

Abstract
The initial reinforcing properties of drugs of abuse, such as cocaine, are largely attributed to their ability to activate the mesolimbic dopamine system. Resulting increases in extracellular dopamine in the nucleus accumbens (NAc) are traditionally thought to result from cocaine’s ability to block dopamine transporters (DATs). Here we demonstrate that cocaine also interacts with the immunosurveillance receptor complex, Toll-Like Receptor 4 (TLR4), on microglial cells to initiate central innate immune signaling. Disruption of cocaine signaling at TLR4 suppresses cocaine-induced extracellular dopamine in the NAc, as well as cocaine conditioned place preference and cocaine self-administration. These results provide a novel understanding of the neurobiological mechanisms underlying cocaine reward/reinforcement that includes a critical role for central immune signaling, and offer a new target for medication development for cocaine abuse treatment.
Drugs of abuse are traditionally thought to produce their initial rewarding/reinforcing effects by enhancing activity of the mesolimbic dopamine system, resulting in increased extracellular dopamine in the nucleus accumbens (NAc)1,2. Cocaine increases dopamine through its pharmacological antagonism of DAT located on dopamine terminals3,4. While attention has primarily focused on neuronal actions, recent evidence suggests that abused drugs, such as cocaine, activate innate immune signaling within the brain5,6. However, it remains unresolved how cocaine engages the brain’s innate immune system, and what pharmacodynamic consequences might result.
The innate immune system of the brain is comprised primarily of microglial cells expressing a variety of pattern-recognition receptors. Of these, the prototypic pattern-recognition receptor, Toll Like Receptor 4 (TLR4) and its cell surface binding protein, MD2, detect a range of substances, including endogenous danger signals (substances released by cellular stress and damage; DAMPs), microbes or invading pathogens (MAMPs/PAMPs), and exogenous small molecules and their metabolites (xenobiotics; XAMPs)7–12. TLR4-induced microglial reactivity causes the release of proinflammatory substances such as interleukin-1 beta (IL-1β)7, triggering agent-specific changes in behavior. Interestingly, cocaine and other abused drugs cause increased proinflammatory immune signaling throughout the brain5,13,14, but the mechanism that produces cocaine-induced central immune proinflammatory signaling is unknown. Although specific mechanisms and functional implications are unclear, proinflammatory central immune signaling has neuroexcitatory effects15,16 that could be relevant to cocaine pharmacodynamics.
We hypothesize that cocaine induces central immune signaling through the TLR4/MD-2 complex, due to the ability of TLR4 to respond to a diverse range of molecules and its importance in innate immune activation. The present series of studies explores this hypothesis using in silico, in vitro and in vivo paradigms to assess cocaine’s interaction with the TLR4 complex, the role of TLR4 signaling in cocaine-induced dopamine increase, and behavioral measures of drug reward and reinforcement. Our findings demonstrate that cocaine induces central immune signaling through activation of TLR4, resulting in proinflammatory signals that contribute to cocaine-induced changes in the mesolimbic dopamine system and cocaine reward. These findings provide evidence requiring a reconceptualization of cocaine neuropharmacology and offer a new target for medication development.
Materials and Methods
Subjects
For rat studies at the University of Colorado Boulder, viral-free adult, male Sprague Dawley rats (275–350 g; Harlan) were pair-housed in standard Plexiglas cages with ad libitum choice food and water and maintained on a 12 h light/dark cycle. Rats were allowed 1 week of acclimation before any procedures. For mouse studies conducted at the University of Colorado Boulder, adult male (25–30 g) C3HeB/FeJ and C3H/HeJ mice (Jackson Laboratories, Bar Harbor, ME) were group-housed until surgery and maintained on a reverse 12 h light/dark cycle with lights on at 7:00 A.M., with ad libitum access to food and water.
For procedures at the National Institute on Drug Abuse, viral-free adult, male ~300g Sprague Dawley rats (Taconic Farms) were single-housed, with food (Scored Bacon Lover Treats, BIOSERV) and water, and allowed at least 1 week acclimation period. After acclimation, weights of rats were maintained at ~320 g by adjusting their daily food ration. The animal housing room was temperature and humidity controlled and maintained on a 12/12 h light/dark cycle with lights on at 07:00 A.M.
Naïve animals were used for each study. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado Boulder or the National Institute on Drug Abuse Intramural Research Program Institutional Animal Care and Use Committee.
Drugs
Cocaine HCl was obtained from the National Institute on Drug Abuse (NIDA; Research Triangle Park, NC and Bethesda, MD, USA) or Sigma-Aldrich (St. Louis, MO). (+)-Naloxone and (+)-naltrexone were synthesized by Dr. Kenner Rice (Chemical Biology Research Branch, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD). Drug doses are reported as free-base where appropriate. LPS-RS (a TLR4 antagonist naturally produced by Rhodobacter sphaeroides), IL1ra, and minocycline were purchased from Sigma (St Louis, MO, USA). Neurotensin was purchased from Bachem (Torrance, CA, USA).
In silico TLR4/MD-2 complex computer modeling
In silico docking simulation methods, identical to those previously described17, used a high-resolution crystalline structure of the dimer of human TLR4 and its coreceptor MD-2, and the software suite AutoDock 4. Briefly, the complexed human TLR4 and MD-2 pdb file was obtained from RCSB Protein Data Bank database (PDBID: 3fxi). Modified pdb files were inputted into AutoDock 4.0 (http://autodock.scripps.edu), hydrogens added, and resaved in pdbqt format. GA and EtG structures were gathered using PubChem isomeric SMILES then converted to .pdb using a structure file generator (http://cactus.nci.nih.gov/services/translate/). All dockings were executed with Lamarkian genetic algorithms.
Biophysical Characterizations
Materials
Detailed descriptions of materials are as previously described10. The murine microglial BV-2 cell line was provided by Dr. Rona Giffard (Stanford University). Insect expression human MD-2-pAcGP67A vector was provided by Dr. Jie-Oh Lee (KAIST, Korea)18 and high 5 insect cell was provided by Dr. Xuedong Liu (University of Colorado, Boulder).
MD-2 expression and purification
MD-2 expression and purification was performed as described previously10,18,19. Briefly, baculovirus was prepared by co-transfection of SF-9 insect cells with MD-2-pAcGP67A vector and bright linearized baculovirus DNA (BD Bioscience, San Diego, CA, USA). After 2–3 rounds of amplification, the MD-2 baculovirus suspension reached a titer of ~108/ml virus particles and was used to transfect high 5 insect cells to express MD-2. MD-2 was secreted into the medium. After 3–4 day transfection, the medium was harvested and subjected to IgG sepharose affinity purification. SDS-PAGE analysis showed that the purity of the prepared protein was >95%
Biophysical ELISA binding assays
Two different ELISAs were performed to investigate the cocaine and MD-2 interaction.
ELISA 1
Indicated concentrations of biotin labeled cocaine aptamer (5’-biotin- GG GAG ACA AGG AAA ATC CTT CAA TGA AGT GGG TCG ACA-3’) and cocaine mix were coated onto the streptavidin coated plates in phosphate buffer solution (PBS, 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.4) at room temperature for 2 h. The wells were washed 3 times with PBST buffer (PBS supplemented with 0.05% Tween-20) and then blocked with 5% bovine serum albumin solution at room temperature for 1 h. After washing with PBST 3 times, indicated concentrations of MD-2-protein A or MD-2-protein A and LPS reaction mix were added and incubated for 0.5–1 h at room temperature. After washing with PBST 5 times, mouse IgG-horseradish peroxidase (HRP) conjugate was diluted (1:4000), added into the wells, and incubated at room temperature for 1 h. After washing with PBST 7 times, 100 µl of TMB reagents were added and incubated at room temperature for 10–30 min. 50 µl of 1 M H3PO4 was subsequently added to stop the color reaction. The absorbance at 450 nm was measured on a Beckman-Coulter DTX 880 micro-plate reader; the reference wavelength was 620 nm.
ELISA 2
Indicated concentration of MD-2-Protein A or BSA was coated onto the polystyrene surface of 96-well ELISA microplate (BD Bioscience, San Jose, CA, USA) in 0.1 M acetate buffer (pH 5.0). The wells were blocked by SuperBlock (PBS) Blocking Buffer (Pierce, Rockford, IL, USA) and washed as described in ELISA 1. Indicated concentration of LPS and/or cocaine were added and incubated at room temperature for 0.5–1 h. After 5 washings, biotin labeled cocaine aptamer and streptavidin coupled HRP conjugate mix were added into the wells and incubated at room temperature for 1 h. After a further 7 washings, the color reaction was developed and measured as described in ELISA 1.
Bis-ANS displacement assay
Fluorescence measurements were performed on a Fluorolog-3 spectrofluorimeter (Horiba Jobin Yvon, Edison, NJ, USA) and were carried out under room temperature in a 2×10 mm quartz cell (Starna Cells, Atascadero, CA, USA). Different concentrations of cocaine were titrated into MD-2 (1.0 µM) and Bis-ANS (1.0 µM) reaction mix. After overnight equilibrium at room temperature, Bis-ANS fluorescence intensity was measured. The excitation wavelength of extrinsic fluorescence probe Bis-ANS was 385 nm; emission at 420–550 nm was recorded. Appropriate controls were subtracted from spectra obtained on the samples. The fluorescence intensity at 478 nm was plotted against cocaine concentration. Ki of cocaine was determined using the equation: Ki = Kapp/(1 + [Bis-ANS]/KD(Bis-ANS -MD-2)).
Collection of tissue micro-punches
After completion of the cocaine and/or (+)-naloxone timecourse injections, rats were euthanized (65 mg/kg sodium pentobarbital, intraperitoneal; Abbott Laboratories, North Chicago, IL, USA), transcardially perfused with ice cold 0.9% saline. Brains were flash frozen in chilled isopentane, frozen on dry ice and stored at −80°C. Brains were cryostat sectioned (30 µm) at −20°C. The location of each region, including the VTA, NAc (predominately the NAc shell) and the ventral medial prefrontal cortex (vmPFC) was determined using a brain atlas (Paxinos and Watson). Circular micro-punches of 0.25 cm in length were taken from each region on both hemispheres using the blunt-end of 18-guage, stainless steel hypodermic tubing. Micro-punches were stored in 1.5 ml microcentrifuge tubes, flash frozen in liquid nitrogen, and stored at −80°C until mRNA extraction. Tissue sections were collected and thaw mounted on glass slides for verification of micropunch location; no micropunches were excluded due to incorrect collection sites.
Quantitative real time reverse-transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was isolated utilizing a standard method of phenol:chloroform. For detailed descriptions of RNA isolation, cDNA synthesis, PCR amplification protocols, and primer sequences, refer to prior publication (Frank et al., 2006). PCR amplification of cDNA was performed using the Quantitect SYBR Green PCR Kit (Qiagen, Valencia, CA). Formation of PCR product was monitored in real time using the MyiQ Single-Color Real-Time PCR Detection System (BioRad, Hercules, CA). Relative gene expression was determined using the 2−ΔΔCT method20.
Isolated Neonatal Microglial Cell Culture
Brains from P0/1 neonatal Sprague–Dawley rat pups were collected and the cortices carefully dissected and the overlying meninges removed. The tissue was then minced with a scalpel blade and digested for 30 min in Liberase and DNAse (0.1 U per brain) at 37°C with agitation. The cells were triturated with a 27 gauge hypodermic needle. MEM (100 U/mL penicillin, 100 µg/mL streptomycin, 0.6% glucose and 2 mM l-glutamine) was added and the cells were centrifuged at 250×g for 5 min at RT. The supernatant was discarded, and the cells were resuspended in 1 ml of MEM media per brain and then filtered through a 70 µm and then 40 µm filters. The cells were plated in 75 cm2 tissue culture flasks at 4×106 cells per flask and were incubated at 37°C and 5% CO2 until confluence was reached (about 10 days). Media was changed every 3 days; the first change was a complete media change and subsequent changes were a 50% media change. Once confluence was reached, microglial cells were shaken from the remaining astrocytes for 90 min at 160 rpm. The media containing the microglia was removed and centrifuged at 300×g for 5 min at RT. The supernatant discarded and the pellet resuspended in 1 ml fresh MEM media. A small aliquot of cells was counted with trypan exclusion and plated in 96-well v-bottom tissue culture plates at 40,000–50,000 cells per well in 100 µl. 24 h later, cell cultures were incubated for 4 h at 37°C with the following drug treatments in media: LPS (0, 0.01, 0.1, 1, 10, or 100 ng/mL) or cocaine (0, 0.01, 0.1, 1, or 10 µM), or cocaine (0, 0.1, or 1) with (+)-naloxone (0, 1, 10, 100 µM). Following incubation, cells were centrifuged at 1000×g for 10 min at 4°C. The supernatant was discarded and cells processed for mRNA gene expression using the SuperScript™ III CellsDirect cDNA Synthesis System (Invitrogen, Carlsbad, CA). Relative gene expression was detected and measured as described above.
In vivo Microdialysis
A detailed description of in vivo microdialysis equipment, surgeries, and procedures was published previously19. Microdialysis guide cannula (CMA Microdialysis) were surgically implanted and aimed at the right or left NAc shell stereotaxic coordinates relative to bregma: anterior/posterior = +1.7 mm; medial/lateral = +/−0.8 mm; relative to dura: dorsal/ventral = −5.6 mm, bite bar = 0; (Paxinos and Watson, 1998) in a counterbalanced fashion.
Rats were placed in separate Plexiglas bowls with ad libitum food and water in the microdialysis testing room. Microdialysis probes were inserted through each guide cannula and artificial CSF perfused through the probes using a CMA infusion pump at a rate of 0.2 µl/min overnight. The next morning, the flow rate was increased to 1.5 µl/min for the duration of the experiment. Two h later, 3 baseline samples were collected and then drug treatments were administered. The sample tubes were changed every 20 min for a total of 4 h (12 samples total) and stored at −80°C until HPLC analysis.
Systemic administration of drugs
All rats received two subcutaneous (s.c.) injections, of either 2.5 mg/kg (+)-naloxone, for a total of 5 mg/kg (+)-naloxone or equivolume saline. 10 min following the first s.c. injection, rats received the second identical s.c. injection along with an intraperitoneal injection of either 10 mg/kg cocaine HCL or saline.
VTA microinjections
For studies requiring an intra-VTA microinjection, rats received a 1 µL of drug (5 µg LPS-RS, 10 µg IL1 receptor antagonist, 10 ng LPS, 10 nMol neurotensin, or sterile saline) 10 min prior to an intraperitoneal administration of 10 mg/kg cocaine or equivolume saline.
Rats were euthanized with intraperitoneal 65 mg/kg sodium pentobarbital (Abbott Laboratories) before brain extraction. Brains were cryostat sectioned and sections containing each rat’s cannula track were mounted on slides and stained with cresyl violet, coverslipped, and viewed under a light microscope. To be included in data analysis, at least 75% of the probe had to be within the NAc shell. Dialysate samples were analyzed using high performance liquid chromatography (HPLC) along with electrochemical detection using a method previously described19.
Conditioned Place Preference
Detailed descriptions including dimensions, etc., of the place preference apparatus were published previously19. Briefly, the place preference apparatus comprised of two distinct conditioning environments with a neutral space in-between. One environment had a floor of metal bars and walls with black and white stripes. The floor of the second environment was a black plate, perforated with evenly spaced holes, and the walls were black with white polka dots. The activity of each rat was recorded using Logitech Quickcam Pro 5000 webcams, which were connected to a computer running AnyMaze (Stoelting), to track and record the time a rat spent in each of the compartments.
An unbiased conditioned place preference protocol was used. On day 1, all rats were placed individually in the conditioned place preference apparatus and allowed to freely explore for 20 min, to assess baseline preferences. Any rat that spent <20% or >80% of the entire time in either environment was removed from the study. Rats were then randomly assigned to treatments and conditioning environment in a counterbalanced fashion. In studies utilizing (+)-naloxone, for each conditioning session, all rats received two s.c. injections, of either 2.5 mg/kg (+)-naloxone, or equivolume saline. Ten min following the first s.c. injection, the second identical s.c. injection was co-administered with an intraperitoneal injection of either 10 mg/kg cocaine HCl or saline. Rats were then immediately placed into their assigned compartment for 30 min. On days 2–4, rats were conditioned twice each day, once in the morning and then in the afternoon, alternating conditioning between the drug-paired compartment and the vehicle-paired compartment. Place preference testing took place on day 5, and was run identically to pre-exposure testing on day 1. All rats were tested in a drug-free state (i.e. they received no injections). Conditioning was calculated as a difference between time spent in the drug-paired environment before and after conditioning. For studies where minocycline was administered, pre-exposure was conducted to assess baseline preferences as described above. For conditioning, rats received 25 mg/kg minocycline dissolved in sterile water or equivolume sterile water via gavage 40 min prior to intraperitoneal administration of 10 mg/kg cocaine HCl or saline. In this case, rats experienced 4 conditioning sessions once daily, on days 2–5, with alternating treatments. Place preference was assessed on day 6 as described above and all rats were tested in a drug free state.
Self-Administration
Self-administration procedures were performed in operant conditioning chambers (Med-Associates, St Albans, VT) equipped with two response levers and an infusion pump system.
After 24–48 h of food restriction, rats were trained with food pellets to lever press with each press producing a pellet, and eventually each five presses (fixed-ratio 5 schedule of reinforcement). Once responses were reliably producing 30 food pellets within a two-hour session, animals were fed ad libitum for at least 1 day before surgery. Catheters were implanted into the jugular vein under halothane anesthesia (1–2.5%) as previously described21.
After recovery from surgery, cocaine self-administration training was conducted in 2 h daily sessions until criteria for stable cocaine self-administration behavior were met. Cocaine injections, delivered over 5 sec concurrent with the illumination of a stimulus light above the active lever, followed by a 15 sec time-out period when the house light remained off and responding produced no consequence. Responses on the second lever produced no consequence. The positions of active and inactive levers were counterbalanced. To assess a full range of cocaine doses in a single session, the final phase of training consisted of separating the session into five sequential 20 min components to deliver cocaine injections in an ascending order as follows: no injection (referred to as extinction), 0.03, 0.09, 0.27, and 0.89 mg/kg/inj, each preceded by a 2 min time-out period. A sample injection of cocaine at the corresponding dose occurred independently of responding at the end of each time-out. Training continued until: 1) a minimum of 5.0 mg/kg cocaine was self-administered within a session with < 20% variation in the total number of injections compared with the previous session, 2) the dose of cocaine that maintained maximal response rates varied by no more than one-half log unit over two consecutive test sessions, and 3) maximal response rates were at least 5-fold higher than response rates maintained during extinction.
TLR4 mutant Mouse Self-Administration
Self-administration procedures were performed in operant conditioning chambers equipped with two nose-poke portals located on opposite walls and an infusion pump system22. Chambers were modified for sucrose self-administration mounting liquid-dispensing spigots above both ports for mice to freely lick.
Mice were implanted with chronic intravenous jugular catheters, as previously described22. Animals were individually housed and returned to the colony room. Seven days after catheterization mice were trained to nose-poke for intravenous cocaine (0.75 mg/kg/infusion) during daily 3-h sessions over 7 days. Nose-pokes into the active port resulted in a cocaine infusion of 50 µL delivered over 4 s on a fixed-ratio 1 schedule of reinforcement. Each reinforced response resulted in a 10 sec timeout period, during which time the active port was illuminated with a white LED. Mice were trained to nose-poke for sucrose (10% in sterile H2O; Sigma-Aldrich, St. Louis, Missouri) during daily 3 h sessions over 7 days; nose-pokes into the active port resulted in a sucrose delivery of 30 µL delivered over 2 sec on a fixed-ratio 1 schedule of reinforcement for mice to freely lick. For both cocaine and sucrose mice, nose pokes into the inactive port were recorded, but had no consequence. If animals’ behavior deviated by >30% of the prior day’s responding, catheter integrity and access to the jugular vein were examined using 10 mg/kg sodium brevital (JHP Pharmaceuticals, Parsippany, New Jersey). If animals did not exhibit sedation within several seconds they were omitted from the study. All genotypes were blind to the investigators.
After 7 days of cocaine or sucrose self-administration, mice were challenged to a progressive ratio schedule in which each successive cocaine or sucrose delivery required an increasing amount of nose-poke responses according to the following progression; 1, 3, 4, 5, 7, 9, 12, 15, 19, 23, 28, 33, etc. Breakpoints were determined as the final ratio of responses/infusion achieved before a 1 h period when no further infusions were earned; immediately afterwards, catheter integrity was examined using 10 mg/kg sodium brevital. If animals did not exhibit sedation within several seconds they were omitted from the study.
HPLC analysis of brain cocaine concentrations in the presence of (+)-naloxone or (+)-naltrexone
Rats were given s.c. injections of either saline, 2.5 mg/kg (+)-naloxone or 2.5 mg/kg (+)-naltrexone, followed 10 min later by a second identical saline/(+)-naloxone/(+)-naltrexone injection paired with an intraperitoneal 10 mg/kg cocaine injection. 5 or 20 min later, rats were sacrificed and brains removed. The hippocampus was collected and analyzed for cocaine concentrations using reverse-phase HPLC coupled with ultraviolet detection, as previously described23.
Pharmacokinetic profile of (+)- naltrexone
A protein precipitation method was used to measure (+)-naltrexone content in collected plasma samples, using a blinded procedure, as previously described24. 20 mL of rat plasma and additional 20 mL of 50% acetonitrile were mixed prior to the precipitation with 75 mL of acetonitrile containing the isotope-labeled naloxone (naloxone-d5, the internal standard, 5 ng/mL). The mixture was vortexed, centrifuged, and the supernatant was then analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). The standard curve range is 1 to 400 ng/mL and the lower limit of quantitation is 1 ng/mL. The LC-MS/MS method was validated with 3 inter-day assays (n = 12) and 1 intra-day assay (n = 6); all precision values (coefficient of variation, CV) and accuracy values (relative error) were within 15%, suggesting that the method was sufficiently reproducible for analysis of study samples.
Caliper Life Sciences NovaScreen compound screening assay
(+)-Naloxone was profiled in a NovaScreen compound screening assay 64 radioligand/enzyme assays at two concentrations: 0.1 µM and 10 µM in duplicate.
DA Transporter, Sigma1 and Sigma2 receptor binding
Detailed descriptions of materials and methods were previously described25,26. For DAT assays, brains from male Sprague-Dawley rats (Bioreclamation, Westbury, NY) were removed, the striata dissected and quickly frozen. Membranes were prepared by homogenizing tissues in 20 volumes (w/v) of ice-cold modified sucrose phosphate buffer (0.32 M sucrose, 7.74 mM Na2HPO4, 2.26 mM NaH2PO4, pH adjusted to 7.4) using a Brinkman Polytron (setting 6 for 20 sec; Kinematica AG, Lucerne, Switzerland) and centrifuged at 30,000 g for 10 min at 4°C. The resulting pellet was resuspended in buffer, recentrifuged, and resuspended in buffer to a concentration of 10 mg/ml. Experiments were conducted in assay tubes containing 0.5 ml sucrose phosphate buffer for 120 min on ice. Each tube contained 0.5nM [3H]WIN35,428 (specific activity 76 Ci/mmol) (PerkinElmer Life and Analytical Sciences, Waltham, MA) and 1 mg of striatal tissue (original wet weight [OWW]). Nonspecific binding was determined using 0.1 mM cocaine HCl (Sigma-Aldrich, St. Louis, MO).
For σR binding, frozen whole guinea pig brains (minus cerebellum) were thawed on ice and homogenized in 10 mM Tris-HCl with 0.32 M sucrose, pH 7.4 (10 ml/g tissue). The homogenate was centrifuged at 1000g for 10 min at 4°C. The supernatant was collected into a clean centrifuge tube, and the remaining pellet was resuspended by vortex in 10 ml buffer (10 mM Tris HCl, pH 8.0) and centrifuged again at 50,000g for 15 min at 4°C. The resulting pellet was resuspended in 50 mM Tris-HCl, pH 8.0 buffer to 80 mg/ml OWW. Ligand binding experiments were conducted in polypropylene assay tubes containing 0.5 ml of 50 mM Tris-HCl buffer, pH 8.0. For σ1R binding, each tube contained 3 nM [3H](+)-pentazocine (PerkinElmer Life and Analytical Sciences) and 8.0 mg tissue, OWW. Nonspecific binding was determined using 10 mM haloperidol. For σ2R binding, each tube contained 3 nM [3H]1,3-di-o-tolylguanidine (DTG) (PerkinElmer Life and Analytical Sciences), 200 nM (+)-pentazocine, and 8 mg tissue, OWW. Nonspecific binding was determined using 100 mM haloperidol. The reaction was started with the addition of tissue, and the tubes were incubated for 120 min at room temperature.
Incubations for all binding assays were terminated by rapid filtration through Whatman GF/B filters (Whatman/GE Healthcare, Maidstone, Kent, United Kingdom), presoaked in polyethylenimine, using a Brandel R48 filtering manifold (Brandel, Gaitherburg, MD). The filters were washed twice with 5 ml ice cold buffer and transferred to scintillation vials. Beckman Ready Safe (3 ml) was added, and the vials were counted the next day using a Beckman 6000 liquid scintillation counter at 50% efficiency (Beckman Coulter, Brea, CA).
All assays were typically conducted in at least three independent experiments, each performed in triplicate. From the displacement data, IC50 values were computed using a nonlinear, least-squares regression analysis, affinities (Ki values) were calculated using the Cheng-Prusoff equation.
Biogenic amine transporter assays
Brains from rats were removed, striatum dissected and quickly frozen. Membranes were prepared by homogenizing tissues in 20 volumes (w/v) of ice cold modified sucrose phosphate buffer and centrifuged. The resulting pellet was resuspended in buffer, recentrifuged and resuspended in buffer to a concentration of 10 mg/ml. Nonspecific binding was determined using 0.1 mM cocaine HCl. Incubations were terminated by rapid filtration. The filters were washed twice with 5ml cold buffer and transferred to scintillation vials. The vials were counted the next day using a Beckman 6000 liquid scintillation counter.
Statistics
Statistical tests were run and graphs created in GraphPad Prism Version 5. Data are presented as mean ± SEM. Appropriate statistical analyses were chosen based on experimental design. The specific statistical analysis used is indicated in the text and in each figure caption for all studies. Bonferroni post-hoc tests were used for one-way ANOVAs, two way ANOVAs, and repeated measures ANOVAs. Significance threshold was set to p< 0.05 for all analyses. Sample sizes, although appropriate for relative studies, were generally too small to test variance; however, in instances where an unpaired two-tailed t-test was used, there were no differences in variances. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported in previous publications9,10,17,19,21,22,24,25,27. Data collection and quantification was performed blinded whenever possible; final analyses were not performed blind to the conditions of the experiments. However, when possible, behavioral analyses and experiments were performed blind to the experimenter.
Supplementary information is available at Molecular Psychiatry’s website.
Results
Cocaine interacts with the TLR4/MD-2 complex
To assess if cocaine possesses relevant physicochemical interactions with the TLR4/MD-2 complex, in silico and in vitro biophysical studies were conducted. In silico, cocaine docked to the same binding domain of MD-2 as the classical TLR4 agonist lipopolysaccharide (LPS)18 (Fig. 1). MD-2 has been identified as a cell surface protein co-receptor required for TLR4 signaling11,12. Importantly, recent in silico evidence showed that the selective TLR4 antagonists, (+)-isomers of naloxone and naltrexone (non-opioid enantiomers of (−)-naloxone and (−)-naltrexone, respectively), also docked to MD-217. Therefore, to assess the possible utility of novel pharmacological blockade of cocaine-MD-2 interactions, docking of (+)-naloxone and (+)-naltrexone was also re-examined. As expected, both compounds docked to this same pocket of MD-2. Importantly, when (+)-naloxone or (+)-naltrexone was pre-docked in silico, subsequent cocaine docking was disrupted, suggesting that cocaine, (+)-naloxone, and (+)-naltrexone have the physicochemical potential to interact with and affect TLR4/MD-2 signaling. The competitive nature of the docking of the (+)-isomers with cocaine indicates their potential as functional antagonists of cocaine-induced TLR4 activation.
Figure 1. Computer in silico modeling of cocaine interactions with the TLR4/MD-2 receptor complex.
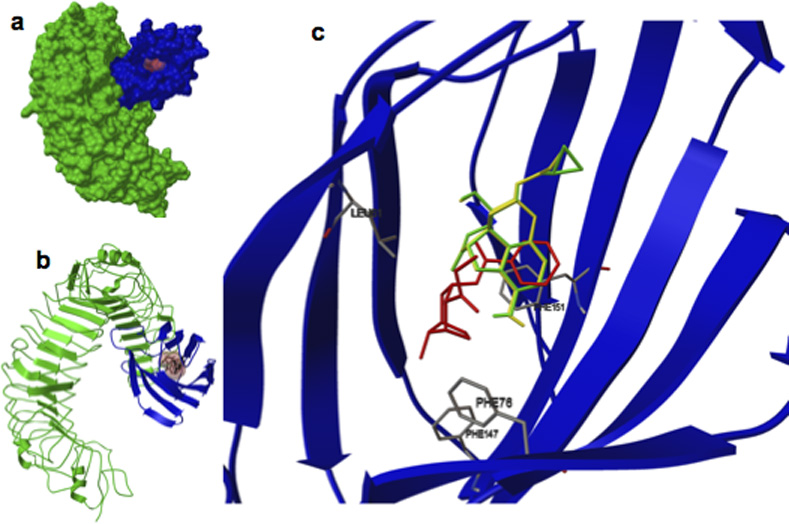
(a) Crystalline structure of TLR4 (green) and MD2 (blue). (b) TLR4 (green) and MD2 (blue), the brown fill is the location that cocaine, and TLR4 antagonists (+)-naloxone and (+)-naltrexone prefer to dock. (c) Magnified image of the MD2 structure (blue) and preferred docking location of cocaine (red), (+)-naltrexone (green), and (+)-naloxone (yellow). Images are compositions of individual docking simulations superimposed and indicate that each compound prefers to dock in the same location. Cocaine docking in the presence (+)-naloxone and (+)-naltrexone causes a fundamental shift in docking conformation.
Based on the in silico results, cocaine’s ability to bind to purified human MD-2 was tested using a biotin-labeled aptamer for cocaine, which was immobilized on a streptavidin-coated plate (Fig. 2a10). Aptamer-immobilized cocaine bound to human MD-2 in a concentration-dependent manner (Fig. 2b). Binding to the negative control, protein A, was negligible. Next, biotin-labeled cocaine aptamer was coated onto a streptavidin plate, bound with cocaine followed by human MD-2, or the negative control, protein A. Cocaine-bound MD-2 was detected by anti-MD2-IgG-HRP conjugate (Fig. 2c). To explore whether cocaine binds competitively to the LPS-binding pocket on MD-2 as suggested by the in silico data, human MD-2 was added to a streptavidin plate coated with biotin labeled cocaine aptamer bound by cocaine and then varying concentrations of LPS were added (Fig. 2d). The resulting decreased signaling indicates that LPS displaced cocaine binding to MD-2. When MD-2 was titrated into streptavidin-coated plates in the presence of cocaine, human MD-2 was captured in a concentration-dependent manner. Protein A binding was again negligible (Supplementary Fig. 1a,b). LPS likewise competed with cocaine for binding to immobilized MD-2 (Supplementary Fig. 1c). Lastly, competitive binding of cocaine to MD-2 was assessed by fluorescence. The fluorescence intensity of Bis-ANS, a molecular probe that binds to the LPS binding pocket of MD-2, increases upon MD-2 binding. Cocaine decreased Bis-ANS fluorescence in a concentration-dependent manner, indicating that it competitively binds MD-2 (Fig. 2e; data fitting to a one-site competitive model gives a Ki of 23.9 ± 5.9 µM). Collectively, in silico and biophysical characterizations demonstrate that cocaine competitively binds to the LPS binding pocket on MD-2, providing compelling support for an interaction with the TLR4/MD-2 complex.
Figure 2. Biophysical characterization of cocaine and MD-2 binding.
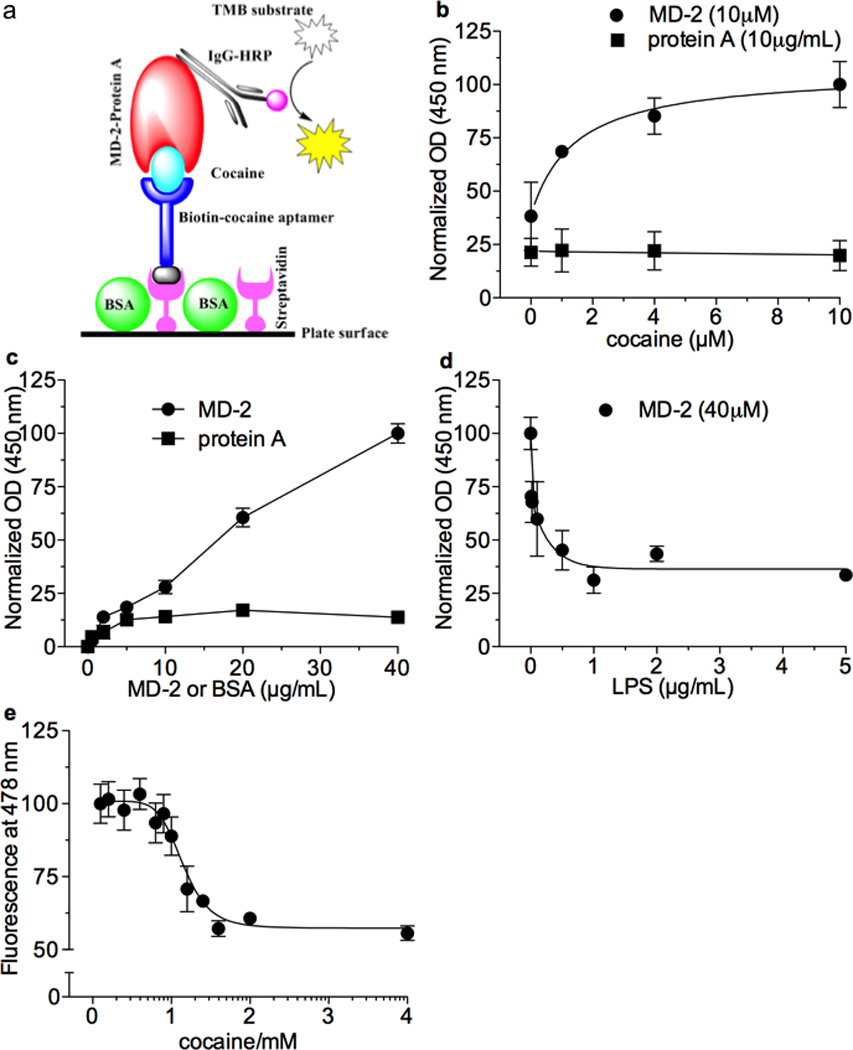
(a) Schematic illustration. Biotin labeled cocaine aptamer was immobilized onto the streptavidin coated plate as the probe for capturing cocaine. Cocaine bound MD-2 was detected by IgG-horseradish peroxidase (HRP) conjugate. It should be noted that the illustration is only a graphic presentation and does not represent the scale of each component. (b) Human MD2 (10 µg/mL) bound varying concentrations of cocaine against 10 µM of biotin labeled cocaine aptamer in a concentration-dependent manner; negative control protein A binding was negligible. c) Varying concentrations of human MD-2 bound cocaine (4 µM) in a concentration-depend manner against a fixed concentration of cocaine-aptamer (4 µM); the negative control protein, BSA, demonstrated negligible binding. (d) LPS concentration-dependently displaced human MD2 (40 µg) binding to cocaine (4 µg) against cocaine aptamer (4µg). (e) Florescent competitive binding assay: Bis-ANS fluorescent signaling increases upon MD-2 binding, cocaine decreased Bis-ANS fluorescence in a concentration-dependent manner, indicating that it competitively binds MD2. Data fitting to a one-site competitive model yields a Ki of 23.9 ± 5.9 µM.
Cocaine-induced proinflammatory signaling in isolated neonatal microglial cells is TLR4-dependent
We next asked whether cocaine could influence mRNA expression of IL-1β in a TLR4-dependent manner in isolated neonatal microglial cells. In order to test for appropriate TLR4 responsiveness, cells were treated with LPS. Following 1 h incubation, LPS concentration-dependently increased mRNA expression of IL-1β (Fig. 3a, one-way ANOVA, F(5,23) = 62.29,p < 0.003). Cocaine treatment also upregulated IL-1β mRNA (Fig. 3b, one-way ANOVA, F(4,19) = 11.56, p = 0.0002), an effect that was blocked by (+)-naloxone co-treatment (Fig. 3c, p < 0.0001; two-way ANOVA, interaction F(6,33) = 19.68, p < 0.0001). Further, (+)-naloxone treatment alone did not alter IL-1β mRNA expression (Fig. 3c, p > 0.05). These findings suggest that cocaine activates microglia through a TLR4-dependent mechanism.
Figure 3. Cocaine-induced signaling in isolated neonatal microglial cells is TLR4 dependent.

a) LPS dose dependently upregulates mRNA expression of IL-1β (p < 0.01 and p <0.001, bonferroni post hoc) in neonatal microglia following 1 hr incubation (one-way ANOVA, F(5,23) = 62.29,p < 0.003).
b) Cocaine (0.1 µM and 1µM) upregulates mRNA expression of IL-1β (p < 0.01, bonferroni post hoc) in neonatal microglia following a 1 hr incubation period (one-way ANOVA, F(4,19) = 11.56, p = 0.0002).
c) (+)-Naloxone (1, 10, 100 µM) suppresses cocaine-induced upregulation of IL-1β mRNA (p < 0.0001; two-way ANOVA, interaction F(6,33) = 19.68, p < 0.0001, bonferroni post hocs). Incubation with (+)-naloxone alone had no effect on IL-1β mRNA expression (p > 0.05).
Cocaine-induced upregulation of interleukin-1 beta mRNA in the VTA is suppressed by systemic (+)-naloxone
Our in vitro data indicate that cocaine interacts with TLR4 to produce a central immune response. We next explored whether in vivo cocaine administration would induce proinflammatory changes in brain regions relevant to cocaine reward. Brains were collected 30 min or 2 h after cocaine (10 mg/kg). Micropunches were collected from the ventral medial prefrontal cortex (vmPFC), NAc (predominately NAc shell), and VTA. mRNA for the proinflammatory cytokine, IL-1β, was measured using quantitative RT-PCR. IL-1β mRNA was reliably increased in the VTA (p < 0.01, Bonferroni post hoc) 2 hr after cocaine. IL-1β mRNA levels in vmPFC and NAc were unaltered at both time points (Fig. 4a; two way ANOVA, effect of brain region (F(2,38) = 3.7, p = 0.03) and time (F(2,38) = 4.5, p = 0.01).
Figure 4. (+)-Naloxone suppresses cocaine-induced upregulation of interleukin-1β mRNA in the VTA.

(a) Cocaine (10 mg/kg, i.p.) induced upregulation of IL1β mRNA in the VTA (p < **0.01) but not the NAc or vmPFC. Two-way ANOVA revealed an effect of region (p = 0.034) and time following cocaine injection (p = 0.017), data are means ± SEMs, n = 5–6/group. (b) Cocaine-induced (10 mg/kg, i.p.) upregulation compared to saline (p = 0.01) of IL1β mRNA within the VTA is attenuated by (+)-naloxone (2.5 mg/kg given in two s.c. injections, with the first injection 10 min prior to the second which is paired with cocaine) administration (p <0.05). Bonferroni post-hocs were preceded by two-way ANOVA, indicating a main effects of cocaine (F(1,17)=4.61, p=0.001) and (+)-naloxone (F(1,17)=4.61, p=0.0447). Data are means ± SEMs, n=5–6/group. (c) Cocaine-induced (10 mg/kg, i.p.) upregulation compared to saline (p < 0.01) of IL1β mRNA within the VTA is attenuated by (+)-naloxone (2.5 mg/kg given in two s.c. injections, with the first injection 10 min prior to the second which is paired with cocaine) administration (p < 0.01). Bonferroni post-hocs were preceded by two-way ANOVA, indicating main effects of cocaine (F(1,16)=5.18, p=0.03) and (+)-naloxone (F(1,16)=16.12, p=0.001). Data are means ± SEMs, n=5/group.
To determine if cocaine-induced upregulation of IL-1β mRNA in the VTA is TLR4-dependent we tested the effects of the TLR4 antagonist19,24,27, (+)-naloxone, administered just prior to cocaine. In this instance, reliable upregulation of IL1β mRNA within the VTA was detected at both 30 min and 2 h following cocaine administration. Cocaine-induced increases of intra-VTA IL1β mRNA (Fig 4b; F(1,17)=14.6, p < 0.01; Fig 4c; F(1,16)=5.18, p = 0.03) were blocked by (+)-naloxone both 30 min (Fig. 4b; F(1,17)=4.7, p < 0.05) and 2 h (Fig. 4c; F(1,16)= 16.1, p = 0.001) after cocaine administration. Further, extensive screening did not identify any off-target effects of (+)-naloxone (Supplementary Tables 1–3) suggesting that the effect of (+)-naloxone is largely a result of its antagonism of TLR4. Together, the data suggest that cocaine initiates proinflammatory central immune signaling in the VTA through a TLR4-dependent mechanism.
TLR4 and IL1β signaling in VTA contribute to cocaine-induced elevations of extracellular NAc dopamine
Given that a single cocaine administration increases IL1β mRNA in the VTA, we next explored how TLR4 activation and IL1β signaling contributes to cocaine-induced alterations in extracellular NAc dopamine concentrations. Using in vivo microdialysis we first tested the effects of the blood-brain barrier permeable TLR4 antagonist, (+)-naloxone 27–31, on cocaine-induced dopamine release in the NAc. Cocaine (10 mg/kg) produced robust increases in extracellular NAc dopamine that were blunted to control levels with (+)-naloxone pretreatment (Fig. 5a; F(24,88)=2.4; p = 0.002). Importantly, (+)-naloxone treatment alone did not influence basal dopamine levels (p > 0.05) suggesting that (+)-naloxone did not independently produce effects on dopamine signaling. It remains possible that TLR4 antagonism nonspecifically disrupts dopamine cell responsiveness, leading to what appeared to be a TLR4-mediated suppression of cocaine-induced elevations in NAc dopamine. To explore this potential alternative, (+)-naloxone was tested in the presence of neurotensin, an endogenous mediator of dopamine transmission that is not known to interact with TLR432. An intra-VTA infusion of neurotensin induced increased NAc dopamine that was unaltered by a pretreatment of (+)-naloxone (p < 0.05, Supplementary Fig. 2a). These findings suggest that suppression of cocaine-induced elevations of NAc dopamine by (+)-naloxone cannot be dismissed as a global disruption of neuronal responsiveness.
Figure 5. Cocaine induced increases of NAc dopamine are dependent on TLR4 and IL-1β signaling within the VTA.

(a) Cocaine (10 mg/kg, s.c.) produces elevated extracellular dopamine in the NAc 40 min (p < 0.0001) and 60 min (p < 0.01) following drug administration (repeated measures two-way ANOVA with bonferroni post-hocs, time and treatment interaction p < 0.001). Administration of (+)-naloxone (2.5 mg/kg in two s.c. injections spaced 10 min apart) blocked this effect; there were no differences between this group and the saline or (+)-naloxone treated rats. Prior to drug treatment, there were no differences in extracellular dopamine concentrations in the NAc shell across all groups. Data are means ± SEMs; n = 4/group. (b) Cocaine (10 mg/kg, i.p.) produces elevated extracellular dopamine in the NAc that sustains for 40–100 min (bonferroni, ****p < 0.00001, **p < 0.01, *p <0.05) following drug administration. Intra-VTA LPS-RS blocked this effect. There were no differences between other treatment groups. Data are means ± SEMs; n = 4/group. (c) Cocaine (10 mg/kg, i.p.) produces elevated extracellular dopamine in the NAc that are significant at 40, 60, and 100 min (bonferroni, ****p < 0.00001, **p < 0.01) following drug administration. Intra-VTA IL1ra suppressed this effect. There were no differences between other treatment groups. Data are means ± SEMs; n = 3–4/group. (d) LPS (10 ng in 1 µL) microinjected into the VTA produces increased extracellular dopamine within the NAc compared to vehicle microinjection-controls (two-way ANOVA, effect of treatment F(1,144)= 35.83, p < 0.0001 and time F(11,144) = 2.89, p = 0.0018) 40 min and 100 min post-microinjection (p < 0.05, bonferroni post-hoc). Data are means ± SEMs; n = 5–7/group.
To explore the importance of TLR4 signaling within the VTA on cocaine-induced elevations of NAc dopamine, rats received either intra-VTA treatments of vehicle or LPS-RS, followed by cocaine (10 mg/kg) or vehicle. The lipophilic nature of (+)-naloxone makes it ideal for distribution following a systemic administration. Unfortunately, this is not conducive to a localized brain infusion. Therefore, the classical TLR4 antagonist, LPS-RS, was selected given its well-characterized specificity for the TLR4 complex and comparative lack of diffusion. Intra-VTA LPS-RS attenuated cocaine-induced extracellular NAc dopamine increases (Fig. 5b; repeated measures two way ANOVA (F(3,131) = 32.0, p < 0.0001), indicating that TLR4 activation in the VTA is necessary for cocaine-induced increases in NAc dopamine. As was the case for (+)-naloxone, intra-VTA LPS-RS had no effect on intra-VTA neurotensin induced elevations of NAc dopamine (Supplementary Fig. 2b).
Since cocaine increased IL-1β mRNA in the VTA, we explored whether cocaine-induced IL-1β signaling in the VTA influences the mesolimbic dopamine system. To test this possibility, intra-VTA IL-1 receptor antagonist (IL-1ra) was administered 10 min prior to cocaine (10 mg/kg) and NAc shell extracellular dopamine quantified. Intra-VTA IL1ra suppressed cocaine-induced elevations of NAc dopamine (Fig. 5c; two way ANOVA, effect of treatment F(3, 104) = 29.5, p < 0.0001). To ensure that the effect of intra-VTA IL1ra is not due to non-specific disruption of neuronal reactivity, the effects of intra-VTA IL-1ra on intra-VTA neurotensin-induced increases of NAc dopamine were assessed. IL1ra did not alter neurotensin-induced dopamine elevations in the NAc (Supplementary Fig. 2c, two way ANOVA, effect of treatment, F(2,82) = 6.7, p = 0.002). Notably, IL1ra infusion into the VTA did not alter basal DA levels in the NAc; this implies that IL1β within the VTA may not be required for homeostatic maintenance of dopamine signaling within the mesolimbic dopamine pathway. Collectively, results from (+)-naloxone, LPS-RS, and IL1ra indicate that TLR4 and IL1β signaling are powerful mediators of cocaine’s effects on the mesolimbic dopamine system.
In order to test whether activation of TLR4 signaling within the VTA is sufficient to produce increased extracellular dopamine concentrations within the NAc, rats received an intra-VTA microinjection of LPS, the potent and well characterized TLR4 agonist. Intra-VTA LPS administration increased of extracellular NAc dopamine levels (Fig. 5d, two-way ANOVA, effect of treatment F(1,144)= 35.83, p < 0.0001 and time F(11,144) = 2.89, p = 0.0018) suggesting that activation of TLR4 signaling within the VTA is sufficient to elevate extracellular dopamine levels within the NAc. Although this is an intriguing phenomenon, it is important to note that the LPS treatment, only partially elevates dopamine concentrations (< 200% increase) compared with systemic cocaine (300% increase). The finding that intra-VTA microinjection of LPS induced an elevation of NAc dopamine supports that TLR4 signaling within the VTA serves an important modulatory role for the mesolimbic dopamine pathway. However, it may also imply that other synergistic mechanisms are required in order to produce as robust an effect as observed with systemic cocaine administration.
TLR4 signaling contributes to behavioral correlates of cocaine reward and reinforcement
The present data demonstrate that cocaine-induced TLR4 signaling has a profound influence on the dopamine system, and is particularly dependent on TLR4-induced IL1β increases in the VTA. To explore whether this effect extends to the behavioral effects of cocaine, we tested (+)-naloxone on cocaine conditioned place preference (CPP). Pretreatment with (+)-naloxone blocked the development of cocaine-induced CPP compared to vehicle (Fig. 6a, two-way ANOVA, F(1,27) = 13.6; p = 0.001). It is important to note that rats were tested for place preference in a drug-free state. Importantly, (+)-naloxone alone produced no appetitive or aversive CPP (p=0.60). Further, we confirmed that brain cocaine concentrations were not altered by similar pretreatment with (+)-naloxone or (+)-naltrexone (Supplementary Fig. 3).
Figure 6. Microglial TLR4 signaling is necessary for the expression of cocaine-induced CPP.
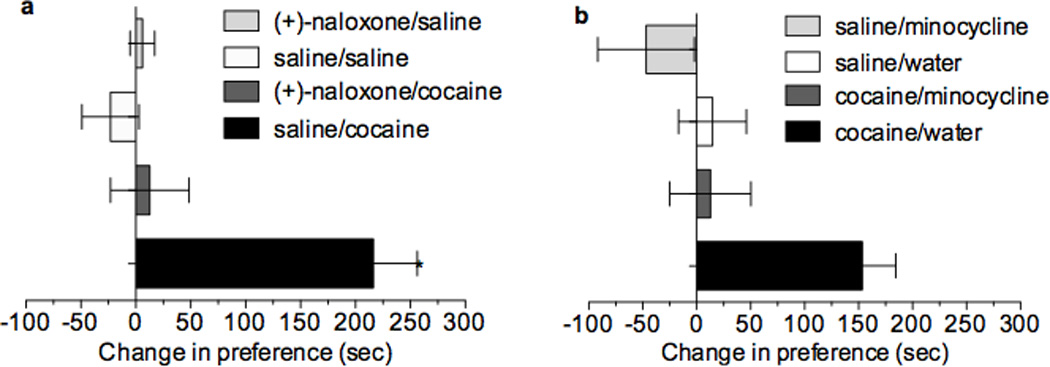
(a) Cocaine (10 mg/kg, i.p.) produces place preference (p<0.0001) and (+)-naloxone (5 mg/kg divided into two s.c. injections spaced 10 min apart) suppresses this effect; no other treatment groups were different from the saline group, nor from one another. Interaction of cocaine and (+)-naloxone (F(1,27) = 13.6, p = 0.001), with a main effect of each (n < 0.01), detected by two-way ANOVA followed by bonferroni, data are mean ± SEMs, n = 7–9/group. (b) Cocaine (10 mg/kg, i.p.) produces significant place preference (p < 0.05, two-way ANOVA with bonferroni). Minocycline (50 mg/kg, gavage, 12 h prior and 25 mg/kg 45 min prior to conditioning) significantly attenuated cocaine-induced (10mg/kg, i.p.) place preference. In the absence of cocaine, neither water nor minocycline altered place preference (main effect of cocaine (p < 0.01) and minocycline (p < 0.01), data are mean ± SEMs, n = 11–12/group).
Although TLR4 is predominately located on microglia33, it is unknown whether it may be expressed on other cell types within the mesolimbic pathway that contribute to drug reward. Our data indicate that in vitro cocaine activates microglial cells through a TLR4-dependent mechanism to produce a proinflammatory response; therefore, if (+)-naloxone blockade of CPP is due to antagonism of TLR4 on microglia, then in vivo blockade of microglial activation should also result in a suppression of cocaine CPP. We assessed the ability of the putative, blood-brain barrier permeable microglial activation inhibitor, minocycline34,35, to alter cocaine-induced CPP. Cocaine produced robust CPP that was suppressed by minocycline pretreatment (Fig. 6b; two-way ANOVA, F(1,42) = 7.7, p = 0.008). Taken with the finding that (+)-naloxone also blocks cocaine CPP, this suggests that cocaine signals through the TLR4 complex produces activation, likely of microglial origin, that contributes to the subjective rewarding effects of cocaine.
Given the robust effects observed using CPP, we next assessed whether TLR4 antagonism would influence cocaine reinforcement in an operant conditioning paradigm. Rats were trained to self-administer cocaine and underwent fixed ratio dose-response testing25. Control animals (vehicle treated) yielded the characteristic inverted U-shaped dose-response (Fig. 7a; F4,20 = 6.7, p = 0.001, one-way repeated measures ANOVA). Pretreatment of (+)-naltrexone suppressed responding for cocaine at the peak cocaine doses (F8,40 = 3.7, p = 0.003; effect of cocaine dose, F(4,40) = 8.3, p < 0.001); in contrast, these doses of (+)-naltrexone failed to suppress responding to food (Fig. 7b; p = 0909). (+)-Naltrexone displays binding and biophysical effects similar to those of (+)-naloxone (Supplementary Figs. 4–5). These findings suggest that pharmacological inhibition of the TLR4 complex impaired cocaine reinforcement.
Figure 7. TLR4 signaling is required for cocaine self-administration.

(a) Response rates maintained by cocaine injections were affected (F4,20 = 6.7, p = 0.001, one-way repeated measures ANOVA ) by dose. The highest rate of responding is maintained by cocaine at a dose of 0.27 mg/kg/injection (**p = 0.002). A pre-session dose of 22.4 mg/kg (+)-naltrexone (s.c.) decreases response rates maintained at 0.27 mg/kg/injection cocaine (***p<0.001). There is an interaction of cocaine dose and (+)-naltrexone dose (F8,40 = 3.7, p = 0.003) and a main effect of cocaine dose (F(4,40) = 8.3, p < 0.001). EXT: extinction; data are means ± SEMs; n = 6/group. (b) In contrast, food-maintained behavior was virtually insensitive to pre-session treatment with (+)-naltrexone (two-way repeated measures ANOVA, p = 0.909). EXT: extinction; data are means ± SEMs; n = 6/group). (c) (+)-Naltrexone was more potent in decreasing responding maintained by the maximal reinforcing doses of cocaine than in decreasing responding maintained by food presentations. Data are means ± SEMs; n = 6/group. (d) C3H/HeJ TLR4 mutant mice do not self-administer cocaine, while their normal C3H/FeJ normal TLR4 counterparts demonstrate normal cocaine self-administration. Repeated measures two-way ANOVA reveals an interaction of genotype and drug (p = 0.0134) and a main effect of both drug (p = 0.0143) and genotype (p = 0.0029). On days 2–7, FeJ mice self-infused more cocaine than HeJ TLR4 mutant mice, whereas HeJ mice demonstrated no difference in cocaine infusions than either FeJ or HeJ saline groups (**p <0.001, ***p<0.0001 bonferroni post-hoc). Data are ± SEMs; n = 6–12/group. (e) Over the 7 days of testing HeJ TLR4 mutant mice self-administer less cocaine (***p = 0.0002, t(17)=4.821, unpaired t-test) and (f) nose-poked less (***p < 0.0001, t(16)=5.421, unpaired t-test) than their FeJ normal TLR4 counterparts. Data are ± SEMs; n = 7–12/group. (g) TLR4 mutant C3H/HeJ mice self-administer sucrose no differently than their normal TLR4 C3H/FeJ counterparts. (h) There are no differences in the daily number of sucrose (10%) deliveries between C3H/FeJs and C3H/HeJs (two-way ANOVA, p = 0.80), (i) the total amount of sucrose earned over the 7 day testing period (two-tailed, unpaired t-test, p = 0.16), or (j) the final ratio of nose-pokes (two tailed, unpaired t-test, p = 0.44). All data are ± SEMs; n = 9/group.
In order to expand the assessment of TLR4-signaling on cocaine reinforcement beyond pharmacological blockade of TLR4, we took a genetic approach using C3H/HeJ mice that possess a point mutation that impedes TLR4-NFκB signaling37–39. Acquisition of cocaine self-administration and performance on a progressive ratio schedule of reinforcement for C3H/HeJ mice was compared to the C3H control substrain, C3H/FeJ. C3H/HeJ TLR4-mutant mice self-administered less cocaine across sessions than their TLR4-intact C3H/FeJ counterparts on both fixed and progressive ratio schedules of reinforcement (Fig. 7c–e; repeated measures two-way ANOVA, interaction of genotype and drug, F(1,25) = 7.1, p = 0.01). No differences in sucrose self-administration were observed between these strains, indicating that there is neither impairment in operant learning nor a generalized disruption of motivated behavior (Figure 7). The parallel results from both the pharmacological and genetic blockade of TLR4 signaling on cocaine self-administration offers profound evidence of the importance of TLR4 signaling in cocaine reinforcement.
Discussion
The present studies demonstrate that cocaine interacts with TLR4 to induce proinflammatory signaling that is necessary for the rewarding effects of cocaine. These findings, combined with previous work, provide the foundation for our recently proposed xenobiotic hypothesis40. This hypothesis suggests that in serving its immune-surveillance role, TLR4 detects and identifies drugs of abuse, such as cocaine, as foreign compounds and initiates proinflammatory immune signaling in response to the perceived threat.
Proinflammatory cytokines are neuroexcitatory in that they upregulate surface expression of AMPA and NMDA receptors, increase conductivity of NMDA receptors15,16,41, increase spontaneous neurotransmitter release15, and increase glutamate transmission42. Nitric oxide, released as part of the proinflammatory cascade, has been shown to inhibit dopamine uptake43, potentially contributing to increased extracellular dopamine concentrations in the NAc. Dopamine neurons express IL-1 receptors44,45 and IL-1β microinfused into rat anterior hypothalamus augmented the release of dopamine46. Here we show that systemic cocaine administration induces upregulation of IL1β mRNA within the VTA and that selective intra-VTA blockade of either TLR4 or IL1β signaling suppresses cocaine-induced increases of dopamine concentrations within the NAc. Further, we show that activation of intra-VTA TLR4 signaling increases NAc dopamine concentrations. Thus, it is possible that cocaine activates TLR4 to induce IL-1β release that in turn enhances dopamine signaling via IL-1 receptors expressed on dopamine neurons in the VTA. Interestingly, dopamine increases resulting from TLR4 activation did not accumulate as rapidly nor to the maximum concentrations compared with systemic administration of cocaine. Further, it has been shown that cocaine-induced dopamine elevations within the NAc are preserved in DAT KO mice47, suggesting that there is an alternate or a second co-mechanism by which cocaine produces high concentrations of dopamine within the NAc.
It might be that the neuroexcitatory influence of IL-1β/TLR4 signaling within the VTA drives the shift from typical phasic dopamine cell firing to burst firing observed in the presence of cocaine. Perhaps this increased dopamine cell firing when paired with blockade of DAT, results in robust increases of extracellular concentrations of dopamine within the NAc, enhancing the rewarding properties of cocaine. Essentially, it appears that in order for cocaine to exert its dramatic effects on the mesolimbic pathway both TLR4 signaling and DAT blockade are necessary.
Recent evidence suggests that proinflammatory TLR4-dependent mechanisms also extend to opioids. We have shown that opioids induce proinflammatory signaling via TLR49,17. Like cocaine, morphine reward and reinforcement are TLR4 dependent19,36. In the case of morphine, it might be that activation of proinflammatory signaling through TLR4, paired with disinhibition of dopamine signaling via mu-opioid receptor stimulation, increases NAc dopamine levels. Based on evidence presented here, it appears that the xenobiotic hypothesis can now be extended to encompass multiple drug classes. This implies that a required synergism between neuronal systems and central proinflammatory immune signaling may be the rule rather than the exception underlying the rewarding and reinforcing effects of opioids, cocaine, and potentially other abused substances, such as methamphetamine6,13 and alcohol14,48.
While our data suggest that microglia are the primary mediators of the central proinflammatory response, there are other related processes that could also influence the rewarding effects of abused drugs. Astrocytes are another non-neuronal cell type in the brain that have been implicated in the effects of abused drugs49. In addition to being immunocompetent and participating in proinflammatory signaling50, astrocytes are important modulators of synaptic activity, formation, function, plasticity, elimination and glutamate transmission51. Although there is some controversy regarding TLR4 expression on astrocytes, under basal conditions, cultured astrocytes express low levels of TLR4 mRNA, which upregulates when exposed to TLR4 ligands52. Importantly, astrocyte functioning is closely tied to microglial activity. TLR4 signaling rapidly triggers a microglial proinflammatory response, subsequently activating astrocytes41,53. This shift in activity can impact neuronal excitability and functioning, as astrocytes begin to release proinflammatory cytokines and glutamate41,54. Alterations in glutamatergic signaling are frequently associated with the neuroplasticity thought to underlie the addictive effects of cocaine55. Additionally, excessive extracellular glutamate can have neurotoxic consequences56. Therefore, cocaine-induced activation of microglial TLR4 may trigger a broader proinflammatory response involving astrocytes, instigating glutamatergic dysregulation.
Microglia and proinflammatory cytokines may also have neurotoxic effects57 and chronic activation of TLR4 by drugs of abuse may have effects on the brain that contributes to drug-induced neuropathologies. Brains of human stimulant users show increased activated microglia6, potentially contributing to decreased numbers of dopamine neurons and cortical deficits observed in chronic psychostimulant users58,59. IL-1β, in particular, contributes to dopamine cell death following TLR4 stimulation with LPS45, and dopamine neurons may be particularly susceptible to microglial-mediated neurotoxicity57. Blockade of proinflammatory signaling through IL1ra administration or TLR4 antagonism has protective effects45,60. Additionally, microglia form positive feedback loops61; when repeatedly activated, microglia can become primed62, so that with subsequent stimulation the proinflammatory response becomes stronger. While the initial proinflammatory central immune signaling paired with actions on neuronal targets might be sufficient to produce increased dopamine signaling associated with reward and reinforcement, repeated exposure to cocaine might begin to prime microglia. Augmentation of proinflammatory responding with each subsequent drug exposure might lead to disruptions in astrocyte modulation of synaptic excitability, driving neuroplasticity, leading to the development of addiction, and eventually triggering neurotoxic levels of proinflammatory and glutamatergic signaling.
It is becoming evident that abused drugs are proinflammatory and drug addiction should be conceptualized within the realm of neuroimmunopharmacology40. The xenobiotic hypothesis of drug abuse incorporates these concepts and suggests that the rewarding, and possibly addictive, effects of abused drugs requires stimulation of both neuronal and glial cell functioning. The data presented here indicate that cocaine-induced activation of TLR4 triggers proinflammatory signaling that is required to produce cocaine-induced neurochemical and behavioral changes. Further, we demonstrate that TLR4 activation within the VTA is sufficient to increase NAc dopamine, indicating that TLR4 signaling potently influences mesolimbic dopamine activity. The role of glial cells, TLR4, and proinflammatory mediators in dopamine cell functioning and toxicity has overarching implications for numerous diseases affecting dopamine systems that could guide the development of pharmacotherapies aiming to treat these pathologies. Altogether, we provide compelling evidence that the xenobiotic hypothesis could encompass both the initial stages of drug use driven by rewarding effects of drugs, as well as the drug-induced changes in the brain associated with chronic use.
Supplementary Material
Acknowledgements
The authors thank the NIDA Addiction Treatment Discovery Program (NIDA ATDP) for data generated through a contract with Caliper Life Sciences. A portion of this work was supported by the intramural research programs of the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism. This work was supported by NIH grants DA029420, DA033358, GM103843 and GM101279, NIDA grant N01DA-9-8883, and DoD grant PR110146. MRH is funded by an Australian Research Council Research Fellowship (DP110100297).
Footnotes
Author contributions
A.L. Northcutt: design, completion and statistical analyses of the conditioned place preference and follow-up control studies; oversight, design, completion and statistical analyses of in vivo microdialaysis and follow-up control studies; design, over-sight, and analysis of neonatal microglial cell culture studies; over-sight and completion of HPLC procedures, quantification, and analysis; design and completion of brain mRNA studies, oversight of RT-PCR procedures, statistical analysis of RT-PCR data, design and completion of pharmacodynamic study; data graphics and figure preparation; manuscript preparation and review
M.R. Hutchinson: all aspects of in silico analyses and data presentation; data graphics; general project oversight; figure and manuscript preparation and review
X. Wang: design, completion and analysis of biophysical ELISA binding assays, Bis-ANS displacement assay; figure design; manuscript review
M.V. Barrata: oversight of design, completion, and statistical analysis of mouse self-administration studies; manuscript preparation and review
T. Hiranita: design, completion, and analysis of rat self-administration studies; figure preparation; manuscript preparation and review
T.A. Cochran: surgical preparation of the in vivo microdialysis animals, microdialysis sample collection, HPLC sample preparation, collection and processing of micropunches for RT-PCR procedures, completion of neonatal microglial cell studies.
M.B. Pomrenze: design, completion, and statistical analysis of mouse self-administration studies, figure preparation, manuscript preparation and review
E.L. Galer: surgical preparation of the in vivo microdialysis animals, microdialysis sample collection, HPLC sample preparation; assistance with tissue collection for HPLC analysis of brain cocaine concentration
T.A. Kopajtic: design and completion of the sigma-1 binding study and striatal membrane transporter binding study; table preparation; manuscript preparation and review
C.M. Li: design, completion, and analysis of pharmacokinetic profiling (+)- naltrexone
J. Amat: training and oversight of HPLC analyses; manuscript review
G.Larson: assistance and oversight of tissue collection; completion of HPLC analysis for brain cocaine concentration study; data analysis; manuscript review
D.C. Cooper: training and oversight of mouse-self administration procedures
Y. Huang: oversight of design and completion of pharmacokinetic profiling (+)- naltrexone
C.E. O’neill: assistance with statistical analysis; figure and manuscript preparation and review
H. Yin: oversight of design and analysis of biophysical ELISA binding assays; Bis-ANS displacement assay; and BV-2 cell culture studies; manuscript review
N.R. Zahniser: oversight of design and analysis of studies assessing brain cocaine concentrations via HPLC; manuscript preparation and review
J. L. Katz: design and oversight of sigma-1, sigma-2 and striatal membrane assays; design and oversight of rat self-administration studies and analysis; figure and table preparation; manuscript drafting and review
K. C. Rice: design, synthesis, purification and verification of (+)-naloxone and (+)-naltrexone; manuscript preparation and review
S.F. Maier: oversight of experimental designs, control studies, and statistics; manuscript drafting and review
R.K. Bachtell: oversight of experimental designs and control studies; oversight of statistical analysis; manuscript drafting, organization, preparation, drafting, and review
L.R. Watkins: general project management, organization and oversight; figure and manuscript drafting, preparation, and review
Author Information
The authors have no competing financial interests for this work.
References
- 1.Pontieri FE, Tanda G, Di Chiara G. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:12304–12308. doi: 10.1073/pnas.92.26.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- 3.Volkow ND, et al. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature. 1997;386:827–830. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- 4.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 5.Cearley CN, Blindheim K, Sorg BA, Krueger JM, Churchill L. Acute cocaine increases interleukin-1beta mRNA and immunoreactive cells in the cortex and nucleus accumbens. Neurochemical research. 2011;36:686–692. doi: 10.1007/s11064-011-0410-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sekine Y, et al. Methamphetamine causes microglial activation in the brains of human abusers. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:5756–5761. doi: 10.1523/JNEUROSCI.1179-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee H, Lee S, Cho IH, Lee SJ. Toll-like receptors: sensor molecules for detecting damage to the nervous system. Current protein & peptide science. 2013;14:33–42. doi: 10.2174/1389203711314010006. [DOI] [PubMed] [Google Scholar]
- 8.Miyake K. Endotoxin recognition molecules, Toll-like receptor 4-MD-2. Seminars in immunology. 2004;16:11–16. doi: 10.1016/j.smim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Hutchinson MR, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain, behavior, and immunity. 2010;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimazu R, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. The Journal of experimental medicine. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes and infection / Institut Pasteur. 2004;6:1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Goncalves J, et al. Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain. Annals of the New York Academy of Sciences. 2008;1139:103–111. doi: 10.1196/annals.1432.043. [DOI] [PubMed] [Google Scholar]
- 14.Coller JK, Hutchinson MR. Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacology & therapeutics. 2012;134:219–245. doi: 10.1016/j.pharmthera.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 15.Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viviani B, et al. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hutchinson MR, et al. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience. 2010;167:880–893. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park BS, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 19.Hutchinson MR, et al. Opioid activation of toll-like receptor 4 contributes to drug reinforcement. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:11187–11200. doi: 10.1523/JNEUROSCI.0684-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Hiranita T, Soto PL, Newman AH, Katz JL. Assessment of reinforcing effects of benztropine analogs and their effects on cocaine self-administration in rats: comparisons with monoamine uptake inhibitors. The Journal of pharmacology and experimental therapeutics. 2009;329:677–686. doi: 10.1124/jpet.108.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pomrenze MB, et al. Cocaine self-administration in mice with forebrain knock-down of trpc5 ion channels. F1000Research. 2013;2:53. doi: 10.12688/f1000research.2-53.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cass WA, Zahniser NR. Cocaine levels in striatum and nucleus accumbens: augmentation following challenge injection in rats withdrawn from repeated cocaine administration. Neuroscience letters. 1993;152:177–180. doi: 10.1016/0304-3940(93)90512-j. [DOI] [PubMed] [Google Scholar]
- 24.Lewis SS, et al. (+)-naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats. The journal of pain : official journal of the American Pain Society. 2012;13:498–506. doi: 10.1016/j.jpain.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hiranita T, et al. Decreases in cocaine self-administration with dual inhibition of the dopamine transporter and sigma receptors. The Journal of pharmacology and experimental therapeutics. 2011;339:662–677. doi: 10.1124/jpet.111.185025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garces-Ramirez L, et al. Sigma receptor agonists: receptor binding and effects on mesolimbic dopamine neurotransmission assessed by microdialysis. Biological psychiatry. 2011;69:208–217. doi: 10.1016/j.biopsych.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchinson MR, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) The European journal of neuroscience. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutchinson MR, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2010;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hutchinson MR, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis SS, et al. (+)-Naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats. Journal of Pain. 2012 doi: 10.1016/j.jpain.2012.02.005. invited revision in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iijima I, Minamikawa J, Jacobson AE, Brossi A, Rice KC. Studies in the (+)-morphinan series. 5. Synthesis and biological properties of (+)-naloxone. Journal of medicinal chemistry. 1978;21:398–400. doi: 10.1021/jm00202a018. [DOI] [PubMed] [Google Scholar]
- 32.Laitinen K, Crawley JN, Mefford IN, De Witte P. Neurotensin and cholecystokinin microinjected into the ventral tegmental area modulate microdialysate concentrations of dopamine and metabolites in the posterior nucleus accumbens. Brain research. 1990;523:342–346. doi: 10.1016/0006-8993(90)91511-e. [DOI] [PubMed] [Google Scholar]
- 33.Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. Journal of neuropathology and experimental neurology. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 34.Bastos LF, de Oliveira AC, Watkins LR, Moraes MF, Coelho MM. Tetracyclines and pain. Naunyn-Schmiedeberg’s archives of pharmacology. 2012;385:225–241. doi: 10.1007/s00210-012-0727-1. [DOI] [PubMed] [Google Scholar]
- 35.Hutchinson MR, et al. Minocycline suppresses morphine-induced respiratory depression, suppresses morphine-induced reward, and enhances systemic morphine-induced analgesia. Brain, behavior, and immunity. 2008;22:1248–1256. doi: 10.1016/j.bbi.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Theberge FR, et al. Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biological psychiatry. 2013;73:729–737. doi: 10.1016/j.biopsych.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodridge HS, et al. Phosphorylcholine mimics the effects of ES-62 on macrophages and dendritic cells. Parasite immunology. 2007;29:127–137. doi: 10.1111/j.1365-3024.2006.00926.x. [DOI] [PubMed] [Google Scholar]
- 38.Kaisho T, Akira S. Dendritic-cell function in Toll-like receptor- and MyD88-knockout mice. Trends in immunology. 2001;22:78–83. doi: 10.1016/s1471-4906(00)01811-1. [DOI] [PubMed] [Google Scholar]
- 39.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 40.Hutchinson MR, Watkins LR. Why is neuroimmunopharmacology crucial for the future of addiction research? Neuropharmacology. 2014;76:218–227. doi: 10.1016/j.neuropharm.2013.05.039. Pt B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watkins LR, Hutchinson MR, Milligan ED, Maier SF. “Listening” and “talking” to neurons: implications of immune activation for pain control and increasing the efficacy of opioids. Brain research reviews. 2007;56:148–169. doi: 10.1016/j.brainresrev.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandolesi G, et al. Interleukin-1beta alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:12105–12121. doi: 10.1523/JNEUROSCI.5369-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pogun S, Baumann MH, Kuhar MJ. Nitric oxide inhibits [3H]dopamine uptake. Brain research. 1994;641:83–91. doi: 10.1016/0006-8993(94)91818-x. [DOI] [PubMed] [Google Scholar]
- 44.Ho A, Blum M. Induction of interleukin-1 associated with compensatory dopaminergic sprouting in the denervated striatum of young mice: model of aging and neurodegenerative disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1998;18:5614–5629. doi: 10.1523/JNEUROSCI.18-15-05614.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Long-Smith CM, Collins L, Toulouse A, Sullivan AM, Nolan YM. Interleukin-1beta contributes to dopaminergic neuronal death induced by lipopolysaccharide-stimulated rat glia in vitro. Journal of neuroimmunology. 2010;226:20–26. doi: 10.1016/j.jneuroim.2010.05.030. [DOI] [PubMed] [Google Scholar]
- 46.Shintani F, et al. Interleukin-1 beta augments release of norepinephrine, dopamine, and serotonin in the rat anterior hypothalamus. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1993;13:3574–3581. doi: 10.1523/JNEUROSCI.13-08-03574.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carboni E, et al. Cocaine and amphetamine increase extracellular dopamine in the nucleus accumbens of mice lacking the dopamine transporter gene. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:141–144. doi: 10.1523/JNEUROSCI.21-09-j0001.2001. RC141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu L, Hutchinson MR, White JM, Somogyi AA, Coller JK. Association of IL-1B genetic polymorphisms with an increased risk of opioid and alcohol dependence. Pharmacogenetics and genomics. 2009;19:869–876. doi: 10.1097/FPC.0b013e328331e68f. [DOI] [PubMed] [Google Scholar]
- 49.Narita M, et al. Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2006;31:2476–2488. doi: 10.1038/sj.npp.1301007. [DOI] [PubMed] [Google Scholar]
- 50.Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- 51.Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bowman CC, Rasley A, Tranguch SL, Marriott I. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- 53.Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochemistry international. 2004;45:397–407. doi: 10.1016/j.neuint.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 54.Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalivas PW, Hu XT. Exciting inhibition in psychostimulant addiction. Trends in neurosciences. 2006;29:610–616. doi: 10.1016/j.tins.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 56.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature reviews. Neuroscience. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 58.Little KY, et al. Decreased brain dopamine cell numbers in human cocaine users. Psychiatry research. 2009;168:173–180. doi: 10.1016/j.psychres.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 59.Bechara A, et al. Decision-making deficits, linked to a dysfunctional ventromedial prefrontal cortex, revealed in alcohol and stimulant abusers. Neuropsychologia. 2001;39:376–389. doi: 10.1016/s0028-3932(00)00136-6. [DOI] [PubMed] [Google Scholar]
- 60.Yang S, et al. Curcumin protects dopaminergic neuron against LPS induced neurotoxicity in primary rat neuron/glia culture. Neurochemical research. 2008;33:2044–2053. doi: 10.1007/s11064-008-9675-z. [DOI] [PubMed] [Google Scholar]
- 61.Watkins LR, Maier SF. The pain of being sick: implications of immune-to-brain communication for understanding pain. Annual review of psychology. 2000;51:29–57. doi: 10.1146/annurev.psych.51.1.29. [DOI] [PubMed] [Google Scholar]
- 62.Dilger RN, Johnson RW. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. Journal of leukocyte biology. 2008;84:932–939. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.