

Abstract
Background
Malaria parasites that infect birds can have narrow or broad host-tropisms. These differences in host specificity make avian malaria a useful model for studying the evolution and transmission of parasite assemblages across geographic ranges. The molecular mechanisms involved in host-specificity and the biology of avian malaria parasites in general are important aspects of malaria pathogenesis that warrant further examination. Here, the transcriptome of the malaria parasite Plasmodium gallinaceum was characterized to investigate the biology and the conservation of genes across various malaria parasite species.
Methods
The P. gallinaceum transcriptome was annotated and KEGG pathway mapping was performed. The ripr gene and orthologous genes that play critical roles in the purine salvage pathway were identified and characterized using bioinformatics and phylogenetic methods.
Results
Analysis of the transcriptome sequence database identified essential genes of the purine salvage pathway in P. gallinaceum that shared high sequence similarity to Plasmodium falciparum when compared to other mammalian Plasmodium spp. However, based on the current sequence data, there was a lack of orthologous genes that belonged to the erythrocyte-binding-like (EBL) and reticulocyte-binding-like homologue (RH) family in P. gallinaceum. In addition, an orthologue of the Rh5 interacting protein (ripr) was identified.
Conclusions
These findings suggest that the pathways involved in parasite red blood cell invasion are significantly different in avian Plasmodium parasites, but critical metabolic pathways are conserved throughout divergent Plasmodium taxa.
Electronic supplementary material
The online version of this article (doi:10.1186/s12936-015-0814-0) contains supplementary material, which is available to authorized users.
Keywords: Plasmodium, Transcriptome, Avian malaria, Invasion, Erythrocytes, ripr
Background
A key determinant of host species susceptibility and host-specificity in malaria infections is the successful interaction between host erythrocyte receptors and parasite invasion ligands [1–4]. For mammalian malaria species, the invasion ligands that are implicated in host-specificity are relatively well characterized and can be grouped into two gene families, the erythrocyte-binding-like (EBL) and reticulocyte-binding-like homologue (RH) genes [5]. EBL and RH genes have been difficult to identify in avian malaria species since the host erythrocytes are nucleated, which in turn leads to methodological challenges for sequencing genetic material from these parasites [6, 7]. Therefore, the invasion pathways and ligands involved in avian malaria parasite host-specificity remain poorly understood. However, insight into the invasion process of avian malaria parasites was recently gained from the sequenced transcriptomes of Plasmodium relictum, a host generalist avian malaria parasite [7], and Plasmodium gallinaceum, a parasite of domestic chickens (Gallus gallus) [6].
Sequencing of the P. gallinaceum transcriptome has led to the identification of several orthologous genes of mammalian Plasmodium spp., two of which are critical for these parasites to invade host erythrocytes, the apical membrane antigen-1 (ama-1) and the rhoptry neck protein 2 (RON2) [6]. Additionally, an orthologue of the merozoite apical erythrocyte binding-ligand (maebl) gene, a member of the EBL family, was also identified and is currently the only known EBL gene expressed in avian Plasmodium [8]. These invasion genes are fairly conserved and show greater sequence similarity to Plasmodium falciparum genes, in comparison to other mammalian Plasmodium spp. The genotypic and biological similarities between P. falciparum and P. gallinaceum [9–13] suggest that knowledge gained from P. gallinaceum may be applicable to P. falciparum or vice versa, and thus highlights the importance of P. gallinaceum as a Plasmodium model. Furthermore, orthologous genes of EBL and RH receptors are encoded in the Gallus gallus genome [14–16]. Among these host receptors are glycophorin C, complement component receptor 1 and basigin, which bind to the EBL/RH proteins EBA-140, Rh4 and Rh5, respectively [3, 14, 15]. It is plausible that avian Plasmodium also utilize these diverse invasion pathways. However, mammalian Plasmodium spp. are generally host-specific as opposed to avian Plasmodium spp.
The host range of avian malaria parasites can include multiple avian host species or can be restricted to a single avian host species [17]. Investigating the genetic determinants of host-specificity in avian malaria parasites may lead to a better understanding of the molecular mechanisms that augment host switching or zoonotic malaria. However, little is known regarding the molecular mechanisms of avian malaria pathogenesis. The aim of this study was to elucidate key features of avian malaria parasite biology by characterizing the P. gallinaceum transcriptome and by identifying orthologous genes that may contribute to the host-specificity of these Plasmodium parasites.
Methods
Sequencing and assembly of the Plasmodium gallinaceum transcriptome
RNA from P. gallinaceum-infected chick blood was obtained for transcriptome sequencing and analysis as previously described [6]. In brief, White Leghorn chickens were infected with erythrocytes containing P. gallinaceum and infections were verified by PCR amplification of the P. gallinaceum cytochrome b gene and microscopy. Total RNA was extracted from infected chick blood, and cDNA libraries were prepared for sequencing on the HiSeq2000 platform. Raw reads were deposited in the NCBI sequence read archive (accession No. SRR1611148). To remove chicken sequences, the total reads were mapped to the Gallus gallus (chicken) genome using Bowtie [18]. The unmapped reads were collected for de novo assembly with or without quality trimming using Trimmomatic/Trinity [19] (Additional files 1, 2, 3, 4); paired end Trimmomatic parameters used were: LEADING:5 TRAILING:5 SLIDINGWINDOW:4:15 MINLEN:36. Lowly supported Trinity transcripts (FPKM <1) were removed using the Trinity RSEM utility and the remaining transcripts were clustered with CD-HIT-EST [20] using a sequence similarity threshold of 97%. Subsequently, a BLASTx query with the assembled P. gallinaceum transcriptomes was performed against the non-redundant protein database to remove any remaining chicken sequences. Queries with hits matching Gallus gallus sequences were removed using a custom BLAST parser script. The de novo transcriptome assemblies (before and after filtering) were evaluated using RSEM-EVAL [21].
Transcriptome characterization and analyses
Transcripts from the filtered assemblies were annotated using Blast2Go [22]. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were inferred and analysed through the KEGG database [23]. A database of P. falciparum EBL and RH gene sequences (Additional file 5) was generated and a tBLASTx query with the assembled P. gallinaceum transcripts was performed. A minimum query coverage of 60% and an E value cut-off of 1 × 10−10 were chosen for identifying putative orthologues. tBLASTx query of the EBL and RH gene sequences were also performed against the P. gallinaceum genome.
Phylogenetic analyses and characterization of Plasmodium gallinaceum orthologues
Sequences were aligned using MUSCLE in SEAVIEW [24]. For the maximum likelihood (ML) and Bayesian inference analysis, the Modeltest Version 3.7 [25] was used to determine the most appropriate nucleotide and amino acid substitution model based on the Akaike Information Criterion of the orthologous genes. The GTR + I + G model was selected for both the ripr and adenosine deaminase (ada) alignments. The GTR + G model was selected for both the hypoxanthine–guanine phosphoribosyl transferase (hgprt) and purine nucleoside phosphorylase (pnp) alignments. The GTR + I model was selected for the nucleoside transporter 1 (nt1) alignment. The WAG + I + G, LG + G, LG + G, WAG + G and CPREV + G amino acid substitution models were selected for the amino acid sequence alignments of ripr, ada, hgprt, pnp, and nt1, respectively. ML methods were implemented in RAxML [26] using 1,000 rapid bootstrap inferences. A Bayesian approach, as implemented in Beast v1.8.0 [27], was used to estimate posterior probabilities of the tree branches. The non-synonymous substitutions per non-synonymous sites divided by the synonymous substitutions per synonymous sites (dN/dS) ratio was analysed in an alignment of multiple Plasmodium spp. ripr gene sequences (see Additional file 5 for the gene IDs and accession numbers) using a sliding window method, as applied in DNAsp [28]. The variance of dN and dS (dN–dS) and standard errors were calculated by the Nei and Gojobori method with the Jukes and Cantor correction, and the significance was assessed using a two-tailed Z test with 1,000 bootstrap pseudosamples in MEGA v5.2.2 [29]. Genetic distances were calculated in MEGA v5.2.2.
Results
Characterization of the Plasmodium gallinaceum transcriptome
Without quality trimming prior to de novo assembly, the de novo transcriptome assembly resulted in 44,256 contigs (including alternatively spliced isoforms) with N50 of 1125 nt and a mean sequence length of 772 nt. After filtering out contigs that were not well supported based on the abundance estimations, transcripts that are redundant or highly similar, and transcripts with BLAST hits matching chicken sequences, the resulting assembly had 17,832 transcripts, with N50 of 1348 nt and a mean sequence length of 893 nt. Filtered assemblies resulted in better RSEM-EVAL scores (Additional file 6) and were used for functional annotation.
A total of 8,550 sequences were annotated with Blast2go (Additional files 7, 8, 9). GO terms were assigned to contigs and the sequences were grouped based on biological processes, molecular functions, and cellular components (Fig. 1). Analysis of all the sequences identified GO annotations for 3,429 sequences in biological processes, 2,160 sequences in molecular functions and 1,737 sequences in cellular components. The sequences associated with cellular components were represented by several sub-categories: cell (43%), organelle (31%) and macromolecular complex (21%). Similarly, the sequences associated with biological processes were distributed primarily across several sub-categories: cellular processes (33%), metabolic processes (32%), single-organism processes (14%), and localization (7%). In contrast, the majority of sequences for molecular components were mainly associated with two sub-categories, catalytic activity (42%) and binding (46%) components.
Fig. 1.
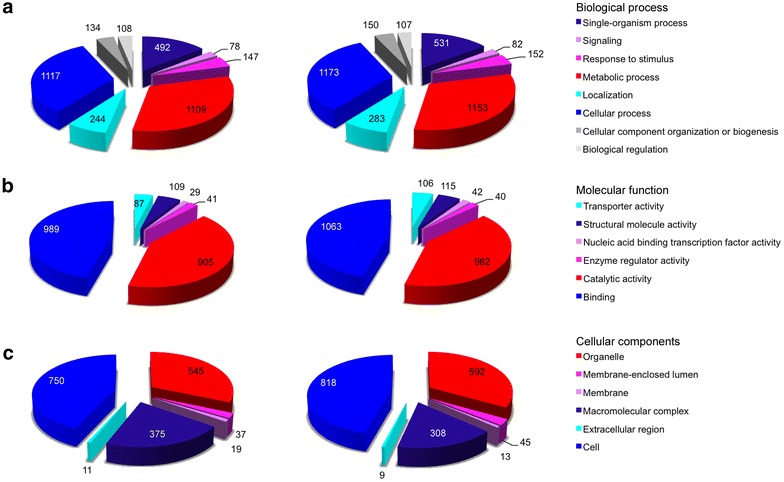
Functional annotations of the assembled sequences without (left) and with (right) quality trimming from the blood stages of Plasmodium gallinaceum. The pie charts show the number of sequences that are grouped into three general categories: biological process (a), molecular function (b) and cellular components (c).
In addition, the P. gallinaceum transcriptome was further characterized by KEGG pathway analysis, which resulted in a total 909 sequences mapping to 71 KEGG pathways (Additional file 10). Thiamine, pyrimidine and purine metabolism comprised the greatest amount of mapped sequences amongst the assigned KEGG pathways. These findings and the fact that Plasmodium parasites are purine auxotrophs prompted a search in the P. gallinaceum transcriptome assembly for genes that play critical roles in purine salvage. A partial cDNA sequence encoding a P. gallinaceum version of the purine salvage enzyme PNP and full-length cDNA sequences encoding the enzymes ADA, HGPRT and NT1 were identified. Phylogenetic analyses consistently showed, with strong posterior probabilities and bootstrap support, that the identified orthologous P. gallinaceum genes are most similar to the P. falciparum genes in these analyses (Fig. 2; Additional file 11).
Fig. 2.
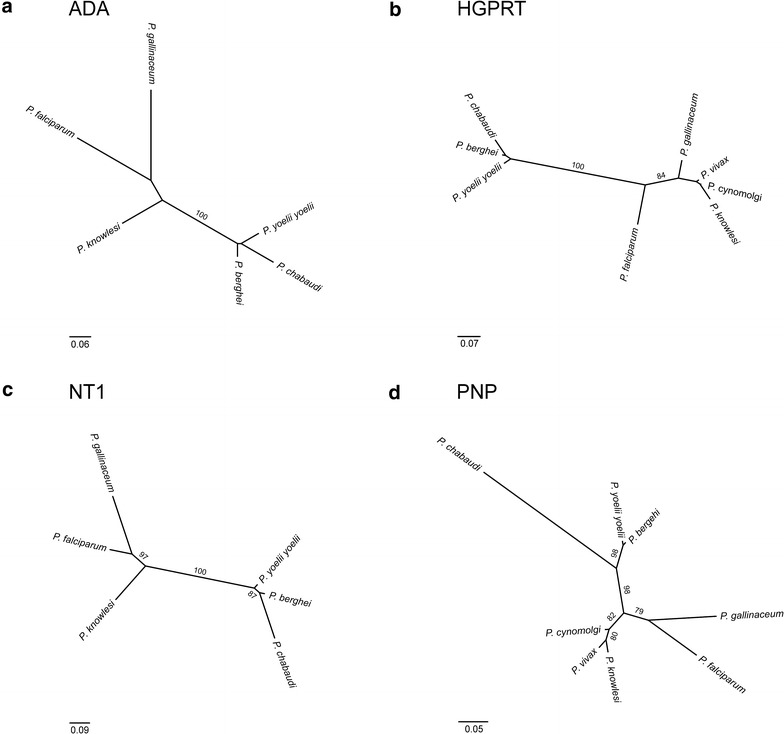
Phylogeny of Plasmodium spp. based on the amino acid sequences of a adenosine deaminase (ADA), b hypoxanthine–guanine phosphoribosyl transferase (HGPRT), c nucleoside transporter 1 (NT1), and d purine nucleoside phosphorylase (PNP). See Additional file 5 for accession numbers.
Quality trimming of the raw sequence reads prior to de novo assembly generated a higher number of contigs, 233,882 in total, with N50 of 532 nt and a mean sequence length of 474 nt. The resulting filtered transcriptome had 32,549 contigs with N50 of 950 nt and a mean sequence length of 678 nt. Quality trimming and filtering of the transcriptome assembly did not significantly change the annotation/mapping and KEGG pathway result (Fig. 1, Additional files 12, 13, 14, 15).
Identification and characterization of ripr in Plasmodium gallinaceum
To identify orthologous EBL and RH genes, BLAST searches were performed against P. falciparum EBL and RH gene sequences using the P. gallinaceum transcriptome sequences as queries. There were no significant BLAST hits that met the selected criteria. However, a partial cDNA sequence, with a length of 3,006 base pairs (bp), encoding an orthologue of ripr (sequence comp28469_c1_seq2, Additional file 1) was identified in P. gallinaceum. To confirm that the ripr sequence is present in the P. gallinaceum genome, a BLAST search against the P. gallinaceum genome was performed using the identified partial cDNA sequence of ripr as a query. The search resulted with a significant hit comprising 100% query coverage and identity to nucleotide (nt) positions 1101-1495 of the contig Pg_2265551.c000013273. The identified genomic sequence was then used to annotate the full-length ripr gene of P. gallinaceum. Furthermore, 6,207 P. gallinaceum RNA-seq reads mapped to the full-length P. gallinaceum ripr sequence with coverage across the entire gene.
The full-length gene and the translated amino acid sequence of P. gallinaceum ripr are 3,051 bp and 1,016 amino acids in length, respectively, and share 69% nucleotide and 54% amino acid sequence identity in a pair-wise comparison to P. falciparum. Phylogenetic analysis of P. gallinaceum and mammalian Plasmodium spp. ripr shows that P. gallinaceum ripr is most similar to that of P. falciparum (Fig. 3a). Sixty-six amino acids at positions 482–547 and eight amino acids at positions 554–561 (relative to P. falciparum RIPR) were absent from P. gallinaceum RIPR (Fig. 3b). Ninety cysteine residues were present in P. gallinaceum RIPR, 87 of which are conserved between P. falciparum and P. gallinaceum. The ten EGF-like domains of RIPR are also present and highly conserved in P. gallinaceum RIPR (Fig. 3c); as observed with P. falciparum RIPR, two EGF-like domains were clustered at the N-terminus, whereas the remaining eight EGF-like domains were clustered around the C-terminus in P. gallinaceum RIPR. dN/dS methods were applied to orthologous Plasmodiumripr sequences to assess diversifying selection on the ripr gene. There were no significant differences between dN and dS when analysing the entire coding region of ripr (dN = 0.50 ± 0.01, dS = 0.67 ± 0.03, P > 0.05). However, sliding window analysis of dN/dS revealed significant signatures of diversifying selection throughout the region encoding the C-terminus of RIPR (Fig. 3d; Additional file 16). In contrast to the C-terminus, the dN/dS ratios were below 1.0 throughout the region encoding the N-terminus, suggesting that the N-terminus of RIPR is under strong purifying selection.
Fig. 3.
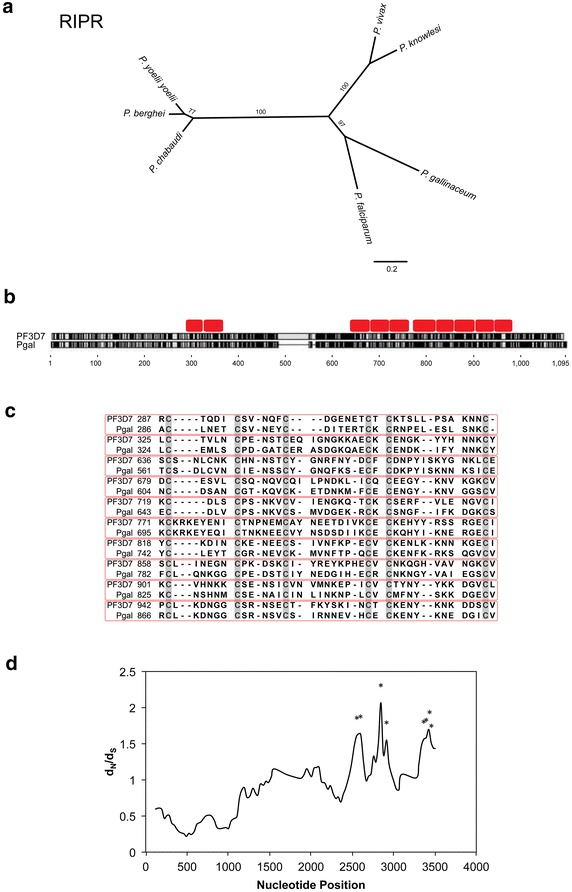
Comparison of Plasmodium rip. a Phylogeny of the Plasmodium based on amino acid sequences of RIPR. b Schematic of P. falciparum and P. gallinaceum RIPR amino acid alignment. Conserved regions are represented by black blocks. Variable regions are represented by grey blocks. Gaps in the alignment are represented by horizontal black lines. Epidermal growth factor-like domains are represented by red blocks. c Alignment of P. falciparum and P. gallinaceum epidermal growth factor-like domains. Individual epidermal growth factor-like domains are outlined in red and are ordered according to location, with the domain located closest to the N-terminus positioned at the top and the domain located closest to the C-terminus positioned at the bottom of the alignment. d Sliding window plot of dN/dS of ripr. The window length is 180 bp with a step size of 90 bp. Asterisks indicate regions with a significant excess of non-synonymous substitutions (P ≤ 0.05).
Discussion
Malaria parasites pose a significant threat to avian wildlife populations, and the emergence of avian malaria in naïve populations can have major ecological consequences [30]. The emergence of avian malaria is attributed to malaria parasites’ diverse host-specificity and geographic dispersal ranges that are facilitated by its avian hosts [31–33]. Large geographic dispersal ranges increase the probability for parasites to encounter new potential hosts, therefore increasing the chances for host-switching events [17, 33–36]. Indeed, host switching is a common phenomena that appears to be a distinct feature of avian malaria parasites [34, 37–40], i.e., mammalian malaria parasites are generally host-specific, whereas avian malaria parasites have a broad host-specificity. One aim of this study was to identify orthologous genes that may play a role in host-specificity, and to subsequently assess the evolution of these genes between mammalian and avian Plasmodium.
Based on the currently available sequence data, avian Plasmodium appear to lack the majority of EBL and RH genes that have been implicated in Plasmodium host-specificity, including eba-175, eba-181, eba-140, Rh1, Rh2a, Rh2b, Rh4,Rh5 [2–5, 41–45]. The only identified gene that exhibits clear orthology was maebl [8]. It is important to note that this finding may be attributed to several possibilities. For instance, the technical challenges and limitations for obtaining sequence data from avian Plasmodium parasites may have resulted in insufficient sequence coverage or sequencing that is not deep enough to detect these genes. An anticipated high quality draft genome, currently being produced as part of the Wellcome Trust Sanger Institute parasite genomics project, may help to resolve these uncertainties. Therefore, these findings do not rule out the presence of EBL and RH orthologues in avian Plasmodium. However, the number of invasion-associated genes also differ when comparing the genomes of Plasmodium reichenowi and P. falciparum [5]; only three of six Rh gene orthologues (Rh2b, Rh4 and Rh5) were found in P. reichenowi [5], and differences in the presence or absence of orthologous RBL and EBL genes were also observed between P. reichenowi, Plasmodium knowlesi and Plasmodium cynomolgi, Plasmodium vivax, and P. falciparum [46–51]. Moreover, the var gene family, which encode immunovariant adhesion proteins, seems to be restricted to primate malaria species [5].
Considering these findings, significant differences between EBL and RBL genes of avian and mammalian Plasmodium species are unlikely haphazard. Rather, it is speculated that avian Plasmodium spp. likely lack members of the EBL and RH family, and that the evolutionary shift of Plasmodium into mammals and the differences in host cell biology (nucleated erythrocytes in birds versus anucleated erythrocytes in mammals) led to the gain and diversification of the genes that modulate red blood cell invasion. In spite of this possibility, ripr is conserved and expressed in P. gallinaceum, suggesting that this gene is of ancient origin. Surprisingly, an orthologue of the RIPR binding partner RH5 was not identified, possibly due to low levels of rh5 transcripts in the sequenced blood stages of P. gallinaceum. However, an orthologue of rh5 appears to be absent from the P. gallinaceum genome as well. In the case of the latter, the putative avian Plasmodium RIPR may not be a functional counterpart of the mammalian Plasmodium spp. RIPR, despite being relatively conserved at the amino acid sequence level. The region encoding the N-terminus of RIPR lacks sequence diversity and appears to have evolved under purifying selection. Conversely, comparisons of the region encoding the RIPR C-terminus revealed evidence for diversifying selection. In this comparison, the observed nucleotide substitutions at the C-terminus may be indicative of interspecies sequence or structural specificity for the respective RH5 binding partners since this region directly interacts with RH5 [52]. Sequencing ripr from other avian Plasmodium spp. will thus be important to further assess the diversity of ripr and the selective pressures acting on ripr in avian malaria parasites. Taken together, these findings suggest that avian Plasmodium may express a novel set of invasion ligands or highly divergent EBL/RH genes that are currently undetectable.
Contrary to the results with EBL/RH genes, BLAST searches for essential genes involved in the purine salvage pathway detected highly conserved orthologues of ADA, HGPRT, NT1, and PNP in P. gallinaceum, highlighting the importance of the Plasmodium purine salvage pathway throughout the intra-erythrocytic growth of Plasmodium. In the intra-erythrocytic stages, Plasmodium parasites grow rapidly and require the salvage of host cell purines to synthesize nucleic acids [53, 54]. To carry out the purine salvage pathway, purine nucleosides must be transported into the parasites. This process is mediated by NT1, an essential purine transporter [55]. Transport of purine nucleosides results in adenosine being converted to hypoxanthine by ADA and PNP [56]. HGPRT can then convert hypoxanthine to inosine monophosphate (IMP), which is used as the precursor for the synthesis of purine nucleotides during nucleic acid synthesis [57]. These critical enzymes are highly expressed and abundant in Plasmodium [56–58]. Inhibiting these enzymes is lethal to Plasmodium [56, 59, 60], which therefore provides promising targets for anti-malarial drugs. Interestingly, Plasmodium purine salvage pathways were observed in initial investigations of the avian malaria parasite Plasmodium lophurae [61]. Thus, the conservation and expression of these enzymes that are essential to this pathway was expected, and affirmed that the de novo assembly generated P. gallinaceum transcripts of good quality.
Conclusions
Phylogenetic analysis of the purine salvage enzymes position P. gallinaceum closest to P. falciparum, and are consistent with several studies based on an assortment of genes [6, 13, 62]. The results here provide sequence data from an additional five nuclear genes to support this hypothesis. However, the theory that human P. falciparum originated from a lateral transfer of an avian parasite, as originally proposed [9], is contentious [63, 64], and additional sequencing from other avian Plasmodium species will be necessary to firmly resolve these relationships. Nevertheless, the genetic and biochemical similarities between P. falciparum and P. gallinaceum are evident, as demonstrated by genomic, biochemical and proteomic approaches [6, 8, 12, 13, 65, 66]. It is clear that improved methods for obtaining pure genetic material of avian malaria will be important for future genomic studies and would advance the field rapidly. As demonstrated previously [6] and reported here, establishing the P. gallinaceum transcriptome database will serve as a valuable fundamental resource for understanding avian malaria parasite biology.
Authors’ contributions
EJL performed the transcriptome characterization and phylogenetic analyses, and wrote the manuscript. HXA assisted with the bioinformatics analyses. SMT wrote the BLAST parsing script. RNMS conceived and participated in the design of the study and the drafting of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by a National Institute of Health Grant SC2AI089120-01A1, the Minority Biomedical Research Support-Research Initiative for Scientific Enhancement Grant R25-GM059298, the Initiative to Maximize Student Development Grant R25-GM103757, and the Genentech Foundation MS Dissertation Scholarship Award. We would like to thank Kevin Chan (Washington University in St. Louis) for his assistance with the bioinformatics analyses. The P. gallinaceum genomic sequence data were provided by the parasite genomics group at the Wellcome Trust Sanger Institute and can be obtained from http://www.sanger.ac.uk/research/projects/parasitegenomics/.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Additional files
Plasmodium gallinaceum de novo transcriptome assembly without quality trimming and no subsequent filtering of transcripts.
Plasmodium gallinaceum de novo transcriptome assembly without quality trimming and with subsequent filtering of transcripts.
Plasmodium gallinaceum de novo transcriptome assembly with quality trimming and no subsequent filtering of transcripts.
Plasmodium gallinaceum de novo transcriptome assembly with quality trimming and with subsequent filtering of transcripts.
Genes and the corresponding GenBank Accession numbers and Gene IDs for the parasite taxa used in the study.
RSEM-eval score of various assemblies.
Annotation file for the assembly without quality trimming.
Mapping file for the assembly without quality trimming.
Sequence table file for the assembly without quality trimming.
KEGG pathway file for the assembly without quality trimming.
Phylogeny of Plasmodium genes.
Annotation file for the assembly with quality trimming.
Mapping file for the assembly with quality trimming.
Sequence table file for the assembly with quality trimming.
KEGG pathway file for the assembly with quality trimming.
Pairwise comparisons of genetic distances, and the proportion of nonsynonymous differences per nonsynonymous site and synonymous differences per synonymous site between Plasmodium species.
Contributor Information
Elvin J Lauron, Email: ejlauron@wustl.edu.
Han Xian Aw Yeang, Email: awyeang@wustl.edu.
Samantha M Taffner, Email: staffner@dom.wustl.edu.
Ravinder N M Sehgal, Email: sehgal@sfsu.edu.
References
- 1.Martin MJ, Rayner JC, Gagneux P, Barnwell JW, Varki A. Evolution of human-chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc Natl Acad Sci USA. 2005;102:12819–12824. doi: 10.1073/pnas.0503819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hayton K, Gaur D, Liu A, Takahashi J, Henschen B, Singh S, et al. Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe. 2008;4:40–51. doi: 10.1016/j.chom.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crosnier C, Bustamante LY, Bartholdson SJ, Bei AK, Theron M, Uchikawa M, et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature. 2011;480:534–537. doi: 10.1038/nature10606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wanaguru M, Liu W, Hahn BH, Rayner JC, Wright GJ. RH5–Basigin interaction plays a major role in the host tropism of Plasmodium falciparum. Proc Natl Acad Sci USA. 2013;110:20735–20740. doi: 10.1073/pnas.1320771110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otto TD, Rayner JC, Böhme U, Pain A, Spottiswoode N, Sanders M, et al. Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nat Commun. 2014;5:4754. doi: 10.1038/ncomms5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauron EJ, Oakgrove KS, Tell LA, Biskar K, Roy SW, Sehgal RN. Transcriptome sequencing and analysis of Plasmodium gallinaceum reveals polymorphisms and selection on the apical membrane antigen-1. Malar J. 2014;13:382. doi: 10.1186/1475-2875-13-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hellgren O, Kutzer M, Bensch S, Valkiūnas G, Palinauskas V. Identification and characterization of the merozoite surface protein 1 (msp1) gene in a host-generalist avian malaria parasite, Plasmodium relictum (lineages SGS1 and GRW4) with the use of blood transcriptome. Malar J. 2013;12:381. doi: 10.1186/1475-2875-12-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez C, Marzec T, Smith CD, Tell LA, Sehgal RNM. Identification and expression of maebl, an erythrocyte-binding gene, in Plasmodium gallinaceum. Parasitol Res. 2013;112:945–954. doi: 10.1007/s00436-012-3211-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waters AP, Higgins DG, McCutchan TF. Plasmodium falciparum appears to have arisen as a result of lateral transfer between avian and human hosts. Proc Natl Acad Sci USA. 1991;88:3140–3144. doi: 10.1073/pnas.88.8.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Escalante AA, Ayala FJ. Evolutionary origin of Plasmodium and other Apicomplexa based on rRNA genes. Proc Natl Acad Sci USA. 1995;92:5793–5797. doi: 10.1073/pnas.92.13.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rathore D, Hrstka SCL, Sacci JB, De La Vega P, Linhardt RJ, et al. Molecular mechanism of host specificity in Plasmodium falciparum infection role of circumsporozoite protein. J Biol Chem. 2003;278:40905–40910. doi: 10.1074/jbc.M306250200. [DOI] [PubMed] [Google Scholar]
- 12.Li F, Patra KP, Vinetz JM. An anti-Chitinase malaria transmission-blocking single-chain antibody as an effector molecule for creating a Plasmodium falciparum-refractory mosquito. J Infect Dis. 2005;192:878–887. doi: 10.1086/432552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pick C, Ebersberger I, Spielmann T, Bruchhaus I, Burmester T. Phylogenomic analyses of malaria parasites and evolution of their exported proteins. BMC Evol Biol. 2011;11:167. doi: 10.1186/1471-2148-11-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lobo C-A, Rodriguez M, Reid M, Lustigman S. Glycophorin C is the receptor for the Plasmodium falciparum erythrocyte binding ligand PfEBP-2 (baebl) Blood. 2003;101:4628–4631. doi: 10.1182/blood-2002-10-3076. [DOI] [PubMed] [Google Scholar]
- 15.Tham W-H, Wilson DW, Lopaticki S, Schmidt CQ, Tetteh-Quarcoo PB, Barlow PN, et al. Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc Natl Acad Sci USA. 2010;107:17327–17332. doi: 10.1073/pnas.1008151107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ensembl. http://useast.ensembl.org/Gallus_gallus/Info/Index
- 17.Lauron EJ, Loiseau C, Bowie RCK, Spicer GS, Smith TB, Melo M, et al. Coevolutionary patterns and diversification of avian malaria parasites in African sunbirds (Family Nectariniidae) Parasitology. 2014;142:635–647. doi: 10.1017/S0031182014001681. [DOI] [PubMed] [Google Scholar]
- 18.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Fillmore N, Bai Y, Collins M, Thomson JA, Stewart R, et al. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2014;15:553. doi: 10.1186/s13059-014-0553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 23.Genome. http://www.genome.jp/kegg/
- 24.Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci CABIOS. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 25.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 26.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 27.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- 29.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Riper C, III, van Riper SG, Goff ML, Laird M. The epizootiology and ecological significance of malaria in Hawaiian land birds. Ecol Monogr. 1986;56:327. doi: 10.2307/1942550. [DOI] [Google Scholar]
- 31.Figuerola J, Green AJ. Haematozoan parasites and migratory behaviour in waterfowl. Evol Ecol. 2000;14:143–153. doi: 10.1023/A:1011009419264. [DOI] [Google Scholar]
- 32.Hubálek Z. An annotated checklist of pathogenic microorganisms associated with migratory birds. J Wildl Dis. 2004;40:639–659. doi: 10.7589/0090-3558-40.4.639. [DOI] [PubMed] [Google Scholar]
- 33.Jenkins T, Thomas GH, Hellgren O, Owens IPF. Migratory behavior of birds affects their coevolutionary relationship with blood parasites. Evol Int J Org Evol. 2012;66:740–751. doi: 10.1111/j.1558-5646.2011.01470.x. [DOI] [PubMed] [Google Scholar]
- 34.Ricklefs RE, Outlaw DC, Svensson-Coelho M, Medeiros MCI, Ellis VA, Latta S. Species formation by host shifting in avian malaria parasites. Proc Natl Acad Sci USA. 2014;111:14816–14821. doi: 10.1073/pnas.1416356111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hellgren O, Waldenström J, Peréz-Tris J, Szöll E, Si O, Hasselquist D, et al. Detecting shifts of transmission areas in avian blood parasites: a phylogenetic approach. Mol Ecol. 2007;16:1281–1290. doi: 10.1111/j.1365-294X.2007.03227.x. [DOI] [PubMed] [Google Scholar]
- 36.Hoberg EP, Brooks DR. A macroevolutionary mosaic: episodic host-switching, geographical colonization and diversification in complex host–parasite systems. J Biogeogr. 2008;35:1533–1550. doi: 10.1111/j.1365-2699.2008.01951.x. [DOI] [Google Scholar]
- 37.Ricklefs RE, Fallon SM. Diversification and host switching in avian malaria parasites. Proc R Soc Lond B Biol Sci. 2002;269:885–892. doi: 10.1098/rspb.2001.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waldenström J, Bensch S, Kiboi S, Hasselquist D, Ottosson U. Cross-species infection of blood parasites between resident and migratory songbirds in Africa. Mol Ecol. 2002;11:1545–1554. doi: 10.1046/j.1365-294X.2002.01523.x. [DOI] [PubMed] [Google Scholar]
- 39.Fallon SM, Bermingham E, Ricklefs RE. Island and taxon effects in parasitism revisited: avian malaria in the Lesser Antilles. Evol Int J Org Evol. 2003;57:606–615. doi: 10.1111/j.0014-3820.2003.tb01552.x. [DOI] [PubMed] [Google Scholar]
- 40.Szymanski MM, Lovette IJ. High lineage diversity and host sharing of malarial parasites in a local avian assemblage. J Parasitol. 2005;91:768–774. doi: 10.1645/GE-417R1.1. [DOI] [PubMed] [Google Scholar]
- 41.Loiseau C, Harrigan RJ, Robert A, Bowie RCK, Thomassen HA, Smith TB, et al. Host and habitat specialization of avian malaria in Africa. Mol Ecol. 2012;21:431–441. doi: 10.1111/j.1365-294X.2011.05341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walther EL, Valkiūnas G, González AD, Matta NE, Ricklefs RE, Cornel A, et al. Description, molecular characterization, and patterns of distribution of a widespread New World avian malaria parasite (Haemosporida: Plasmodiidae), Plasmodium (Novyella) homopolare sp. nov. Parasitol Res. 2014;113:3319–3332. doi: 10.1007/s00436-014-3995-5. [DOI] [PubMed] [Google Scholar]
- 43.Mayer DCG, Mu J-B, Kaneko O, Duan J, Su X, Miller LH. Polymorphism in the Plasmodium falciparum erythrocyte-binding ligand JESEBL/EBA-181 alters its receptor specificity. Proc Natl Acad Sci USA. 2004;101:2518–2523. doi: 10.1073/pnas.0307318101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayer DCG, Mu J-B, Feng X, Su X, Miller LH. Polymorphism in a Plasmodium falciparum erythrocyte-binding ligand changes its receptor specificity. J Exp Med. 2002;196:1523–1528. doi: 10.1084/jem.20020750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chattopadhyay D, Rayner JC, McHenry AM, Adams JH. The structure of Plasmodium falciparum EBA175 ligand domain and the molecular basis of host specificity. Trends Parasitol. 2006;22:143–145. doi: 10.1016/j.pt.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gunalan K, Gao X, Liew KJL, Preiser PR. Differences in erythrocyte receptor specificity of different parts of the Plasmodium falciparum reticulocyte binding protein homologue 2a. Infect Immun. 2011;79:3421–3430. doi: 10.1128/IAI.00201-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rayner JC, Huber CS, Galinski MR, Barnwell JW. Rapid evolution of an erythrocyte invasion gene family: the Plasmodium reichenowi reticulocyte binding like (RBL) genes. Mol Biochem Parasitol. 2004;133:287–296. doi: 10.1016/j.molbiopara.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 48.Semenya AA, Tran TM, Meyer EV, Barnwell JW, Galinski MR. Two functional reticulocyte binding-like (RBL) invasion ligands of zoonotic Plasmodium knowlesi exhibit differential adhesion to monkey and human erythrocytes. Malar J. 2012;11:228. doi: 10.1186/1475-2875-11-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okenu DMN, Meyer EV-S, Puckett TC, Rosas-Acosta G, Barnwell JW, Galinski MR. The reticulocyte binding proteins of Plasmodium cynomolgi: a model system for studies of P. vivax. Mol Biochem Parasitol. 2005;143:116–120. doi: 10.1016/j.molbiopara.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 50.Meyer EVS, Semenya AA, Okenu DMN, Dluzewski AR, Bannister LH, Barnwell JW, et al. The reticulocyte binding-like proteins of P. knowlesi locate to the micronemes of merozoites and define two new members of this invasion ligand family. Mol Biochem Parasitol. 2009;165:111–121. doi: 10.1016/j.molbiopara.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002;419:512–519. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- 52.Chen L, Lopaticki S, Riglar DT, Dekiwadia C, Uboldi AD, Tham W-H, et al. An EGF-like protein forms a complex with PfRh5 and is required for invasion of human erythrocytes by Plasmodium falciparum. PLoS Pathog. 2011;7:e1002199. doi: 10.1371/journal.ppat.1002199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Koning HP, Bridges DJ, Burchmore RJS. Purine and pyrimidine transport in pathogenic protozoa: from biology to therapy. FEMS Microbiol Rev. 2005;29:987–1020. doi: 10.1016/j.femsre.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 54.Hyde JE. Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr Drug Targets. 2007;8:31–47. doi: 10.2174/138945007779315524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El Bissati K, Zufferey R, Witola WH, Carter NS, Ullman B, Ben Mamoun C. The plasma membrane permease PfNT1 is essential for purine salvage in the human malaria parasite Plasmodium falciparum. Proc Natl Acad Sci USA. 2006;103:9286–9291. doi: 10.1073/pnas.0602590103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Madrid DC, Ting L-M, Waller KL, Schramm VL, Kim K. Plasmodium falciparum purine nucleoside phosphorylase is critical for viability of malaria parasites. J Biol Chem. 2008;283:35899–35907. doi: 10.1074/jbc.M807218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Raman J, Ashok CS, Subbayya SI, Anand RP, Selvi ST, Balaram H. Plasmodium falciparum hypoxanthine guanine phosphoribosyltransferase. FEBS J. 2005;272:1900–1911. doi: 10.1111/j.1742-4658.2005.04620.x. [DOI] [PubMed] [Google Scholar]
- 58.Reyes P, Rathod PK, Sanchez DJ, Mrema JE, Rieckmann KH, Heidrich HG. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol Biochem Parasitol. 1982;5:275–290. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- 59.Shi W, Ting L-M, Kicska GA, Lewandowicz A, Tyler PC, Evans GB, et al. Plasmodium falciparum purine nucleoside phosphorylase: crystal structures, immucillin inhibitors, and dual catalytic function. J Biol Chem. 2004;279:18103–18106. doi: 10.1074/jbc.C400068200. [DOI] [PubMed] [Google Scholar]
- 60.Queen SA, Jagt DL, Reyes P. In vitro susceptibilities of Plasmodium falciparum to compounds which inhibit nucleotide metabolism. Antimicrob Agents Chemother. 1990;34:1393–1398. doi: 10.1128/AAC.34.7.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walsh CJ, Sherman IW. Purine and pyrimidine synthesis by the avian malaria parasite, Plasmodium lophurae. J Protozool. 1968;15:763–770. doi: 10.1111/j.1550-7408.1968.tb02209.x. [DOI] [PubMed] [Google Scholar]
- 62.Silva JC, Egan A, Friedman R, Munro JB, Carlton JM, Hughes AL. Genome sequences reveal divergence times of malaria parasite lineages. Parasitology. 2011;138:1737–1749. doi: 10.1017/S0031182010001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roy SW, Irimia M. Origins of human malaria: rare genomic changes and full mitochondrial genomes confirm the relationship of Plasmodium falciparum to other mammalian parasites but complicate the origins of Plasmodium vivax. Mol Biol Evol. 2008;25:1192–1198. doi: 10.1093/molbev/msn069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu W, Li Y, Learn GH, Rudicell RS, Robertson JD, Keele BF, et al. Origin of the human malaria parasite Plasmodium falciparum in gorillas. Nature. 2010;467:420–425. doi: 10.1038/nature09442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patra KP, Johnson JR, Cantin GT, Yates JR, Vinetz JM. Proteomic analysis of zygote and ookinete stages of the avian malaria parasite Plasmodium gallinaceum delineates the homologous proteomes of the lethal human malaria parasite Plasmodium falciparum. Proteomics. 2008;8:2492–2499. doi: 10.1002/pmic.200700727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Langer RC, Li F, Popov V, Kurosky A, Vinetz JM. Monoclonal antibody against the Plasmodium falciparum chitinase, PfCHT1, recognizes a malaria transmission-blocking epitope in Plasmodium gallinaceum ookinetes unrelated to the chitinase PgCHT1. Infect Immun. 2002;70:1581–1590. doi: 10.1128/IAI.70.3.1581-1590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]