutations in next-generation sequence data. Genome Biol. 2012;13:R34. doi: 10.1186/gb-2012-13-5-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Smith SA. Optimizing de novo assembly of short-read RNA-seq data for phylogenomics. BMC Genomics. 2013;14:328. doi: 10.1186/1471-2164-14-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vodovar N, Goic B, Blanc H, Saleh MC. In silico reconstruction of viral genomes from small RNAs improves virus-derived small interfering RNA profiling. J Virol. 2011;85:11016–21. doi: 10.1128/JVI.05647-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorzer I, Guelly C, Trajanoski S, Puchhammer-Stockl E. The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. J Virol Methods. 2010;169:248–52. doi: 10.1016/j.jviromet.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 34.Runckel C, Westesson O, Andino R, Derisi JL. Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog. 2013;9:e1003164. doi: 10.1371/journal.ppat.1003164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyerhans A, Vartanian JP, Wain-Hobson S. DNA recombination during PCR. Nucleic Acids Res. 1990;18:1687–91. doi: 10.1093/nar/18.7.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu X, Wu L, Huang H, McDonel PE, Palumbo AV, Tiedje JM, Zhou J. Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl Environ Microbiol. 2001;67:880–7. doi: 10.1128/AEM.67.2.880-887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lahr DJ, Katz LA. Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase. Biotechniques. 2009;47:857–66. doi: 10.2144/000113219. [DOI] [PubMed] [Google Scholar]
- 39.Williams R, Peisajovich SG, Miller OJ, Magdassi S, Tawfik DS, Griffiths AD. Amplification of complex gene libraries by emulsion PCR. Nat Methods. 2006;3:545–50. doi: 10.1038/nmeth896. [DOI] [PubMed] [Google Scholar]
- 40.Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14:618–30. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- 41.Wu NC, De La Cruz J, Al-Mawsawi LQ, Olson CA, Qi H, Luan HH, Nguyen N, Du Y, Le S, Wu TT, Li X, Lewis MJ, Yang OO, Sun R. HIV-1 quasispecies delineation by tag linkage deep sequencing. PLoS One. 2014;9:e97505. doi: 10.1371/journal.pone.0097505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Litovchick A, Dumelin CE, Habeshian S, Gikunju D, Guie MA, Centrella P, Zhang Y, Sigel EA, Cuozzo JW, Keefe AD, Clark MA. Encoded Library Synthesis Using Chemical Ligation and the Discovery of sEH Inhibitors from a 334-Million Member Library. Sci Rep. 2015;5:10916. doi: 10.1038/srep10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.