

ABSTRACT
Pupylation is a posttranslational modification peculiar to actinobacteria wherein proteins are covalently modified with a small protein called the prokaryotic ubiquitin-like protein (Pup). Like ubiquitination in eukaryotes, this phenomenon has been associated with proteasome-mediated protein degradation in mycobacteria. Here, we report studies of pupylation in a streptomycete that is phylogentically related to mycobacteria. We constructed mutants of Streptomyces coelicolor lacking PafA (Pup ligase), the proteasome, and the Pup-proteasome system. We found that these mutants share a high susceptibility to oxidative stress compared to that of the wild-type strain. Remarkably, we found that the pafA null mutant has a sporulation defect not seen in strains lacking the Pup-proteasome system. In proteomics experiments facilitated by an affinity-tagged variant of Pup, we identified 110 pupylated proteins in S. coelicolor strains having and lacking genes encoding the 20S proteasome. Our findings shed new light on this unusual posttranslational modification and its role in Streptomyces physiology.
IMPORTANCE The presence of 20S proteasomes reminiscent of those in eukaryotes and a functional equivalent of ubiquitin, known as the prokaryotic ubiquitin-like protein (Pup), in actinobacteria have motivated reevaluations of protein homeostasis in prokaryotes. Though the Pup-proteasome system has been studied extensively in mycobacteria, it is much less understood in streptomycetes, members of a large genus of actinobacteria known for highly choreographed life cycles in which phases of morphological differentiation, sporulation, and secondary metabolism are often regulated by protein metabolism. Here, we define constituents of the pupylome in Streptomyces coelicolor for the first time and present new evidence that links pupylation and the oxidative stress response in this bacterium. Surprisingly, we found that the Pup ligase has a Pup-independent role in sporulation.
INTRODUCTION
Protein homeostasis is a tightly regulated and critically important phenomenon in bacterial physiology (1). It is thought to be particularly complex in actinobacteria, which have both the canonical proteolytic machinery of prokaryotes and 20S proteasomes analogous to those of eukaryotes that are accompanied by a functional equivalent of ubiquitin known as the prokaryotic ubiquitin-like protein (Pup) (2–7). Pupylation, the covalent attachment of Pup to proteins, was the first protein-protein modification discovered in prokaryotes. This posttranslational modification has been extensively studied in mycobacteria, wherein its mechanism and physiological role are analogous to those of ubiquitination in eukaryotes (4–7). Through related mechanisms, both ubiquitin and Pup are covalently attached to lysine residues of proteins via the ATP-dependent activation of a constituent carboxylate moiety (3). While ubiquitin is attached via a C-terminal glycine residue (2), Pup is linked via the side chain of a glutamate residue at its carboxy terminus (4–8). In mycobacteria, this residue is the product of hydrolytic deamidation of a glutamine side chain which is catalyzed by an enzyme called Dop (deamidase of Pup) (5–11). The enzyme that catalyzes the ATP-dependent activation of the deamidated side chain of Pup and its subsequent ligation to lysine residues of selected proteins is called either Pup ligase or PafA (proteasomal accessory factor A) (5, 8–11). As is the case for the major physiological role of ubiquitination, experimental evidence acquired in studies of mycobacteria suggests that Pup-tagged proteins are unfolded by the proteasomal ATPase MPa (also known as ARC) and translocated into the barrel-shaped core of the 20S proteasome wherein hydrolytic degradation occurs (4, 12). Though pupylation has been closely tied to degradation of proteins by the 20S proteasome, it is not yet clear if this phenomenon has other roles, like controlling protein subcellular localizations, associations, and activities, as is the case for ubiquitination.
In Mycobacterium tuberculosis, genes encoding Pup, mediators of pupylation (Dop and PafA), and components of the proteasome are dispensable for viability yet essential for survival under conditions of nitrosative stress (13–15). The latter observations have been invoked to explain the capacity of M. tuberculosis to evade the innate immune response and persist in the host (13, 14). The genetic analyses of the Pup-proteasome system in mycobacteria have been complemented by biochemical analyses wherein pupylated proteins (i.e., the pupylome) have been identified in M. tuberculosis and Mycobacterium smegmatis (16–19). In these mycobacterial species, as many as 55 pupylated proteins have been identified in cell lysates. The Pup modification was observed on proteins of nearly all structural and functional classes (16).
While the relevance of the Pup-proteasome system to the pathogenicity of M. tuberculosis has made it the focus of much attention, we and others (20, 21) have been intrigued by the homologous system in Streptomyces bacteria. These actinobacteria are best known as producers of antibiotics and for their complex life cycle, during which both morphological differentiation and sporulation occur (22–24). Because protein metabolism plays key regulatory roles in several aspects of Streptomyces physiology (23), we predicted that the Pup-proteasome system would be a compelling subject for research. For our studies, we chose Streptomyces coelicolor, which is the model organism of the genus. It has genes whose products are homologous to proteins of the Pup-proteasome system in M. tuberculosis (Fig. 1). The genes encoding the 20S proteasome components (PrcA and PrcB) and Pup are clustered in a genetic locus referred to as the Pup-proteasome system, pps. Though the cognate proteins share at least 60% identity with those in M. tuberculosis, a remarkable and perhaps meaningful difference is that Pup in S. coelicolor has a glutamate residue rather than a glutamine at its carboxy terminus (Fig. 1B) (25). This substitution obviates the need for the Dop-catalyzed deamidation of Pup (which precedes pupylation in most other bacteria) and thus leaves open questions about the function of the Dop in S. coelicolor. As was the case in M. tuberculosis, it has been reported that the prc genes are not essential for viability (20, 21). However, proteomic analyses of the S. coelicolor prc null strain revealed a marked overproduction of proteins that mediate stress responses (21). While the physiological significance of the prc locus has been assessed for S. coelicolor (21), the genes encoding Pup and the enzymes that catalyze pupylation have not been characterized. To broaden our understanding of the Pup-proteasome system in S. coelicolor, we constructed and characterized S. coelicolor strains lacking genes encoding the 20S proteasome, the Pup-proteasome system, and Pup ligase. In addition to phenotypic analyses of these pupylation-deficient strains, we performed proteomic experiments to identify pupylated proteins in S. coelicolor by expressing a gene encoding an affinity-tagged allele of Pup in both proteasome (prc) null and wild-type strains of S. coelicolor.
FIG 1.

Pup-proteasome system of S. coelicolor. (A) Gene organization. SCO1640 is 74% identical to PafA from M. tuberculosis. SCO1643 and SCO1644 are 54% and 50% identical to PrcA and PrcB from M. tuberculosis. SCO1647 is 62% identical to Dop. (B) Alignment of Pup amino acid sequences from S. coelicolor (Sco), M. tuberculosis (Mtb), and M. smegmatis (Msm). (C) Schematic illustrating the affinity-tagged Pup attached to a protein. The slash indicates the tryptic digestion site on Pup. In the proteomic analyses, proteins were considered pupylation targets only if matching peptides having the GGE stem peptide (243 Da) were identified.
MATERIALS AND METHODS
Detailed procedures regarding cloning, strain construction, and the proteomic analyses can be found in the supplemental material.
Strains, media, and culture conditions.
Streptomyces coelicolor strains were grown at 30°C on mannitol soy flour medium (SFM), Difco nutrient agar medium, yeast extract-malt extract medium (YEME), or tryptone soya broth (TSB) (26). SFM was used for conjugations between S. coelicolor and Escherichia coli and for generating spore stocks. E. coli strains DH5α and ET12567/pUZ8002 were grown in L broth at 37°C for routine subcloning. For the selection of E. coli, ampicillin, apramycin, chloramphenicol, hygromycin, and kanamycin were employed at 100, 50, 25, 80, and 50 μg/ml, respectively. Nalidixic acid was used at 20 μg/ml to counterselect E. coli in conjugations with S. coelicolor. Apramycin and hygromycin were used at 50 μg/ml for the selection of S. coelicolor exconjugants.
Construction of pps (SCO1643-46), prc (SCO1634-44), and pafA (SCO1640) null mutants.
Strains of S. coelicolor M145 in which either the pps locus (SCO1643-46), the prc locus (SCO1643-44), or the pafA gene (SCO1640) was replaced with an apramycin (apr) resistance cassette were constructed via PCR-targeted mutagenesis (27). Gene replacements were confirmed by genomic DNA isolation and amplification of the region by PCR. Complementation of the pafA null strain was performed in two ways. In one strategy, the null strain was conjugated with derivatives of the integrative plasmid pMS81 harboring the respective genetic loci, including their promoters. In the other strategy, the null strain was conjugated with the integrative plasmid pIJ10257 harboring the genes of interest under the control of the constitutive ermE* promoter.
Scanning electron microscopy.
Wild-type, pafA null strains, and pafA overexpression strains of S. coelicolor were grown on SFM for 4 days. Biomass (i.e., spores and aerial hyphae) on the surfaces of the confluent lawns was harvested by adherence to double-sided tape. In turn, the tape was affixed to microscopy stubs for imaging. After desiccation for 48 h in the presence of Drierite, the samples were coated with Au by low-vacuum sputter coating. The samples were then imaged on a Hitachi 2700 scanning electron microscope equipped with a lanthanum hexaboride gun. Images were collected and analyzed with a Quartz PCI digital imaging system. The microscope was set to a band contrast of 8, a working distance of 15 mm, and 8 kV. The images were collected for 80 s with magnifications of ×1,500, ×6,000, and ×15,000 for collection at 20 nm, 5 nm, and 2 nm, respectively.
Oxidative-stress assay.
In assessments of the strains' sensitivities to oxidative stress, equal numbers of spores (1 × 108) of the wild type, the Δprc::apr, Δpps::apr, and ΔpafA::apr strains, and the ΔpafA::apr strain with wild-type pafA (i.e., the complemented null strain) were plated on either SFM or Difco nutrient agar. To the center of each freshly inoculated plate was added a sterilized and dry paper disc (6-mm diameter) that had been submerged in a 40% solution of cumene hydroperoxide in distilled water (dH2O). After growth at 30°C for 48 h on Difco nutrient agar or 72 h on SFM, the size of the cleared zone was measured. These experiments were replicated 4 times, and the average sizes of the zones of inhibition are reported.
Construction of His-tagged Pup.
To facilitate the isolation and identification of pupylated proteins in cell lysates from the S. coelicolor strains of interest, an affinity-tagged version of Pup was constructed. Using a subcloning strategy described in the supporting material, the wild-type pup gene in the pps locus was replaced with an allele encoding Pup with a hexahistidine affinity tag fused to its N terminus. Using the aforementioned plasmid as the template in a PCR, we amplified a fragment containing the engineered pup gene and the downstream open reading frame (encoding a hypothetical protein, SCO1645) in an effort to generate a plasmid from which the engineered Pup and not the prc genes could be expressed. These strategies were employed to ensure that the engineered pup gene would be transcribed from the native pup promoter. Plasmids harboring either the entire engineered pup gene in its native context within the pps locus (pJS873) or the engineered pup gene alone (pJS875) were cloned into the integrative plasmid pMS81 and introduced into the S. coelicolor pps null strain by conjugation. The former strain was employed for studies of pupylation in a mimic of wild-type S. coelicolor, whereas the latter was used to characterize pupylation in the absence of a functional proteasome.
Isolation of pupylated proteins.
S. coelicolor pps null strains harboring either pJS873 (pps locus with an embedded pup allele encoding His-tagged Pup) or pJS875 (only pup allele encoding His-tagged Pup) were grown to stationary phase in TSB. Cultures of wild-type S. coelicolor and the pafA null strain (both lacking the His-tagged Pup) were grown in parallel for control experiments. Growth of the cultures was assessed via measurements of dry cell weights at various time points over a period of 36 h after spore germination. In duplicate experiments, mycelia were harvested at single time points in early exponential (14 h post-spore germination), late exponential (18 h post-spore germination), and stationary (24 h post-spore germination) phases by centrifugation at 4,000 rpm for 10 min. (The growth rates were measured by quantification of dry cell weights.) The mycelial pellets were resuspended in 1.4 ml of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole [pH 8.0]) and 4.5 μl of 1:9 benzonase buffer (50 mM Tris-HCl, 20 mM NaCl, 20 mM MgCl2 [pH 8.0]). For lysis, lysozyme was added to a final concentration of 1 mg/ml. After incubation on ice at 37°C, mycelia were lysed by sonication at 60% power three times at 30-s intervals with 2 min of rest between each sonication. The lysates were clarified at 4°C by centrifugation at 13,000 rpm for 15 min and applied to a Qiagen nickel-nitrilotriacetic acid (Ni-NTA) column under nondenaturing conditions. By following the manufacturer's protocol, the column was washed and the bound proteins were eluted. The eluted proteins were finally digested with trypsin. Lysates from two different cultures were subjected to the affinity purification method. In duplicate experiments, the wild type and the ΔpafA::apr strains grown to stationary phase were also subjected to identical proteomic analyses without the Ni-NTA chromatography.
Protein identification via MS and bioinformatics.
To identify proteins isolated from the lysates via Ni-NTA chromatography, the eluates were digested with trypsin and the resulting peptides were loaded onto a reverse-phase column and separated via an Agilent 1200 high-performance liquid chromatograph (HPLC). The peptides were identified by high-resolution tandem mass spectrometry (MS/MS) analyses using a coupled LTQ Orbitrap Velos mass spectrometer housed at the Brown University Center for Genomics and Proteomics. The mass spectral data were searched using the MASCOT software algorithm against the S. coelicolor protein database. In the bioinformatic identifications of the peptide fragments, trypsin specificity with two missed cleavage sites was allowed and the MS mass tolerance was 7 ppm, while the MS/MS tolerance was 0.5 Da. A total of 857 proteins were identified among all the biological samples. Using the target decoy method, the false-discovery rates were calculated for each sample. For 9 of the 12 samples, they ranged between 0 and 6%. The remaining samples had false-discovery rates of 8.3, 11.5, and 12.5%. Nevertheless, the iterative nature of data analysis required for identification of pupylated proteins negated the significance of the false-discovery rates. Specifically, our criterion for the assignment of any of the 857 proteins as a pupylation target was the identification of a constituent peptide having a lysine reside and a 243-Da mass difference due to the presence of a characteristic tryptic fragment of Pup. This Gly-Gly-Glu (GGE) fragment corresponds to the last three residues at the C terminus of Pup and is linked to lysine residues via an isopeptide linkage (Fig. 1C). Identifications were contingent on at least one unique peptide spectrum match (PSM) in the molecular weight search (MOWSE) with a protein score cutoff of 1.
RESULTS AND DISCUSSION
Phenotypic analyses of S. coelicolor pps, prc, and pafA null mutants.
To visually assess the significance of pupylation and proteasome-mediated degradation in morphological differentiation and secondary metabolism, the prc (Δprc::apr), pafA (ΔpafA::apr), and pps (Δpps:apr) null strains were grown on both soya flour media (SFM) and Difco nutrient agar (26). The former is a minimal medium optimized for spore formation in S. coelicolor, while the latter is a nutrient-rich medium that promotes fast growth without sporulation (26). The production of the pigmented secondary metabolites by S. coelicolor can be easily visualized on both media (26). The prc and the pps null strains were virtually indistinguishable from the wild-type strain on both SFM and Difco nutrient agar (see the supplemental material). These observations indicate that pupylation and proteasome-mediated degradation of proteins are dispensable for secondary metabolism and sporulation in S. coelicolor. In stark contrast, we found that the pafA null strain exhibited defects in both secondary metabolism and morphological differentiation. On both SFM and Difco nutrient agar, the pafA null strain overproduced the blue secondary metabolite actinorhodin (see Fig. S1 in the supplemental material). Another phenotype of the pafA null strain was that its confluent lawns lacked the gray sporulation pigment after 4 days of growth on SFM, whereas lawns of the wild-type strain were completely gray at the same time point. Curiously, we found that the pafA null strain's sporulation defect was apparent only when the spores were plated at low densities (<105 CFU of spores spread over 25 ml of SFM in a 100-mm diameter petri dish). To determine if the absence of the gray spore pigment reflected a metabolic defect or an aberration in sporulation, we analyzed the surfaces of confluent lawns of the pafA null strain by scanning electron microscopy and observed elongated and nonsegmented hyphae with very few spores (Fig. 2; see also Fig. S7 to S9 in the supplemental material). Conversely, electron micrographs of the gray, confluent lawns of the wild-type strain featured spores of uniform shape and size (Fig. 2; see also Fig. S10 to S12). The apparent sporulation defect of the pafA null strain was corroborated by our findings that confluent lawns of the pafA mutant yielded a tenth the number of spores typically recovered from those of wild-type S. coelicolor. Interestingly, a peculiarity of the spores from the pafA null strain was that their viability was significantly compromised by brief exposure to heat (see Fig. S2). Importantly, the pafA null strain's defects in antibiotic production and sporulation could be suppressed completely by provision of plasmids harboring the wild-type pafA gene under the control of either its own promoter or the constitutive ermE* promoter (see Fig. S1 and S13 to S15).
FIG 2.

Scanning electron micrographs of the pafA null (left) and wild-type S. coelicolor (right) grown for 72 h on SFM at 30°C. The pafA null strain has elongated and nonsegmented hyphae with very few spores.
Our finding that the pafA null strain had defects in secondary metabolism and sporulation that were not exhibited by either the prc or pps null strains suggested that PafA may have physiological roles either in addition to or instead of its predicted role in proteasome-mediated protein degradation. To substantiate the predicted functional relationship between the pafA and the prc genes, we performed experiments that would reveal whether or not the cognate null strains shared phenotypes. Specifically, we assessed the sensitivities of the prc, pps, and pafA null strains to oxidative stress (i.e., exposure to cumene hydroperoxide). These experiments were selected because the 20S proteasome is reported to play an important role in the oxidative stress response (21). Indeed, the prc null strain is hypersensitive to cumene hydroperoxide (21). As is consistent with the prediction that the pafA, pps, and prc genes have related physiological roles, we found that the strains lacking these genes are equally more sensitive to cumene hydroperoxide than the wild-type strain (Fig. 3; see also Fig. S3 in the supplemental material). The oxidant hypersusceptibilities of the pafA null strains was suppressed completely by provision of a plasmid harboring the wild-type pafA gene under the control of its native promoter (Fig. 3; see also Fig. S4). While the pafA and pps null strains' shared susceptibility to oxidative stress functionally links the corresponding genes, the pafA null mutant's defect in sporulation suggests the intriguing possibility that PafA has a role beyond pupylation.
FIG 3.
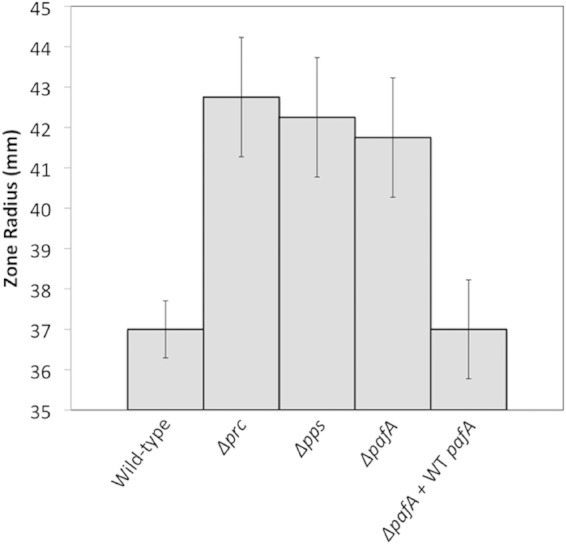
Oxidative-stress sensitivity assay. Surface-grown cultures of the wild-type, Δprc, Δpps, ΔpafA, and complemented ΔpafA strains on Difco nutrient agar were exposed to a localized 40% concentration of cumene hydroperoxide, and the radii of the zones of growth inhibition were measured. The average radii of the zones of inhibition measured in four different experiments are shown. The error bars reflect the standard deviations of the measurements in those experiments. An expanded table reflecting assays performed on SFM can be found in Fig. S3 in the supplemental material.
Identification of S. coelicolor pupylated proteins.
In analogy to published approaches used to characterize the pupylomes of M. smegmatis and M. tuberculosis (17–19, 22), we engineered a gene encoding a variant of S. coelicolor Pup with an N-terminal hexahistidine tag that when expressed in S. coelicolor strains of interest would enable pupylated proteins to be captured from lysates via Ni affinity chromatography for easy identification via proteomic methods. Further, we designed our experiments to limit artifacts that could complicate the interpretation of proteomic data. First, to promote native pupylations, we expressed the gene encoding the affinity-tagged Pup from its native promoter rather than one used for ectopic expression. Second, to ensure homogeneity of pupylation, we expressed the engineered pup gene only in strains in which the native pup gene had been deleted. With these considerations, a plasmid harboring only the engineered pup gene was introduced into the pps null mutant (having the intact pafA gene) to yield a strain that could pupylate proteins but lacked the capacity to degrade them. Likewise, a plasmid harboring the engineered pup gene within the pps locus was introduced into the pps null mutant (having the intact pafA gene) to generate a strain that could pupylate and degrade proteins in a fashion analogous to the wild-type strain. As an indication of the functionality of the engineered Pup, we found that the latter strain did not exhibit the cumene hydroperoxide hypersusceptibility of the parent pps null strain (see Fig. S4 in the supplemental material).
The two aforementioned S. coelicolor strains were used in experiments to assess pupylation within the proteome. They were grown in duplicate in tryptone soya broth, and the mycelia were harvested by centrifugation at single time points during early exponential, late exponential, and stationary phases of growth. Lysates of the mycelia were applied to an Ni-NTA column to capture proteins that had been posttranslationally modified with the affinity-tagged Pup. Through sequential tryptic digests and peptide mass fingerprinting of the eluted proteins, we identified peptides derived from 857 different proteins, of which 110 were pupylated (see the supplemental material). Our definition of a protein as a pupylation target was contingent on the observation of a constituent peptide having both a lysine residue and a 243-Da mass shift attributed to the GGE tripeptide remnant of the trypsin-catalyzed cleavage of Pup (Fig. 1C). A complete list of pupylated proteins identified in the experiments is presented in Fig. 4. Control experiments were performed in the same fashion with stationary-phase cultures of wild-type and pafA null strains lacking the engineered pup gene (see the supplemental material). From the wild-type strain, we identified 22 pupylated proteins, 19 of which were also isolated from the strains harboring the engineered Pup. As expected, we identified no pupylated proteins in lysates from the strain lacking Pup ligase. Notably, we identified twice the greatest number of pupylated proteins that have been reported for any actinobacterium (i.e., M. tuberculosis) (16). This observation could reflect the fact that the genome of S. coelicolor is nearly twice the size of those of M. tuberculosis and M. smegmatis.
FIG 4.

Venn diagram showing proteins isolated from lysates of S. coelicolor strains expressing gene encoding N-terminally His-tagged Pup. The growth phase at which the pupylated proteins were isolated is indicated by the symbols. The set on the left shows the proteins isolated from a strain with a functional proteasome (Wild-type*). The set on the right shows the proteins isolated from a strain lacking a functional proteasome. The union of the two sets shows the 16 proteins that were captured from the strains with and without a functional proteasome at the same time point(s). As indicated, 2-amino-3-ketobutyrate coenzyme A ligase, tellurium resistance protein, and zinc protease were isolated from both strains, but at different time points.
An inherent challenge in the identification of all pupylated proteins in S. coelicolor or any other actinobacterium is that pupylation is a reversible phenomenon (28). This consideration led us to carry out the proteomic analyses on lysates from mycelia in different phases of growth. In lysates of mycelia in the early exponential phase, we identified 41 and 27 pupylated proteins from the strains with and without a functional proteasome, respectively. Ten of the proteins were common to both strains at this time point. Fewer pupylated proteins were recovered and identified in lysates from mycelia in the late exponential phase of growth. Indeed, 23 were recovered from the strain with a functional proteasome and 21 from the strain without. Only 3 proteins were in common between these two strains. In lysates of mycelia in stationary phase, 29 proteins were identified in the strain with a functional proteasome and 17 in the strain without. Six proteins were observed in lysates from the two strains harvested in stationary phase. Collectively, 19 of the 110 pupylated proteins were found in strains having and lacking a functional proteasome; 16 of these shared proteins were isolated at the same time point(s) in both strains (Fig. 4). While our findings of some proteins in strains with and without a functional proteasome could imply that proteins are pupylated and not degraded, our inability to quantify protein abundance precludes our capacity to support or refute this conjecture. Though the reversibility of pupylation complicates comprehensive definition of the total pupylome in S. coelicolor, our reliance on lysine residues and a 243-Da mass shift gives us a high degree of confidence in the identities of the pupylated proteins.
In an effort to correlate pupylation with the bacterium's physiology, we examined the predicted functions of the pupylated proteins. We found that the pupylated proteins varied significantly with respect to physiological function and subcellular localization. Based on the annotated functions, we estimate that 50% of the proteins are involved with intermediary metabolism, 14% have roles in nucleic acid metabolism, 16% underlie protein metabolism, 18% have predicted functions in regulation, and 5% are putative mediators of stress responses and toxin resistance. With respect to subcellular localization, we estimate that 8 of the 110 proteins are likely to be localized to the cell wall. Collectively, these statistics are largely consistent with those reported for mycobacteria (16–19).
Conclusion.
As in the studies of pupylation in mycobacteria (16–19), we found a large and functionally diverse set of pupylated proteins in S. coelicolor. Likewise, we found it difficult to assess the functional significance of this posttranslational modification in S. coelicolor. Pupylation could be a response to stochastic damage of proteins by oxidants and/or other forces. Alternatively, the capture of proteins in the strain harboring a functional proteasome could represent a snapshot of progressing degradation and/or situations wherein pupylation affects protein activity or localization rather than half-life. In any case, the findings reported herein are new insights into protein homeostasis in Streptomyces bacteria. Indeed, we uncovered evidence indicating that pupylation is a posttranslational modification in Streptomyces bacteria. Further, our finding that disruptions of genes encoding Pup ligase (pafA) and the Pup-proteasome render S. coelicolor highly sensitive to cumene hydroperoxide strongly suggests that pupylation is important for the oxidative stress response. More surprisingly, our observations that the pafA null strain has defects in both sporulation and secondary metabolism, whereas the pps and prc null strains do not, indicate that PafA may have an activity other than pupylation in Streptomyces bacteria. Collectively, these studies provide new perspectives on posttranslational modifications, stress responses, and sporulation in S. coelicolor, which are likely to translate to other sporogenic actinobacteria.
Although our study has shed light on the basic aspects of the Pup-proteasome system, numerous questions remain. For example, how does Pup ligase recognize and posttranslationally modify such a diverse set of proteins with Pup? In eukaryotes, multiple ubiquitin ligases are required to effect ubiquitination of proteome constituents. Further, are there additional signals beyond Pup that are required to target proteins for proteolysis? In addition, does pupylation, like ubiquitylation, influence the subcellular localizations, associations, and/or activities of proteins? Efforts to answer these questions are under way in these laboratories.
Supplementary Material
ACKNOWLEDGMENTS
J.K.S. is the recipient of an NSF CAREER award. C.L.C. was supported by a dissertation fellowship from the Brown University Department of Chemistry. M.S.F. was supported by an undergraduate teaching and research assistantship from Brown University and by the Royce Fellowship Program.
We thank James Clifton and Geoff Williams for their expert technical support and help with proteomic analyses and microscopy, respectively.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00302-15.
REFERENCES
- 1.Guo MS, Gross CA. 2014. Stress-induced remodeling of the bacterial proteome. Curr Biol 24(10):R424–R434. doi: 10.1016/j.cub.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciechanover A. 1994. The ubiquitin-proteasome proteolytic pathway. Cell 79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 3.Hochstrasser M. 2009. Origin and function of ubiquitin-like proteins. Nature 458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burns K, Darwin K. 2012. Pupylation: proteasomal targeting by a protein modifier in bacteria. Methods Mol Biol 832:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samanovic M, Li H, Darwin K. 2013. The pup-proteasome system of Mycobacterium tuberculosis, p 267–295. In Dougan DA. (ed), Regulated proteolysis in microorganisms. Springer, Dordrecht, The Netherlands. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearce M, Mintseris J, Ferreyra J, Gygi S, Darwin K. 2008. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns K, Liu W, Boshoff H, Dorrestein P, Barry C. 2009. Proteasomal protein degradation in mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem 284:3069–3075. doi: 10.1074/jbc.M808032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forer N, Korman M, Elharar Y, Vishkautzan M, Gur E. 2013. Bacterial proteasome and PafA, the pup ligase, interact to form a modular protein tagging and degradation machine. Biochemistry 52:9029–9035. doi: 10.1021/bi401017b. [DOI] [PubMed] [Google Scholar]
- 9.Festa R, Pearce M, Darwin K. 2007. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J Bacteriol 189:3044–3050. doi: 10.1128/JB.01597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guth E, Thommen M, Weber-Ban E. 2011. Mycobacterial ubiquitin-like protein ligase PafA follows a two-step reaction pathway with a phosphorylated pup intermediate. J Biol Chem 286:4412–4419. doi: 10.1074/jbc.M110.189282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber-Ban E. 2009. Bacterial ubiquitin-like modifier pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol 16:647–651. doi: 10.1038/nsmb.1597. [DOI] [PubMed] [Google Scholar]
- 12.Striebel F, Hunkeler M, Summer H, Weber-Ban E. 2010. The mycobacterial Mpa–proteasome unfolds and degrades pupylated substrates by engaging Pup's N-terminus. EMBO 29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darwin K, Ehrt S, Gutierrez-Ramos J, Weich N, Nathan C. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 14.Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. 2007. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med 13:1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sassetti C, Boyd D, Rubin E. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 16.Festa R, McAllister F, Pearce M, Mintseris J, Burns K, Gygi S, Darwin H. 2010. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis. PLoS One 5:e8589. doi: 10.1371/journal.pone.0008589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poulsen C, Akhter Y, Jeon A, Ulms G, Meyer H, Stefanski A, Stuhler K, Wilmanns M, Song Y. 2010. Proteome-wide identification of mycobacterial pupylation targets. Mol Syst Biol 6:386. doi: 10.1038/msb.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watrous J, Burns K, Liu W, Patel A, Hook V, Bafna V, Barry C, Bark S, Dorrestein P. 2010. Expansion of the mycobacterial “PUPylome”. Mol BioSyst 6:376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tung C. 2012. PupDB: a database of pupylated proteins. BMC Bioinformatics 13:40. doi: 10.1186/1471-2105-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagy I, Tamura T, Vanderleyden J, Baumeister W, De Mot R. 1998. The 20S proteasome of Streptomyces coelicolor. J Bacteriol 180:5448–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Mot R, Schoofs G, Nagy I. 2007. Proteome analysis of Streptomyces coelicolor mutants affected in the proteasome system reveals changes in stress-responsive proteins. Arch Microbiol 188:257–271. doi: 10.1007/s00203-007-0243-8. [DOI] [PubMed] [Google Scholar]
- 22.Chater K, Biró S, Lee K, Palmer T, Schrempf H. 2010. The complex extracellular biology of Streptomyces. FEMS Microbiol Rev 34:171–198. doi: 10.1111/j.1574-6976.2009.00206.x. [DOI] [PubMed] [Google Scholar]
- 23.Chater K, Chandra G. 2008. The use of the rare UUA codon to define “Expression Space” for genes involved in secondary metabolism, development and environmental adaptation in Streptomyces. J Microbiol 46:1–11. doi: 10.1007/s12275-007-0233-1. [DOI] [PubMed] [Google Scholar]
- 24.Flärdh K, Buttner M. 2009. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat Rev Microbiol 7:36–49. doi: 10.1038/nrmicro1968. [DOI] [PubMed] [Google Scholar]
- 25.Bentley S, Chater K, Cerdeno-Tarrage A, Challis G, Thomson N, James K, Harris D, Quail M, Keiser H, Harper D, Bateman A, Brown S, Chandra G, Chen C, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Honsby T, Howarth S, Huang C, Kieser T, Larke L, Murphy L, Oliver K, ONeil S, Rabbinowitsch E, Rajandream M, Rutherford K, Rutter S, Seeger K, Saunder D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell B, Parkhill J, Hopwood D. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 26.Kieser T, Bibb M, Buttner M, Chater K, Hopwood D. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, England. [Google Scholar]
- 27.Gust B, Challis G, Fowler K, Kieser T, Chater K. 2003. PCR targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A 100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burns K, Cerda-Maira F, Wang T, Li H, Bishal W, Darwin K. 2010. “Depupylation” of prokaryotic ubiquitin-like protein from mycobacterial proteasome substrates. Mol Cell 39:821–828. doi: 10.1016/j.molcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.