

Abstract
Artificial selection of high yield crops and better livestock is paramount importance in breeding programs. Selection of elite parents with preferred traits from a phalanx of inbred lines is extremely laborious, time-consuming and highly random. General combining ability (GCA) was proposed and has been widely used for the evaluation of parents in hybrid breeding for more than half a century. However, the genetic and molecular basis of GCA has been largely overlooked. Here, we present two pleotropic QTLs are accounting for GCA of days to heading (DTH), plant height (PH) and spikelet per panicle (SPP) using an F2-based NCII design, the BC3F2 population as well as a set of nearly isogenic lines (NILs) with five testers. Both GCA1 and GCA2 were loss-of-function gene in low-GCA parent and gain-of-function gene in high-GCA parent, encoding the putative Pseudo-Response Regulators, OsPRR37 and Ghd7, respectively. Overexpression of GCA1 in low-GCA parent significantly increases GCA effects in three traits. Our results demonstrate that two GCA loci associate with OsPRR37 and Ghd7 and reveal that the genes responsible for important agronomic traits could simultaneously account for GCA effects.
One of the most fundamental functions of living organisms is to pass on to their progeny advantageous traits that ensure positive interactions with their environment, whereas any unfit traits are gradually eliminated by natural environmental pressures. This is the well-known evolutionary theory of natural selection1. Similarly, artificial selection is a process whereby humans select preferred traits during an organism’s breeding. To meet the increasing global demand for food, artificial selection of high-yield crops and improved livestock has been widely applied and is of paramount importance to hybrid breeding programs. However, the selection of parents from a suite of inbred lines is extremely laborious, time-consuming and random. In addition, parents with excellent agronomic traits do not always pass on those traits to their progeny. Thus, Sprague and Tatum proposed the concept of general combining ability (GCA) to evaluate breeding parents2. GCA has been widely and successfully used as a metric for selecting elite parents in conventional livestock and crop breeding programs.
The first research regarding the genetic variance of GCA and specific combining ability (SCA) was reported by Griffing (1956) using a diallelic cross-mating design. Since then, it has become possible to produce precise estimates of GCA and SCA3. As GCA variation is derived from the additive effect of traits with high heritability, alleles accounting for this additive effect are believed to easily accumulate in progeny as a result of artificial selection. By contrast, SCA is characterised by non-additive effects, including dominance, over-dominance and epistasis, which are considered difficult to resolve in progeny due to their low heritability and their interactions with the environment4. Until now, most studies of combining ability have focused on the identification of promising parents5,6,7,8,9,10. However, the genetic and molecular basis of GCA has been largely overlooked. It is important to uncover the genetic basis of GCA to broaden understanding of the molecular mechanisms underlying GCA and improve the selection of elite varieties in hybrid breeding.
Recently, GCA has been used as one of the most reliable ways to predict the performance of hybrids. Several prediction methodologies based on molecular markers and genomic, transcriptomic and metabolic prediction methods have been developed11,12,13,14. A GCA-based model with SCA estimates based on molecular marker data has shown high prediction efficacy11. Furthermore, a haplotype blocks-based model for hybrid performance prediction has been developed. A comparison between single AFLP marker- and GCA-based approaches has shown that the hybrid performance prediction based on GCA effects was effective for inter-group hybrids with a predominance of GCA over SCA12.
More recent studies have attempted to uncover the genetic basis of GCA by employing a quantitative trait locus (QTL) mapping approach15,16,17. Qu et al. identified the genetic loci for combining ability that correspond to agronomic traits through QTL mapping using three testcross populations and a backcross recombination inbred line (BCRIL) in rice. The characteristics of the QTLs for combining ability were found to be similar to those of the QTLs for BCRIL performance16. Qi et al. identified several genetic loci of GCA and SCA in maize using four testers from different heterotic groups and introgression lines (ILs)17. Their studies revealed that GCA is a quantitative trait and is controlled by genes that are similar to those of most mapped QTLs. Moreover, our recent studies revealed that the phenotypic predisposition of the parent in F1 hybrid is correlated with transcriptome preference of the positive GCA parent18. However, the genes responsible for GCA remain to be uncovered.
In the present work, we identified 13 QTLs that account for the GCA of three agronomically important traits associated with rice grain yield by using an F2-based NCII design with five testers. We also finely mapped two major pleotropic QTLs—GCA1 and GCA2—through the BC3F2 population and a set of nearly isogenic lines (NILs). We revealed that GCA1 and GCA2 encode the putative Pseudo-Response Regulator OsPRR37 and Ghd7, respectively. Furthermore, we confirmed that GCA1 functions as a positive regulator not only of the agronomic traits per se but also of GCA by virtue of the overexpression of GCA1 (GCA1OX) in rice. Additional experiments revealed that GCA1 is located in the nucleus and that the loss of the 234 amino acids at its C terminus does not affect its nuclear localisation. Expression profiling of GCA1 indicated that it was constitutively expressed in all developmental stages and tissues and was highly expressed in leaves, young panicles and stem. Our results revealed that the genes for important agronomic traits could simultaneously account for the GCA effects. These findings provide new insight into the molecular genetic basis of GCA and the relationship between important agronomic traits and the effects of GCA.
Results
Variance analysis of combining ability for F2 population
To evaluate the GCA effects of DTH, PH and SPP, we used a diallelic cross of six elite rice varieties. We found that Guangluai #4 (GL) exhibited the lowest GCA effects, whereas Teqing (TQ) exhibited the preferred positive GCA effects for three agronomically important traits (see Supplementary Table S1 online). Therefore, GL and TQ were chosen to generate an F2 population for genetic mapping. To identify the effects of GCA and SCA in the F2 population, variance analysis of GCA and SCA for the three agronomic traits DTH, PH and SPP was carried out on 139 individual plants in the F2 population. The mean squares for the GCA effects from the F2 individuals and testers as well as the SCA effects from progeny of F2 individuals and testers (Testers×F2) were found to be significant for all the three agronomic traits (Table 1). The results showed that the variance components of GCA (Vgca) ranged from 68.6%–96.18%, whereas the variance components of SCA (Vsca) ranged from 6.46%–31.4%, indicating that the GCA variance is substantially larger than the SCA variance (Table 1). These results suggested that both additive and non-additive effects contribute to the genetic variation observed among the hybrids and that the additive effect plays a predominant role in controlling the genetic variation of the three traits, whereas non-additive effects play a minor role.
Table 1. Variance analysis of GCA and SCA of agronomic traits.
| Trait | Testers | F2 | Testers×F2 | Error | Vgca | Vsca |
|---|---|---|---|---|---|---|
| DTH | 243557** | 1328** | 151** | 13 | 93.5% | 6.5% |
| PH | 310630** | 324** | 108** | 18 | 96.2% | 3.8% |
| SPP | 183481** | 2391** | 959** | 227 | 68.6% | 31.4% |
**Significance at the level of P < 0.01.
Identification of QTLs for GCA
Our previous results showed that the GCA effects made a major contribution to the performance of hybrid offspring. To genetically map the GCA loci for the three traits, the GCA values from a segregation population are required as phenotypes for genetic mapping. We first evaluated the GCA effects in 139 individuals derived from the F2 population. As shown in Table 2, the GCA effects among 139 individual plants in the F2 population ranged from −17.8 to 30.6 for DTH, −10.1 to 9.2 for PH and −22.0 to 15.7 for SPP. These results demonstrated that the values of GCA in the F2 population were typical normal distributions, and the mean values of GCA for the three traits were almost zero, suggesting that the GCA effects are quantitative traits. These results indicated that this F2 population was suitable for further QTL analysis of GCA.
Table 2. Performance of GCA in the F2 population.
| GCA Trait | GCA range | GCA mean | Skewness | Kurtosis |
|---|---|---|---|---|
| DTH | −17.8 to 30.6 | 7.19E-07 | 0.8 | 1.1 |
| PH | −10.1 to 9.2 | 1.44E-06 | 0.0 | −0.2 |
| SPP | −22.0 to 15.7 | 5.76E-06 | −0.3 | 0.3 |
We then further mapped the QTLs of GCA for the three agronomic traits. A total of 139 individual plants from the F2 population and 141 molecular markers (SSRs, InDels and SNPs) were used to construct a linkage map with coverage of approximately 1,763 cM (see Supplementary Fig. S1 online). The GCA values of 139 individual plants from the F2 population were obtained from total of 695 TC progenies (see the Method section). A total of 13 major QTLs for GCA of the three agronomic traits were identified on five chromosomes based on a cut-off LOD score ≥ 3.0 (Table 3). Five QTLs for the GCA of DTH were identified on chromosomes 1, 6 and 7, and four of them showed high LOD scores (LOD score > 10). In particular, two QTLs on chromosome 7 explained 29.9% and 34.9% of the variance in the GCA of DTH with additive effects of 6.6 and 7.1 in the F2-based TC population, respectively. The LOD peaks of two QTLs were localised at two distant intervals—RM3670-RM6449 and S77-RM1306—indicating that two different loci control the GCA of DTH on chromosome 7. For the GCA of PH, three QTLs were identified on chromosomes 7 and 11. Two QTLs on chromosome 7 explained 42.4% and 16.8% of the GCA variance, respectively. The LOD peaks of the two QTLs for GCA of PH were located within the identical intervals occupied by the other two QTLs for the GCA of DTH, respectively. Furthermore, five QTLs of the GCA for SPP were detected on chromosomes 1, 3, 7 and 11. Surprisingly, we found that two QTLs on chromosome 7 explained 26.3% and 38.2% of the variance in the GCA of SPP, respectively, which coincided with the two QTL intervals of the GCA for DTH and PH. These results suggested that the two clusters of QTLs on chromosome 7 are pleotropic QTLs that simultaneously affect the GCA variation for all the three traits. Therefore, we designated the pleotropic locus on the telomere region as GCA1 and the other locus on the centromere region as GCA2 (Fig. 1a,b). Given that GCA1 and GCA2 on chromosome 7 explained the highest variance of GCA for three traits, we focused on these two pleotropic QTLs for further study.
Table 3. QTL analysis of GCA for three agronomic traits.
| GCA Trait | Chr. | Position (cM) | Left marker | Right marker | LOD | PVE (%) | Add | Dom | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Marker name | Position (bp) | Marker name | Position (bp) | |||||||
| DTH | 1 | 183 | S14 | 22,622,816 | S15 | 24,792,180 | 3.21 | 1.71 | 1.38 | −1.49 |
| DTH | 6 | 63 | RM3431 | 8,756,801 | RM3438 | 7,477,690 | 16.52 | 10.99 | −4.53 | −0.80 |
| DTH | 6 | 91 | RM584 | 3,416,638 | RM5815 | 2,928,145 | 11.21 | 6.72 | −3.30 | −1.43 |
| DTH | 7 | 85 | RM3670 | 13,386,382 | RM6449 | 15,357,245 | 32.41 | 29.86 | 6.55 | −0.02 |
| DTH | 7 | 213 | S77 | 29,253,591 | RM1306 | 28,894,398 | 36.90 | 34.87 | 7.13 | −0.84 |
| PH | 7 | 86 | RM3670 | 13,386,382 | RM6449 | 15,357,245 | 21.09 | 42.41 | 3.70 | −0.50 |
| PH | 7 | 213 | S77 | 29,253,591 | RM1306 | 28,894,398 | 10.20 | 16.80 | 2.28 | −0.51 |
| PH | 11 | 0 | RM6327 | 364,257 | RM6335 | 479,545 | 4.27 | 6.38 | −1.44 | −0.19 |
| SPP | 1 | 73 | RM10043 | 645,573 | RM1 | 5,050,749 | 7.42 | 13.06 | 3.24 | 0.80 |
| SPP | 3 | 153 | RM416 | 31,248,603 | RM3867 | 31,540,070 | 5.27 | 6.91 | 2.41 | −0.90 |
| SPP | 7 | 83 | RM214 | 12,730,966 | RM3670 | 13,386,382 | 16.56 | 26.29 | 4.57 | −0.32 |
| SPP | 7 | 221 | ID710 | 28,724,269 | ID711 | 28,829,798 | 20.82 | 38.17 | 5.51 | −1.08 |
| SPP | 11 | 58 | RM7463 | 10,095,283 | RM26567 | 12,830,466 | 4.58 | 5.90 | 2.37 | −0.31 |
The GCA traits (DTH, PH and SPP) used for QTL mapping represent the GCA effect of days to heading, plant height and spikelets per panicle, respectively. Position (cM) indicates the LOD peak position on the genetic linkage map. Position (bp) indicates the physical position of QTL flanking markers on corresponding chromosome (Chr.). PVE (%) represents the percentage of total phenotypic variance explained by the QTL. Add and Dom indicate the additive effect and dominant effect, respectively.
Figure 1. Mapping of the GCA1 and GCA2 on chromosome 7 and fine mapping of GCA1.

(a) Two pleotropic GCA QTLs for DTH, PH and SPP were simultaneously detected near the centromere region and the telomere region, respectively, on chromosome 7. (b) Linkage map of chromosome 7. (c) Fine mapping of GCA1 with recombinants from BC3F2-GCA1 population. The numbers under each molecular marker indicate the number of recombination events detected between GCA1 and individual markers. (d) The protein structure of GCA1 as predicted by PROSITE (http://prosite.expasy.org/)43. The structures of full length (GCA1) and truncated (tGCA1) GCA1 protein were presented. The numbers below each domain show the predicted start and end positions. “aa” = amino acids.
Fine mapping of the QTLs for GCA
To precisely map GCA1 and GCA2, two BC3F2 populations containing GCA1 (BC3F2-GCA1) and GCA2 (BC3F2-GCA2) were developed by continuous backcrossing using GL as the recurrent parent via marker-assisted selection (MAS) with the flanking SSR markers RM3670 and RM1306, respectively (see Supplementary Fig. S2a online). The characterisation of agronomic trait performance indicated that DTH in dominant individuals was increased by 14.2 days in the BC3F2-GCA1 population and 12.1 days in the BC3F2-GCA2 population compared with the corresponding recessive individuals under natural short day (NSD) conditions. The DTH of dominant individuals from the BC3F3-GCA1 and BC3F3-GCA2 populations was increased by 23.3 days and 20.9 days compared with those of corresponding recessive individuals under natural long day (NLD) conditions, respectively. Simultaneously, other two traits, PH and SPP, were significantly increased in the dominant individuals of BC3F3-GCA1 and BC3F3-GCA2 populations (Table 4). Bioinformatics analysis revealed that the GCA2-linked marker was located approximately 2 Mb from the pleotropic gene Ghd719. Further sequence analysis of Ghd7 in two parental lines revealed that the null allele type Ghd7-0 was present in GL, whereas the functional Ghd7 was present in TQ. Taking together, these results suggested that Ghd7 is GCA2, located near the centromere, and contributed to the GCA variations of DTH, PH and SPP. Moreover, these results showed that GCA1 was located at the telomere region approximately 20 Mb away from Ghd7, suggesting that it may be a new locus. Therefore, we subsequently concentrated on this locus for further fine mapping in the BC3F2-GCA1 population.
Table 4. Trait performance of BC3F2 plants and BC3F3 families with recessive (Rec) and dominant (Dom) genotypes under NSD and NLD conditions.
| Season | Genotypes | DTH | PH | SPP |
|---|---|---|---|---|
| Mean ± SD | Mean±SD | Mean ± SD | ||
| 2012H | BC3F2-GCA1(Rec) | 77.9 ± 1.5 | 74.0 ± 3.5 | 154.3 ± 10.1 |
| BC3F2-GCA1(Dom) | 92.1 ± 2.2 | 80.3 ± 3.0 | 188.4 ± 13.0 | |
| P-value | 8.48E-25 | 3.35E-07 | 3.03E-11 | |
| BC3F2-GCA2(Rec) | 83.0 ± 2.2 | 71.3 ± 1.9 | 121.0 ± 14.5 | |
| BC3F2-GCA2(Dom) | 95.1 ± 1.6 | 80.2 ± 2.8 | 171.9 ± 15.3 | |
| P-value | 1.98E-21 | 8.28E-14 | 7.83E-13 | |
| 2012W | BC3F3-GCA1(Rec) | 63.6 ± 2.5 | 92.8 ± 3.8 | 148.4 ± 10.3 |
| BC3F3-GCA1(Dom) | 86.9±2.9 | 112.4±4.0 | 194.8±12.6 | |
| P-value | 3.64E-25 | 2.25E-17 | 2.84E-15 | |
| BC3F3-GCA2(Rec) | 65.0 ± 1.2 | 94.9 ± 3.8 | 136.4 ± 14.9 | |
| BC3F3-GCA2(Dom) | 85.9 ± 1.6 | 108.4 ± 3.0 | 185.1 ± 13.9 | |
| P-value | 3.10E-35 | 5.61E-15 | 5.64E-13 |
The mean value±SD (standard deviation) was calculated using 20 plants for each genotype under natural short-day (NSD) conditions in the spring of 2012, Hainan (2012H) and natural long-day (NLD) conditions in the summer of 2012, Wuhan (2012W). The P-value was calculated with One-tailed t-test between the genotypes.
To obtain sufficient recombinant plants for fine mapping of GCA1, we first used the molecular markers encompassing the GCA1 region to genotype plants in the BC3F2-GCA1 population. Two polymorphic SSR markers (RM22140 and RM1306) and two newly developed InDel markers (ID77 and ID710) were used to screen 3,127 individuals from BC3F2-GCA1 population. A total of 23 recombinant individuals were identified, and their GCA values for three agronomic traits were obtained by crossing to the tester varieties (see Supplementary Table S2 online). We found that the GCA values of the three agronomic traits from those recombinant individuals were highly correlated based on the linkage analysis. Further genetic analysis of the GCA effects of the 23 recombinants indicated that the GCA1 interval mapped to the region extending from RM1306 to the end of chromosome, which represents a physical distance of 802 kb. To precisely locate GCA1 within this region, three SNP markers (S77, S722 and S726) newly developed were used for genotyping within the 802 kb region. Finally, the location of GCA1 was narrowed to a region of approximately 443 kb extending from S77 to the end of chromosome 7 (Fig. 1c and more details can be found as Supplementary Table S2 online).
Determining the candidate gene for GCA1
To determine the candidate gene responsible for GCA1, bioinformatics tools were applied to predict the candidate genes that encompassed the GCA1 region. A total of 65 predicted genes covered the GCA1 region, according to the database of the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/). The expression patterns of those genes in both parents were obtained by RNA sequencing data20. Three criteria were used to select candidate genes for further sequencing analysis: 1) the difference of the expression level between high GCA and low GCA parents is >or ≅2.0; 2) the expression level of the gene is high; 3) the known functional importance of predicted protein. Five genes—LOC_Os07g49150 (a putative 26S protease regulatory subunit), LOC_Os07g49230 (a putative ubiquitin-activating enzyme), LOC_Os07g49460 (a response regulator receiver domain containing protein), LOC_Os07g49520 (a putative 2-oxoglutarate dehydrogenase E1 component) and LOC_Os07g49530 (a putative MYB family transcription factor)—satisfied the criteria and were selected for further PCR-based sequencing (see Supplementary Table S3 online). Sequencing results showed that an 8 bp deletion in the tenth exon of LOC_Os07g49460 in GL produced a premature stop codon at the position 1525 bp from the translation start site, whereas no sequence differences were found in ORF regions of the other four genes between GL and TQ. Further studies revealed that LOC_Os07g49460 encodes the known protein OsPRR37, an orthologue of Arabidopsis Pseudo-Response Regulator 7 (PRR7). The 8 bp deletion in the OsPRR37 in GL resulted in a 234 amino acid truncation at the C terminus, which led to the loss of the CCT domain, whereas the TQ allele has an entire protein with 742 amino acids (Fig. 1d). Recently, OsPRR37 was reported to regulate heading date, grain number and plant height as a pleiotropic gene and may contribute to rice cultivation at a wide range of latitudes21,22,23,24. Thus, the OsPRR37 gene was considered as the candidate gene for GCA1.
Confirmation of the gene corresponds to GCA1
To verify whether the candidate gene OsPPR37 is corresponding to GCA1, the overexpression transgenic lines (GCA1OX) carrying 35S:OsPRR37 were generated. The delayed flowering phenotype was observed in positive transgenic plants. The DTH, PH and SPP of homozygous GCA1OX−5 plants were increased by 32.5 days, 23.9 cm and 62.6 spikelets, respectively, under natural long day (NLD) condition compared with negative plants (Fig. 2 and more details can be found as Supplementary Table S4 online). We further found that the mRNA level of OsPRR37 in positive GCA1OX lines was significantly higher than that in NIL (GCA1+/+) (Fig. 3a). The GCA effects of homozygous GCA1OX plants in T1 families, NILs (GCA1+/+ and GCA1−/−) and NILs (GCA2+/+ and GCA2−/−), as well as the parents GL and TQ were evaluated using TC progenies derived from five testers (Table 5). The results indicated that the GCA effects of three independent GCA1OX transgenic lines were significantly higher than those of the transgenic recipient parent GL for all the three agronomic traits, suggesting that overexpression of OsPRR37 significantly increased the GCA effects. Simultaneously, the GCA effects of the three agronomic traits in GL, NIL(GCA1−/−) and NIL(GCA2−/−) lines were significantly lower than that in TQ, NIL(GCA1+/+) and NIL(GCA2+/+) lines. These results confirmed that OsPRR37 was the gene corresponding to GCA1. Again, the GCA effects of NIL (GCA2−/−) and NIL (GCA2+/+) supported that Ghd7 corresponds to GCA2. These results indicated that GCA1 and GCA2 significantly increase the GCA of DTH, PH and SPP.
Figure 2. Agronomic trait performance of nearly isogenic lines (NILs) and overexpression line GCA1OX−5.

(a) Agronomic traits of different genotypes (GCA1−/− or negative (Neg), GCA1+/+ or positive homozygote (Pos) for NILs and GCA1OX−5, respectively) under natural long-day (NLD) conditions. Means±standard derivations (SD) were obtained from 20 plants for each genotype (more details can be found as Supplementary Table S4 online). One-tailed t-test was used to reveal the significant difference of the agronomic traits between different genotypes of the NILs and GCA1OX−5. DTH: days to heading; PH: plant heght; SPP: spikelets per panicle; “**” indicates significance at the level of P < 0.01. (b–e) Comparisons of plants and panicles of NILs and GCA1OX−5 with different GCA1 genotypes. Scale bar = 10 cm.
Figure 3. Expression pattern of GCA1.

(a) Diurnal expression of in NIL(GCA1+/+) and GCA1OX lines. The rice Actin1 gene was used as the internal control, and the receptor parent GL was used as the negative control with the TQ-specific qRT-PCR primer. Values are shown as the mean ± SD from three independent experiments. The open and filled bars at top represent the light and dark periods, respectively. The time points below the X-axis show the time course during the day at Wuhan in the summer of 2014. (b–d) GUS staining of various organs at different stages in GCA1 promoter-GUS transgenic plants.(b) Germinating seeds, scale bar = 2 mm; (c) seedlings at 14 days after germination, scale bar = 1 cm; (d) young panicle with intact stem, scale bar = 1 cm.
Table 5. GCA estimation of NILs and GCA1 OX lines with five testers.
| Genotype | GCADTH | GCAPH | GCASPP |
|---|---|---|---|
| GL | −9.4 | −7.6 | −10.7 |
| TQ | 1.4 | 10.3 | 20.6 |
| NIL(GCA1−/−) | −8.4 | −5.9 | −8.6 |
| NIL(GCA1+/+) | 9.1 | 2.8 | −1.3 |
| NIL(GCA2−/−) | −10.1 | −8.7 | −10.0 |
| NIL(GCA2+/+) | 5.2 | 3.2 | -0.3 |
| GCA1OX-5 | 4.4 | 2.7 | 2.1 |
| GCA1OX-6 | 4.2 | 0.4 | 5.7 |
| GCA1OX-9 | 3.6 | 2.9 | 2.7 |
Two NIL sets [NIL (GCA1+/+ or GCA1−/−) and NIL (GCA2+/+ or GCA2−/−)], three GCA1OX lines (GCA1OX-5, GCA1OX−6 and GCA1OX−9) and two parental lines (GL and TQ) were used for GCA estimation. GCADTH, GCAPH and GCASPP represent the corresponding GCA values that were estimated from the whole TC progeny derived from five testers.
GCA1 exhibits a constitutive and diurnal expression pattern
To understand the expression profiles of GCA1, the transgenic plants carrying a GUS reporter gene driven by the GCA1 promoter were generated. The results revealed that GCA1 was constitutively expressed in all tissues and at all developmental stages (Fig. 3b–d). The most abundant expression of GCA1 was found in the leaf, young panicle, stem and internode, implying that GCA1 could function in panicle development and stem elongation, consistent with the GCA effects of DTH, PH and SPP. The expression profile of GCA1 in NIL (GCA1+/+) plants exhibited a diurnal pattern (Fig. 3a). The expression level of GCA1 in NIL (GCA1+/+) increased from dawn to noon and then gradually decreased until midnight, with a peak expression level at noon under NLD conditions. Surprisingly, the overexpression of GCA1 driven by the 35S promoter in GCA1OX lines also exhibited a diurnal pattern, but the diurnal expression pattern was altered and peaked around dusk (at 20:00 for GCA1OX−5 and 16:00 for GCA1OX−9), with the lowest expression levels observed in the morning (at 8:00 for both GCA1OX lines). These results imply that the mRNA level of GCA1 itself could be regulated by unknown diurnal signal(s).
Previous research has reported that AtPRR7, an orthologue of GCA1, has a potential bipartite nuclear localisation signal at the C terminus (amino acids 680 to 696). The localisation of the PRR7-GUS fusion was observed to be nuclear in leek epidermal cells25. To investigate whether the full-length GCA1 and the truncated GCA1 (tGCA1) had the same nuclear localisation, we monitored the localisation of sGFP-GCA1 and sGFP-tGCA1 via a rice protoplast transient assay. The results showed that both sGFP-GCA1 and sGFP-tGCA1 localised in the nucleus, whereas sGFP alone was detected throughout the cytoplasm (see Supplementary Fig. S3 online). These results showed that GCA1 protein functions in the nucleus, but the truncation of the 234 amino acids at the C terminus does not disturb its nuclear localisation.
Discussion
In this study, we identified 13 QTLs for the GCA of three important agronomic traits that were mainly associated with grain yield in rice. We identified two major pleotropic QTLs—GCA1 and GCA2—that corresponded to OsPRR37 and Ghd7, respectively, and functioned as positive regulators of GCA for DTH, PH and SPP and these traits per se. Our findings revealed that the genes responsible for GCA effects correspond to the genes controlling important agronomic traits. To take another example in this study, two QTLs for the GCA of DTH were found to be located between RM3431 and RM3438 and between RM584 and RM5815 on chromosome 6. The physical regions represented by these SSR markers are close to the physical positions of the genes Hd1 and Hd3a that control flowering time26,27, indicating that Hd1 and Hd3a may be responsible for the QTL loci for corresponding GCA effects. These results suggested that QTLs for GCA are directly associated with QTLs for these important agronomic traits.
Previous studies have reported that GCA1 and/or GCA2 were widely found in elite rice varieties19,21,23,24,28. For example, the modern elite parents, Minghui 63, Teqing, Huanghuazhan, Guichao #2, Peiai 64S contain both GCA1 and GCA2 while 93-11 contains GCA2. It is apparently the result of domestic selection during cross-breeding. This pattern explains that high GCA-effect parents could easily pass on the corresponding traits to their hybrid progenies by simultaneously integrating the GCA controlling genes into the genome of hybrid progenies during cross-breeding program. It is well explained why elite varieties could be easily developed from parents with higher GCA effects. Our findings help remove the mystery of GCA that has existed since 1942, even though this trait has been widely used for crop and livestock improvements.
GCA effects have widely been applied by breeders to evaluate breeding parents. The consensus of opinion is that elite breeding parents possess preferred GCA effects, which are characteristics of wide adaptability, high hereditary capacity and elite agronomic traits. Our data show that GCA1 and GCA2 encode OsPRR37 and Ghd7, respectively, which have both been reported to be associated with agronomic traits for high grain yield and adaptability at different latitudes19,21,23,24. This finding has further confirmed that GCA-controlling genes in elite breeding parents are associated with wide adaptability, important agronomic traits and the capacity to pass on these traits to their hybrid progenies. Our results elucidate why GCA has been effective as a criterion for evaluating the breeding parents. Together, these results also provide new insight into the molecular genetic basis of GCA and the relationship between important agronomic traits and GCA effects. The updated concept of GCA revealed in our findings will provide a useful tool for evaluating and selecting elite parents for molecular design breeding in the future.
In our study, we found that the GCA effect of SPP in NIL(GCA1+/+) was lower than that of GCA1OX transgenic lines, and the GCA effect of SPP in NIL(GCA1+/+) and NIL(GCA2+/+) was even lower than zero (Table 5). According to the processing of GCA effect data, GCA value is a relative estimate and the sum of GCA effects for all varieties or inbred lines evaluated must be equal or nearly zero. Thus, the negative value of GCA effect of SPP in NIL(GCA1+/+) and NIL(GCA2+/+) was partly caused by the extremely high GCA effect of SPP in parent TQ. Alternatively, the negative value of the GCA effect of SPP in NIL(GCA1+/+) does not mean inconsistency between NIL(GCA1+/+) and GCA1OX transgenic lines. As in this respect, the GCA effects of NIL(GCA1+/+) and GCA1OX transgenic lines showed the same trends when comparing to the GCA effects of both NIL(GCA1−/−) or GL. For the other explanation, the GCA difference could be caused by the different expression pattern of GCA1 between NIL(GCA1+/+) and GCA1OX transgenic lines (Fig. 3a). If it is case, however, it will be an interesting issue remains to know that how the expression pattern of GCA1 affect GCA effect in future.
Previous studies have reported that GCA1 belongs to the PRR family, identified as circadian clock-associated PRR homologues of Arabidopsis. Each of the family members contains two domains: the Pseudo-Receiver domain at the N terminus and a CCT domain at the C terminus29,30,31. More recently, studies have revealed that the GCA1 homologue PRR7, together with PRR5 and PRR9, acts as a transcriptional repressor by binding the promoters of the core clock genes CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) and LATE ELONGATED HYPOCOTYL (LHY) in Arabidopsis32,33. In our study, the diurnal expression pattern of GCA1 and the nuclear localisation of the GCA1 protein indicated that GCA1 might have a similar function as a circadian-regulated transcription factor in rice. Interestingly, GCA2 also possesses a CCT domain exhibiting a diurnal expression pattern and nuclear localisation19. Our study confirms that elimination of the 234 amino acids at the C terminus, which contains the CCT domain, does not alter the nuclear localisation of GCA1, indicating that the nuclear localisation signal does not reside in the C terminus. Recently, it has been shown that the CCT domain is crucial for interactions between Arabidopsis PRR5 and target DNA in vivo34. Therefore, we hypothesize that the low GCA-effect phenotype can be attributed to the loss of the CCT domain at the C terminus, leading to a loss of capacity for interacting with its target nuclear DNA. Therefore, CCT domain could play critical role on the GCA effects for those traits. GCA1 and GCA2 might function as transcription factors of various downstream genes in the nucleus by regulating their diurnal expression patterns or expression levels.
Methods
Plant materials and the F2 population
Estimation of the GCA effects of elite rice varieties was conducted with a diallelic cross design using the Yuetai B (YB), Zhenshan 97 B (ZS), Aijiaonante (AJ), Guangluai #4 (GL), 93-11(L6) and Teqing (TQ) varieties. An F2 population with 139 individuals derived from GL×TQ was used in this study. Five varieties—ZS, AJ, GL, L6 and Aizizhan (AZ)—were used as testers to cross to individual plants from the F2 population based on the NCII mating design in Lingshui, Hainan Province, in the spring of 2009. The 139 F2 plants were crossed with the five testers to evaluate GCA effects and were planted in Wuhan, Hubei Province, in the summer of 2009. Twenty plants of each testcross (TC) progeny were planted in a 16.5 cm × 20 cm area following a randomised complete block design with three replicates.
Development of the BC3F2 population for fine mapping
Individual plants from the F2 population containing the QTL region of interest were continually backcrossed to GL. The BC3F1 plants were selected by marker-assisted selection (MAS), and the GCA effects of the recombinant plants from BC3F2 were evaluated according to the agronomic traits of the TC progeny (Supplementary Fig. S2a online). The agronomic traits of the individual plants in TC populations were recorded. All TC progeny were planted in the same manner as the TC populations of F2 plants.
Measurement of agronomic traits and statistical analyses
For primary mapping, data for the agronomic traits days to heading (DTH), plant height (PH) and spikelets per panicle (SPP) were collected for between 10 and 15 plants from each plot. For fine mapping, the accurate GCA effects of three traits were measured for five plants of non-segregated lines and for between 10 and 15 plants of segregated lines derived from the TC progeny in each replication. Variance analysis and evaluation of the GCA were carried out as previously described35. The GCA effects were calculated with the following formula:
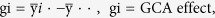 |
where 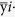 = average of the five TC progenies between individual plant i and the five testers and
= average of the five TC progenies between individual plant i and the five testers and 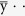 = overall mean of the five TC populations.
= overall mean of the five TC populations.
The variance analysis and effect evaluation of GCA were processed with DPS software version 7.0536. The descriptive statistics—Student’s t-test of the agronomic traits—was performed in SPSS statistics software version 17.0 (SPSS Inc., Chicago, IL, USA).
QTL mapping
A total of 141 polymorphic molecular markers were used to construct the linkage map using plants from the F2 population, including 123 SSRs (http://www.gramene.org/), 14 SNPs and four InDel markers developed from previous genome sequencing data for GL and TQ20. Conventional polymerase chain reaction (PCR) and polyacrylamide gel electrophoresis (PAGE) techniques were used for SSR and InDel genotyping. For SNP genotyping, the MALDI-TOF MS method was used to identify SNP polymorphisms in the F2 population as described previously37. Genetic linkage map construction and QTL mapping analysis were conducted with QTL IciMapping software version 3.138. An LOD score of 3.0 was used to indicate the presence of QTLs for each trait. The segregation of the traits for each BC3F3 family and the corresponding TC progeny was used to distinguish the genotype of the GCA locus in each recombinant plant of BC3F2.
Plasmid construction and transformation
The coding sequence (CDS) of the candidate gene GCA1 was confirmed by PCR-based sequencing of the exons in genomic DNA and cDNA obtained by the mRNA reverse transcription of TQ. The CDS (2,229 bp) and the 3′UTR (623 bp) were artificially synthesised by Gene-Script (Nanjing, China) and were inserted into the binary vector pCAMBIA1300. A CaMV 35S promoter was then fused to the 5′ end of the CDS, resulting in the overexpression construct pOsPMP625. To generate the GCA1 promoter-GUS construct, a 1,884 bp promoter region was amplified by PCR and inserted upstream of the 5′ end of the reporter gene beta-glucuronidase (GUS) in the binary vector pCAMBIA1300, designated pOsPMP626. All transgenic plants were generated by introduction into GL through Agrobacterium-mediated transformation.
Evaluation of the GCA effect
NILs corresponding to different genotypes GCA1 and GCA2 were obtained from homozygous BC4F2 plants with more than 90% of the genetic background derived from the GL line, as determined by genome-wide SSR and InDel markers (see Supplementary Fig. S2b,c online). Homozygous NIL (GCA1+/+ or GCA1−/−) and NIL (GCA2+/+or GCA2−/−) were determined by using appropriate molecular markers. The homozygous GCA1OX plants from the T1 generation were determined by qPCR as described previously39. Two NIL sets [NIL (GCA1+/+ or GCA1−/−) and NIL (GCA2+/+ or GCA2−/−)], three independent homozygous GCA1OX lines and the parent lines (GL and TQ) were crossed to five tester varieties [ZS, AJ, L6, YB and Guichao #2 (GC2)] at Lingshui, Hainan Province, in the spring of 2014. The GCA effects and the traits of these lines were evaluated.
RNA extraction and quantitative RT-PCR
The uppermost fully expanded leaves from at least three individual plants were harvested at 45 days after germination under natural long day (NLD) conditions. Total RNA was extracted from the pooled leaf materials using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions and then treated with RNase-free DNase I (New England Biolabs, Hitchin, UK). First-strand cDNA was synthesised using M-MLV reverse transcriptase and oligo (dT) primer (Promega, Madison, WI, USA) as described by the manufacturer. Quantitative RT-PCR was carried out in a total volume of 10 μl, including 2 μl of cDNA template (5- to 10-fold dilutions), 0.5 μl of 10 μM gene-specific primer and 5 μl of SYBR Green qPCR Supermix-UDG with ROX Reference Dye (Invitrogen, Carlsbad, CA, USA). The qRT-PCR was conducted on a StepOne System (ABI) using the following parameters: 95°C for 10 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The relative gene expression levels were calculated according to the ΔΔCT method described previously40. The rice Actin1 gene was used as an internal control to normalise the data.
GUS staining assay
A GUS staining assay was performed as described in a previous study41. Briefly, fresh tissue from the transgenic plants was washed three times with a staining solution containing 50 mM NaPO4 buffer (pH 7.0), 2 mM K3Fe(CN)6, 2 mM K4Fe(CN)6 and 0.2% Triton X-100 on ice and then incubated with staining solution containing 1 mM X-Gluc at 37 °C overnight. The coloured tissues were photographed with a camera (EOS 500D; Canon).
Subcellular localisation
The CDS of full-length GCA1 and truncated GCA1 (tGCA1) were amplified using appropriate primers based on the sequences in TQ and GL, respectively (Supplementary Table 4). Isolated GCA1 and tGCA1 were inserted into the pHBT-sGFP(S65T) construct at the C terminal of sGFP without a stop codon between sGFP and GCA1 or tGCA1 to create the in-frame fusion constructs sGFP-GCA1 and sGFP-tGCA1, respectively. Rice protoplasts were used for determining subcellular localisation. Protoplast isolation and PEG-mediated transfection were carried out as described in a previous study42. The nuclear marker AtH2B fused to a CFP construct (H2B-CFP) driven by CaMV 35S promoter was co-transformed with sGFP-GCA1 and sGFP-tGCA1. To avoid fluorescence artefacts that could disturb localisation, an optimised concentration of plasmid DNA (i.e., 15 μg in total) was used for the transient expression assay. Subcellular localisation was visualised on an Olympus Fluoview 1000 laser scanning microscope (http://www.olympus-global.com). Triplicates of each transient expression experiment were performed to obtain a robust result.
Additional Information
How to cite this article: Liu, C. et al. OsPRR37 and Ghd7 are the major genes for general combining ability of DTH, PH and SPP in rice. Sci. Rep. 5, 12803; doi: 10.1038/srep12803 (2015).
Supplementary Material
Acknowledgments
We thank Wuhan Biorun Bio-Tech Co., Ltd. performed transgenic rice. This research is supported by grants from the Ministry of Science and Technology of China (2006CB100105 and 2011CB100102).
Footnotes
Author Contributions C.L. performed most of the experiments and wrote the manuscript; G.S. conducted F2 population and primary Q.T.L. analysis; Y.Z. helped C.L. phenotypic data collection and subcellular localisation; X.Q., Z.L. and Z.G. helped C.L. field trial and crossing; D.J. managed all field trial. D.Y. designed the experiments and wrote the manuscript. All the authors discussed results and contributed to the manuscript.
References
- Darwin C. R. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. London: John Murray (1859). [PMC free article] [PubMed] [Google Scholar]
- Sprague G. F. & Tatum L. A. General vs specific combining ability in single cross of corn. J. Amer. Soc. Agron. 34, 923–932 (1942). [Google Scholar]
- Griffing B., Concept of general and specific combining ability in relation to diallel cross systems. Aust.J.Biol.Sci. 9, 463–493 (1956). [Google Scholar]
- Zuo-mei L. On the combining ability selection in hybrid rice breeding. Rice Science 13, 1–5 (1999). [Google Scholar]
- Cross H. Z. Diallel analysis of duration and rate of grain filling of seven inbred lines of corn. Crop Sci 15, 532–535 (1975). [Google Scholar]
- Jamison M. G., White J. M., Vinson W. E. & Hinkelmann K. Diallel analysis of growth traits in mice. Genetics 81, 369–76 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahangar L., Ranjbar G. A. & Nouroozi M. Estimation of combining ability for yield and yield components in rice (Oryza sativa L.) cultivars using diallel cross. Pak J Biol Sci 11, 1278–81 (2008). [DOI] [PubMed] [Google Scholar]
- Vieira R. A. et al. Diallel analysis of leaf disease resistance in inbred Brazilian popcorn cultivars. Genet Mol Res 8, 1427–36 (2009). [DOI] [PubMed] [Google Scholar]
- Gao W., Baars J. J., Dolstra O., Visser R. G. & Sonnenberg A. S. Genetic variation and combining ability analysis of bruising sensitivity in Agaricus bisporus. PLoS One 8, e76826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend T. et al. The use of combining ability analysis to identify elite parents for Artemisia annua F1 hybrid production. PLoS One 8, e61989 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag T. A., Melchinger A. E., Sorensen A. P. & Frisch M. Prediction of single-cross hybrid performance for grain yield and grain dry matter content in maize using AFLP markers associated with QTL. Theor Appl Genet 113, 1037–47 (2006). [DOI] [PubMed] [Google Scholar]
- Schrag T. A. et al. Prediction of single-cross hybrid performance in maize using haplotype blocks associated with QTL for grain yield. Theor Appl Genet 114, 1345–55 (2007). [DOI] [PubMed] [Google Scholar]
- Schrag T. A. et al. Prediction of hybrid performance in maize using molecular markers and joint analyses of hybrids and parental inbreds. Theor Appl Genet 120, 451–61 (2010). [DOI] [PubMed] [Google Scholar]
- Riedelsheimer C. et al. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat Genet 44, 217–20 (2012). [DOI] [PubMed] [Google Scholar]
- Ai-zhi L. et al. Conversion of the statistical combining ability into a genetic concept. J Integr Agr 11, 43–52 (2012). [Google Scholar]
- Qu Z. et al. QTL mapping of combining ability and heterosis of agronomic traits in rice backcross recombinant inbred lines and hybrid crosses. PLoS One 7, e28463 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H. et al. Identification of combining ability loci for five yield-related traits in maize using a set of testcrosses with introgression lines. Theor Appl Genet 126, 369–77 (2013). [DOI] [PubMed] [Google Scholar]
- Song G. et al. The phenotypic predisposition of the parent in F1 hybrid is correlated with transcriptome preference of the positive general combining ability parent. BMC Genomics 15, 297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W. et al. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40, 761–7 (2008). [DOI] [PubMed] [Google Scholar]
- Song G. et al. Global RNA sequencing reveals that genotype-dependent allele-specific expression contributes to differential expression in rice F1 hybrids. BMC Plant Biol 13, 221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B. H. et al. Natural variation in OsPRR37 regulates heading date and contributes to rice cultivation at a wide range of latitudes. Mol Plant 6, 1877–88 (2013). [DOI] [PubMed] [Google Scholar]
- Liu T., Liu H., Zhang H. & Xing Y. Validation and characterization of Ghd7.1, a major quantitative trait locus with pleiotropic effects on spikelets per panicle, plant height, and heading date in rice (Oryza sativa L.). J Integr Plant Biol 55, 917–27 (2013). [DOI] [PubMed] [Google Scholar]
- Yan W. et al. Natural variation in Ghd7.1 plays an important role in grain yield and adaptation in rice. Cell Res 23, 969–71 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H. et al. Days to heading 7, a major quantitative locus determining photoperiod sensitivity and regional adaptation in rice. Proc Natl Acad Sci USA 111, 16337–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski K. A. & Quail P. H. Arabidopsis PSEUDO-RESPONSE REGULATOR7 is a signaling intermediate in phytochrome-regulated seedling deetiolation and phasing of the circadian clock. Plant Cell 15, 2654–65 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M. et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12, 2473–2484 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S. et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol 43, 1096–105 (2002). [DOI] [PubMed] [Google Scholar]
- Lu L., Yan W., Xue W., Shao D. & Xing Y. Evolution and association analysis of Ghd7 in rice. PLoS One 7, e34021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre E. M., Harmer S. L., Harmon F. G., Yanovsky M. J. & Kay S. A. Overlapping and distinct roles of PRR7 and PRR9 in the Arabidopsis circadian clock. Curr Biol 15, 47–54 (2005). [DOI] [PubMed] [Google Scholar]
- Farré E. M. & Kay S. A. PRR7 protein levels are regulated by light and the circadian clock in Arabidopsis. The Plant Journal 52, 548–560 (2007). [DOI] [PubMed] [Google Scholar]
- Murakami M., Tago Y., Yamashino T. & Mizuno T. Comparative overviews of clock-associated genes of Arabidopsis thaliana and Oryza sativa. Plant Cell Physiol 48, 110–21 (2007). [DOI] [PubMed] [Google Scholar]
- Nakamichi N. et al. PSEUDO-RESPONSE REGULATORS 9, 7, and 5 are transcriptional repressors in the Arabidopsis circadian clock. Plant Cell 22, 594–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Kim J. & Somers D. E. Transcriptional corepressor TOPLESS complexes with pseudoresponse regulator proteins and histone deacetylases to regulate circadian transcription. Proc Natl Acad Sci USA 110, 761–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamichi N. et al. Transcriptional repressor PRR5 directly regulates clock-output pathways. Proc Natl Acad Sci USA 109, 17123–17128 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo H. The Analysis of combining ability in p×q mating pattern (in Chinese). Journal of Jiangsu Agricultural College 3, 51–57 (1982). [Google Scholar]
- Tang Q. Y. & Zhang C. X. Data Processing System (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci 20, 254–60 (2013). [DOI] [PubMed] [Google Scholar]
- Storm N., Darnhofer-Patel B., van den Boom D. & Rodi C. P. MALDI-TOF mass spectrometry-based SNP genotyping. Methods Mol Biol 212, 241–62 (2003). [DOI] [PubMed] [Google Scholar]
- Li H., Ye G. & Wang J. A modified algorithm for the improvement of composite interval mapping. Genetics 175, 361–74 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Jiang D. & Yang D. Fast-tracking determination of homozygous transgenic lines and transgene stacking using a reliable quantitative real-time PCR assay. Appl Biochem Biotechnol 175, 996–1006 (2015). [DOI] [PubMed] [Google Scholar]
- Livak K. J. & Schmittgen T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods 25, 402–8 (2001). [DOI] [PubMed] [Google Scholar]
- Jefferson R. A. The GUS reporter gene system. Nature 342, 837–8 (1989). [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods 7, 30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C.J.A. et al. New and continuing developments at PROSITE. Nucleic Acids Res 41, 344–347 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.