

Fibronectin adhesion stimulation of focal adhesion kinase (FAK)–Src–PI3K is an upstream regulatory branch of the Hippo pathway and stimulates the activity and nuclear localization of YAP in a Lats-dependent manner.
Abstract
The Hippo pathway is involved in the regulation of contact inhibition of proliferation and responses to various physical and chemical stimuli. Recently, several upstream negative regulators of Hippo signaling, including epidermal growth factor receptor ligands and lysophosphatidic acid, have been identified. We show that fibronectin adhesion stimulation of focal adhesion kinase (FAK)-Src signaling is another upstream negative regulator of the Hippo pathway. Inhibition of FAK or Src in MCF-10A cells plated at low cell density prevented the activation of Yes-associated protein (YAP) in a large tumor suppressor homologue (Lats)–dependent manner. Attachment of serum-starved MCF-10A cells to fibronectin, but not poly-d-lysine or laminin, induced YAP nuclear accumulation via the FAK–Src–phosphatidylinositol 4,5-bisphosphate 3-kinase (PI3K) signaling pathway. Attenuation of FAK, Src, PI3K, or PDK1 activity blocked YAP nuclear accumulation stimulated by adhesion to fibronectin. This negative regulation of the Hippo pathway by fibronectin adhesion signaling can, at least in part, explain the effects of cell spreading on YAP nuclear localization and represents a Lats-dependent component of the response to cell adhesion.
Introduction
Contact inhibition of proliferation (CIP) was originally defined as inhibition of cell division when cells reach their stationary density despite periodic nutrient renewal (McClatchey and Yap, 2012). In a dynamic tissue microenvironment, however, CIP is determined not only by postconfluent cell density but also by the quantitative interplay between cell–cell contacts, mitogens, and ECM. Increased cell–cell contact elevates the threshold level of EGF to overcome CIP (Kim et al., 2009). In addition, matrix stiffening dramatically reduces the threshold for responding to EGF (Kim and Asthagiri, 2011). The balance among these environmental cues is crucial in development, tissue regeneration, and organ size control.
The Hippo pathway has been implicated in the regulation of CIP (Gumbiner and Kim, 2014; Johnson and Halder, 2014). This growth inhibitory signaling pathway consists of a highly conserved kinase cascade leading to the activation of Lats (large tumor suppressor homologue) kinases, which control the nuclear exclusion and inactivation of transcriptional coactivator YAP (Yes-associated protein) and its paralogue TAZ (transcriptional coactivator with PDZ-binding motif). When YAP/TAZ are translocated into the nucleus, they interact with TEAD (TEA domain family member) DNA-binding transcription factors to transcribe growth-promoting and antiapoptotic genes (Zhao et al., 2008). YAP/TAZ are also known to interact with other transcription factors including p73, ErbB4, Smads, and FBJ murine osteosarcoma viral oncogene homologue to activate various target genes (Basu et al., 2003; Komuro et al., 2003; Varelas et al., 2010; Shao et al., 2014). Several physiological upstream regulators created by cell–cell contact (cadherin–catenin complex, polarity proteins, and tight junction proteins) are known to positively regulate the Hippo pathway (Kim et al., 2011; Gumbiner and Kim, 2014). The physical properties of cells, such as cell shape, ECM elasticity, and cytoskeletal tension, also play a role in controlling the Hippo pathway (Halder et al., 2012; Gumbiner and Kim, 2014). This mechanotransduction pathway may regulate YAP/TAZ activity independently of the Lats kinases, but through Rho-Rock–dependent actomyosin contractility (Dupont et al., 2011; Aragona et al., 2013; Calvo et al., 2013; Low et al., 2014).
Recently, mitogens including insulin, EGF, lysophosphatidic acid (LPA), and sphingosine 1-phosphate as well as proteases such as thrombin have been identified as negative regulators of the Hippo pathway leading to YAP/TAZ nuclear activity (Miller et al., 2012; Mo et al., 2012; Straßburger et al., 2012; Yu et al., 2012; Fan et al., 2013). We previously reported that treatment with EGF, LPA, or serum inhibits Hippo signaling through the activation of the PI3K (phosphatidylinositol 4,5-bisphosphate 3-kinase)–PDK1 (3-phosphoinositide–dependent protein kinase 1) pathway (Fan et al., 2013). PDK1 forms a complex with the Hippo signaling core complex, and EGF signaling blocks the complex formation in a PI3K–PDK1-dependent manner, leading to the activation of YAP by dephosphorylation and nuclear accumulation. We wondered whether other classes of upstream regulators of PI3K–PDK1 signaling could similarly regulate the Hippo pathway. In this study, we identified the stimulation of FAK–Src–PI3K by adhesion to fibronectin as an upstream regulatory branch of the Hippo pathway, which controls the activity and subcellular localization of YAP in a Lats-dependent manner.
Results
PI3K, PDK1, and Src control YAP subcellular localization
In our previous study, we found that PI3K–PDK1 signaling in response to growth factors inhibits the Hippo pathway (Fan et al., 2013). PI3K and PDK1 inhibitors prevented growth factor–stimulated YAP nuclear localization in confluent MCF-10A cells at low concentrations expected for specific effects on these enzymes (Fan et al., 2013). In subconfluent MCF-10A cells, YAP is also localized in the nucleus even under starvation conditions without any growth factors, which is enhanced by the depletion of upstream Hippo pathway activator Nf2 (Neurofibromin 2, also known as Merlin; Fig. 1 A). Treatment of serum-starved, subconfluent MCF-10A with PI3K or PDK1 inhibitor caused the cytoplasmic localization of YAP (Fig. 1 A). This phenomenon was dependent on Lats kinases because Lats1/2 depletion blocked the effects of PI3K or PDK1 inhibitors on cytoplasmic localization of YAP (Fig. 1 A). This suggests the presence of some upstream PI3K regulators other than soluble mitogenic growth factors that negatively regulate the Hippo signaling pathway in subconfluent MCF-10A cells. To identify other potential regulators, we first tested whether inhibitors of other signaling molecules affect YAP localization in serum-starved, low cell density MCF-10A cells.
Figure 1.

PI3K, PDK1, and Src regulation of nuclear YAP via Lats in serum-starved, subconfluent cells. (A) PI3K and PDK1 inhibitors relative to Lats. MCF-10A cells transfected with control, Nf2, or Lats1/2 siRNAs were serum starved and treated with DMSO (solvent control), 10 µM wortmannin (PI3K inhibitor), or 5 µM BX-795 (PDK1 inhibitor) for 30 min. On-target plus nontargeting pool was used as a control siRNA. Localization of endogenous YAP was identified by immunofluorescence staining. (B) SFK inhibitors and YAP localization. Serum-starved, low cell density MCF-10A cells were incubated with SFK inhibitors (10 µM each of PP2, dasatinib, SKI-1, and SU6656) for 30 min. 10 µM each of PP3 and imatinib were used as controls. YAP subcellular localization was determined by immunofluorescence staining. Alexa Fluor 594 secondary antibody was used for SU6656, which has high background green fluorescence. (C) Biochemical effects of Src inhibition. PP3- or PP2-treated MCF-10A cells were analyzed by Western blot using anti-YAP and anti–phospho-YAP (S127) antibodies. Phosphorylated YAP was detected by mobility shift on Phos-tag SDS-PAGE. (D) SFK inhibitors relative to Lats. MCF-10A cells transfected with control, Nf2, or Lats1/2 siRNA were serum starved and treated with 10 µM PP3 or PP2. After 30 min, cells were fixed for immunofluorescence staining with anti-YAP antibody. (E) Depletion of individual SFK. MCF-10A cells were transfected with control, Src, Fyn, or Yes siRNA. After serum starvation, subcellular localization of endogenous YAP was identified by immunofluorescence staining and quantified based on the criteria shown under the graph. More than 120 cells from four random views were quantified. (F) Src knockdown relative to Lats. MCF-10A cells were transfected with control, Src, Lats1/2, or combined siRNA of Src and Lats1/2. Cells were serum starved for 24 h before fixation and stained with anti-YAP antibody. (A, B, and D–F) One of three independent results is presented. Bars, 25 µm.
Src has been known to act as an upstream regulator of PI3K (Pleiman et al., 1994; Lu et al., 2003). Interestingly, the Src family kinase (SFK) inhibitor PP2 blocked the nuclear localization of YAP within 30 min as effectively as PI3K and PDK1 inhibitors (Fig. 1 B). Additional SFK inhibitors, including SKI-1, SU6656, and the dual Src/Abl kinase inhibitor dasatinib, were also tested. PP3, an analogue of PP2, and imatinib, an Abl kinase inhibitor, were used as controls. Inhibition of endogenous SFK activity with these inhibitors for 30 min blocked YAP nuclear accumulation in serum-starved, low cell density MCF-10A cells (Fig. 1 B). Treatment with PP2 also increased the phosphorylation of YAP on its Lats phosphorylation site S127 as indicated by phospho-YAP (S127)–specific antibody and a decreased mobility on Phos-tag SDS-PAGE gel (Fig. 1 C; Kinoshita et al., 2006). Knockdown of Lats1/2 kinases abolished the effect of PP2 on the cytoplasmic localization of YAP (Fig. 1 D) in contrast to upstream Hippo pathway activator Nf2. These indicate that the effect of SFK activity in regulating YAP localization depends on Lats, the core kinase of the Hippo pathway.
The SFKs consist of nine family members, three of them—Src, Fyn, and Yes—being ubiquitously expressed (Thomas and Brugge, 1997). To identify which of these three ubiquitous SFK members is involved in the regulation of Hippo signaling, the expression of individual family members Src, Fyn, and Yes were knocked down by siRNA transfection (Fig. S1). Importantly, the knockdown of Src alone strongly prevented the nuclear localization of YAP in serum-starved, low cell density MCF-10A cells, whereas depletion of Yes had minimal effects (Fig. 1 E). The impact of Fyn depletion on the nuclear localization of YAP was less effective than Src knockdown, presumably because of its comparatively lower expression level in MCF-10A cells (Fig. S1 A). Similar to SFK inhibitor treatment, this phenomenon was Lats dependent because the knockdown of Lats1/2 prevented the effects of Src depletion on cytoplasmic retention of YAP (Fig. 1 F). This suggests that Src is the predominant SFK that inhibits the Hippo signaling pathway in MCF-10A cells.
Src controls YAP activity
For gain-of-function experiments, we generated inducible MCF-10A and HEK-293T cell lines expressing constitutively active chicken-Src (Y527F; CA-Src; Sandilands et al., 2004) under control of doxycycline-inducible promoter because Src function is sensitive to the level of expression (Li et al., 2002). Also, inducible expression avoided problems of cell detachment due to overexpression of Src by transient transfection. As expected, induced expression of CA-Src-GFP in MCF-10A cells increased the level of total tyrosine-phosphorylated proteins (Fig. 2 A) and activated downstream components of the PI3K–AKT pathway (Fig. S1 B). It induced the phosphorylation of p85, the regulatory subunit of PI3K, at residue Y458. It also increased the phosphorylation of AKT at two key residues, S473 at the C terminus and T308, which is known to be phosphorylated by active PDK1. Treatment with the SFK inhibitor PP2 completely prevented the activation of the PI3K–AKT pathway by the expression of CA-Src-GFP in MCF-10A cells (Fig. S1 B).
Figure 2.

Activation of Src increases YAP nuclear activity. (A) Biochemical effects of Src expression. Doxycycline-inducible expression of constitutively activated chicken-Src (Y527F, CA-Src)-GFP fusion protein in MCF-10A cells. Cells were treated with the indicated amount of doxycycline (Dox) for 12 h in complete medium and serum starved for an additional 24 h in the presence of doxycycline. Cells were lysed and subjected to Western blot analysis with the indicated antibodies. We performed two independent experiments. Tet, tetracycline (or doxycycline) inducible. (B) CA-Src expression and YAP localization. Doxycycline-inducible CA-Src-GFP–expressing MCF-10A cells were treated with 1 µg/ml doxycycline for 12 h and serum starved for 24 h in starvation medium containing doxycycline. Cells were fixed and immunofluorescence stained with anti-YAP antibody. Induction and membrane localization of CA-Src-GFP fusion protein were detected as green fluorescence. The results represent at least three independent experiments. (C) Reporter assay. Doxycycline-inducible HEK-293T cells expressing myr-GFP or CA-Src-GFP were transfected with HIP-flash or HOP-flash reporters. Luciferase activity was measured in the absence or presence of doxycycline. Data were obtained from three independent experiments. Error bars represent standard deviation. (D) Biochemical effects of CSK depletion. MCF-10A cells transfected with control (Ctrl) or CSK siRNAs were serum starved for 24 h. Cell lysates were resolved in regular or Phos-tag SDS-PAGE gels and subjected to Western blotting with the indicated antibodies. Bar, 25 µm.
Induced expression of CA-Src increased the level of unphosphorylated YAP on its Lats target site in a dose-dependent manner, indicated by the decreased level of phospho-S127 and faster migration in Phos-tag SDS-PAGE (Fig. 2 A). CA-Src-GFP induction also stimulated the nuclear accumulation of endogenous YAP (Fig. 2 B), consistent with inhibition of the Hippo pathway. To monitor the Hippo pathway, we developed a HIP/HOP-flash reporter assay, HIP (Hippo-YAP signaling incompetent promoter; negative control)-flash and HOP (Hippo-YAP signaling optimal promoter)-flash reporters (Fig. S2 A), and confirmed the specificity of reporter gene expression (Fig. S2, B–D). As shown in Fig. 2 C, induced expression of CA-Src-GFP, but not membrane-targeted GFP, selectively activated HOP-flash activity in HEK-293T cells. Collectively, these results show that catalytically active Src promotes the transcriptional activity of YAP through dephosphorylation of S127, its Lats target site, and its nuclear accumulation.
Src kinase activity is tightly regulated by its conformational status. In its inactive, “closed” state, Src kinase activity is repressed through intramolecular interactions due to the phosphorylation of C-terminal regulatory Y530 residue by C-terminal Src kinase (CSK; Okada, 2012). Depletion of CSK increased the activation of Src detected by increased Y416 autophosphorylation (Fig. 2 D). CSK knockdown also increased the level of unphosphorylated YAP (Fig. 2 D). To inhibit endogenous SFK activity, we overexpressed CSK. Normally, recruitment of CSK to the plasma membrane is controlled by CSK-binding proteins such as PAG (also known as Cbp; Okada, 2012), but instead we targeted it to the membrane via a myristoyl group. Similar to treatment with SFK inhibitors (Fig. 1 B), induced expression of myristoylated CSK-GFP (myr-CSK-GFP) in serum-starved, low cell density MCF-10A cells increased the cytoplasmic localization of YAP (Fig. S3). The function of CSK depends on its kinase activity, as the expression of kinase-dead myr-CSK-K222R-GFP had no effect on the cellular localization and phosphorylation status of YAP in low cell density MCF-10A cells (Fig. S3). These data indicate that control of endogenous Src activity regulates YAP localization.
We wished to investigate the effect of Src on the Mst-dependent phosphorylation of Lats. Overexpression of Nf2 in HEK-293T cells has been shown to promote Lats1 phosphorylation by enhancing the complex formation of Lats1 with the Sav1–Mst1/2 complex on the plasma membrane (Yin et al., 2013). Transient transfection of HEK-293T cells confirmed that Lats1 hydrophobic motif (T1079) was phosphorylated by the expression of Sav1–Mst2 and was enhanced by Nf2 coexpression. Expression of CA-Src-GFP significantly inhibited the Mst2 phosphorylation on Lats1 even in the presence of Nf2 (Fig. 3 A). We also found that overexpression of Src-GFP or CA-Src-GFP decreased the binding between Sav1 and Lats1 (Fig. 3 B). These results indicate that the active Src negatively regulates the phosphorylation and activity of Lats1 as well as the Sav1–Lats1 interaction.
Figure 3.

Regulation of Lats1 and the Hippo complex by Src activity. (A) Inhibition of Mst2-dependent phosphorylation of Lats1 by active Src. HEK-293T cells expressing the indicated constructs were harvested at 24 h after transfection. Exogenous Flag-Lats1 protein was immunoprecipitated using anti-flag affinity gels and subjected to Western blotting with the indicated antibodies. Blots represent three independent results. (B) Src disrupts Sav1 binding to Lats1. Exogenous protein-expressing HEK-293T cells were harvested in NP-40 buffer. Sav1–Lats1 complex was coimmunoprecipitated using an anti-HA antibody. One of three independent results is presented. (C) Size-exclusion chromatography. Doxycycline-inducible TurboRFP- or myr-CSK-GFP–expressing MCF-10A cells in low cell density (∼30%) were treated with 2 µg/ml doxycycline for 12 h in complete medium and starved for 24 h before harvest. Cytosolic proteins were fractionated by HPLC gel filtration chromatography on a Superose 12 column. Each fraction and corresponding input samples (Inp) were analyzed by SDS-PAGE and Western blotting with the indicated antibodies. IB, immunoblot; IP, immunoprecipitation; WCL, whole cell lysate.
Regulation of Lats phosphorylation and activity depends on formation of a protein complex with Sav1 and other proteins. To determine whether Hippo pathway protein complexes are affected by Src activity, cytosolic fraction of serum-starved, low cell density MCF-10A cells that express TurboRFP or myr-CSK-GFP were fractionated by HPLC gel filtration chromatography (Fig. 3 C). The expression of myr-CSK-GFP increased the level of Lats1 protein in fractions (fractions 16 and 17) corresponding to >670 kD in which a portion of phospho-Lats1 (S909) was also detected. The expression of myr-CSK-GFP also increased the level of PDK1 and Sav1 in these fractions (Fig. 3 C). This result implies that inhibition of SFK activity by CSK expression increases the Hippo complex formation.
FAK controls the Hippo pathway via Src–PI3K–PDK1
FAK functions upstream of Src and PI3K signaling during cell–ECM adhesion (Cabodi et al., 2010). Autophosphorylation of FAK on the Y397 residue is triggered by many stimuli, including integrin receptor binding to ECM and mechanical stretching (Provenzano and Keely, 2011). The phosphorylated Y397 residue creates a high affinity binding site for SH2-containing proteins, such as Src and PI3K, which activate a downstream signaling cascade (Cabodi et al., 2010). To test whether FAK is involved in regulating the Hippo signaling pathway, serum-starved, subconfluent MCF-10A cells were treated with two different FAK inhibitors for 30 min. PF-573228 is a selective inhibitor of FAK (Slack-Davis et al., 2007), whereas PF-562271 is a dual inhibitor of FAK and PYK2 (Roberts et al., 2008). Both inhibitors effectively prevented FAK autophosphorylation at the Y397 residue, an indicator of FAK activity, in a dose-dependent manner (Fig. 4 A). They also increased phosphorylation of YAP with a similar dose dependence as the phosphorylation of FAK (Fig. 4 A). Moreover, treatment with these FAK inhibitors increased cytoplasmic localization of YAP (Fig. 4 B). Depletion of Lats kinases blocked cytoplasmic accumulation of YAP caused by FAK inhibition (Fig. 4 B). As an alternative molecular approach to interfere with endogenous FAK, inducible FRNK (FAK-related nonkinase)-expressing MCF-10A cells were generated. FRNK is an autonomously expressed splicing variant of FAK that contains the noncatalytic C-terminal domain of FAK (Richardson et al., 1997) and is known to act as a dominant-negative regulator of endogenous FAK and Pyk2 activity (Sieg et al., 1999; Govindarajan et al., 2000). Induced expression of FRNK blocked FAK activation in response to cell attachment to fibronectin (Fig. 4 C). Similar to results in the previous study (Zhao et al., 2012), suspension of MCF-10A cells phosphorylated YAP, whereas attachment to fibronectin-coated coverslips induced dephosphorylation of YAP (Fig. 4 C). Expression of FRNK inhibited dephosphorylation and nuclear localization of YAP in MCF-10A cells attached to fibronectin (Fig. 4, C and D). These results suggest that FAK plays an important role in the regulation of YAP phosphorylation and localization in a Lats-dependent manner.
Figure 4.

FAK regulates the phosphorylation and subcellular localization of YAP via Lats kinases. (A) Dose dependence of FAK inhibitors. Serum-starved, subconfluent MCF-10A cells were incubated for 30 min at the indicated concentrations of the two FAK inhibitors. Cell lysates were separated by SDS-PAGE or Phos-tag SDS-PAGE gels and immunoblotted with the indicated antibodies. γ-Tubulin was used as a loading control. We performed two independent experiments. (B) FAK inhibitors and YAP localization. MCF-10A cells transfected with control or Lats1/2 siRNA were serum starved and treated with DMSO, 5 µM PF-573228, or 10 µM PF-562271 for 30 min. Endogenous YAP was immunofluorescence stained with anti-YAP antibody. One of three independent results is presented. (C) Biochemical effects of FRNK expression. Doxycycline-inducible FRNK-GFP–expressing MCF-10A cells were treated with 1 µg/ml doxycycline (Dox) for 12 h and serum starved for 24 h in starvation medium containing doxycycline. Cells were detached, held in suspension for 30 min (Sus), and then replated on fibronectin (FN)-coated coverslips in starvation medium for 2 h. The result shown is one of two independent results. (D) FRNK expression and YAP localization. MCF-10A cells were either uninduced or induced to express FRNK-GFP with 1 µg/ml doxycycline for 12 h and serum starved for 24 h in starvation medium containing doxycycline. Cells were dissociated and seeded on fibronectin-coated coverslips in starvation medium. Induction of FRNK-GFP and localization to focal adhesions were detected as green fluorescence. More than 150 cells from four random views were quantified, and data represent one of three independent results. Tet, tetracycline (or doxycycline) inducible. Bars, 25 µm.
To determine a functional order for FAK, Src, and PI3K–PDK1 in the regulation of the Hippo signaling pathway, inhibitors were added to the CA-Src–expressing MCF-10A cells (Fig. 5). Induced expression of CA-Src led to the nuclear localization of YAP (Fig. 2 B and Fig. 5). As before, treatment with Src, PI3K, or PDK1 inhibitors completely prevented the effect of CA-Src on nuclear YAP. The FAK inhibitor, however, did not block the effect of CA-Src even though it completely prevented the nuclear localization of YAP in MCF-10A cells without CA-Src expression (Fig. 5). These results imply that FAK acts as an upstream effector of Src in the regulation of YAP activity, and PI3K and PDK1 act as downstream effectors. Thus, the FAK–Src–PI3K–PDK1 pathway is responsible for YAP nuclear activity in serum-starved, subconfluent MCF-10A cells.
Figure 5.

PI3K–PDK1, but not FAK, act downstream of Src. MCF-10A cells expressing doxycycline-inducible CA-Src-GFP fusion protein were treated with 1 µg/ml doxycycline (Dox) in complete medium for 12 h and starved for 24 h in starvation medium containing doxycycline. Indicated inhibitors were added to culture medium and incubated for 30 min before fixation. Subcellular localization of YAP was determined by immunofluorescence staining. One of three independent results is presented. Tet, tetracycline (or doxycycline) inducible. Bar, 25 µm.
PI3K–PDK1 also mediate nuclear localization of YAP in confluent MCF-10A cells in response to mitogens such as EGF and LPA (Fan et al., 2013), and we therefore wished to test the roles of Src and FAK in their effects. The effect of mitogens on YAP localization can also be observed when added to serum-starved, subconfluent MCF-10A cells (Fig. 6 A). Pretreatment of cells with FAK inhibitor did not prevent YAP nuclear accumulation stimulated by EGF or LPA, suggesting that FAK is not required for receptor tyrosine kinase and G protein–coupled receptor regulation of the Hippo pathway (Fig. 6 A). Inhibition of Src kinase by siRNA-mediated depletion, myr-CSK-GFP expression, or dasatinib treatment prevented the EGF effect on nuclear localization of YAP (Fig. 6, A–C). This suggests that Src mediates EGF receptor (EGFR)–dependent regulation of the Hippo pathway, even though we previously reported that the Src inhibitor PP2 did not block the effect of EGF on YAP (Fan et al., 2013). In contrast, Src knockdown or induced expression of myr-CSK-GFP did not prevent the YAP nuclear accumulation induced by LPA signaling by G protein–coupled receptor (Fig. 6, A and B), indicating that Src does not act downstream of LPA. Because PI3K inhibitors block YAP nuclear accumulation caused by LPA treatment (Fan et al., 2013), LPA appears to activate YAP nuclear accumulation through PI3K in a Src-independent manner.
Figure 6.

Roles of Src and FAK in the regulation of nuclear YAP by EGF or LPA. (A) Src depletion and FAK inhibition. MCF-10A cells transfected with control or Src siRNA were serum starved for 24 h and treated with EGF or LPA for 30 min. For FAK inhibitor treatment experiment, serum-starved, subconfluent MCF-10A cells were pretreated for 30 min with DMSO or 5 µM PF-573228, followed by a 30-min EGF or LPA treatment. YAP subcellular localization was determined by immunofluorescence staining. One of three independent results is presented. (B) CSK expression to inhibit Src activity. Paired with uninduced cells, doxycycline (Dox)-induced MCF-10A cells expressing myr-CSK-GFP fusion protein were serum starved for 24 h. Cells were treated with EGF or LPA for 30 min before fixation. Subcellular localization of YAP was determined by immunofluorescence staining. One representative out of three independent experiments is shown. Tet, tetracycline (or doxycycline) inducible. (C) Src inhibitors. Serum-starved, subconfluent MCF-10A cells were pretreated with PP3, PP2, imatinib, or dasatinib for 30 min. Cells were then treated with 20 ng/ml EGF for 30 min before fixation. Endogenous YAP was immunofluorescence stained. The result represents three independent experiments. Bars, 25 µm.
Adhesion to fibronectin activates YAP via FAK–Src–PI3K–PDK1
FAK-Src signaling is often stimulated by integrins in focal adhesions when cells spread on a substrate, as in this study. To directly test whether integrin activation of FAK controls the Hippo signaling pathway, we examined the subcellular localization of YAP in MCF-10A cells attached to fibronectin-, poly-d-lysine–, or laminin-coated surfaces. To minimize the actions of soluble factors that inactivate the Hippo pathway, it is critical to treat MCF-10A cells with serum starvation and amphiregulin-blocking antibody (Zhang et al., 2009; Fan et al., 2013) for 24 h before cells are detached to perform the adhesion assay. Suspended cells were allowed to adhere and spread on coverslips coated with fibronectin, poly-d-lysine, or laminin in starvation medium. Plating on a fibronectin-coated coverslip led to a significant increase of nuclear localization and dephosphorylation of YAP compared with plating on a poly-d-lysine– or laminin-coated coverslip (Fig. 7 A and see Fig. 8 B).
Figure 7.

Attachment to fibronectin-coated coverslips induces YAP nuclear accumulation via the FAK–Src–PI3K–PDK1 pathway. (A) Attachment to fibronectin, poly-d-lysine, or laminin. Serum-starved MCF-10A cells were dissociated with Accutase and sparsely seeded on fibronectin (FN)-, poly-d-lysine (PL)–, or laminin (LN)-coated coverslips in starvation medium. Cells were incubated for 2 h before fixation. Subcellular localization of YAP was identified by immunofluorescence staining and quantified based on the criteria shown in Fig. 1 E. More than 170 cells from eight random views were quantified. DAPI staining was used to locate the nucleus. We performed three independent experiments. (B) Effects of inhibitors and growth factors on attachment-induced YAP nuclear localization. Serum-starved MCF-10A cells were detached and seeded on fibronectin- or poly-d-lysine–coated coverslips in starvation medium. After 3 h of incubation, cells were treated with the indicated inhibitors or mitogens for 30 min. Subcellular localization of YAP was identified by immunofluorescence staining. More than 120 cells from eight random views were quantified and confirmed in three independent experiments. Bars, 25 µm.
We next examined the effects of roles of signaling mediators on the subcellular localization of YAP in MCF-10A cells attached to fibronectin-coated coverslips. FAK, SFK, PI3K, and PDK1 inhibitors all decreased the nuclear localization of YAP stimulated by attachment to fibronectin-coated surfaces to levels similar to poly-d-lysine attachment (Fig. 7 B). In contrast, treatment with EGF or LPA increased the nuclear localization of YAP in MCF-10A cells attached to a poly-d-lysine–coated coverslip (Fig. 7 B). Notably, LPA treatment resulted in an additional increase of nuclear YAP in MCF-10A cells attached to a fibronectin-coated coverslip. To determine the role of SFK in the regulation of YAP cellular localization by ECM–cell binding, doxycycline-induced CA-Src-GFP–, myr-CSK-GFP–, or myr-GFP (control)–expressing MCF-10A cells were attached to fibronectin-, poly-d-lysine–, or laminin-coated coverslips. Induced expression of CA-Src-GFP led to the increase of nuclear localization and dephosphorylation of YAP in MCF-10A cells attached to poly-d-lysine– or laminin-coated coverslips (Fig. 8, A and B). In contrast, inhibition of endogenous SFK activity by myr-CSK-GFP prevented the nuclear localization and dephosphorylation of YAP in fibronectin-attached MCF-10A cells (Fig. 8, A and B). Collectively, these data suggest that fibronectin–integrin signaling controls the Hippo pathway through FAK–Src–PI3K–PDK1 (Fig. 9).
Figure 8.
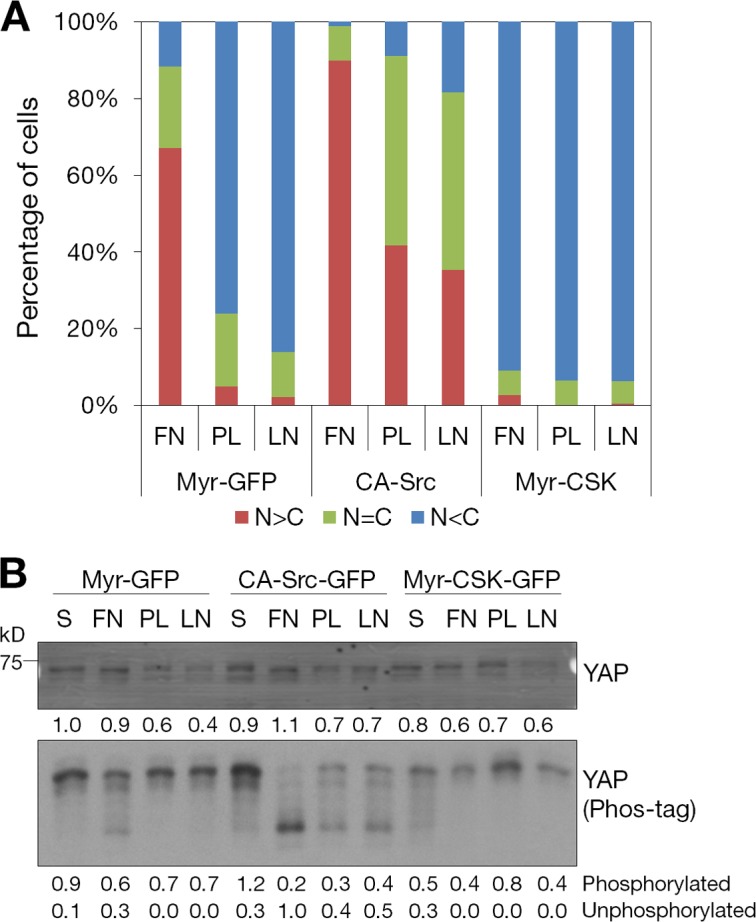
Role of Src in the regulation of YAP by ECM adhesion. (A) Effects of Src on cell–matrix adhesion regulation of YAP subcellular localization. Doxycycline-inducible myr-GFP– (control), CA-Src-GFP–, or myr-CSK-GFP–expressing MCF-10A cells were treated with 1 µg/ml doxycycline for 12 h and maintained in starvation medium containing doxycycline for an additional 24 h. Cells were dissociated and seeded on fibronectin (FN)-, poly-d-lysine (PL)–, or laminin (LN)-coated coverslips in starvation medium. Subcellular localization of YAP was identified by immunofluorescence staining, and >200 cells from eight random views were quantified and confirmed in two independent experiments. (B) Effects of myr-GFP, CA-Src-GFP, and myr-CSK-GFP expression on YAP phosphorylation. Cells described in A were lysed with 2× SDS buffer and separated by SDS-PAGE or Phos-tag SDS-PAGE gels and immunoblotted with anti-YAP antibody.
Figure 9.
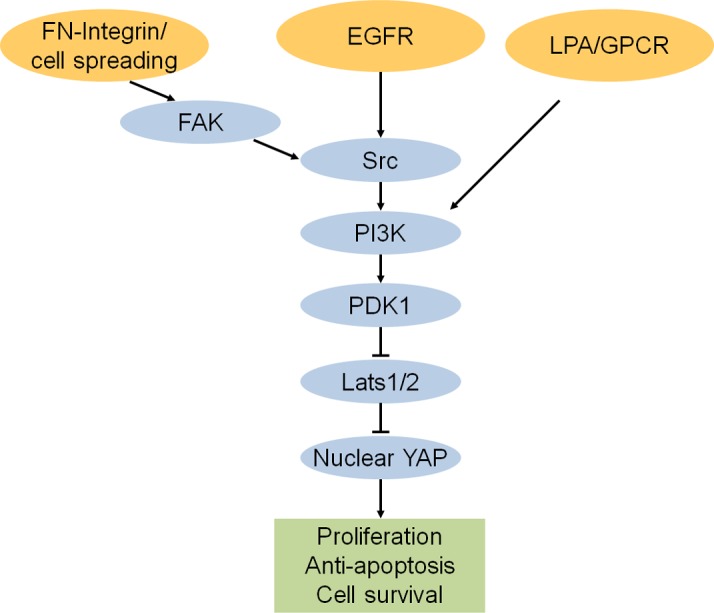
Upstream negative regulators of the Hippo pathway. Soluble growth factors including EGF and LPA inactivate the growth-inhibitory Hippo pathway through the stimulation of PI3K–PDK1 signaling. EGFR, but not LPA, signaling depends on Src kinase. Integrin receptors bind to fibronectin (FN), and mechanical stimulus triggers FAK-Src signaling and leads to the activation of YAP in a PI3K–PDK1-dependent manner.
Discussion
In this study, we find that an integrin signaling pathway regulates the Hippo pathway and nuclear localization of YAP. Previous work demonstrated that PI3K–PDK1 signaling activity counters Hippo signaling and activates nuclear YAP by inhibiting Lats kinase activity in response to soluble mitogenic growth factors (Straßburger et al., 2012; Fan et al., 2013; Gumbiner and Kim, 2014). Our observation in the current study that PI3K and PDK1 inhibitors cause Lats-dependent YAP nuclear exclusion in highly spread subconfluent cells, even in the complete absence of soluble mitogens, suggested that PI3K–PDK1 might have similar roles in integrin adhesion signaling. This was confirmed by the evidence for a role of FAK-Src signaling in Lats-dependent regulation of nuclear YAP, because FAK-Src activation is a major mechanism of integrin signaling, including activation of PI3K in response to adhesion to fibronectin (Tilghman and Parsons, 2008; Hynes, 2009; Schwartz, 2010). Moreover, adhesion to fibronectin was found to specifically enhance YAP nuclear localization dependent on the FAK–Src–PI3K–PDK1 signaling pathway. Thus, the well-known mitogenic integrin signaling pathway appears to operate in parallel with mitogenic growth factors to counteract the growth inhibitory Hippo pathway.
Other studies have also provided evidence for a role of integrins in regulation of YAP activity. The expression of constitutively active β1-integrin mutants in membrane type 1 matrix metalloproteinase–null skeletal stem cells triggers FAK activation and increases nuclear localization of YAP/TAZ (Tang et al., 2013). Also, mechanical stimulation of osteoblasts induced activation of YAP and was dependent on Src-JNK activity but was prevented in integrin αv–deficient osteoblasts (Kaneko et al., 2014). Furthermore, homozygous knockout of YAP in mice causes defects in axial development and yolk sac vasculature at embryonic day 8.5 that are very similar to mouse embryos carrying mutations influencing fibronectin–integrin α5β1 interactions as well as Src, Yes, and Fyn triple-knockout mouse embryos (Morin-Kensicki et al., 2006). We suggest that these integrin-YAP relationships may be mediated by the FAK–Src–PI3K–PDK1 pathway via Lats as described in our study.
One earlier study did not detect a role for integrin-FAK signaling in the regulation of nuclear YAP in response to cell spreading, arguing instead for a distinct cell shape/actin/Rho signaling pathway (Zhao et al., 2012). In that study, FAK inhibitor treatment was not found to inhibit cell attachment–induced YAP dephosphorylation. Using their reported concentrations of the same FAK inhibitor (1 µM PF-562271), we observed a similar result regarding YAP phosphorylation status (Fig. 4 A, eighth lane). However, increased amounts of PF-562271 in doses that inhibit FAK autophosphorylation dramatically increase the phosphorylation of YAP (Fig. 4 A). Also, the more selective FAK inhibitor PF-573228 leads to YAP phosphorylation at substantially lower concentrations (Fig. 4 A). Moreover, expression of dominant-negative FAK protein FRNK prevents fibronectin-induced dephosphorylation and nuclear accumulation of YAP, demonstrating that the Hippo pathway is controlled by the activation of FAK (Fig. 4, C and D). FAK inhibitors increased the cytoplasmic localization of YAP in a Lats-dependent manner (Fig. 4 B), indicating that they act through the Hippo pathway. In the earlier study (Zhao et al., 2012), detecting the effect of fibronectin adhesion on YAP may have been missed because it requires very careful control of cell-plating conditions to avoid residual mitogens, including blocking autocrine-secreted amphiregulin with neutralizing antibodies (Zhang et al., 2009; Fan et al., 2013). Under the conditions of our experiments, the effects of FAK inhibitors and cell adhesion were consistent, as were the roles of Src and PI3K signaling.
In the context of integrin-FAK signaling, we find that Src controls YAP activity in a Lats-dependent manner. Depletion of Lats1/2 kinases prevents YAP nuclear exclusion caused by SFK inhibitors (Fig. 1 D). Also, SFK inhibitor PP2 increases the phosphorylation of YAP on S127 residue (Fig. 1 C), a well-known Lats target phosphorylation site that induces 14-3-3 binding and cytoplasmic retention (Zhao et al., 2007), whereas the activation of Src, through overexpression of active Src or CSK depletion, decreases the phosphorylation of YAP (Fig. 2, A and D) and Mst-dependent phosphorylation of Lats (Fig. 3 A). In contrast, expression of membrane-targeted CSK increased the YAP nuclear exclusion (Fig. S3). Src appears to exert its effects on Lats and YAP via PI3K and PDK1 because PI3K or PDK1 inhibitor treatment prevents the effect of CA-Src on nuclear YAP. The stimulation of PDK1 by EGF and LPA signaling was previously found to dissociate the Hippo pathway complex and activate YAP (Fan et al., 2013). Our findings also indicate that Src stimulation of the PI3K–PDK1 pathway causes YAP nuclear accumulation because of its effects on the core Hippo pathway complex and Lats activity.
A recent study found that Src and Yes directly bind, phosphorylate, and regulate the activity of YAP rather than control the Hippo pathway during epithelial regeneration in inflammatory diseases (Taniguchi et al., 2015). However, we failed to detect the tyrosine phosphorylation of YAP by Src-GFP or CA-Src-GFP expression in HEK-293T cells but confirmed the reduction of YAP phosphorylation on S127 residue (Fig. S4 A). Interestingly, tyrosine residues of Mob1, Sav1, Nf2, and Lats1, but not Mst2, were phosphorylated by the expression of CA-Src-GFP in HEK-293T cells (Fig. S4, B and C). Although we cannot rule out the possibility that Src also interacts with other Hippo pathway components (Sudol, 1994; Zaidi et al., 2004; Rosenbluh et al., 2012; Enomoto and Igaki, 2013; Taniguchi et al., 2015), especially in other contexts, the sum of our findings demonstrates that Src affects YAP in MCF-10A cells through the Hippo complex and Lats activity.
MCF-10A cell attachment to fibronectin and collagen I (unpublished data), but not laminin or poly-d-lysine, induces nuclear localization of YAP (Fig. 7 A). In these experiments, substrate rigidity and mitogen starvation conditions are the same after attachment to different substrate-coated coverslips. Spreading on fibronectin, but not poly-d-lysine, is known to increase the phosphorylation of FAK and paxillin (Weiger et al., 2009). Laminin and fibronectin differentially activate Rho family GTPases through discrete signaling pathways involving the α3β1 and α5β1 integrins (Gu et al., 2001). Thus, our findings indicate that extracellular matrix composition delivers distinct signals to control the Hippo pathway.
The fibronectin–integrin signaling pathway identified in this study may contribute, at least in part, to the observed mechanotransduction mechanism that regulates the Hippo pathway and/or YAP nuclear activity. Cell shape and the actin cytoskeleton are critical regulators of YAP nuclear activity (Dupont et al., 2011; Sansores-Garcia et al., 2011; Wada et al., 2011; Aragona et al., 2013; Kim et al., 2013). Spread, flattened cells under tension have nuclear YAP, whereas round cells on soft substrates exhibit cytoplasmic YAP (Dupont et al., 2011; Wada et al., 2011), and loss of actin regulatory proteins leads to increased YAP activity (Sansores-Garcia et al., 2011; Aragona et al., 2013). Integrins are well-known mechanotransducers that link to the actin cytoskeleton and cause cell spreading and increased tension (Schwartz, 2010; Weber et al., 2011), and therefore it makes sense that FAK-Src–mediated integrin signaling could be involved. However, in some cases, the effect of cell shape and perturbation of the actin cytoskeleton affects YAP independent of Lats activity or the Hippo signaling cascade (Dupont et al., 2011; Aragona et al., 2013), so there may also be a shape-dependent signaling mechanism distinct from the pathway we describe. Rho signaling has been implicated in the actin/tension-dependent shape pathway (Takeichi, 2014), but the detailed mechanism of YAP regulation is not yet understood. Importantly, Lats-dependent mechanisms of cell shape/cytoskeletal regulation of nuclear YAP have also been reported (Sansores-Garcia et al., 2011; Wada et al., 2011; Halder et al., 2012; Zhao et al., 2012; Kim et al., 2013). We propose that the activation of PI3K–PDK1 by integrin-FAK-Src signaling is a component of the reported Lats-dependent cell shape/mechanotransduction perhaps in parallel with the reported Lats-independent regulation of YAP by cell shape. It is worth noting that PI3K signaling is also known to regulate Rho/Rac signaling (Welch et al., 2003; Di Paolo and De Camilli, 2006), so it is possible that there is overlap between the two mechanisms.
In summary, we demonstrate that the adhesion of cells to fibronectin functions as a negative upstream regulator of the Hippo pathway via the FAK–Src–PI3K–PDK1 pathway. Our results provide an explanation for the effects of the fibronectin–integrin interaction on YAP nuclear localization and can account, at least in part, for the Lats-dependent components of mechanotransduction.
Materials and methods
Cell culture
MCF-10A human mammary epithelial cells (a gift from Joan S. Brugge, Harvard Medical School, Boston, MA) were cultured in DMEM/F12 medium supplemented with 5% of horse serum, 0.5 µg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 µg/ml insulin, and 20 ng/ml recombinant human EGF (Debnath et al., 2003). MCF-10A cells were passaged every 2–3 d and cultured for no more than 25 passages. HEK-293T cells were purchased from ATCC and cultured in DMEM/F12 medium supplemented with 10% FBS, sodium pyruvate, and nonessential amino acids.
Reagents
siRNA oligonucleotides specific to human Src (GAGAACCUGGUGUGCAAAG), Fyn (GCUCUGAAAUUACCAAAUC), Yes (GAAGGACCCUGAUGAAAGA), CSK (L-003110-00-0005), Nf2 (L-003917-00-0005), Lats1 (L-004632-00-0005), Lats2 (L-003865-00-0005; GE Healthcare), and RNAiMax (Life Technologies) were used to knock down the expression of genes that we investigated. On-target plus nontargeting pool (D-001810-10-05; GE Healthcare) was used as a control siRNA. Antibodies used for immunofluorescence staining or Western blot include YAP (1:100 dilution for immunofluorescence staining, 1:1,000 dilution for Western blotting; mouse; no. 63.7), CSK (1:1,000 dilution; rabbit; no. C-20), Sav1 (1:1,000 dilution; mouse; no. JJ-6), FAK (1:1,000 dilution; rabbit; no. C-20), GFP (1:2,000 dilution; mouse; no. B-2), c-Myc (1:1,000 dilution; mouse; no. 9E10; Santa Cruz Biotechnology, Inc.), Src (1:1,000 dilution; mouse; no. 2110), phospho-PI3K (1:1,000 dilution; rabbit; no. 4228), AKT (1:1,000 dilution; mouse; no. 2920), pAkt-T308 (1:1,000 dilution; rabbit; no. 4056), pAkt-S473 (1:1,000 dilution; rabbit; no. 3787), pSrc-Y416 (1:1,000 dilution; rabbit; no. 2113), pYAP-S127 (1:1,000 dilution; rabbit; no. 4911), pLats1-T1079 (1:1,000 dilution; rabbit; no. 8654), pLats1-S909 (1:1,000 dilution; rabbit; no. 9157), PDK1 (1:1,000; rabbit; no. 3062), Fyn (1:1,000; rabbit; no. 4023; Cell Signaling Technology), pFAK-Y397 (1:1,000 dilution; rabbit; no. F7926), Flag (1:1,000 dilution; mouse; no. F1804), γ-tubulin (1:3,000 dilution; rabbit; no. T3559), β-actin (1:5,000 dilution; mouse; no. A5316; Sigma-Aldrich), HA (1:1,000 dilution; rabbit; no. PRB-101P; Covance), Yes (1:1,000 dilution; mouse; no. 610375; BD), p85α (1:1,000 dilution; rabbit; no. 04–403), and phospho-tyrosine (1:1,000 dilution; mouse; no. 05–321; EMD Millipore). The following inhibitors were used: SFK inhibitors SKI-1 and SU6656, Src/Abl inhibitor dasatinib monohydrate, Abl inhibitor imatinib mesylate, PDK1 inhibitor BX-795, FAK inhibitors PF-573228 and PF-562271 (Santa Cruz Biotechnology, Inc.), SFK inhibitor PP2 and its inactive analogue PP3 (EMD Millipore), and PI3K inhibitor wortmannin (Sigma-Aldrich). Unless indicated otherwise, all inhibitors were used at a concentration of 10 µM, except for PF-573228 and BX-795 (5 µM). Flag-YAP and Flag-Lats1, under a cytomegalovirus (CMV) promoter, were gifts from Marius Sudol (Weis Center for Research, Danville, PA). HA-mob1, HA-Nf2, HA-Sav1, and Myc-Mst2 in the pcDNA3 backbone were gifts from Kun-Liang Guan (University of California, San Diego, La Jolla, CA). Src-GFP (GFP-tagged chicken c-src under a CMV promoter), CA-Src-GFP (GFP-tagged chicken c-src mutant; Y527F), and DN-Src-GFP (GFP-tagged deletion mutant of the chicken c-src; amino acids 1–251) were gifts from Margaret Frame (University of Edinburgh, Edinburgh, Scotland, UK).
Construction of plasmids
To generate doxycycline-inducible lentiviral vector, coding sequence of TurboRFP in TRIPZ (GE Healthcare) was replaced with GFP, and the following genes were cloned to generate GFP-fusion proteins. The constitutively active Src was from CA-Src-GFP vector. Myr-CSK was amplified from the human Orfeome library (Lamesch et al., 2007) with primers containing a myristoylation signal peptide sequence of chicken c-src (amino acids 1–16). Lysine 222 residue of CSK was mutated to arginine using the QuikChange II Site-Directed Mutagenesis kit (Agilent Technologies) to create the kinase-dead form of CSK (K222R). FRNK was amplified (amino acids 693–1,053) from the chicken FAK expression vector under a CMV promoter (a gift from James Casanova, University of Virginia, Charlottesville, VA). Induced expression of target genes was validated in HEK-293T cells by transient transfection and doxycycline treatment.
Construction of reporters
To measure the transcriptional activity of TEAD-YAP, we developed a HIP/HOP-flash luciferase reporter system. HOP-flash contains multiple copies of wild-type TEAD-binding sites with minimal promoter and a luciferase reporter gene. HIP-flash was generated as a negative control for HOP-flash activity. TEAD-binding sites in the promoter of CTGF (Zhao et al., 2008) were multimerized (Nakajima and Yaoita, 1997) using primers 5′-GGTGGGGAGGAATGCGAGGAATGTCCCTGTTTGTGGTGGGGA-3′ and 5′-ACAAACAGGGACATTCCTCGCATTCCTCCCCACCACAAACAG-3′ and then were cloned into pCR-bluntII-Topo vector (Life Technologies). Mutated TEAD-binding sites were multimerized using primers 5′-GGTGGGGAGagATGCGAGagATGTCCCTGTTTGTGGTGGGGA-3′ and 5′-ACAAACAGGGACATctCTCGCATctCTCCCCACCACAAACAG-3′. Wild-type and mutated TEAD-binding sites are underlined, and mutated sequences are marked in lowercase letters. DNA fragments with eight TEAD-binding sites or seven mutant TEAD-binding sites were ligated into pGL3-basic vector (Promega) together with minimal promoter of TOP-flash reporter (Korinek et al., 1997), creating HOP-flash and HIP-flash reporters, respectively.
Generation of doxycycline-inducible cell lines
Lentiviral doxycycline-inducible vectors were cotransfected with envelope vector pMD2.G (Addgene plasmid 12259) and packaging vector psPAX2 (Addgene plasmid 12260) into HEK-293T/17 cells using Lipofectamine 2000 (Life Technologies). After 18 h of transfection, the medium was replaced with UltraCULTURE medium (Lonza) supplemented with l-glutamine and antibiotics. Starting the next day, the lentivirus-containing supernatants were collected every 12 h for 3 d and stored at 4°C. After spin-down at 2,000 rpm for 5 min to remove cell debris, supernatants were filtered using a 0.45-µm pore size filter. About 30 ml of filtered supernatants were then transferred into ultracentrifuge tubes containing 5 ml of 20% sucrose cushion. Lentiviral particles were concentrated by centrifugation at 100,000 g for 2 h at 4°C and finally resuspended in 1× HBSS buffer. MCF-10A cells were incubated overnight with lentiviral particles and 6 µg/ml polybrene (EMD Millipore) and replaced with fresh medium. Antibiotics selection was started 3 d after transduction with 2 µg/ml puromycin and maintained on selection medium for an additional 7 d. After selection, cells were treated with 2 µg/ml doxycycline for 24 h, and MCF-10A cells with high levels of GFP were collected by FACS cell sorting. Cells were then maintained in the absence of doxycycline.
Mitogen and inhibitor treatment
For the inhibitor treatment experiments, 6 × 104 MCF-10A cells were seeded into 1 well of 24-well plates (∼10% cell density). After 24 h, cells were serum starved in DMEM/F12 medium supplemented with 1 µg/ml amphiregulin-blocking antibody (AF-262; R&D Systems) for 12–24 h. The blocking antibody to amphiregulin, an EGFR ligand secreted by MCF-10A cells (Zhang et al., 2009), was added to the starvation medium to prevent non–cell-autonomous activation of EGFR signaling. 3 h before the treatment with mitogens or inhibitors, the culture medium was replaced with fresh starvation medium to remove secreted soluble factors from cells. Inhibitors were added for 30 min. For the cotreatment experiments, cells were pretreated with the indicated inhibitors for 30 min and then treated with 20 ng/ml EGF (PeproTech) or 25 µM LPA (Sigma-Aldrich). Doxycycline-inducible MCF-10A cell lines were induced by 1 µg/ml doxycycline for 12 h in complete medium and starved for another 24 h with 1 µg/ml amphiregulin-blocking antibody and 1 µg/ml doxycycline. We performed at least two independent experiments.
Immunofluorescence staining and luciferase assay
MCF-10A cells seeded in a 24-well plate or in fibronectin-, poly-d-lysine–, or laminin-coated coverslips were used in immunofluorescence staining. Cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature (or overnight at 4°C) and washed three times for 5 min each in 100 mM glycine containing PBS, followed by permeabilization with 0.1% Triton X-100 in PBS for 10 min. After blocking with 3% nonfat dry milk in PBS for 1 h, cells were incubated with primary antibody diluted in 1% BSA/PBS overnight at 4°C. After washing with PBS, cells were incubated with Alexa Fluor 488– or 594–conjugated secondary antibodies (Life Technologies) for 1 h and washed with PBS. Cell nuclei were counterstained and mounted with a mounting medium with DAPI (Vectashield; Vector Laboratories). Immunofluorescence staining images were collected at room temperature using a 20× objective (LUCPlanFLN; NA 0.45; Olympus) on an inverted microscope (IX71; Olympus) coupled to a camera (ORCA-R2 C10600-10B; Hamamatsu Photonics). Images were acquired, normalized, analyzed by SlideBook 5.0 (Intelligent Imaging Innovations), and assembled for illustration using Photoshop (Adobe). For luciferase reporter assay, myr-GFP– or CA-Src-GFP–inducible HEK-293T cells were seeded in 24-well plates the day before transfection. HIP-flash or HOP-flash reporter and indicated plasmids were cotransfected with Lipofectamine 2000 (Life Technologies). 24 h after transfection, myr-GFP– or CA-Src-GFP–inducible HEK-293T cells were incubated in 0.1% FBS containing DMEM/F12 medium with or without 1 µg/ml doxycycline for 16 h. Luciferase activity was measured with a luciferase assay system (Promega).
HPLC gel filtration chromatography
Doxycycline-inducible TurboRFP or myr-CSK-GFP–expressing MCF-10A cells were seeded in two 150-mm-diameter plastic culture dishes at low cell density (∼30%) and treated with 2 µg/ml doxycycline for 12 h in complete medium. Cells were then starved for 24 h in DMEM/F12 medium containing 2 µg/ml doxycycline. Cells were washed with ice-cold HBSS and resuspended in S100/P100 buffer (10 mM Hepes, pH 7.4, 1 mM EDTA, and 150 mM NaCl) supplemented with protease and phosphatase inhibitors. Cells were passed repeatedly through a 27-gauge needle and then subjected to low speed centrifugation at 21,130 g for 5 min. Supernatants were further ultracentrifuged at 100,000 g for 1 h. The resulting supernatants were collected and concentrated using Microcon centrifugal filters (EMD Millipore). The concentrated supernatants were fractionated by gel filtration HPLC on a Superose 12 column (GE Healthcare) as follows: 0.15 ml of the cytosolic fraction was loaded onto the column at a flow rate of 0.5 ml/min, the absorbance at 280 nm was monitored, and 0.5-ml fractions were collected. The proteins were incubated with 2 ml of chilled acetone at −20°C for 2 h and precipitated by centrifugation at 12,000 g for 5 min. The proteins were resuspended in SDS-DTT-urea buffer (6 M urea, 1% wt/vol DTT, 62.5 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, and 0.01% bromophenol blue) and subjected to Western blotting.
Immunoprecipitation and immunoblotting
For immunoprecipitation, HEK-293T cells were washed with ice-cold HBSS and lysed with radioimmunoprecipitation assay buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 2 mM EDTA, 1 mM Na3VO4, 10 mM NaF, and protease inhibitors) or NP-40 buffer (150 mM NaCl, 1.0% NP-40, 50 mM Tris, pH 8.0, 1 mM Na3VO4, 10 mM NaF, and protease inhibitors) at 24 h after transfection. Cell lysates were incubated with the indicated immunoprecipitation antibodies and Sepharose 4 Fast Flow Protein A/G beads (GE Healthcare) or anti-flag M2 affinity gels (Sigma-Aldrich). For regular Western blotting, cells were washed with ice-cold HBSS and directly lysed with 2× SDS buffer (0.12 M Tris, pH 6.8, 4% SDS, 20% glycerol, and 10% 2-mercaptoethanol) after brief sonication to reduce viscosity. For regular SDS-PAGE, the lysates were separated by 5–17% gradient SDS-PAGE gel and transferred to polyvinylidene difluoride membrane (EMD Millipore) using a semidry transfer apparatus (Bio-Rad Laboratories). Phos-tag SDS-PAGE gel consisted of a stacking gel (4.5% wt/vol acrylamide, 125 mM Tris, pH 6.8, and 0.1% SDS) and a separating gel (7% wt/vol acrylamide, 380 mM Tris, pH 8.8, 15 µM Phos-tag acrylamide, 30 µM MnCl2, and 0.1% SDS). A 37.5:1 wt/vol mixture of acrylamide/bis-acrylamide (Bio-Rad Laboratories) was used as an acrylamide solution. Gel electrophoresis was performed at 10 mA with SDS running buffer (25 mM Tris, 192 mM glycine, and 0.1% wt/vol SDS) until the dye reached the bottom. After electrophoresis, acrylamide gel was equilibrated two times with transfer buffer (25 mM Tris, 192 mM glycine, 0.1% wt/vol SDS, and 10% methanol) containing 10 mM EDTA for 10 min, followed by washing two times with transfer buffer without EDTA. Wet transfer was performed to transfer proteins onto polyvinylidene difluoride membrane (EMD Millipore). The blot was sequentially incubated with primary antibody and HRP-conjugated secondary antibody. The HRP reaction was performed with chemiluminescent HRP substrate (EMD Millipore) and exposed to x-ray film (Genesee Scientific) or detected using an imaging system (LAS-3000; Fujifilm). Proteins were also detected with an infrared imaging system (Odyssey; LI-COR Biosciences) using IRDye-labeled secondary antibody (LI-COR Biosciences). Western blot images were assembled for illustration using Photoshop (Adobe). Band intensity was quantified using ImageJ (National Institutes of Health).
Attachment of cells to coated coverslips
Coverslips were acid-washed and coated with 20 µg/ml fibronectin (Sigma-Aldrich), 50 µg/ml poly-d-lysine, or 20 µg/ml laminin. MCF-10A cells were starved for 24 h with DMEM/F12 medium containing 1 µg/ml amphiregulin-blocking antibody. Cells were dissociated with 2 ml Accutase (Life Technologies) and centrifuged at 1,000 rpm for 5 min. Cells were then washed two times with DMEM/F12 medium to remove residual Accutase. 6 × 104 MCF-10A cells were held in suspension by rotating on a tube rotator at 37°C for 30 min. Cells were then allowed to attach to coated coverslips in starvation medium and incubated at 37°C for 2–3 h. For Western blotting, the suspended cells were seeded on 25-mm-diameter fibronectin-, poly-d-lysine–, or laminin-coated coverslips in starvation medium for 2 h and directly lysed with 2× SDS buffer.
Online supplemental material
Fig. S1 shows the validation of siRNA depletion of SFKs and the activation of downstream PI3K signaling by Src expression. Fig. S2 shows a diagram of HIP/HOP-flash reporters and the validation of the specificity of reporter gene expression in response to YAP/TAZ activity. Fig. S3 shows the cytoplasmic localization of YAP by the induced expression of myr-CSK. Fig. S4 shows the tyrosine phosphorylation of Hippo pathway components by the expression of Src kinase. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201501025/DC1.
Supplementary Material
Acknowledgments
We thank Martha Spano for generous technical help in HPLC gel filtration chromatography and the University of Virginia Flow Cytometry Core Facility for cell sorting.
The work described in this article was supported by National Institutes of Health grant R01 GM098615 (to B.M. Gumbiner).
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- CIP
- contact inhibition of proliferation
- CMV
- cytomegalovirus
- CSK
- C-terminal Src kinase
- EGFR
- EGF receptor
- FRNK
- FAK-related nonkinase
- HIP
- Hippo-YAP signaling incompetent promoter
- HOP
- Hippo-YAP signaling optimal promoter
- Lats
- large tumor suppressor homologue
- LPA
- lysophosphatidic acid
- PI3K
- phosphatidylinositol 4,5-bisphosphate 3-kinase
- SFK
- Src family kinase
- TAZ
- transcriptional coactivator with PDZ-binding motif
- TEAD
- TEA domain family member
- YAP
- Yes-associated protein
References
- Aragona M., Panciera T., Manfrin A., Giulitti S., Michielin F., Elvassore N., Dupont S., and Piccolo S.. 2013. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 154:1047–1059. 10.1016/j.cell.2013.07.042 [DOI] [PubMed] [Google Scholar]
- Basu S., Totty N.F., Irwin M.S., Sudol M., and Downward J.. 2003. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell. 11:11–23. 10.1016/S1097-2765(02)00776-1 [DOI] [PubMed] [Google Scholar]
- Cabodi S., del Pilar Camacho-Leal M., Di Stefano P., and Defilippi P.. 2010. Integrin signalling adaptors: not only figurants in the cancer story. Nat. Rev. Cancer. 10:858–870. 10.1038/nrc2967 [DOI] [PubMed] [Google Scholar]
- Calvo F., Ege N., Grande-Garcia A., Hooper S., Jenkins R.P., Chaudhry S.I., Harrington K., Williamson P., Moeendarbary E., Charras G., and Sahai E.. 2013. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15:637–646. 10.1038/ncb2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J., Muthuswamy S.K., and Brugge J.S.. 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 30:256–268. 10.1016/S1046-2023(03)00032-X [DOI] [PubMed] [Google Scholar]
- Di Paolo G., and De Camilli P.. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature. 443:651–657. 10.1038/nature05185 [DOI] [PubMed] [Google Scholar]
- Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature. 474:179–183. 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- Enomoto M., and Igaki T.. 2013. Src controls tumorigenesis via JNK-dependent regulation of the Hippo pathway in Drosophila. EMBO Rep. 14:65–72. 10.1038/embor.2012.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R., Kim N.G., and Gumbiner B.M.. 2013. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. USA. 110:2569–2574. 10.1073/pnas.1216462110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan G., Eble D.M., Lucchesi P.A., and Samarel A.M.. 2000. Focal adhesion kinase is involved in angiotensin II-mediated protein synthesis in cultured vascular smooth muscle cells. Circ. Res. 87:710–716. 10.1161/01.RES.87.8.710 [DOI] [PubMed] [Google Scholar]
- Gu J., Sumida Y., Sanzen N., and Sekiguchi K.. 2001. Laminin-10/11 and fibronectin differentially regulate integrin-dependent Rho and Rac activation via p130Cas-CrkII-DOCK180 pathway. J. Biol. Chem. 276:27090–27097. 10.1074/jbc.M102284200 [DOI] [PubMed] [Google Scholar]
- Gumbiner B.M., and Kim N.G.. 2014. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 127:709–717. 10.1242/jcs.140103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G., Dupont S., and Piccolo S.. 2012. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 13:591–600. 10.1038/nrm3416 [DOI] [PubMed] [Google Scholar]
- Hynes R.O. 2009. The extracellular matrix: not just pretty fibrils. Science. 326:1216–1219. 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R., and Halder G.. 2014. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 13:63–79. 10.1038/nrd4161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K., Ito M., Naoe Y., Lacy-Hulbert A., and Ikeda K.. 2014. Integrin αv in the mechanical response of osteoblast lineage cells. Biochem. Biophys. Res. Commun. 447:352–357. 10.1016/j.bbrc.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., and Asthagiri A.R.. 2011. Matrix stiffening sensitizes epithelial cells to EGF and enables the loss of contact inhibition of proliferation. J. Cell Sci. 124:1280–1287. 10.1242/jcs.078394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Kushiro K., Graham N.A., and Asthagiri A.R.. 2009. Tunable interplay between epidermal growth factor and cell–cell contact governs the spatial dynamics of epithelial growth. Proc. Natl. Acad. Sci. USA. 106:11149–11153. 10.1073/pnas.0812651106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N.G., Koh E., Chen X., and Gumbiner B.M.. 2011. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA. 108:11930–11935. 10.1073/pnas.1103345108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Kim M., Lee S., Kuninaka S., Saya H., Lee H., Lee S., and Lim D.S.. 2013. cAMP/PKA signalling reinforces the LATS–YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J. 32:1543–1555. 10.1038/emboj.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E., Kinoshita-Kikuta E., Takiyama K., and Koike T.. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics. 5:749–757. 10.1074/mcp.T500024-MCP200 [DOI] [PubMed] [Google Scholar]
- Komuro A., Nagai M., Navin N.E., and Sudol M.. 2003. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J. Biol. Chem. 278:33334–33341. 10.1074/jbc.M305597200 [DOI] [PubMed] [Google Scholar]
- Korinek V., Barker N., Morin P.J., van Wichen D., de Weger R., Kinzler K.W., Vogelstein B., and Clevers H.. 1997. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 275:1784–1787. 10.1126/science.275.5307.1784 [DOI] [PubMed] [Google Scholar]
- Lamesch P., Li N., Milstein S., Fan C., Hao T., Szabo G., Hu Z., Venkatesan K., Bethel G., Martin P., et al. 2007. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 89:307–315. 10.1016/j.ygeno.2006.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Okura M., and Imamoto A.. 2002. Focal adhesions require catalytic activity of Src family kinases to mediate integrin-matrix adhesion. Mol. Cell. Biol. 22:1203–1217. 10.1128/MCB.22.4.1203-1217.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low B.C., Pan C.Q., Shivashankar G.V., Bershadsky A., Sudol M., and Sheetz M.. 2014. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 588:2663–2670. 10.1016/j.febslet.2014.04.012 [DOI] [PubMed] [Google Scholar]
- Lu Y., Yu Q., Liu J.H., Zhang J., Wang H., Koul D., McMurray J.S., Fang X., Yung W.K., Siminovitch K.A., and Mills G.B.. 2003. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J. Biol. Chem. 278:40057–40066. 10.1074/jbc.M303621200 [DOI] [PubMed] [Google Scholar]
- McClatchey A.I., and Yap A.S.. 2012. Contact inhibition (of proliferation) redux. Curr. Opin. Cell Biol. 24:685–694. 10.1016/j.ceb.2012.06.009 [DOI] [PubMed] [Google Scholar]
- Miller E., Yang J., DeRan M., Wu C., Su A.I., Bonamy G.M., Liu J., Peters E.C., and Wu X.. 2012. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 19:955–962. 10.1016/j.chembiol.2012.07.005 [DOI] [PubMed] [Google Scholar]
- Mo J.S., Yu F.X., Gong R., Brown J.H., and Guan K.L.. 2012. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 26:2138–2143. 10.1101/gad.197582.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin-Kensicki E.M., Boone B.N., Howell M., Stonebraker J.R., Teed J., Alb J.G., Magnuson T.R., O’Neal W., and Milgram S.L.. 2006. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol. Cell. Biol. 26:77–87. 10.1128/MCB.26.1.77-87.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K., and Yaoita Y.. 1997. Construction of multiple-epitope tag sequence by PCR for sensitive Western blot analysis. Nucleic Acids Res. 25:2231–2232. 10.1093/nar/25.11.2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M. 2012. Regulation of the SRC family kinases by Csk. Int. J. Biol. Sci. 8:1385–1397. 10.7150/ijbs.5141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiman C.M., Hertz W.M., and Cambier J.C.. 1994. Activation of phosphatidylinositol-3′ kinase by Src-family kinase SH3 binding to the p85 subunit. Science. 263:1609–1612. 10.1126/science.8128248 [DOI] [PubMed] [Google Scholar]
- Provenzano P.P., and Keely P.J.. 2011. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J. Cell Sci. 124:1195–1205. 10.1242/jcs.067009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A., Malik R.K., Hildebrand J.D., and Parsons J.T.. 1997. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Mol. Cell. Biol. 17:6906–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts W.G., Ung E., Whalen P., Cooper B., Hulford C., Autry C., Richter D., Emerson E., Lin J., Kath J., et al. 2008. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 68:1935–1944. 10.1158/0008-5472.CAN-07-5155 [DOI] [PubMed] [Google Scholar]
- Rosenbluh J., Nijhawan D., Cox A.G., Li X., Neal J.T., Schafer E.J., Zack T.I., Wang X., Tsherniak A., Schinzel A.C., et al. 2012. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 151:1457–1473. 10.1016/j.cell.2012.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilands E., Cans C., Fincham V.J., Brunton V.G., Mellor H., Prendergast G.C., Norman J.C., Superti-Furga G., and Frame M.C.. 2004. RhoB and actin polymerization coordinate Src activation with endosome-mediated delivery to the membrane. Dev. Cell. 7:855–869. 10.1016/j.devcel.2004.09.019 [DOI] [PubMed] [Google Scholar]
- Sansores-Garcia L., Bossuyt W., Wada K., Yonemura S., Tao C., Sasaki H., and Halder G.. 2011. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 30:2325–2335. 10.1038/emboj.2011.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.A. 2010. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2:a005066 10.1101/cshperspect.a005066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D.D., Xue W., Krall E.B., Bhutkar A., Piccioni F., Wang X., Schinzel A.C., Sood S., Rosenbluh J., Kim J.W., et al. 2014. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 158:171–184. 10.1016/j.cell.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieg D.J., Hauck C.R., and Schlaepfer D.D.. 1999. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J. Cell Sci. 112:2677–2691. [DOI] [PubMed] [Google Scholar]
- Slack-Davis J.K., Martin K.H., Tilghman R.W., Iwanicki M., Ung E.J., Autry C., Luzzio M.J., Cooper B., Kath J.C., Roberts W.G., and Parsons J.T.. 2007. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 282:14845–14852. 10.1074/jbc.M606695200 [DOI] [PubMed] [Google Scholar]
- Straßburger K., Tiebe M., Pinna F., Breuhahn K., and Teleman A.A.. 2012. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Dev. Biol. 367:187–196. 10.1016/j.ydbio.2012.05.008 [DOI] [PubMed] [Google Scholar]
- Sudol M. 1994. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 9:2145–2152. [PubMed] [Google Scholar]
- Takeichi M. 2014. Dynamic contacts: rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell Biol. 15:397–410. 10.1038/nrm3802 [DOI] [PubMed] [Google Scholar]
- Tang Y., Rowe R.G., Botvinick E.L., Kurup A., Putnam A.J., Seiki M., Weaver V.M., Keller E.T., Goldstein S., Dai J., et al. 2013. MT1-MMP-dependent control of skeletal stem cell commitment via a β1-integrin/YAP/TAZ signaling axis. Dev. Cell. 25:402–416. 10.1016/j.devcel.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K., Wu L.W., Grivennikov S.I., de Jong P.R., Lian I., Yu F.X., Wang K., Ho S.B., Boland B.S., Chang J.T., et al. 2015. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 519:57–62. 10.1038/nature14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S.M., and Brugge J.S.. 1997. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 13:513–609. 10.1146/annurev.cellbio.13.1.513 [DOI] [PubMed] [Google Scholar]
- Tilghman R.W., and Parsons J.T.. 2008. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. Semin. Cancer Biol. 18:45–52. 10.1016/j.semcancer.2007.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X., Samavarchi-Tehrani P., Narimatsu M., Weiss A., Cockburn K., Larsen B.G., Rossant J., and Wrana J.L.. 2010. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-β-SMAD pathway. Dev. Cell. 19:831–844. 10.1016/j.devcel.2010.11.012 [DOI] [PubMed] [Google Scholar]
- Wada K., Itoga K., Okano T., Yonemura S., and Sasaki H.. 2011. Hippo pathway regulation by cell morphology and stress fibers. Development. 138:3907–3914. 10.1242/dev.070987 [DOI] [PubMed] [Google Scholar]
- Weber G.F., Bjerke M.A., and DeSimone D.W.. 2011. Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 124:1183–1193. 10.1242/jcs.064618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiger M.C., Wang C.C., Krajcovic M., Melvin A.T., Rhoden J.J., and Haugh J.M.. 2009. Spontaneous phosphoinositide 3-kinase signaling dynamics drive spreading and random migration of fibroblasts. J. Cell Sci. 122:313–323. 10.1242/jcs.037564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch H.C., Coadwell W.J., Stephens L.R., and Hawkins P.T.. 2003. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 546:93–97. 10.1016/S0014-5793(03)00454-X [DOI] [PubMed] [Google Scholar]
- Yin F., Yu J., Zheng Y., Chen Q., Zhang N., and Pan D.. 2013. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 154:1342–1355. 10.1016/j.cell.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.X., Zhao B., Panupinthu N., Jewell J.L., Lian I., Wang L.H., Zhao J., Yuan H., Tumaneng K., Li H., et al. 2012. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 150:780–791. 10.1016/j.cell.2012.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S.K., Sullivan A.J., Medina R., Ito Y., van Wijnen A.J., Stein J.L., Lian J.B., and Stein G.S.. 2004. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J. 23:790–799. 10.1038/sj.emboj.7600073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Ji J.Y., Yu M., Overholtzer M., Smolen G.A., Wang R., Brugge J.S., Dyson N.J., and Haber D.A.. 2009. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat. Cell Biol. 11:1444–1450. 10.1038/ncb1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Wei X., Li W., Udan R.S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L., et al. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21:2747–2761. 10.1101/gad.1602907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J.D., Wang C.Y., Chinnaiyan A.M., et al. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22:1962–1971. 10.1101/gad.1664408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Li L., Wang L., Wang C.Y., Yu J., and Guan K.L.. 2012. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 26:54–68. 10.1101/gad.173435.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.