

ABSTRACT
All methanofuran structural variants contain a basic core structure of 4-[N-(γ-l-glutamyl)-p-(β-aminoethyl)phenoxymethyl]-(aminomethyl)furan (APMF-Glu) but have different side chains depending on the source organism. Recently, we identified four genes (MfnA, MfnB, MfnC, and MfnD) that are responsible for the biosynthesis of the methanofuran precursor γ-glutamyltyramine and 5-(aminomethyl)-3-furanmethanol-phosphate (F1-P) from tyrosine, glutamate, glyceraldehyde-3-P, and alanine in Methanocaldococcus jannaschii. How γ-glutamyltyramine and F1-P couple together to form the core structure of methanofuran was previously unknown. Here, we report the identification of two enzymes encoded by the genes mj0458 and mj0840 that catalyze the formation of F1-PP from ATP and F1-P and the condensation of F1-PP with γ-glutamyltyramine, respectively, to form APMF-Glu. We have annotated these enzymes as MfnE and MfnF, respectively, representing the fifth and sixth enzymes in the methanofuran biosynthetic pathway to be identified. Although MfnE was previously reported as an archaeal adenylate kinase, our present results show that MfnE is a promiscuous enzyme and that its possible physiological role is to produce F1-PP. Unlike other enzymes catalyzing coupling reactions involving pyrophosphate as the leaving group, MfnF exhibits a distinctive α/β two-layer sandwich structure. By comparing MfnF with thiamine synthase and dihydropteroate synthase, a substitution nucleophilic unimolecular (SN-1) reaction mechanism is proposed for MfnF. With the identification of MfnE and MfnF, the biosynthetic pathway for the methanofuran core structure APMF-Glu is complete.
IMPORTANCE This work describes the identification of the final two enzymes responsible for catalyzing the biosynthesis of the core structure of methanofuran. The gene products of mj0458 and mj0840 catalyze the formation of F1-PP and the coupling of F1-PP with γ-glutamyltyramine, respectively, to form APMF-Glu. Although the chemistry of such a coupling reaction is widespread in biochemistry, we provide here the first evidence that such a mechanism is used in methanofuran biosynthesis. MfnF belongs to the hydantoinase A family (PF01968) and exhibits a unique α/β two-layer sandwich structure that is different from the enzymes catalyzing similar reactions. Our results show that MfnF catalyzes the formation of an ether bond during methanofuran biosynthesis. Therefore, this work further expands the functionality of this enzyme family.
INTRODUCTION
Methanofuran is the first in a series of coenzymes involved in the biochemical reduction of carbon dioxide to methane (1–3). This process, known as methanogenesis, is carried out only by the methanogenic archaea, which produce more than 400 million tons of methane each year as an essential part of the global carbon cycle (4). Methanofuran is the initial C1 acceptor molecule involved in the first two-electron reduction of carbon dioxide to produce the formamide derivative of methanofuran, where the formate is attached to the amino group of methanofuran through methanogenesis (5). This is one of the few known pathways for carbon dioxide fixation in nature. In addition, methylotrophic bacteria also use methanofuran as a coenzyme to oxidize formaldehyde to formic acid (6, 7). The chemical structure of methanofuran varies among different methanogens (8); each currently known methanofuran molecule contains the basic core structure of 4-[N-(γ-l-glutamyl)-p-(β-aminoethyl)phenoxymethyl]-2-(aminomethyl)furan (APMF-Glu), but isolated analogs have different attached side chains (8). Recently, our laboratory identified a new methanofuran structure in Methanocaldococcus jannaschii, which contains a long γ-glutamyl tail with 7 to 12 γ-linked glutamates (9) (Fig. 1). Although the function of methanofuran has been known for many years, its biosynthetic pathway has not been fully elucidated.
FIG 1.

Proposed biosynthetic pathway of methanofuran in M. jannaschii.
Recently, we discovered the route for the production of 4-(hydroxymethyl)-2-furancarboxaldehyde-phosphate (4-HFC-P) from glyceraldehyde-3-phosphate (10). The resulting 4-HFC-P undergoes a transamination reaction to produce 5-(aminomethyl)-3-furanmethanol-phosphate (F1-P) (10, 11), a precursor of the furan moiety of methanofuran (Fig. 1). We previously demonstrated that the mj0050 gene encodes a tyrosine decarboxylase that produces tyramine from tyrosine (12). A tyramine-glutamate ligase (the gene product of mj0815) catalyzes the ATP-dependent addition of one glutamate to tyramine via a γ-linked amide bond (13) (Fig. 1). To produce APMF-Glu, we propose that one enzyme is required to catalyze the conversion of F1-P to F1-PP, where the pyrophosphate group serves as a better leaving group in the subsequent condensation reaction. Another enzyme then catalyzes the condensation between F1-PP and γ-glutamyltyramine.
The homologs of mj0458 and mj0840 are widely distributed among the methanofuran-containing organisms, including all methanogenic archaea and some methylotrophic bacteria. Comparative genomic analysis shows that in some methanogens and methylotrophs the homologs of the mj0458 and mj0840 genes appear in the neighborhood of mj0815 (Fig. 2), which encodes the enzyme that catalyzes the ATP-dependent addition of one glutamate to tyramine, producing γ-glutamyltyramine. Therefore, the gene products of mj0458 and mj0840 are likely to be involved in methanofuran biosynthesis. We reported that mj0458 encodes a second type of archaeal adenylate kinase that catalyzes phosphoryl group transfer from one molecule of ATP to one molecule of AMP, generating two molecules of ADP (14). A similar phosphoryl transfer reaction is expected to generate F1-PP from F1-P and ATP. It is possible that the gene product of mj0458 is a promiscuous enzyme, catalyzing both reactions. The gene product of mj0840 is an uncharacterized protein. PSI-blast analysis shows that it belongs to the hydantoinase A family (PF01968) and contains a sugar kinase domain. However, the four histidines (15) that are found in hydantoinase, which are catalytically essential, are not conserved in the proteins encoded by mj0840 and its homologues, indicating that the gene product of mj0840 is unlikely to function as a hydantoinase.
FIG 2.

Clustering of MfnE (mj0458) and MfnF (mj0840) genes with the methanofuran biosynthetic related MfnD (mj0815) gene in some methanogen and methylotroph genomes.
To test whether the gene products of mj0458 and mj0840 are involved in methanofuran biosynthesis, we cloned and heterologously expressed both genes in Escherichia coli. Here, we report that the purified protein expressed from the M. jannaschii gene locus mj0458 catalyzes the ATP-dependent phosphoryl transfer reaction to generate F1-PP from F1-P. The purified protein expressed from the M. jannaschii gene locus mj0840 catalyzes the condensation reaction between F1-PP and γ-glutamyltyramine, likely via a substitution nucleophilic unimolecular (SN-1) reaction mechanism. These are the fifth and sixth enzymes that we have identified in the methanofuran biosynthetic pathway; therefore, we have annotated the gene products of mj0458 and mj0840 as MfnE and MfnF, respectively.
MATERIALS AND METHODS
Chemicals.
All reagents were purchased from Sigma-Aldrich unless otherwise specified.
Chemical synthesis of F1-P from 4-HFC-P.
The chemical synthesis of F1-P started from 4-HFC-P, whose synthesis has been previously described (11), with the following changes. To synthesize 4-HFC-P, 4-HFC (68 mg, 0.53 mmol) was dissolved in 2 ml of acetonitrile to which was added 120 μl of trichloroacetonitrile (1.2 mmol) and tetrabutylammonium phosphate (300 mg, 0.88 mmol). This procedure was patterned after a previously described method (16). After 4 h (room temperature), the solvent was evaporated with a stream of nitrogen gas, and the sample mixed with 2 ml of water and cooled overnight at 3°C. The resulting trichloroacetamide crystals were separated by filtration, and the resulting solution was passed through a Dowex 50 NH4+ column (0.5 by 2 cm) to remove the tetrabutylammonium cation. The reaction mixture containing 4-HFC-P was then evaporated to dryness and dissolved in 3 ml of concentrated NH4OH into which 11 mg of NaBH4 was dissolved. At first, the sample was clear, but after a few minutes it became cloudy. After 2 h at room temperature the solution became clear again, and there was no borohydride present, as detected by a lack of hydrogen production upon acidification of a small volume of the reaction mixture (2 to 3 μl). The reaction mixture was then evaporated to remove ammonia and placed in 2 ml of water, followed by pH adjustment to <4 using 1 M HCl. The resulting sample was then placed on a Dowex 50W8-H+ column (0.8 by 4 cm) that was washed with 2 ml of 20 mM HCl. The F1-P was retained on the column under these conditions. The elution of F1-P was begun by passing 2 ml of water through the column and completed with an additional elution using 3 M NH4OH. TLC analysis with ninhydrin detection of amines showed that the water fraction contained F1-P as the only detectable compound. The F1-P was further purified by preparative TLC using the solvent system (acetonitrile, water, and 88% formic acid [40:20:10, vol/vol/vol]), where it had an Rf of 0.21. The ammonia fraction contained most of the F1-P and also ammonium-containing salts that was detected by the ninhydrin. The total yield of F1-P was 18%. Both F1 and F1-P showed an absorbance maximum at 215 nm, similar to 2,5-dimethylfuran. The concentration of F1-P was estimated based on the extinction coefficient of 2,5-dimethylfuran (ε215 = 7,900 M−1 cm−1) (17).
Analysis of F1 and its derivatives.
Direct analysis of F1 by high-pressure liquid chromatography (HPLC) is difficult due to the short wavelength at which F1 absorbs (215 nm), which is common to many compounds, and the fact that F1 is not retained by reverse-phase HPLC columns. In the present study, F1, F1-P, and F1-PP were each converted to the 7-nitrobenzofurazan (NBD) derivative, as previous reported (10). NBD-F1-P was purified by preparative TLC using the solvent system (5% formic acid in acetonitrile), where NBD-F1-P had an Rf of 0.15. After removal from the TLC plate, the structure of the NBD-derivatives were then confirmed using HPLC (see below) and liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS) analysis (10).
HPLC analysis of NBD-F1, NBD-F1-P, and NBD-F1-PP derivatives.
Chromatographic separation of NBD derivatives was performed on a Shimadzu HPLC system (UFLC) equipped with a C18 reverse-phase column (Kromasi 100-5-C18; 4.6 by 250 mm). The elution profile consisted of 5 min at 95% sodium acetate buffer (25 mM, pH 6.0, in the presence of 0.02% NaN3) and 5% methanol (MeOH), followed by a linear gradient to 45% sodium acetate buffer–55% MeOH over 35 min at 1.0 ml/min. F1-NBD derivatives were detected by fluorescence using an excitation wavelength of 480 nm and an emission wavelength of 542 nm. Under these conditions, NBD derivatives were eluted in the following order: NBD-F1-PP (14.5 min), NBD-F1-P (17.0 min), and NBD-F1 (20.9 min).
HPLC analysis of ATP, ADP, and AMP.
The separation and analysis of ATP, ADP, and AMP in the sample were measured by using a gradient ion pairing method performed on a Shimadzu UFLC System equipped with a C18 reverse-phase column (ODS; 250 by 4.6 mm, 5-μm particle size) and a photodiode array detector. The elution profile consisted of 5 min at 100% buffer A (0.1 M KH2PO4, 10 mM [CH3(CH2)3]4N(Br), pH 3.0) and 0% methanol, followed by a linear gradient to 25% buffer A–75% methanol over 20 min at 1.0 ml/min. ATP, ADP, and AMP were detected by the absorbance at 260 nm. Under these conditions, ATP eluted at 10.8 min, ADP eluted at 5.7 min, and AMP eluted at 4.1 min.
LC-MS analysis of APMF-Glu.
Analysis was performed with an AB Sciex 3200 Q TRAP mass spectrometer system with an Agilent 1200 series liquid chromatograph. A Zorbax (100 by 4.0 mm, 2.6-μm particle size) column was used, and the injection volume was 15 μl. Solvent A was water with 25 mM ammonium acetate, and solvent B was methanol. The flow rate was 0.5 ml/min. A linear gradient elution was used in the following manner (time, percent solvent B): 0.01 min, 5%; 10 min, 65%; 15 min, 65%; and 15.01 min, 5%. The column effluent was passed through a variable wavelength detector set from 200 to 800 nm and then into the Turbo Spray ion source. ESI was used at −4,500 V and a temperature of 600°C. The curtain gas, gas 1, and gas 2 flow pressures were 35, 70, and 60 lb/in2, respectively. The desolvation, entrance, and collision cell entrance potentials were −40, −12, and −22.5 V, respectively.
Cloning of M. jannaschii mj0458 and mj0840 genes and expression of their gene products.
The mj0458 and mj0840 genes were amplified from M. jannaschii genomic DNAs. The primers used for mj0458 were mj0458-Fwd (5′-GGTGGTCATATGCATATAGTAAAAATTGG-3′) and mj0458-Rev (5′-GATCGGATCCTTATATTTTATCTATTCC-3′). The primers used for mj0840 were mj0840-Fwd (5′-GTGTTTGATGTTAATGGGAATTTTTTAACTTCAGAAG-3′) and mj0840-Rev (5′-CTTCTGAAGTTAAAAAATTCCCATTAACATCAAACAC-3′). PCR amplifications were performed at 55°C as the annealing temperature. The PCR products were purified, digested with NdeI and BamHI restriction enzymes, and then ligated into compatible sites in plasmid pT7-7 to make the recombinant plasmids pmj0458 and pmj0840. The sequences were verified by sequencing at the University of Iowa DNA core facility. The resulting plasmids were used to transform the Escherichia coli BL21-Codon Plus (DE3)-RIL (Stratagene). Transformed cells were grown in lysogeny broth (200 ml) supplemented with 100 μg of ampicillin/ml at 37°C with shaking until an optical density at 600 nm of 1.0 was reached. Recombinant protein production was induced by the addition of lactose to a final concentration of 28 mM. After an additional 4 h of culture at 37°C, the cells were harvested by centrifugation (4,000 × g, 5 min) and frozen at −20°C. SDS-PAGE analysis of the total cellular proteins confirmed the induction of the gene products of mj0458 and mj0840 by the appearance of intense protein bands at the expected 24- and 37-kDa molecular sizes, respectively.
Purification of the gene products of recombinant mj0458 and mj0840.
The frozen E. coli cell pellet (∼0.4 g [wet weight] from 200 ml of growth medium) was suspended in 3 ml of extraction buffer (50 mM N-[tris(hydroxymethyl)methyl]2-amionoethanesulfonic acid (TES), 10 mM MgCl2, and 20 mM dithiothreitol at pH 7.0) and lysed by sonication. The crude lysate was then treated by benzonase nuclease (250 U). The protein products from mj0458 and mj0840 were found to remain soluble after heating to 80°C. Therefore, purification of the gene products of mj0458 and mj0840 started by heating the resulting cell extracts for 10 min at 80°C, followed by centrifugation (16,000 × g, 10 min). This process allowed purification of the desired enzymes from the majority of E. coli proteins, which denature and precipitate under these conditions. The supernatant of the gene product of mj0458 was then pooled and dialyzed against buffer containing 50 mM TES and 10 mM MgCl2 at pH 7.0. Further purification of the gene product of mj0840 was performed by anion-exchange chromatography of the 80°C soluble fractions on a MonoQ HR column (1 by 8 cm; Amersham Bioscience) using a linear gradient from 0 to 1 M NaCl in 25 mM Tris buffer (pH 7.5) over 82 min at a flow rate of 1 ml/min.
Enzymatic assay of MfnE (gene product of mj0458).
To test whether MfnE could catalyze the formation of F1-PP from F1-P and ATP, we incubated 80 μM F1-P and 500 μM ATP in the presence of 3.7 μM MfnE at 70°C for 60 min in 50 mM TES buffer in the presence of 5 mM Mg2+ and 5 mM K+ at pH 7.0. The amino compounds in the reaction mixture were then converted to the NBD derivatives and analyzed by HPLC as described above.
To measure the MfnE activity, the reaction was conducted in a 100-μl reaction volume containing 1.9 μM MfnE, 200 μM F1-P or AMP, and 500 μM ATP in 50 mM 4-morpholineethanesulfonic acid (MES) buffer in the presence of 5 mM Mg2+ and 5 mM K+ at pH 7.0 for 20 min at 80°C. After incubation, the reactions were quenched by the addition of 10 μl of 1 M HCl, the precipitated protein was removed by centrifugation (16,000 × g, 10 min), and the resulting sample was neutralized by adding 8.3 μl of 1.5 M Tris buffer (pH 8.8) (the standard enzymatic assay).
Metal and pH dependence of the MfnE-catalyzed reaction.
For the metal-dependent study, the standard enzymatic assays were performed, including a 5 mM concentration of a cation (Mg2+, Mn2+, Ni2+, Co2+, Cu2+, Zn2+, or K+) or 10 mM EDTA in a standard assay. To investigate the influence of pH on catalytic ability, the specific activity at various pHs was measured. The standard enzymatic assay was conducted in 25 mM citrate buffer (pH 4.0 to 6.0), 25 mM MES buffer (pH 6.0 to 7.0), and 25 mM Tricine–3-(cyclohexylamino)1-propanesulfonic acid (CAPS)–TES buffer (pH 6.7 to 11.5). The resulting samples were analyzed by HPLC.
Standard enzymatic assay of MfnF (gene product of mj0840).
The standard assay for measuring MfnF enzymatic activity was conducted at 50°C for 40 min in a 120-μl reaction volume containing 5.2 μg of MfnF (40 μl), 40 μl of MfnE catalyzed reaction mixture (containing ∼100 μM F1-PP), and 40 μl of MfnD reaction mixture (13) (containing ∼1 mM γ-glutamyltyramine and ∼4 mM tyramine) in 50 mM TES buffer in the presence of 5 mM Mg2+ and 5 mM K+ at pH 7.0. After incubation, the reaction mixtures were quenched by the addition of 10 μl of 1 M HCl, the precipitated protein was removed by centrifugation (16,000 × g, 10 min), and the resulting sample was neutralized by adding 8.3 μl of 1.5 M Tris buffer (pH 8.8). The samples were then analyzed by LC-ESI-MS in positive-ion mode.
RESULTS
Synthesis and analysis of F1-P derivatives.
F1-P was chemically synthesized from 4-HFC-P and converted into the NBD derivative. HPLC with fluorescence detector analysis showed that NBD-F1-P eluted as a single peak at 17.0 min (Fig. 3). After treatment with 1 μl of phosphatase (0.2 U/liter), NBD-F1-P was converted to NBD-F1 (20.9 min), which coeluted with NBD-F1 (10). LC-ESI-MS analysis of NBD-F1-P showed a single peak with MH+ = 371.4 m/z and (M − H)− = 369.4 m/z. Tandem MS (MS/MS) of the (M − H)− = 369.4 ion showed fragments at 79, 97, 179, 289, and 323 (Fig. 4A).
FIG 3.

HPLC analysis of NBD-F1, NBD-F1-P, and NBD-F1-PP. (A) An 80 μM sample of synthetic F1-P was converted to NBD-F1-P treated with phosphatase (37°C, 20 min) to produce NBD-F1 to confirm the structure of F1-P. (B) NBD derivatives of a reaction mixture, including 80 μM F1-P and 500 μM ATP in the absence of MfnE (70°C, 60 min). (C) NBD derivatives of a reaction mixture containing 80 μM F1-P and 500 μM ATP in the presence of 3.7 μM MfnE (70°C, 60 min). The reactions in panels B and C were carried out in 50 mM TES buffer in the presence of 5 mM Mg2+ and 5 mM K+ at pH 7.0. The NBD derivatives were detected by fluorescence using an excitation wavelength of 480 nm and an emission wavelength of 542 nm.
FIG 4.

(A) MS/MS spectrum of NBD-F1-P (M − H)− = 369.4 m/z ion with the expected fragments at 79, 97, 179, 289, and 323 m/z. The proposed structures of the expected fragments are shown on the right. (B) MS/MS spectrum of NBD-F1-PP (M − H)− = 449.5 m/z ion generated from the phosphoryl transfer reaction between ATP and F1-P catalyzed by MfnE with the expected fragments at 79, 159, 177, and 403 m/z. The proposed structures of the expected fragment are shown on the right.
Recombinant expression, purification, and analysis of the gene products of mj0458 and mj0840.
Two genes at the loci of mj0458 and mj0840 were cloned and overexpressed in E. coli. Their gene products were purified as described above. The purified proteins migrated as single bands and were >90% pure with apparent molecular masses of 24 kDa (gene product of mj0458) and 37 kDa (gene product of mj0840) (Fig. 5). The identities of the purified proteins were also confirmed by matrix-assisted laser desorption ionization–MS analysis of the tryptic digested protein band from the SDS gel based on a previously described procedure (18).
FIG 5.

Purification of mj0458 gene product (A) and mj0840 gene product (B). In panel B, lanes 7 to 9 represent the target protein in the fractions eluting at 350 to 460 mM NaCl from a MonoQ anion-exchange column. The target protein was ca. 90% pure in the fraction in lane 9.
Promiscuity of the gene product of mj0458 (MfnE).
We had reported that the gene product of mj0458 encodes an adenylate kinase that catalyzes the transfer of a phosphoryl group from ATP to AMP, producing two molecules of ADP (14). In addition to the gene product of mj0458 (MfnE), it is known that another archaeal adenylate kinase in M. jannaschii is the product of the mj0479 gene (19, 20). However, it was unexpected that methanogens would contain two distinct adenylate kinases. Incubation of the gene product of mj0458 with F1-P in the presence of ATP clearly showed the formation of F1-PP and ADP. A control experiment containing the same concentration of substrates in the absence of MfnE showed none of the expected product (data not shown). Generation of ADP from the MfnE reaction was detected by ion pairing HPLC, and the formation of F1-PP was confirmed using HPLC and LC-ESI-MS after conversion to NBD-F1-PP. The NBD-F1-PP was observed as a new HPLC peak eluting at 14.5 min with a corresponding decrease in the intensity of the NBD-F1-P (17.0 min) peak (Fig. 3). LC-ESI-MS analysis of the NBD derivative of the MfnE reaction mixture showed a single peak with MH+ = 451.5 m/z and (M − H)− = 449.5 m/z. MS/MS of the (M − H)− = 449.5 ion showed fragments at 79, 159, 177, 385, 403, and 431 (Fig. 4B), which are consistent with NBD-F1-PP's molecular weight and structure. These results clearly showed MfnE to catalyze the phosphoryl-transfer reaction between ATP and F1-P producing ADP and F1-PP.
The pH-dependent study of MfnE activity showed a pH optimum at 7.0. We also observed that MfnE exclusively used Mg2+ as a cofactor to facilitate phosphoryl group transfer. The addition of 10 mM EDTA to the reaction mixture resulted in complete loss of MfnE activity. The activity of MfnE at 80°C was 0.16 μmol/mg/min using F1-P as the substrate and 0.072 μmol/mg/min when AMP was used as the substrate (both of these values are the extent of substrate conversion to product for each incubation). The adenylate kinase activity of MfnE 80°C is 500-fold less than that of the gene product of mj0479, which encodes the first archaeal adenylate kinase identified in M. jannaschii (19). In addition, The homologs of mj0458 in some methanogens and methylotrophs cluster with MfnD (mj0815), which encodes the enzyme that catalyzes γ-glutamyltyramine formation during methanofuran biosynthesis (13). These results strongly suggest that the possible physiological function of MfnE is to catalyze the formation of F1-PP involved in methanofuran biosynthesis.
The gene product of mj0840 (MfnF) catalyzes the formation of APMF-Glu.
The gene product of mj0840 was annotated as a hypothetical protein in M. jannaschii (21). Incubation of the purified gene product of mj0840 (MfnF) with γ-glutamyltyramine and F1-PP, which are generated from the MfnE and MfnD catalyzed reactions, respectively, clearly showed the formation of APMF-Glu. This was confirmed with mass spectral data showing a single peak with the expected (M − H)− = 374.2 m/z and MH+ = 376.2 m/z. The extracted ion chromatograph of the product with MH+ = 376.2 m/z eluted as a single peak at 18 min (Fig. 6A and B). The identity of APMF-Glu was also supported by MS/MS data (Fig. 6C) with the MH+ = 376.2 ion producing expected fragments at 110, 213, 230, and 267. Also, incubation of γ-glutamyltyramine plus F1-PP in the absence of MfnF showed that no APMF-Glu was formed under the same conditions (data not shown). When incubating γ-glutamyltyramine and F1-P in the presence of MfnF, a trace of APMF-Glu was observed in LC/MS (Fig. 6A), but the intensity was <2% of that when F1-PP was used as the substrate. In addition, MfnF could not use tyramine as a substrate to condense with F1-PP, since no APMF [(M − H)− = 245.1 m/z and MH+ = 247.1 m/z] was detected from the mass spectral data, a finding consistent with our previous hypothesis that condensation of the tyramine moiety with F1-PP must occur after formation of γ-glutamyltyramine (13).
FIG 6.

LC-MS/MS analysis of APMF-Glu in the positive ion mode. (A and B) Extracted ion chromatographs of MH+ = 376.2 m/z ion from the control experiment (A) (incubation of F1-P with γ-glutamyltyramine in the presence of MfnF) and from the reaction mixture of MfnF containing F1-PP generated from the MfnE reaction and γ-glutamyltyramine (B). (C) MS/MS spectrum of MH+ = 376.2 m/z ion with the expected fragments at 110, 213, 230, and 267 m/z. The proposed structures of observed fragments are shown at the bottom of the figure.
DISCUSSION
Methanofuran is the initial C1 acceptor molecule in the formation of methane through methanogenesis. Several structurally different methanofurans are currently known, with the nature of the differences residing in modifications of the side chain attached to the basic core APMF-Glu structure found in all methanofurans (9). With the discovery of MfnA, MfnB, MfnC, and MfnD, the biosynthetic pathway of APMF-Glu was proposed (Fig. 1). In this pathway, at least two enzymes are required to condense F1-P with γ-glutamyltyramine moiety. One enzyme is required to catalyze the conversion of F1-P to F1-PP, where the pyrophosphate group serves as a better leaving group for the subsequent condensation reaction. The other enzyme catalyzes the condensation between F1-PP and γ-glutamyltyramine to produce APMF-Glu. Such a coupling mechanism is a common strategy used in the biosynthesis of coenzymes such as folate (22) and thiamine (23), as well as terpenes and steroids (24–26).
During thiamine biosynthesis, ThiD (4-amino-5-hydroxymethyl-2-methylpyrimidine phosphate [HMP-P] kinase) phosphorylates HMP-P to 4-amino-5-hydroxymethyl-2-methylpyrimidine pyrophosphate (HMP-PP), and then another enzyme, thiamine phosphate synthase (ThiE), catalyzes the coupling reaction of HMP-PP and 4-methyl-5-hydroxyethylthiazole phosphate (23). During folate biosynthesis, the formation of 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (H2HMP-PP) is catalyzed via a one-step pyrophosphoryl transfer reaction directly from ATP to 6-hydroxymethyl-7,8-dihydropterin, and then dihydropteroate synthase catalyzes the condensation of H2HMP-PP and p-aminobenzoic acid (22). Similarly, to condense the F1-P moiety and γ-glutamyltyramine in the methanofuran biosynthesis pathway, a kinase is likely involved in transferring a phosphoryl group to form F1-PP before the condensation step. This assumption is based on the facts that (i) the pyrophosphate group serves as a better leaving group compared to a phosphate group because pyrophosphate is a stronger acid compared to a phosphate group and (ii) pyrophosphate complexes with magnesium ions to form a ubiquitous leaving group.
We previously reported that MfnE encodes a second type of archaeal adenylate kinase (14). In addition to MfnE, it has been established that another archaeal adenylate kinase in M. jannaschii is the product of the mj0479 gene (19, 20). It is unlikely that methanogens would contain two distinct adenylate kinases. Our results clearly show that the promiscuous MfnE also catalyzes the formation of F1-PP from F1-P and ATP. The gene locus mj0458 is in the neighborhood of MfnD (encoded by mj0815) and MfnF (encoded by mj0840), which are known to be involved in methanofuran biosynthesis. We propose that the physiological function of MfnE is to catalyze F1-PP formation during methanofuran biosynthesis. However, to approve our hypothesis, future work will focus on screening the F1-PP kinase activity in other M. jannaschii kinases and further characterization of MfnE.
Many promiscuous enzymes that have been found in archaea (27, 28). M. jannaschii is an autotrophic archaea with a small genome (21). Therefore, promiscuity of enzymes can provide an obvious advantage, allowing it to react with a broader range of substrates to maximize its catalytic versatility using limited enzyme resources (29). Thus, the promiscuous MfnE may also perform a function similar to that of other adenylate kinases, which regulate the ATP/ADP balance in the cell (14).
MfnF catalyzes the formation of an ether bond (C-O), instead of the C-N or C-C bond, as found in folate, thiamine, terpene, and steroid biosynthesis. The other well-known enzyme that catalyzes C-O bond formation is geranylgeranylglycerol phosphate synthase (GGGPS) (30). This enzyme catalyzes the formation of an ether bond between glycerol-1-phosphate and polyprenyl diphosphates, which is essential for biosynthesis of archaeal membrane lipid (30). GGGPS exhibits an α/β TIM barrel structure (31), as do thiamine synthase (32) and dihydropteroate synthase (33, 34).
PSI-blast analysis shows that MfnF belongs to the hydantoinase A family (PF01968) and contains a sugar kinase domain. However, the catalytically essential histidines (15) in hydantoinase are not conserved in MfnF. The crystal structure of the MfnF homolog in Methanococcus maripaludis, which was solved by A. P. Kuzin et al. (PDB 3CET), exhibits a distinctive α/β two-layer sandwich structure (Fig. 7), unlike the α/β TIM barrel structure observed in thiamine synthase (32), dihydropteroate synthase (33, 34), and GGGPS (31). In the active sites of both thiamine synthase and dihydropteroate synthase, the Mg2+ used to stabilize the leaving pyrophosphate is ligated by two aspartic acid residues (33, 35). Such a pyrophosphate stabilizing interaction is similar to that found in farnesyl pyrophosphate synthase (24), aristolochene synthase (25), and pentalenene synthase (26), which catalyze the formation of an allylic carbocation from a pyrophosphate ester. In addition, serine and/or threonine residues are hydrogen bonded to the oxygen atom of the pyrophosphate to activate the pyrophosphate as a leaving group (35). Sequence alignment and structural analysis of MfnF show a highly conserved motif, D135XGSTTXD142, which is likely involved in metal and pyrophosphate binding during catalysis. In addition, a strictly conserved Arg158 likely plays a role in stabilizing pyrophosphate as a leaving group (Fig. 7).
FIG 7.
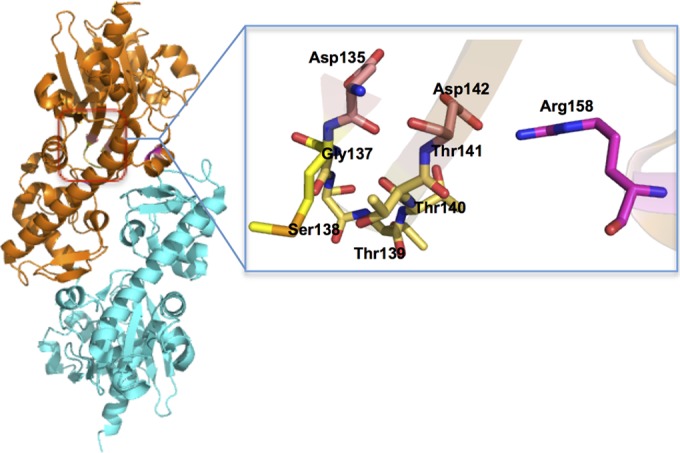
Overall dimer structure of MfnF (left) and possible pyrophosphate and metal-binding pocket (right).
In all examples where pyrophosphate serves as a leaving group, it has been proposed that the reaction mechanism follows an SN-1 mechanism that proceeds via formation of a cationic intermediate before nucleophilic attack by the other substrate (24–26, 33, 35). Therefore, it is reasonable to assume that MfnF catalyzes a reaction following a similar mechanism (Fig. 8). In this mechanism, pyrophosphate is first removed from F1-PP, stabilized by the metal ion (likely Mg2+) and Arg158 (Fig. 7), where the resulting cationic intermediate species (F1+) is resonance stabilized with the positive charge delocalized over the furan ring. The hydroxyl group of γ-glutamyltyramine finally attacks F1+ at the carbocation carbon atom to generate the product APMF-Glu.
FIG 8.

Proposed catalytic mechanism of MfnF.
In summary, we describe here two enzymes, MfnE and MfnF, that are responsible for the formation of APMF-Glu with the first enzyme (MfnE) catalyzing the phosphorylation of F1-P to F1-PP and the second enzyme (MfnF) catalyzing the coupling of F1-PP with γ-glutamyltyramine to produce the core structure of methanofuran. Although such coupling reactions are ubiquitous in biochemistry, we provide here the first evidence that such a mechanism is used in methanofuran biosynthesis. Since methanofuran in M. jannaschii contains 7 to 12 γ-linked glutamates, we propose that once the core structure APMF-Glu is synthesized, another enzyme catalyzes polyglutamylation, producing the final methanofuran molecule (Fig. 1). However, we have yet to identify the enzyme catalyzing this polyglutamylation.
ACKNOWLEDGMENTS
We thank Walter Niehaus for invaluable discussion and Janet Webster for editing the manuscript. We also thank W. Keith Ray and Kim C. Harich for performing the MS experiments. The MS resources are maintained by the Virginia Tech Mass Spectrometry Incubator, a facility operated in part through funding by the Fralin Life Science Institute at Virginia Tech and the Agricultural Experiment Station Hatch Program (CRIS project VA-135981).
This study was supported by National Science Foundation grant MCB1120346.
REFERENCES
- 1.Ferry JG. 1999. Enzymology of one-carbon metabolism in methanogenic pathways. FEMS Microbiol Rev 23:13–38. doi: 10.1111/j.1574-6976.1999.tb00390.x. [DOI] [PubMed] [Google Scholar]
- 2.Deppenmeier U. 2002. The unique biochemistry of methanogenesis. Prog Nucleic Acids Res Mol Biol 71:223–283. [DOI] [PubMed] [Google Scholar]
- 3.Leigh JA, Rinehart KL, Wolfe RS. 1984. Structure of methanofuran, the carbon-dioxide reduction factor of Methanobacterium thermoautotrophicum. J Am Chem Soc 106:3636–3640. doi: 10.1021/ja00324a037. [DOI] [Google Scholar]
- 4.Neue H. 1993. Methane emissions from rice fields. Bioscience 43:466–476. doi: 10.2307/1311906. [DOI] [Google Scholar]
- 5.Leigh JA, Rinehart KL Jr, Wolfe RS. 1985. Methanofuran (carbon dioxide reduction factor), a formyl carrier in methane production from carbon dioxide in Methanobacterium. Biochemistry 24:995–999. doi: 10.1021/bi00325a028. [DOI] [PubMed] [Google Scholar]
- 6.Chistoserdova L, Vorholt JA, Thauer RK, Lidstrom ME. 1998. C1 transfer enzymes and coenzymes linking methylotrophic bacteria and methanogenic Archaea. Science 281:99–102. doi: 10.1126/science.281.5373.99. [DOI] [PubMed] [Google Scholar]
- 7.Pomper BK, Vorholt JA. 2001. Characterization of the formyltransferase from Methylobacterium extorquens AM1. Eur J Biochem 268:4769–4775. doi: 10.1046/j.1432-1327.2001.02401.x. [DOI] [PubMed] [Google Scholar]
- 8.White RH. 1988. Structural diversity among methanofurans from different methanogenic bacteria. J Bacteriol 170:4594–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen KD, White RH. 2014. Identification of structurally diverse methanofuran coenzymes in Methanococcales that are both N-formylated and N-acetylated. Biochemistry 53:6199–6210. doi: 10.1021/bi500973h. [DOI] [PubMed] [Google Scholar]
- 10.Miller D, Wang Y, Xu H, Harich K, White RH. 2014. Biosynthesis of the 5-(aminomethyl)-3-furanmethanol moiety of methanofuran. Biochemistry 53:4635–4647. doi: 10.1021/bi500615p. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Jones MK, Xu H, Ray WK, White RH. 2015. Mechanism of the enzymatic synthesis of 4-(hydroxymethyl)-2-furancarboxaldehyde-phosphate (4-HFC-P) from glyceraldehyde-3-phosphate catalyzed by 4-HFC-P synthase. Biochemistry 54:2997–3008. doi: 10.1021/acs.biochem.5b00176. [DOI] [PubMed] [Google Scholar]
- 12.Kezmarsky ND, Xu H, Graham DE, White RH. 2005. Identification and characterization of a l-tyrosine decarboxylase in Methanocaldococcus jannaschii. Biochim Biophys Acta 1722:175–182. doi: 10.1016/j.bbagen.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Xu H, Harich KC, White RH. 2014. Identification and characterization of a tyramine-glutamate ligase (MfnD) involved in methanofuran biosynthesis. Biochemistry 53:6220–6230. doi: 10.1021/bi500879h. [DOI] [PubMed] [Google Scholar]
- 14.Grochowski LL, Censky K, Xu H, White RH. 2012. A new class of adenylate kinase in methanogens is related to uridylate kinase. Arch Microbiol 194:141–145. doi: 10.1007/s00203-011-0759-9. [DOI] [PubMed] [Google Scholar]
- 15.Xu Z, Liu Y, Yang Y, Jiang W, Arnold E, Ding J. 2003. Crystal structure of d-hydantoinase from Burkholderia pickettii at a resolution of 2.7 angstroms: insights into the molecular basis of enzyme thermostability. J Bacteriol 185:4038–4049. doi: 10.1128/JB.185.14.4038-4049.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lira LM, Vasilev D, Pilli RA, Wessjohann LA. 2013. One-pot synthesis of organophosphate monoesters from alcohols. Tetrahedron Lett 54:1690–1692. doi: 10.1016/j.tetlet.2013.01.059. [DOI] [Google Scholar]
- 17.Delmelle M. 1978. Investigation of retinal as a source of singlet oxygen. Photochem Photobiol 27:731–734. doi: 10.1111/j.1751-1097.1978.tb07671.x. [DOI] [Google Scholar]
- 18.Miller D, O'Brien K, Xu H, White RH. 2014. Identification of a 5′-deoxyadenosine deaminase in Methanocaldococcus jannaschii and its possible role in recycling the radical S-adenosylmethionine enzyme reaction product 5′-deoxyadenosine. J Bacteriol 196:1064–1072. doi: 10.1128/JB.01308-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rusnak P, Haney P, Konisky J. 1995. The adenylate kinases from a mesophilic and three thermophilic methanogenic members of the Archaea. J Bacteriol 177:2977–29781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferber DM, Haney PJ, Berk H, Lynn D, Konisky J. 1997. The adenylate kinase genes of M. voltae, M. thermolithotrophicus, M. jannaschii, and M. igneus define a new family of adenylate kinases. Gene 185:239–244. [DOI] [PubMed] [Google Scholar]
- 21.Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, Kerlavage AR, Dougherty BA, Tomb JF, Adams MD, Reich CI, Overbeek R, Kirkness EF, Weinstock KG, Merrick JM, Glodek A, Scott JL, Geoghagen NS, Venter JC. 1996. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 22.Hanson AD, Gregory JF III. 2011. Folate biosynthesis, turnover, and transport in plants. Annu Rev Plant Biol 62:105–125. doi: 10.1146/annurev-arplant-042110-103819. [DOI] [PubMed] [Google Scholar]
- 23.Jurgenson CT, Begley TP, Ealick SE. 2009. The structural and biochemical foundations of thiamin biosynthesis. Annu Rev Biochem 78:569–603. doi: 10.1146/annurev.biochem.78.072407.102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. 1996. Regulation of product chain length by isoprenyl diphosphate synthases. Proc Natl Acad Sci U S A 93:15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starks CM, Back K, Chappell J, Noel JP. 1997. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science 277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 26.Lesburg CA, Zhai G, Cane DE, Christianson DW. 1997. Crystal structure of pentalenene synthase: mechanistic insights on terpenoid cyclization reactions in biology. Science 277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 27.Jia B, Cheong GW, Zhang S. 2013. Multifunctional enzymes in archaea: promiscuity and moonlight. Extremophiles 17:193–203. doi: 10.1007/s00792-012-0509-1. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Xu H, White RH. 2014. β-Alanine biosynthesis in Methanocaldococcus jannaschii. J Bacteriol 196:2869–2875. doi: 10.1128/JB.01784-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen RA. 1976. Enzyme recruitment in evolution of new function. Annu Rev Microbiol 30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 30.Koga Y, Morii H. 2007. Biosynthesis of ether-type polar lipids in archaea and evolutionary considerations. Microbiol Mol Biol Rev 71:97–120. doi: 10.1128/MMBR.00033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Payandeh J, Fujihashi M, Gillon W, Pai EF. 2006. The crystal structure of (S)-3-O-geranylgeranylglyceryl phosphate synthase reveals an ancient fold for an ancient enzyme. J Biol Chem 281:6070–6078. [DOI] [PubMed] [Google Scholar]
- 32.Chiu HJ, Reddick JJ, Begley TP, Ealick SE. 1999. Crystal structure of thiamin phosphate synthase from Bacillus subtilis at 1.25-Å resolution. Biochemistry 38:6460–6470. doi: 10.1021/bi982903z. [DOI] [PubMed] [Google Scholar]
- 33.Yun MK, Wu Y, Li Z, Zhao Y, Waddell MB, Ferreira AM, Lee RE, Bashford D, White SW. 2012. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 335:1110–1114. doi: 10.1126/science.1214641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hampele IC, D'Arcy A, Dale GE, Kostrewa D, Nielsen J, Oefner C, Page MG, Schonfeld HJ, Stuber D, Then RL. 1997. Structure and function of the dihydropteroate synthase from Staphylococcus aureus. J Mol Biol 268:21–30. doi: 10.1006/jmbi.1997.0944. [DOI] [PubMed] [Google Scholar]
- 35.Peapus DH, Chiu HJ, Campobasso N, Reddick JJ, Begley TP, Ealick SE. 2001. Structural characterization of the enzyme-substrate, enzyme-intermediate, and enzyme-product complexes of thiamin phosphate synthase. Biochemistry 40:10103–10114. doi: 10.1021/bi0104726. [DOI] [PubMed] [Google Scholar]