

Abstract
Hemolytic uremic syndrome (HUS) is a clinical syndrome characterized by the triad of thrombotic microangiopathy, thrombocytopenia, and acute kidney injury. Hemolytic uremic syndrome represents a heterogeneous group of disorders with variable etiologies that result in differences in presentation, management and outcome. In recent years, better understanding of the HUS, especially those due to genetic mutations in the alternative complement pathway have provided an update on the terminology, classification, and treatment of the disease. This review will provide the updated classification of the disease and the current diagnostic and therapeutic approaches on the complement-mediated HUS in addition to STEC-HUS which is the most common cause of the HUS in childhood.
Keywords: Acute kidney injury, hemolytic anemia, complement, STEC, thrombocytopenia, thrombotic microangiopathy
Introduction
Hemolytic uremic syndrome (HUS) is a clinical syndrome characterized with the triad of microangiopathy hemolytic anemia, thrombocytopenia and acute renal damage. It is one of the most common causes of acute renal damage in children. The clinical findings in hemolytic uremic syndrome occur as a result of thrombotic microangiopathy (TMA). The pathological lesion is thickening of arterioles and capillary walls, endothelial swelling and detachment. Fibrin and platelet-rich thrombi lead to obstruction in the vascular lumen. Many tissues and organs including mainly the kidney are affected (1).
Thrombotic microangiopathies represent a group of distinct disorders with different etiologies and pathogenesis. There are two main disease groups: HUS and thrombotic thrombocytopenic purpura (TTP) (1, 2). These two diseases had been used interchangably for long years and attempted to be differentiated with their clinical properties. Current knowledge indicates that both diseases have their own etiologies and pathogenesis. Thrombotic thrombocytopenic purpura occurs in deficiency of a metalloproteinase (ADAMTS 13) which cleaves von Willebrand factor in plasma or in presence of antibody against this. It is differentiated from HUS by decreased ADAMSTS 13 activity (<10%) (3). Shiga-toxin (Stx) producing Escherichia Coli (STEC) is the most common cause of HUS, called as STEC-HUS. In the childhood, complement-related HUS constitutes the majority of HUS cases other than STEC-HUS (4).
In recent years, better understanding of the HUS, especially those due to genetic mutations in the alternative complement pathway have provided an update on the terminology, classification, and treatment of the disease. In this article, especially STEC-HUS and complement-related HUS will be mentioned.
Definition and classification
Traditionally, HUS was classified as diarrhea positive HUS (D+HUS) and diarrhea negative HUS (D-HUS) and D+HUS was considered equivalent to typical HUS and D-HUS was considered equivalent to atypical HUS. However, the classification of D+HUS and D-HUS has been quitted, because diarrhea is the triggering factor in 25–30% of atypical HUS cases.
Hemolytic uremic syndrome has different etiological classifications in the literature (2, 5–7). Some of these classifications overlap with TMA classifications in terms of underlying diseases. In clinical practice, the commonly used classification is typical and atypical HUS classification. Typical HUS is used for STEC-HUS. On the other hand, some controversies related with the definition of atypical HUS (aHUS) exist in the literature. Until recently, the definition of aHUS has been used for all HUS cases other than STEC-HUS. In recent years, it has started to be used only for complement-related HUS by some authors. In the review which was published at the time of writing of this article and which had the characteristics of being an international consensus report for treatment of aHUS, the classification of HUS was updated and it was stated that limitation of the definition of aHUS only to complement-related HUS was still controversial (8). In this report, it was recommended that the definition of aHUS should be limited to the cases of HUS excluding HUS “secondary” to malign diseases, autoimmune diseases, drugs, organ transplantation and HIV infection (“HUS with coexisting diseases”). The TMA and HUS classifications which were included in this article published in April 2015 are shown in Table 1.
Table 1.
Classification of thrombotic microangiopathies (TMAs) (8)
Thrombotic Thrombocytopenic Purpura (TTP)
|
| HELLP syndrome |
HUS
|
APS: antiphospholipid syndrome; CNI: calcineurin inhibitors; DGKE: diacyl glycerol kinase epsilon; HIV: human immunodeficiency virus; HUS: hemolytic uremic syndrome; STEC: shiga-toxin producing E. coli; SLE: systemic lupus erythematosus
STEC-HUS
Epidemiology
STEC-HUS is responsible of 90% of childhood HUS cases. The incidence has been reported to be 2–3/100 000 children. It mainly affects children other than epidemias. It most commonly occurs in children aged 5–6 years (4, 9). It is observed more commonly in summer months and rural areas. It occurs after acute gastroenteritis which develops with enterohemorrhagic E.coli (EHEC) which produces Shiga-toxin or Shigella dysenteria. The most common agent is enterohemorrhagic E.coli (70%). The most common serotype responsible of the disease is E.coli O157:H7 (9). On the other hand, the great EHEC outbreak which affected Germany and 15 European countries in 2011 occured because of E. coli O104:H4 serotype. In this outbreak, a total of 3842 people were affected, HUS developed in 845 and 52 mortalities were reported. In contrary to the previous outbreaks, more than 80% of the subjects were adults (the median age: 42) and most patients had a high socioeconomical level (10, 11). In Australia, an outbreak which caused to hemorrhagic colitis and HUS occured with E. coli O111 serotype (12, 13). Cases of HUS have also been reported with different serotypes (O26, O145, O103) less commonly (14). Shigella dysenteria Type 1 is responsible of a small portion of STEC-HUS cases; it is observed in India, Bangladesh and South Africa. It causes to a more severe disease picture and the risk of progression to chronic renal disease is higher (15).
Pathogenesis
Cattle and sheep are the main sources for EHEC. Infection in humans generally occurs as a result of consumption of food contaminated with animal stool. The most common causes include undercooked meat, unpasteurized milk and dairy products, fruit juices, water, fruits and vegetables. Transmission by contact with animals, direct transmission from human to human and transmission from mother to baby is possible (16–18). It is most commonly observed in summar and autums months (9).
Following intake of contaminated food, the bacteria enters the intestines. In the intestines, Stx (STx1 and Stx 2) is secreted by E. coli. These exotoxins are absorbed in the epithelium of the gastrointestinal system and reach the target organ. They are bound to globotriaosylceramide (Gb3) which a glycopeptide surface receptor in the epitehlium of the target organ, inhibits protein synthesis and leads to endothelial damage, cell death, increase in inflammatory response and thrombocyte activation (see Figure 1) (19, 20). It is thought that the toxin also triggers action on P-selectin which is an adhesion molecule and complement regulating molecules (19).
Figure 1.
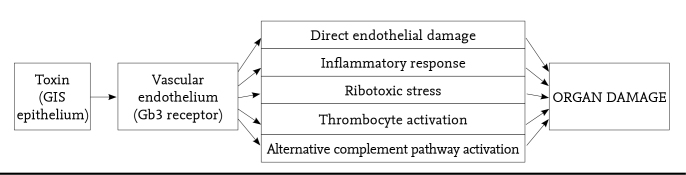
The effects of Stx in STEC-HUS caused by enterohemorrhagic E. coli (20)
GIS: gastrointestinal system; Gb3: globotriaosylceramide; Stx: shiga-toxin
Clinical and laboratory findings
Diarrhea begins approximately 3–8 days after intake of contaminated food. Diarrhea is watery in the beginning and become bloody later. Abdominal pain, nausea and vomiting accompany diarrhea. Fever is observed less commonly. HUS develops in 5–15% of the cases following enterohemorrhagic E. coli diarrhea. The risk factors for development of HUS include the serotype of bacteria (O157:H7), type of toxin (Stx 2), age of the patient (<5 years), use of antibiotic and antimotility drugs, fever, severe diarrhea, female gender, increased leukocyte count and genetic factors (7, 9, 21). However, there is no sufficient evidence for any of them.
Hematological findings and renal findings constitute the main clinical picture of the disease. Patients present with the findings created by this clinical picture. Palor, malaise, loss of appetite, nausea and vomiting may be observed. Decreased urine output may be recognized by some patients or edema may be a chief complaint. Laboratory findings related with the classical triad of the disease (microangiopathic hemolytic anemia, thrombocytopenia and acute renal damage) are present.
Microangiopathic hemolytic anemia: Anemia develops as a result of mechanical damage to erythrocytes while passing through the renal capillaries obstructed with microthrombi and fragmentation of erythrocytes. Anemia has the following properties:
Hemoglobin <10 g/dL, frequently <8 g/dL
Negative Coombs test
Increased reticulocyte count
Increased serum lactic dehydrogenase (LDH) level
Decreased serum haptoglobulin level
Fragmented erythrocytes on peripheral blood smear (helmet cells, schystocytes)
Thrombocytopenia: The platelet count is below 150 000/mm3 (commonly <40 000/mm3). Bleeding is observed rarely.
Acute renal damage: There may be different degrees of renal involvement. Hematuria, proteinuria and increase in serum creatinin are the most important indicators of renal damage. Increased blood pressure is another important finding. It occurs as a result of increased intravascular volume or ischemia due to TMA. Oliguria or oligoanuria may occur. Dialysis is needed becasue of severe renal damage in more than half of the cases (22, 23). Renal pathology is related with glomerular TMA. Preglomerular arterioles and glomerular capillaries are affected. Glomerular TMA is characterized with thickening in the capillary wall, swelling in the endothelial cells and obstruction in the capillary lumen. Rarely, cortical necrosis related with ischemia may be observed (24).
Findings related with the other organs/systems: In hemolytic uremic syndrome, any tissue or organ may be affected in relation with TMA. In some patients, multiple organ involvement may be observed and the possibility of mortality is high. In 25–30% of the cases, central nervous system involvement are present. Lethargy, irritability and seizure are the most common findings. Less frequently coma, stroke, hemiparesia, brain edema and cortical blindness may be observed (9, 22). Severe hypertension may lead to central nervous system findings. Gastrointestinal involvement is common; hemorrahgic colitis, ileum/colon perforation, rectal prolapsus, cholestasis, pancreatitis, transient diabetes and peritonitis may occur (25). Increased troponin I level may be observed in relation with myocardial ischemia (26).
Diagnosis
The diagnosis is made clinically with hemolytic anemia, thrombocytopenia and renal damage which occur suddenly in a patient with a history of diarrhea in the last two weeks. For a definite diagnosis STEC infection should be proven (demonstration of Stx with stool serologic tests or stool cultures). Renal biopsy is not necessary for making a definite diagnosis.
Differential diagnosis
The differential diagnosis should be made with the other conditions causing to TMA.
Disseminated intravascular coagulation (DIC): prothrombin and partial thromboplastin times (PT and a PTT) are prolonged. Fibrinogen, Factor V and Factor VIII levels are decreased. It is differentiated from HUS with these findings.
Thrombotic thrombocytopenic purpura: the diagnosis is made with the finding of a ADAMTS 13 activity of <10%.
Complement-related HUS: It may occur at any age starting from the neonatal period. It is differentiated from STEC-HUS with its recurrent character and familial history. Serologic and genetic tests related with the alternative complement system [serum complement 3 (C3) level, complement factor H and I level, demonstration of anti-factor H antibody, genetic mutations of the proteins related with the complement system] are valuable in the diagnosis. Decreased seum C3 level is an important finding, but a normal level does not exclude complement-related HUS. A slight decrease in C3 may be observed in some cases of STEC-HUS.
Penumococcus-related HUS: Patients affected have a history of severe pneumococcus infection. The diagnosis is made definitely with growth of Streptococcus Pneumoniae in blood, pleural fluid or cerebrospinal fluid cultures or demonstration of T antigen in the erythrocytes.
H1N1 and Influenza-related HUS: the diagnosis is made by demonstration of the agent with culture and polymerase chain reaction (PCR).
Hereditary cobalamin metabolism disorder: although it may occur at any age, it should be especially considered in the differential diagnosis in cases of HUS which occur in the neonatal period and early infancy. The blood homocysteine level is increased and methionine level is decreased. Blood and/or urinary metyl-malonic acid levels are increased (organic acid chromatography). The definite diagnosis is made by demonstration of MMACHC mutation.
Secondary HUS: History of an accompanying condition, drug usage or presence of non-STEC infection suggests the diagnosis of secondary HUS.
Treatment
There is no specific therapy. Priority should be given to supportive treatment.
Supportive treatment: Adjustment of fluid and electrolyte balance, control of blood pressure and regulation of dialysis and hematological variables constitute the essentials of supportive treatment.
Fluid and electrolyte treatment: This is evaluated according to the fluid status and renal function of the patient. In the begining of the disease, vomiting, diarrhea and decreased oral intake may lead to dehydratation. In this case, fluid support with appropriate electrolyte content is given. In presence of hypertension and edema, fluid restriction is applied. In patients with oliguria, edema and hypertension, the daily fluid which should be given is calculated as follows=insensible losses (400 cc/m2/day)+urinary output (mL/h) + additional losses. In patients with severe fluid loading, furosemid may be tried (2 mg/kg/dose); treatment is not continued in cases where no treatment response is obtained. Fluid loading unresponsive to diuretic treatment and electrolyte disorders unresponsive to drug treatment require dialysis treatment.
Hypertension: Primarily, increased fluid loading should be corrected. The first-line drugs in antihypertensive treatment in the acute period of the disease are calcium channel blockers; nifedipin at a dose of 0.25 mg/ kg or amlodipin at a dose of 0.1 mg/kg can be intitated. In urgent cases of increased blood pressure including hypertensive encephalopathy, intravenous treatment is preferred. Sodium nitroprusside (0.5–8 μg/kg/min intravenous infusion) and esmolol (50–200 μg/kg/min intravenous infusion) which are available in our country may be used for this objective. Angiotensin converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB) should be avoided in the acute phase.
Dialysis: Symptomatic uremia (uremic encephalopathy, pericarditis, hemorrhage), azotemia (BUN≥80–100 mg/dL), severe fluid loading unresponsive to diuretics (hypertension, heart failure), electrolyte and acid-base disorders unresponsive to drug medical therapy (K+>6.5 mEq/L, Na<120 mEq/L, pH<7.1) and inability to provide treatment and nutrition because of fluid restriction are the indications of dialysis treatment.
Anemia: Erythrocyte transfusion is recommended in patients with a hemoglobin level of <6 g/dL. Erythrocyte transfusion may be needed at higher hemoglobin levels (<7 g/dL) in symptomatic patients. In patients with hyperpotassemia and fluid loading, erythrocyte transfusion should be performed during dialysis. Considering the possibility of future renal transplantation leukocyte filter should be used to decrease the risk of alloimmunization.
Thrombocytopenia: Platelet transfusion is recommended only for patients with life threatening bleeding or in the preperation period of surgery.
Plasma treatment: There is no sufficient evidence indicating that administration of plasma or plasma exchange is beneficial in STEC-HUS (27). Plasma exchange may be useful in patients with neurological involvement (23).
Eculizumab: Eculizumab is a monoclonal C5 antibody which inhibits complement activation and used in treatment of complement-related HUS. It can be used in cases of STEC-HUS with neurological involvement (28).
The last two therapies will be explained in detail in the part of “complement-related HUS”.
Prognosis
The mortality rate is 3–5% in the acute phase. The mortality is generally related with non-renal causes. The mortality risk is higher in patients with severe neurological involvement. Dialysis is required in approximately 2/3 of the cases in the acute phase. The rate of progression to end stage renal disease (ESRD) is approximately 10% (29). Long term complications including hypertension, proteinuria, chronic renal disease and insulin dependent diabetes may develop in approximately 1/3 of the patients. A long period of oliguria-anuria (>2 weeks) and presence of global/segmental sclerosis and interstitial fibrosis in renal biopsy are the best findings indicating a poor prognosis for renal involvement (27). Recurrence after renal transplantation is not expected in STEC-HUS.
COMPLEMENT - RELATED HUS
In the childhood, complement-related HUS constitutes the majority of HUS cases excluding STEC-HUS. It occurs as a result of uncontrolled activation of alternative complement pathway. It is a rare disease. Although it may occur at any age from the neonatal period to adulthood, it mostly affects children and adolescents. In the childhood, it is observed with a equal frequency in girls and boys. The first attack occurs below the age of 2 years in approximately 70% of the cases and below the age of 6 months in 25% (30). It may occur sporadically or it may be familial. The prognosis of the disease is poor; end stage renal disease develops in approximately 50% of the patients (31).
Etiology and Pathogenesis
The majority of complement-related HUS occur as a result of genetic mutation of complement factors, whereas a small portion (5–6%) occur as a result of antibodies produced against complement proteins (31–33).
Complement factor H (CFH), complement factor I (CFI), membrane cofactor protein (MCP or CD46) and thrombomodulin (THBD) which are regulatory proteins of the alternative complement pathway and C3 and complement factor B (CFB) which are C3 convertase proteins are involved in the pathogenesis of the disease (6). Mutations in the genes encoding these proteins lead to uncontrolled activation in the alternative complement pathway (6, 32, 34). Activation of the complement system leads to overproduction in membrane attack complex (C5b-C9 complex) and C5a. Membrane attack complex causes to endothelial cell death, edema and cell growth. This triggers increase in prothrombotic substances in the subendothelial area, activation of the coagulation system and fibrin deposition. C5a is a strong anaphlatoxin and causes to chemotaxis, leukocyte and endothel activation. Vascular permeability increases. Cellular fragmentation, inflammation and thrombosis result in obstruction in the microvascular area (6, 7, 32, 34).
Genetic Mutations
Factor H mutations: These mutations are the mutations of the gene encoding complement factor H which is one of the regulatory proteins of the alternative complement system. This the most common genetic mutation found in cases of complement-related HUS (20–30%). It may occur at any age from the neonatal period. It is most commonly observed in infancy and early childhood (6). The patients with factor H mutation have the poorest prognosis among the cases of complement-related HUS; mortality or progression to ESRD occurs with a rate of 50–70% in the first one year (35).
It has both autosomal recessive and autosomal dominant types. More than hundered mutations have been defined until now. Serum CFH levels are very low in some mutations and normal or slightly low in some other mutations. Serum C3 levels are found to be low in 30–50% of the cases (31, 35, 36). Therefore, normal serum CFH and C3 levels do not exclude the possibility of CFH mutation.
Membrane cofactor protein (MCP or CD46) mutations: These mutations are the mutations of CD46 which is one of the regulatory proteins of the alternative complement system and also known as MCP. More than fourty mutations have been defined. Most of the mutations are heterozygous. Some are homozygous or compound heterozygous. MCP mutation is found in 5–15% of the cases of complement-related HUS. It is observed more commonly in children compared to adults. Serum C3 level is decreased in less than 1/3 of the cases. Renal disease is the group with the best prognosis. The risk of recurrence is low after renal transplantation (30, 31, 36).
Factor I mutations: Complement factor I is the cofactor of MCP and factor H. CFI mutations have been found in 4–10% of the cases of complement-related HUS. There are about 40 mutations most of which are heterozygous. Serum C3 level is found to be decreased in approximately 1/3 of the cases. The prognosis of the disease is better compared to CFH mutations, but worse compared to MCP mutations. Progression to ESRD is 50–60% in the first two years (30, 31, 37).
C3 mutations: Heterozygous C3 mutations are responsible of 2–10% of the cases of complement-related HUS. Complement C3 constitutes the active parts of C3 convertase together with CFB. Complemet 3 mutations cause to continuous activation of C3 convertase. This activation can not be controlled by the regulatory proteins of the alternative complement system. Serum C3 level is decreased in approximately half of the patients. This decreased level continues during the attack intervals. The prognosis of the disease is poor; the rate of progression to ESRD is high (60–80%). The recurrence rate after renal transplantation is 40–70% (6, 31, 38, 39).
Factor B mutations: They are observed with a lower rate compared to the other mutations (1–4%). Complement factor B mutations lead to continuous activation of the alternative complement pathway. Serum C3 level is decreased. ESRD develops in 70% of the cases. Although a limited number of patients has been reported, recurrence resulting in graft loss has been observed in all patients after transplantation (31, 36, 40).
Thrombomodulin mutations: Thrombomodulin is a cofactor enabling CFI-mediated C3b inactivation. The mutated gene causes to complement activation. Heterozygous thrombomodulin mutation is found in 3–5% of the cases of complement-related HUS. Serum C3 is decreased in approximately 60% of the patients (31, 41).
Complement factor H antibodies
Complement factor H antibodies are found to be positive in approximately 6–10% of the cases of aHUS. These antibodies prevent CHF from binding to C3b and cellular surface. In most cases, serum CFH-related protein (CFHR) 1 and 3 are decreased. In these cases, CFHR1 and CFHR3 homozygous mutations are present. The serum CFH level may be normal or decreased (20%); the C3 level is decreased in approximately half of the cases. It is observed most commonly in children aged between 5 and 13 years. The risk of progression to end stage renal disease is 30–40% (33, 42).
Clinical findings
There is a triggering infection in more than half of the cases. This is mostly an upper respiratory tract infection or acute diarrhea. Onset with diarrhea has been reported with a rate of 23–28% in cases of aHUS (30, 31, 36).
Generally, there is a sudden onset. Patients may present with complaints including palor, malaise, restlessness, vomiting and sometimes edema. As in all cases of HUS, there are clinical and laboratory findings related with the damage to the tissues, organs and systems caused by TMA. The majority of the patients show the findings of the classical HUS triad including microangiopathic hemolytic anemia, thrombocytopenia and acute renal damage (see. STEC-HUS). Hypertension is common and severe; heart failure and neurological complications may be observed. Extrarenal findings occur in 20% of the cases; the most common extrarenal finding is central nervous system involvement; multiple organ involvement is present in 5% of the cases (30, 31).
Diagnosis
The diagnosis is made by clinical and laboratory findings of microangiopathic hemolytic anemia, thrombocytopenia and acute renal damage and demonstration of the disruption in regulation of the complement system. Decreases serum C3 level is a warning finding. C4 is normal in all patients. The definite diagnosis is made by serological and genetic tests related with the complement system. Serum CFH, CFI and CFB levels, anti-factor H antibody measurement and genetic mutations of the complement proteins are the tests performed for this objective. However, these tests are not widely used and easily applicable.
The disease which cause to complement-related HUS show differences in terms of age of onset, laboratory findings and prognosis. These differences give an idea both in terms of the diagnosis and prognosis, but the definite diagnosis is made by demonstration of the genetic mutations of the proteins in the complement system as mentioned before. The clinical and laboratory properties of the diseases causing to complement-related HUS are summarized in Table 2 (6, 8, 43).
Table 2.
| Subgroup | Frequency (%) | Age of onset | C3 | ESRD(%) | Recureence (%) | Recurrence after transplantation |
|---|---|---|---|---|---|---|
| CFH | 20–30 | <2 years | N / ↓ | 50–70 | 50 | 75–90% |
| CFI | 4–10 | < 2 years | N / ↓ | 50–60 | 10–30 | 45–80% |
| MCP | 5–15 | >1 years | N / ↓ | <20 | 70–90 | <20% |
| C3 | 2–10 | Any age | ↓ | 60–80 | 50 | 40–70% |
| CFB | 1–4 | 1 month | ↓ | 50–70 | Yes | 100% |
| THBD | 3–5 | 6 month | N / ↓ | 50–60 | 30 | 1 patient |
| Anti-CFH | 6 | 5–13 years | N / ↓ | 30–40 | 10–60 | Yes |
C3: complement 3; CFB: complement factor B; CFH: complement factor H; CFI: complement factor I; HUS: hemolytic uremic syndrome; MCP: membrane cofactor protein; ESRD: end stage renal disease; THBD: thrombomodulin
Differential diagnosis
Differential diagnosis should be made with the other HUS types and TTP. The cause in 90% of the cases of HUS in the childhood is STEC-HUS. There is no suspicion in terms of the diagnosis in cases with a history of diarrhea in the prodromal period where STEC infection has been proven. However, it should be kept in mind that a history of diarrhea is present in approximately 1/3 of the cases of aHUS.
aHUS should be considered in patients who had a HUS attack before, who have a positive familial history, who were diagnosed with HUS below the age of 6 months and who have had no history of diarrhea before the HUS attack (in the last 15 days) (see Table 3). The approach for patients who are thought to have aHUS is summarized in Table 4.
Table 3.
Subjects with suspicious atypical HUS
| A history of diarrhea in the last 15 days |
| Age <6 months |
| Recurrent HUS |
| Suspicious previous HUS |
| Familial history of HUS |
| Recurrent HUS after transplantation |
| Complement 3 (C3) level ↓ |
Table 4.
Tests used in the differential diagnosis of atypical HUS
| Primary step |
| Screening for STEC in diarrhea (+) subjects→negative→low probability of STEC-HUS |
| ADAMTS13 activity→normal→TTP is excluded |
| PT, aPTT, fibrinogen→normal→DIC is excluded |
| Pneumococcus antigen and Streptococcus pneumoniae in cultures→negative→Pneumococcus-related HUS is excluded |
| H1N1 and influenza→normal→H1N1 and influenza-related HUS is excluded |
| Blood homocysteine, urinary and blood MMA→normal→hereditary cobalamin defect is excluded |
| Serum C3 and C4→C3↓ and C4 normal→alternative complement pathway defect is considered |
| Secondary step |
| Anti-CFH antibodies→positive→anti-CFH antibody HUS |
| CFH, CFI, CFB levels |
| CFH, CFI, MCP, C3 and CFB mutations |
| Tertiary Step |
| Wide mutation screening |
PTT partial thromboplastin time; C3: complement 3; C4: complement 4; CFB: complement factor B; CFH: complement factor H; CFI: complement factor I; DIC: disseminated intravascular coagulation; HUS: hemolytic uremic syndrome; MCP: membrane cofactor protein; MMA: metyl malonic acid; PT: prothrombin time; STEC: Shiga-toxin producing E. coli; TTP: thrombotic microangiopathy
Treatment
Supportive treatment is explained in detail in the part of “STEC-HUS”. Here, plasma treatment and eculizumab treatment which are specific therapies for the disease will be explained.
Plasma treatment: Plasma treatment is used as the first-line therapy in patients with aHUS. Fresh frozen plasma contains normal amounts of CFH, CFI, CFB and C3. It is possible to remove mutant CFH, CFI, CFB and C3 and anti-factor H antibodies from the plasma with plasma exchange. Fluid loading complications related to high amounts of plasma transfusion can be prevented with plasma exchange.
Initiation of plasma treatment is recommended as soon as the diagnosis is made in non-infectious HUS cases. Plasma treatment is recommended as transfusion of fresh frozen plasma (FFP) (10–20 mL/kg) or plasma exchange (60–75 mL/kg). The primary treatment is plasma exchange. If this can not be applied, treatment is initiated with transfusion of fresh frozen plasma. This is administered for at least 5 days (every day initially) and completed to 2 weeks. Afterwards, it can be continued for months by tapering.
Despite appropriate plasma treatment, the rates of full or parital remission are low and the risk of ESRD is high (31). Response to plasma treatment varies according to the involved complement component. Response to plasma treatment is better in patients with complement factor H mutation compared to patients with MCP mutaiton (35).
Eculizumab: Eculizumab is a recombinant monoclonal C5 antibody; it stops complement activation in the last phase by inhibiting production of C5a and C5b by binding to C5. Although it has been used in treatment of paroxysmal nocturnal hemoglobinuria, it has been started to be used only recently in treatment of HUS. Positive results including both hematological remission and increase in the glomerular filtration rate have been reported in patients who are unresponsive to plasma treatment or who have partially controlled disease under plasma treatment (44).
Considering the poor prognosis of the disease, limited effects of plasma treatment and requirement for an intravenous access for plasma exchange, tendencies to administer eculizumab treatment as the first-line therapy especially in children have increased. Thus, eculizumab treatment was recommended as the first-line therapy in children with aHUS in the international consensus report published very recently (8). In patients in whom eculizumab treatment can not be initiated in 24–48 hours, plasma transfusion or plasma exchange is recommended. The eculizumab doses recommended for treatment of aHUS are shown in Table 5.
Table 5.
Eculizumab treatment doses recommended for atypical HUS cases (8)
| Body weight | Initial treatment | Maintenance treatment |
|---|---|---|
| >40 kg | 900 mg weekly 4 doses | 1200 mg, at the 5th week 1200 mg every 2 weeks |
| 30 – 40 kg | 600 mg weekly 2 doses | 900 mg at the 3rd week 900 mg every 2 weeks |
| 20 – 30 kg | 600 mg weekly 2 doses | 600 mg at the 3rd week 600 mg every 2 weeks |
| 10– 20 kg | 600 mg weekly 1 doses | 300 mg at the 2nd week 300 mg every 2 weeks |
| 5– 10 kg | 300 mg weekly 1 doses | 300 mg at the 2nd week 300 mg every 3 weeks |
All patients receiving eculizumab treatment should be vaccinated against Neisseria meningitidis, Streptococcus penumoniae and Haemophilus influenza type B. Prophylactic treatment with penicilin should be given in patients who are too young to be vaccinated with meningococcus vaccine or in whom treatment has had to be initiated before vaccination.
Liver-kidney transplantation: Factor H is a protein which is primarily synthesized in the liver. Liver transplantation alone or liver-kidney transplantation in combination can be performed in patients with known CFH or CFI mutation (45).
Kidney transplantation: Kidney transplantation can be performed in patients who have developed end stage renal diseases. However, recurrence rates range between 20% and 100% depending on the type of mutation. The possibility of graft loss is very high in cases of recurrence. The lowest risk in terms of recurrence after transplantation is observed in patients with MCP mutation (35, 43, 46). Therefore, it has been recommended that genetic mutations be determined before transplantation and renal transplantation be performed with prophylactic eculizumab treatment in cases of risk (8).
Immunosupressive treatment: In cases of HUS with anti-factor H antibody, corticosteroids, cyclophosphamide and rituximab therapies are recommended in addition to plasma exchange (8).
Footnotes
Peer-review: Externally peer-reviewed.
Conflict of Interest: No conflict of interest was declared by the author.
Financial Disclosure: The author declared that this study has received no financial support.
References
- 1.Rosove M. Thrombotic microangiopathies. Semin Arthritis and Rheumatism. 2014;43:797–805. doi: 10.1016/j.semarthrit.2013.11.004. http://dx.doi.org/10.1016/j.semarthrit.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Nester CM, Thomas CP. Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? Hematology Am Soc Hematol Educ Program. 2012;2012:617–25. doi: 10.1182/asheducation-2012.1.617. [DOI] [PubMed] [Google Scholar]
- 3.Cataland SR, Wu HM. Atypical hemolytic uremic syndrome and thrombotic thrombocytic purpura: Clinically differentiating the thrombotic microangiopathies. Eur J Intern Med. 2013;24:486–91. doi: 10.1016/j.ejim.2013.05.007. http://dx.doi.org/10.1016/j.ejim.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035–50. doi: 10.1681/ASN.2004100861. http://dx.doi.org/10.1681/ASN.2004100861. [DOI] [PubMed] [Google Scholar]
- 5.Besbas N, Karpman D, Landau D, et al. European Paediatric Research Group for HUS A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006;70:423–31. doi: 10.1038/sj.ki.5001581. http://dx.doi.org/10.1038/sj.ki.5001581. [DOI] [PubMed] [Google Scholar]
- 6.Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60. doi: 10.1186/1750-1172-6-60. http://dx.doi.org/10.1186/1750-1172-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salvadori M, Bertoni E. Update on hemolytic uremic syndrome: diagnostic and therapeutic recommendations. Worl J Nephrol. 2013;6:56–76. doi: 10.5527/wjn.v2.i3.56. http://dx.doi.org/10.5527/wjn.v2.i3.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loirat C, Fakhouri F, Ariceta G, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2015. Apr 11, [Epub ahead of print] http://dx.doi.org/10.1007/s00467-015-3076-8. [DOI] [PubMed]
- 9.Tarr PI, Gordon CA, Chandler WL. Shiga-toxin producing Escherichia coli and hemolytic uremic syndrome. Lancet. 2005;365:1073–86. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 10.Ruggenenti P, Remuzzi G. A German outbreak of hemolytic uremic syndrome. Lancet. 2011;378:1057–8. doi: 10.1016/S0140-6736(11)61217-8. http://dx.doi.org/10.1016/S0140-6736(11)61217-8. [DOI] [PubMed] [Google Scholar]
- 11.Robert Koch Institut Final presentation and evaluation of epidemiological findings in the EHEC O104:H+ outbreak Germany 2011; September 2011; Available from: http://www.rki.de/EN/Home/EHECfinalreport.pdf. Erişim tarihi: Erişim tarihi: 03.03.2015. [Google Scholar]
- 12.Elliott EJ, Robins-Browne RM, O’Loughlin EV, et al. Nationwide study of haemolytic uraemic syndrome: clinical, microbiological, and epidemiological features. Arch Dis Child. 2001;8:125–31. doi: 10.1136/adc.85.2.125. http://dx.doi.org/10.1136/adc.85.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piercefield EW, Bradley KK, Coffman RL, Mallonee SM. Hemolytic Uremic Syndrome after an Escherichia coli O111 Outbreak. Arch Intern Med. 2010;170:1656–63. doi: 10.1001/archinternmed.2010.346. http://dx.doi.org/10.1001/archinternmed.2010.346. [DOI] [PubMed] [Google Scholar]
- 14.Tozzi AE, Caprioli A, Minelli F, et al. Shiga toxin-producing Escherichia coli infections associated with hemolytic uremic syndrome, Italy, 1988–2000. Emerg Infect Dis. 2003;9:106–8. doi: 10.3201/eid0901.020266. http://dx.doi.org/10.3201/eid0901.020266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhimma R, Rollins NC, Coovadia HM, Adhikari M. Post-dysenteric hemolytic uremic syndrome in children during an epidemic of Shigella dysentery in Kwazulu/ Natal. Pediatr Nephrol. 1997;11:560–4. doi: 10.1007/s004670050338. http://dx.doi.org/10.1007/s004670050338. [DOI] [PubMed] [Google Scholar]
- 16.Bell BP, Goldoft M, Griffin PM, et al. A multistate outbreak of Escherichia coli O157:H7-associated bloody diarrhea and hemolytic uremic syndrome from hamburgers. The Washington experience. JAMA. 1994;272:1349–53. http://dx.doi.org/10.1001/jama.1994.03520170059036. [PubMed] [Google Scholar]
- 17.Vaillant V, Espié E, de Valk H, et al. Undercooked ground beef and person-to-person transmission as major risk factors for sporadic hemolytic uremic syndrome related to Shiga-toxin producing Escherchia coli infections in children in France. Pediatr Infect Dis J. 2009;28:650–3. doi: 10.1097/INF.0b013e3181993731. http://dx.doi.org/10.1097/INF.0b013e318199373. [DOI] [PubMed] [Google Scholar]
- 18.Ulinski T, Lervat C, Ranchin B, Gillet Y, Floret D, Cochat P. Neonatal hemolytic uremic syndrome after mother-to-child transmission of Escherichia coli O157. Pediatr Nephrol. 2005;20:1334–5. doi: 10.1007/s00467-005-1871-3. http://dx.doi.org/10.1007/s00467-005-1871-3. [DOI] [PubMed] [Google Scholar]
- 19.Noris M, Mescia F, Remuzzi G. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol. 2012;8:622–33. doi: 10.1038/nrneph.2012.195. http://dx.doi.org/10.1038/nrneph.2012.195. [DOI] [PubMed] [Google Scholar]
- 20.Trachtman H, Austin C, Lewinski M, Stahl RA. Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat Rev Nephrol. 2012;8:658–69. doi: 10.1038/nrneph.2012.196. http://dx.doi.org/10.1038/nrneph.2012.196. [DOI] [PubMed] [Google Scholar]
- 21.McLaine PN, Rowe PC, Orrbine E. Experiences with HUS in Canada: what have we learned about childhood HUS in Canada? Kidney Int Suppl. 2009;112:S25–8. doi: 10.1038/ki.2008.614. http://dx.doi.org/10.1038/ki.2008.614. [DOI] [PubMed] [Google Scholar]
- 22.Gerber A, Karch H, Allerberger F, Verweyen HM, Zimmerhackl LB. Clinical course and the role of shiga toxin-producing Escherichia coli infection in the hemolytic-uremic syndrome in pediatric patients, 1997–2000, in Germany and Austria: a prospective study. J Infect Dis. 2002;186:493–500. doi: 10.1086/341940. http://dx.doi.org/10.1086/341940. [DOI] [PubMed] [Google Scholar]
- 23.Kielstein JT, Beutel G, Fleig S, et al. Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant. 2012;27:3807–15. doi: 10.1093/ndt/gfs394. http://dx.doi.org/10.1093/ndt/gfs394. [DOI] [PubMed] [Google Scholar]
- 24.Johnson S, Taylor CM. Hemolytic uremic syndromes. In: Avner ED, Harmon WE, Niaudet P, Yoshikawa N, editors. Pediatric Nephrology. 6th ed. Berlin-Heidelberg: Springer-Verlag; 2009. pp. 1155–80. http://dx.doi.org/10.1007/978-3-540-76341-3_48. [Google Scholar]
- 25.de Buys Roessingh AS, de Lagausie P, Baudoin V, Loirat C, Aigrain Y. Gastrointestinal complications of post-diarrheal hemolytic uremic syndrome. Eur J Pediatr Surg. 2007;17:328–34. doi: 10.1055/s-2007-965013. http://dx.doi.org/10.1055/s-2007-965013. [DOI] [PubMed] [Google Scholar]
- 26.Askiti V, Hendrickson K, Fish AJ, Braunlin E, Sinaiko AR. Troponin I levels in a hemolytic uremic syndrome patient with severe cardiac failure. Pediatr Nephrol. 2004;19:345–8. doi: 10.1007/s00467-003-1343-6. http://dx.doi.org/10.1007/s00467-003-1343-6. [DOI] [PubMed] [Google Scholar]
- 27.Scheiring J, Andreoli SP, Zimmerhackl LB. Treatment and outcome of Shiga-toxin-associated hemolytic uremic syndrome (HUS) Pediatr Nephrol. 2008;23:1749–60. doi: 10.1007/s00467-008-0935-6. http://dx.doi.org/10.1007/s00467-008-0935-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364:2561–3. doi: 10.1056/NEJMc1100859. http://dx.doi.org/10.1056/NEJMc1100859. [DOI] [PubMed] [Google Scholar]
- 29.Garg AX, Suri RS, Barrowman N, et al. Long-term renal prognosis of diarrhea-associate haemolytic uraemic syndrome. A systemic review, meta-analysis and metaregression. JAMA. 2003;290:1360–70. doi: 10.1001/jama.290.10.1360. http://dx.doi.org/10.1001/jama.290.10.1360. [DOI] [PubMed] [Google Scholar]
- 30.Sellier-Leclerc AL, Fremeaux-Bacchi V, Macher MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18:2392–400. doi: 10.1681/ASN.2006080811. http://dx.doi.org/10.1681/ASN.2006080811. [DOI] [PubMed] [Google Scholar]
- 31.Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5:1844–59. doi: 10.2215/CJN.02210310. http://dx.doi.org/10.2215/CJN.02210310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geerdink LM, Westra D, van Wijk JA, et al. Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol. 2012;27:1283–91. doi: 10.1007/s00467-012-2131-y. http://dx.doi.org/10.1007/s00467-012-2131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dragon-Durey MA, Loirat C, Cloarec S, et al. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:555–63. doi: 10.1681/ASN.2004050380. http://dx.doi.org/10.1681/ASN.2004050380. [DOI] [PubMed] [Google Scholar]
- 34.Kavanagh D, Goodship T. Genetics and complement in atypical HUS. Pediatr Nephrol. 2010;25:2431–42. doi: 10.1007/s00467-010-1555-5. http://dx.doi.org/10.1007/s00467-010-1555-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–79. doi: 10.1182/blood-2005-10-007252. http://dx.doi.org/10.1182/blood-2005-10-007252%3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loirat C, Noris M, Fremeaux-Bacchi V. Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2008;23:1957–72. doi: 10.1007/s00467-008-0872-4. http://dx.doi.org/10.1007/s00467-008-0872-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kavanagh D, Kemp EJ, Mayland E, et al. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:2150–5. doi: 10.1681/ASN.2005010103. http://dx.doi.org/10.1681/ASN.2005010103. [DOI] [PubMed] [Google Scholar]
- 38.Frémeaux-Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948–52. doi: 10.1182/blood-2008-01-133702. http://dx.doi.org/10.1182/blood-2008-01-133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119:4182–91. doi: 10.1182/blood-2011-10-383281. http://dx.doi.org/10.1182/blood-2011-10-383281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roumenina LT, Jablonski M, Hue C, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood. 2009;114:2837–45. doi: 10.1182/blood-2009-01-197640. http://dx.doi.org/10.1182/blood-2009-01-197640. [DOI] [PubMed] [Google Scholar]
- 41.Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:345–57. doi: 10.1056/NEJMoa0810739. http://dx.doi.org/10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dragon-Durey MA, Sethi SK, Bagga A, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–7. doi: 10.1681/ASN.2010030315. http://dx.doi.org/10.1681/ASN.2010030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–87. doi: 10.1056/NEJMra0902814. http://dx.doi.org/10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 44.Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368:2169–81. doi: 10.1056/NEJMoa1208981. http://dx.doi.org/10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 45.Taylor CM, Machin S, Wigmore SJ, et al. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol. 2010;148:37–47. doi: 10.1111/j.1365-2141.2009.07916.x. http://dx.doi.org/10.1111/j.1365-2141.2009.07916.x. [DOI] [PubMed] [Google Scholar]
- 46.Loirat C, Niaudet P. The risk of recurrence of hemolytic uremic syndrome after renal transplantation in children. Pediatr Nephrol. 2003;18:1095–101. doi: 10.1007/s00467-003-1289-8. http://dx.doi.org/10.1007/s00467-003-1289-8. [DOI] [PubMed] [Google Scholar]