

ABSTRACT
Escherichia coli's DNA polymerase IV (Pol IV/DinB), a member of the Y family of error-prone polymerases, is induced during the SOS response to DNA damage and is responsible for translesion bypass and adaptive (stress-induced) mutation. In this study, the localization of Pol IV after DNA damage was followed using fluorescent fusions. After exposure of E. coli to DNA-damaging agents, fluorescently tagged Pol IV localized to the nucleoid as foci. Stepwise photobleaching indicated ∼60% of the foci consisted of three Pol IV molecules, while ∼40% consisted of six Pol IV molecules. Fluorescently tagged Rep, a replication accessory DNA helicase, was recruited to the Pol IV foci after DNA damage, suggesting that the in vitro interaction between Rep and Pol IV reported previously also occurs in vivo. Fluorescently tagged RecA also formed foci after DNA damage, and Pol IV localized to them. To investigate if Pol IV localizes to double-strand breaks (DSBs), an I-SceI endonuclease-mediated DSB was introduced close to a fluorescently labeled LacO array on the chromosome. After DSB induction, Pol IV localized to the DSB site in ∼70% of SOS-induced cells. RecA also formed foci at the DSB sites, and Pol IV localized to the RecA foci. These results suggest that Pol IV interacts with RecA in vivo and is recruited to sites of DSBs to aid in the restoration of DNA replication.
IMPORTANCE DNA polymerase IV (Pol IV/DinB) is an error-prone DNA polymerase capable of bypassing DNA lesions and aiding in the restart of stalled replication forks. In this work, we demonstrate in vivo localization of fluorescently tagged Pol IV to the nucleoid after DNA damage and to DNA double-strand breaks. We show colocalization of Pol IV with two proteins: Rep DNA helicase, which participates in replication, and RecA, which catalyzes recombinational repair of stalled replication forks. Time course experiments suggest that Pol IV recruits Rep and that RecA recruits Pol IV. These findings provide in vivo evidence that Pol IV aids in maintaining genomic stability not only by bypassing DNA lesions but also by participating in the restoration of stalled replication forks.
INTRODUCTION
In the natural environment, Escherichia coli and other bacteria are exposed to exogenous and endogenous agents that cause DNA damage and mutations. To cope with these challenges and maintain genomic integrity, many bacteria activate a DNA repair system known as the SOS response. In E. coli, the SOS response involves the induction of over 40 genes that include those encoding three DNA polymerases (Pol) that assist in repairing DNA damage: Pol II, Pol IV, and Pol V (1). Pol II (encoded by the polB gene), a member of the B family of DNA polymerases, is an accurate polymerase that can bypass certain types of DNA lesions, a phenomenon known as translesion synthesis (TLS) (2, 3). Pol II is also required for efficient restart of replication when replication forks are stalled at UV lesions (4). Pol IV (encoded by dinB) and Pol V (encoded by umuDC) are error-prone Y family DNA polymerases apparently specialized to perform TLS (5, 6). Both Pol IV and Pol V have open active sites that can accommodate bulky lesions, and both enzymes lack 3′-5′ proofreading exonucleases; thus, on undamaged DNA both Pol IV and V synthesize DNA with low fidelity and processivity (5, 6).
While Pol V can replicate past a wide variety of chemical- and radiation-induced DNA lesions, its levels of expression and activity are tightly regulated, presumably to prevent unwanted mutagenic activity. In the absence of DNA damage Pol V is almost undetectable, and it is not induced until late during the SOS response (5, 6). In contrast, Pol IV can bypass only a limited repertoire of DNA lesions. Pol IV appears to be specialized for bypassing adducts to the N2 position of guanines, such as benzo(α)pyrene, nitrofurazone, and 4-nitroquinoline-1-oxide (NQO) (3, 7, 8). Pol IV is relatively abundant (250 molecules per cell) even in non-SOS-induced cells (9). These high levels suggest that Pol IV may gain access to replicating DNA, and indeed, Pol IV can displace the replicase DNA polymerase III from the sliding clamp in vitro (10, 11). Pol IV can also elongate from misaligned primer/template termini (12). These phenotypes suggest an important role for Pol IV in rescuing stalled replication forks during replication.
After SOS induction, the levels of Pol IV increase 10-fold to about 2,500 molecules per cell (9). In stationary-phase cells, Pol IV is induced 3-fold under the control of the general stress response sigma factor RpoS (13). Thus, Pol IV is clearly the most abundant DNA polymerase in cells under a variety of stress conditions.
In normally growing cells, Pol IV contributes only modestly to spontaneous mutagenesis on the chromosome (14–17). However, overexpression of Pol IV results in a 100-fold or more increase in growth-dependent spontaneous mutations (12, 18, 19). Pol IV is responsible for both adaptive and nonadaptive mutations in stationary-phase cells (13, 15, 20, 21), and under certain stress conditions, Pol IV participates in error-prone double-stand break (DSB) repair (22, 23). In addition, Pol IV-deficient E. coli cells have decreased fitness in long-term stationary-phase cultures (24).
The expression and mutagenic activity of Pol IV are controlled by several cellular factors in addition to the SOS response regulators, LexA and RecA. These additional regulators include RpoS, mentioned above (13); polyphosphate, which mediates a nutrient limitation response (25); GroE, which is part of the heat shock response (26); and HU, which modulates chromosome structure (27). Pol IV activity and processivity in vitro are enhanced by interacting with the beta clamp (28), and its mutagenic activity is also modulated by UmuD, RecA, and the transcription factor NusA (29–31).
Recently, we found that Pol IV physically interacts with Rep DNA helicase in vitro (32). This interaction stimulates the helicase activity of Rep and the polymerase activity of Pol IV (32). In vivo, Rep is required for the full adaptive mutation activity of Pol IV (32). Rep is an SFI class helicase with 3′-5′ polarity and is required for normal progression of the replication fork (33–35). Rep facilitates the loading of the replicative helicase DnaB to restart collapsed replication forks and, in addition, participates in a PriA-independent, PriC-dependent process that can also restart replication forks (36–38).
Here, we report the construction of fluorescently tagged Pol IV fusions and their localization as foci in response to DNA damage and DNA double-strand breaks. We further report that Pol IV colocalizes with and recruits Rep helicase to sites of DNA damage. In addition, Pol IV colocalizes with RecA foci that form in response to DNA damage and at double-strand breaks.
MATERIALS AND METHODS
Bacterial strains and media.
All the bacterial strains used are E. coli K-12 derivatives and are listed in Table 1. Genetic manipulations were performed using standard procedures, as described previously (39); Table 1 lists the donor and recipient strains used for P1vir transductions. Cultures were grown at 37°C or 30°C in Luria-Bertani (LB) broth or minimal medium (M9-0.1% glycerol, glucose, or lactose) (39). When required, minimal medium was supplemented with 100 μg/ml proline. Antibiotics were added to LB broth at the following concentrations: carbenicillin (Carb), 100 μg/ml; kanamycin (Kn), 30 μg/ml; chloramphenicol (Cm), 10 μg/ml; tetracycline (Tc), 20 μg/ml; nalidixic acid (Nal), 40 μg/ml; and NQO, 12 or 20 μM. The primers used in this study are listed in Table 2. Strain constructions were confirmed by PCR amplification and DNA sequencing.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant genotype and/or description | Recipient | Source of allele | Reference |
|---|---|---|---|---|
| Strains | ||||
| IL-03 | zdi-3628::lacZo(240X)::Kn | 44 | ||
| SS3085 | ygaD1::Kn recA4155-gfp901 | 48 | ||
| AR30 | ΔdinB::Zeo | 82 | ||
| RH4695 | Δsrl::Tn10 | 83 | ||
| FC36 | F−ara Δ(gpt-lac) 5thi Rifr (Pro−) | 60 | ||
| FC40 | FC36/F′ Φ(lacI33-lacZ) Pro+ | 60 | ||
| FC1373 | F− Φ(lacI33-lacZ) dTn10 Tcs; revertible Lac− and Tcs on the chromosome | 19 | ||
| FC1577 | FC36 ΔumuDC | This study | ||
| PF1716 | FC36 Δsrl::Tn10 | FC36 | RH4695 | This study |
| PFB236 | FC36 ΔdinB::Zeo | FC36 | AR30 | This study |
| PFB243 | FC40; ΔdinB::Zeo on chromosome and episome | 25 | ||
| PFB1041 | FC36 lacZo::Kn (lacO array at 1.803 Mb) | FC36 | IL-03 | This study |
| PFB1073 | FC36 I-SceI (I-SceI recognition site at 1.642 Mb) | FC36 | pPFV408 | This study |
| PFB1081 | FC36 I-SceI lacZo::Kn | PFB1073 | PFB1041 | This study |
| PFB1103 | FC36 I-SceI lacZo::Kn ΔdinB::Zeo | PFB1081 | PFB236 | This study |
| PFB1137 | FC36 I-SceI ygaD1::Kn recA4155-gfp901 | PFB1073 | SS3085 | This study |
| PFB1182 | FC36 I-SceI ΔumuDC | PFB1073 | FC1577 | This study |
| PFB1195 | FC36 I-SceI lacZo::Kn ΔumuDC | PFB1182 | FC1577 | This study |
| PFB1200 | FC36 I-SceI ΔumuDC ΔdinB::Zeo | PFB1182 | PFB236 | This study |
| PFB1201 | FC36 I-SceI lacZo::Kn ΔumuDC ΔdinB::Zeo | PFB1200 | PFB1041 | This study |
| PFB1209 | FC36 ygaD1::Kn recA4155-gfp901 | FC36 | SS3085 | This study |
| PFB1213 | FC36 ygaD1::cat–I-SceI recA4155-gfp901 | PFB1209 | pWRG100 | This study |
| PFB1215 | FC36 I-SceI lacZo::Kn Δsrl::Tn10 | PFB1081 | PF1716 | This study |
| PFB1216 | FC36 I-SceI lacZo::Kn ΔdinB::Zeo Δsrl::Tn10 | PFB1103 | PF1716 | This study |
| PFB1220 | FC36 recA4155-gfp901 | PFB1213 | FC36 | This study |
| PFB1224 | FC36 I-SceI lacZo::Kn recA4155-gfp901 | PFB1215 | PFB1220 | This study |
| PFB1225 | FC36 I-SceI lacZo::Kn ΔdinB::Zeo recA4155-gfp901 | PFB1216 | PFB1220 | This study |
| PFB1248 | FC36 FRT–cat–FRT–I-SceI (I-SceI recognition site at 1.801 Mb) | FC36 | pPFV427 | This study |
| PFB1263 | FC36 ΔdinB::Zeo ΔumuDC | PFB236 | FC1577 | This study |
| PFB1265 | FC36 I-SceI (1.801 Mb) lacZo::Kn | B1041 | PFB1248 | This study |
| PFB1271 | FC36 I-SceI (1.801 Mb) lacZo::Kn ΔdinB::Zeo | PFB1265 | PFB243 | This study |
| PFB1277 | FC36 lacZo::Kn ΔdinB::Zeo ΔumuDC | PFB1263 | PFB1041 | This study |
| PFB1279 | FC36 I-SceI (1.801 Mb) lacZo::Kn ΔdinB::Zeo ΔumuDC | PFB1277 | PFB1248 | This study |
| PFB1284 | FC1373 ΔdinB::Zeo | FC1373 | PFB243 | This study |
| Plasmids | ||||
| pACYC184 | Cloning vector; low copy number; orip15A Cmr Tcr | 43 | ||
| pBAD24 | Arabinose-inducible expression vector; high copy number; oripMB1 Ampr | 45 | ||
| pBAD33 | Arabinose-inducible expression vector; low copy number; orip15A Cmr | 45 | ||
| pWSK29 | Cloning vector; low copy number; oripSC101 Ampr | 41 | ||
| pKD3 | Template plasmid for gene disruption; resistance gene (cat) is flanked by FRT sites; oriR6Kγ Ampr Cmr | 47 | ||
| pKD13 | Template plasmid for gene disruption; resistance gene (kan) is flanked by FRT sites; oriR6Kγ Ampr Knr | 47 | ||
| pKD32 | Template plasmid for gene disruption; resistance gene (cat) is flanked by FRT sites; oriR6Kγ Ampr Cmr | 47 | ||
| pKD46 | Lambda red expression under the control of arabinose-inducible promoter; oriR101 repA101(Ts) origin; Ampr | 47 | ||
| pLAU53 | pBAD24 with LacI-ECFP and TetR-EYFP; Ampr | 44 | ||
| pCP20 | Plasmid containing yeast Flp recombinase gene (FLP); oriR101 repA101(Ts) Ampr Cmr | 84 | ||
| pWRG99 | pKD46 with I-SceI endonuclease under the control of tetracycline-inducible promoter (PtetA); Ampr | 46 | ||
| pWRG100 | pKD3 with I-SceI recognition site; oriR6γ Cmr Ampr | 46 | ||
| pVS132 | EYFP on pTrc99A; oripMB1 Ampr | 40 | ||
| pPYG768 | pWSK29 with DinB+ under its native promoter | 18 | ||
| pPFB913 | pWSK29 with 100-bp upstream DinB-12L-EYFP | This study | ||
| pPFB914 | pACYC184 with Rep-mCh | This study | ||
| pPFB1035 | pBAD33 with LacI-ECFP and PtetA I-SceI endonuclease | This study | ||
| pPFB1162 | pWSK29 with 100-bp upstream mCh-DinBE104A | This study | ||
| pPFB1173 | pWSK29 with 100-bp upstream DinBE104A-12L-EYFP | This study | ||
| pPFB1178 | pWSK29 with 16L-EYFP | This study | ||
| pPFB1179 | pWSK29 ΔlacZ | This study | ||
| pPFB1188 | pWSK29 with 450-bp upstream DinB-20L-EYFP | This study | ||
| pPFV404 | mCh-10L on pMp92 | D. B. Kearns, personal communication | ||
| pPFV407 | pWSK29 with 100-bp upstream mCh-10L-DinB | This study | ||
| pPFV408 | pKD13 with I-SceI recognition site | This study | ||
| pPFV427 | pKD32 with I-SceI recognition site | This study |
Amp, ampicillin; Zeo, zeomycin; kan, kanamycin resistance gene; cat, chloramphenicol resistance gene.
TABLE 2.
Oligonucleotides used in the study
The underlined sequences indicate restriction enzyme sites.
Plasmid construction.
The plasmids used in this study are listed in Table 1. The plasmid constructions were confirmed by restriction digestion and sequencing. To construct a fluorescently labeled Pol IV protein under the control of the endogenous dinB promoter on a low-copy-number vector, the dinB gene with 100 bp upstream from the dinB start codon was amplified from genomic DNA of E. coli strain FC36 using the primer pair 5′ NcoI NheI 100 bp US DinB and 3′ BamHI no stop DinB. The PCR product was digested with restriction enzymes NcoI and BamHI and ligated to pVS132 (40), which carries enhanced yellow fluorescent protein (EYFP), thereby placing EYFP at the C terminus of DinB, linked by 12 amino acids (DinB-12L-EYFP). The resultant plasmid was used as the template to amplify DNA encoding DinB-12L-EYFP using primers 5′ SalI 100 bp US DinB and EYFP Rev HindIII. This PCR fragment was cloned into SalI/HindIII-digested pWSK29 (41), yielding plasmid pPFB913 carrying DinB-20L-EYFP.
Overlap PCR extension was used to construct an active-site mutation, E104A (12, 42), in DinB-12L-EYFP (yielding DinBE104A-12L-EYFP). Using pPFB913 as the template, the DNA sequence upstream of the region of dinB that encodes amino acid E104 was amplified with the primer pair 5′ SalI 100 bp US DinB and GC-E104A rev, and the dinB DNA sequence downstream of E104 including EYFP was amplified with the primer pair GC-E104A Fwd and EYFP Rev HindIII. A third round of overlap PCR with the primer pair 5′ SalI 100 bp US DinB and EYFP Rev HindIII yielded the DNA fragment encoding DinBE104A-EYFP. This fragment was digested with SalI and HindIII and ligated into the SalI/HindIII-digested pWSK29, yielding plasmid pPFB1173 carrying DinBE104A-12L-EYFP. Loss of function of the mutant DinB was confirmed by NQO sensitivity (see below).
To generate a DinB-EYFP fusion with a longer linker, the primer pair 5′ BamHI 16 aa Linker EcoRI Fwd and EYFP Rev HindIII was used to amplify the EYFP gene from pPFB913, thereby placing a 20-amino-acid linker at the N terminus of EYFP. The PCR product was cloned into BamHI/HindIII-digested pWSK29, yielding plasmid pPFB1178. The dinB coding sequence with 450 bp upstream of the dinB start codon was amplified from plasmid pPYG768 with primers pJP anti-sense and 3′ BamHI no stop DinB. This PCR product was digested with SacI and BamHI and ligated to SacI/BamHI-digested plasmid pPFB1178, yielding plasmid pPFB1188 carrying DinB-20L-EYFP.
To construct an mCherry (mCh) fusion to the N terminus of Pol IV, the dinB gene with 100 bp upstream of the dinB start codon was amplified from the genomic DNA of FC36 using the primer pair 5′ EcoRI DinB and 3′ HindIII stop DinB. The PCR product was cloned into pWSK29 using EcoRI and HindIII. The mCherry gene with a 10-amino-acid linker was amplified from plasmid pPFV404 using the primer pair 5′ SalI 100bp US DinB mCherry and 3′ EcoRI Linker. This PCR product was cloned into the above-described plasmid (pWSK29/DinB) using SalI and EcoRI, yielding plasmid pPFV407 carrying mCh-10L-DinB.
Overlap PCR was used to construct an active-site mutant of mCh-10L-DinB. Partial dinB coding sequences were amplified from the template plasmid pPFV407 using the primers pJP sense and GC-E104A Rev and GCE104A Fwd and pJP anti-sense. The PCR products were used as the templates for a third round of overlap PCR with the primer pair 5′ SalI 100bp US DinB and 3′ HindIII stop DinB. The resultant fragment was digested with SalI and HindIII and cloned into SalI/HindIII-digested pWSK29, yielding plasmid pPFB1162 carrying mCh-10L-DinBE104A.
A Lac− vector control for adaptive-mutation assays was generated by digesting pWSK29 with PvuII to remove the lacZ gene and its promoter, followed by religation, yielding plasmid pPFB1179.
To construct an mCherry fusion to the C terminus of Rep, the chromosomal rep gene with its promoter was amplified from genomic DNA of FC36 using the primers 5′ SalI 100bp US Rep and 3′ BamHI Rep no stop and cloned into SalI/BamHI-digested pACYC184 (43). The mCherry sequence was amplified from pPFV404 with the primers 5′ BamHI 12 Link mCherry and 3′ HindIII mCherry stop and ligated into the BamHI/HindIII-digested pACYC184-Rep plasmid, yielding plasmid pPFB914 carrying Rep-mCh.
To generate a plasmid carrying both LacI-ECFP (enhanced cyan fluorescent protein) and the I-SceI endonuclease, the DNA encoding LacI-ECFP was amplified from pLAU53 (44) using the primers 5′ KpnI LacI Fwd and 3′ XbaI stop ECFP and cloned in KpnI/XbaI-digested pBAD33 (45), placing LacI-ECFP under the control of the arabinose promoter. The DNA encoding the I-SceI endonuclease under the control of a tetracycline-inducible promoter (PtetA) was amplified from plasmid pWRG99 (46) using the primers 5′ SalI tetR Fwd and 3′ SphI I-SceI Rev. This PCR product was cloned into the pBAD33-LacI-ECFP plasmid using SalI and SphI, yielding plasmid pPFB1035. The functionality of the I-SceI enzyme on pPFB1035 was confirmed by transforming it into PFB1073 and assaying for sensitivity to anhydrotetracycline (AHT), a tetracycline analogue that is a gratuitous inducer of PtetA.
Insertion of the I-SceI endonuclease recognition site and the lacO array onto the chromosome.
To create I-SceI endonuclease recognition site insertion cassettes, the I-SceI recognition site was cloned outside the FLP recognition target (FRT) sites in plasmids pKD13 (Knr) and pKD32 (Cmr) (47). Primers pKD13 P1 Fwd and pKD13-I-SceI site-BamHI-Rev (encoding the I-SceI recognition site) were used to amplify the Knr cassette from pKD13; the PCR product was digested with BglII and BamHI and cloned back into pKD13. The resultant plasmid, pPFV408, was confirmed to contain the cassette FRT–kan–FRT–I-SceI by sequencing. To clone the I-SceI site into pKD32, plasmids pPFV408 and pKD32 were digested with XbaI. The larger digestion product (∼2.5 kb) of pPFV408 containing pKD13 backbone and the I-SceI site was ligated with the ∼1,000-bp XbaI digestion product of pKD32, which consisted of part of the Cmr gene cassette. The resultant plasmid, pPFV427, was confirmed to contain the cassette FRT–cat–FRT–I-SceI by sequencing.
λ Red recombination-mediated recombineering (47) was used to insert the I-SceI recognition site in the chromosomal ydfU-rem intergenic region (at 1.642 Mb). A PCR product that included sequences upstream and downstream of the ydfU-rem intergenic region, the Knr cassette flanked by FRT sites, and the I-SceI site was generated using pPFV408 as the template and the primers 5′ Homo ydfU rem Wanner P1 and 3′ Homo ydfU rem Wanner P4. This product was transformed into FC36 carrying pKD46, selecting for Knr transformants, which were single-colony purified and grown at 42°C to eliminate the plasmid. The Knr gene was removed using Flp recombinase (47), leaving behind a Kn scar and the I-SceI site, which was confirmed by sequencing. The functionality of the I-SceI recognition site was confirmed by amplifying the I-SceI insertion site region flanked by 150 bp upstream and downstream with the primers 5′ US 150 bp ydfU-rem Fwd and 3′ DS 150bp ydfU-rem Rev and digesting the PCR product with I-SceI endonuclease (Fermentas, St. Leon-Rot, Germany).
To insert a lacO array at 1.803 Mb on the chromosome, a P1vir lysate of E. coli strain IL-03 (44) was used to transfer lacO [lacZo(240X)::Kn] (selecting for Knr transductants) into FC36, yielding strain PFB1041, and into PFB1073 (FC36::I-SceI), yielding strain PFB1081, with the lacO array 161 kb from the I-SceI endonuclease recognition site.
To place the recA4155-gfp (green fluorescent protein [GFP]) and the lacO array in the same strain, ygaD1::Kn near the recA4155-gfp allele (48) was replaced with the wild-type ygaD+ gene using scarless recombineering (46). Plasmid pWRG100 (46) was used as the template to amplify the cat–I-SceI cassette with primers ygaD-left-pWRG100-Fwd and recA-right-pWRG100-Rev, and the PCR product was transformed into strain PFB1209, selecting for Cmr and screening for Kns, creating strain PFB1213 with ygaD1::Kn replaced with cat–I-SceI near the recA4155-gfp allele. Five hundred base pairs containing the ygaD+ gene was amplified from FC36 genomic DNA using the primers ygaD1 F0010 and recAstartRV, and the PCR product was transformed into PFB1213 carrying plasmid pWRG99 (46). Transformants were selected on LB plus Carb (50 μg/ml) plus AHT (500 ng/ml) and screened for loss of Cmr. This procedure restored the ygaD+ gene near the recA4155-gfp allele, creating strain PFB1220. The presence of the ygaD+ gene in PFB1220 was confirmed by sequencing. Strains PFB1081 and PFB1103 were transduced with P1vir lysate of strain PFB1716 (Δsrl::Tn10), selecting for Tcr and screening for Srl−, to generate strains PFB1215 and PFB1216, respectively. The recA4155-gfp allele from PFB1220 was transduced into PFB1215 and PF1216, selecting for Srl+ and screening for Tcs, creating strains PFB1224 (carrying the I-SceI site, lacO array, and RecA-GFP) and PFB1225 (like PFB1224 but with the ΔdinB mutation).
NQO sensitivity assays.
NQO sensitivity assays were performed as described previously (49, 50). The NQO stock was 100 mM in N,N-dimethylformamide (Sigma-Aldrich). Cultures of each strain were grown overnight in LB broth plus appropriate antibiotics at 37°C with aeration. Each culture was diluted 10−1 to 10−6 by serial 10-fold dilutions in sterile 0.85% saline solution, and 10 μl of each dilution was spotted on LB agar plates with and without 12 μM NQO. The plates were incubated in the dark at 37°C for 18 to 24 h and then photographed.
Determination of growth-dependent mutation rates.
Cultures of strains carrying a mutant tetA allele on the chromosome (51) were grown overnight at 37°C in LB broth plus appropriate antibiotics and diluted 105-fold into the same medium, and 100-μl aliquots were distributed into 45 wells of a 96-well microtiter plate, which was then incubated for 24 h at 37°C with aeration. Aliquots from nine cultures for each strain were combined into three samples, and appropriate dilutions of these were plated on LB agar plates to determine the total cell numbers. One hundred microliters of each culture was spread onto LB agar plates supplemented with 20 μg/ml Tc, the plates were incubated at 37°C for 24 h, and Tcr colonies were counted. Mutation rates and appropriate confidence limits were calculated using the Ma-Sandri-Sarkar maximum-likelihood method (52) implemented by the FALCOR Web tool (http://www.mitochondria.org/protocols/FALCOR.html; 53).
Adaptive-mutation assays.
Small-scale adaptive-mutation assays were performed as described previously (54). For each strain, four independent cultures were grown in liquid M9-glycerol minimal medium plus appropriate antibiotics (at half the concentrations added to LB broth) for 24 to 48 h at 37°C with aeration until the cultures reached saturation, and 10 μl of each culture was spread on a quadrant of an M9 lactose minimal medium agar plate. The plates were incubated at 37°C, and the Lac+ revertant colonies were counted every day. Lac+ colonies appearing on days 3 to 5 were considered to be due to adaptive mutations.
Immunoblot detection of DinB and DinB-EYFP.
Immunoblotting, as previously described (13, 55), was used to determine the levels of Pol IV in SOS-induced cells. Briefly, overnight cultures in LB broth plus appropriate antibiotics were diluted 1:500 in the same medium and grown with shaking at 37°C. At an optical density at 600 nm (OD600) of 0.5, Nal was added at 40 μg/ml, and the cultures were incubated for an additional hour. Cells from 1 ml of each culture were harvested by centrifugation, resuspended in 1× SDS-PAGE sample loading buffer, and boiled for 15 min or frozen for subsequent processing. The total amount of protein in the samples was determined by Bradford assay (Bio-Rad Laboratories), and an aliquot of each sample containing ∼40 μg protein was used for immunoblotting with rabbit anti-Pol IV polyclonal antibody, as previously described (13). The protein bands on the immunoblots were visualized by chemiluminescence (CSPD; Roche). For a loading control, immunoblots were stripped with BlotFresh Western blot stripping reagent, version II (SignaGen Laboratories), and probed with mouse anti-GroEL monoclonal antibody (Enzo Life Sciences). Densitometric analysis was performed using ImageJ, version 1.46r (http://imagej.nih.gov/ij/). The relative intensity of DinB or DinB fusion proteins was determined by dividing the intensity of DinB or the DinB fusion band by the intensity of the GroEL band.
Microscopy.
For localization studies, phase and fluorescence microscopy were performed with a Nikon Eclipse 80i microscope (Nikon Instruments) equipped with a Nikon Plan Apo 100× phase-contrast objective and a CoolSNAP HQ2 camera (Photometrics, AZ) and using Metamorph image capture software (Molecular Devices, Sunnyvale, CA). The following microscope settings were used to visualize fluorescence signals: DAPI (4′,6-diamidino-2-phenylindole), UV-2E/C DAPI filter cube (excitation filter, 340 to 380 nm; barrier filter, 435 to 485 nm); EYFP, C-FL HYQ fluorescein isothiocyanate (FITC) filter cube (excitation filter, 490 to 510 nm; barrier filter, 520 to 550 nm); ECFP, C-FL HYQ CFP filter cube (excitation filter, 426 to 446 nm; barrier filter, 460 to 500 nm); GFP, C-FL HYQ FITC filter cube (excitation filter, 460 to 500 nm; barrier filter, 515 to 550 nm); mCherry and propidium iodide, C-FL HYQ Texas Red filter cube (excitation filter, 532 to 587 nm; barrier filter, >590 nm). The images were exported as TIFF files and processed and analyzed using ImageJ software.
For photobleaching, a Nikon Eclipse Ti inverted microscope (Nikon Instruments) was used in epifluorescence illumination mode with a Plan Apo VC 1.49-numerical-aperture (NA) 100× oil immersion objective and a yellow fluorescent protein (YFP)-HYQ barrier filter and YFPHQ emission filter. Images were acquired at 0.013 μm per pixel in continuous-frame-transfer mode by a 1,024- by 1,024-pixel Hammatsu ORCA-flash 4.0 sCMOS camera (Hamamatsu Corporation, Hamamatsu, Japan) controlled by Nikon NIS Element AR software (Nikon Instruments) in the continuous-acquisition mode with exposure times of 300 ms for a duration of 120 to 180 s at a frame transfer rate of 100 frames/s. The images were processed and analyzed using ImageJ software.
Exposure to Nal or NQO and colocalization studies.
Strains carrying plasmids expressing fusion proteins were grown overnight in LB broth with appropriate antibiotics, diluted 1:500 in the same medium, and incubated with aeration at 37°C, except strains with the recA4155-gfp allele, which were incubated at 30°C. When a culture reached an OD600 of ∼0.5, Nal (40 μg/ml) or NQO (20 to 160 μM) was added, and the culture was incubated further. Aliquots (100 μl) were drawn every 30 min for 2 to 4 h, washed once with 0.85% saline, and resuspended at 10× in 0.85% saline. Three microliters of the cell suspension was spotted on a glass slide and immobilized with a poly-l-lysine-treated coverslip for microscopy.
Nucleoid staining was performed with DAPI (2 μg/ml). To estimate the viability of cells following Nal treatment, appropriate dilutions of suspensions of treated and untreated cells at various time intervals were plated on LB agar plates, and colonies were counted after 24 h at 37°C. The fraction of dead cells among Nal-treated cells was determined using the LIVE/DEAD Bac Light bacterial viability kit (Invitrogen, Inc.). The cells were suspended in 1 ml of saline solution to a final OD600 of 1.0, stained with propidium iodide according to the manufacturer's directions, spotted on a glass slide, and immobilized with a poly-l-lysine-treated coverslip for microscopy.
Analysis of the number of DinB-EYFP molecules per focus.
Cells of strain PFB243/pPFB1188 (ΔdinB/pDinB-20L-EYFP) were grown overnight at 37°C to saturation in LB broth plus Carb. The culture was diluted 1:500 in the same medium and grown with aeration at 37°C to an OD600 of 0.5. Nal was added at 40 μg/ml, and the culture was incubated for an additional hour. A 1-ml aliquot of cells was centrifuged, washed twice with 1× PBS, and resuspended in 100 μl 1× PBS. Three to 5 μl of the cell suspension was spotted on a glass slide, covered with a 1.5-mm coverslip, and subjected to photobleaching as described above.
To obtain the intensity drop that corresponds to the photobleaching of a single DinB-EYFP molecule (the unitary step size), we modified previously published methods (56, 57). A square 7- by 7-pixel area region of interest (ROI) was defined around each DinB-EYFP focus (ROI-focus), and an ROI of the same size was drawn in a region adjacent to the focus to determine the background cytoplasmic fluorescence (ROI-cyto). For each ROI-focus photobeaching trace, the values from the initial intensity to the minimum-intensity plateau (usually at 250 frames equaling 75 s of exposure) were filtered using the edge-preserving Chung-Kennedy algorithm (58) written in Mathematica (Mathematica 10; Wolfram Research, Inc.). All pairwise differences of these values were computed (i.e., the value of every data point was subtracted from the value of each data point that followed in the filtered trace), and the resulting values were distributed into a histogram with 1,024 bins to give the pairwise difference distribution function (PDDF). The bin values were then normalized by the number of pairwise differences [n(n − 1)/2, where n is the number of data points in each trace] to give the fraction of the total differences in each bin. The number of peaks in this histogram represents the number of photobleaching steps in the trace, and the distance between the peaks represents the intensity drop of the unitary step. To better identify the frequency of the peaks, a Fourier transform was performed on the normalized histogram bin values (using the function Fourier followed by Power in Mathematica), and the dominant peak was taken as the number of photobleaching steps in the trace (μ in Table S1 in the supplemental material). The maximum value of the PDDF, which is equal to the difference between the maximum and minimum values of the fitted Chung-Kennedy step curve (delta in Table S1 in the supplemental material), was divided by μ to give the unitary step size. The intensity of each focus (I-focus in Table S1 in the supplemental material) was calculated by first fitting a smooth exponential decay curve to the original values of the ROI-focus and ROI-cyto photobleaching traces (using NonlinearModelFit in Mathematica) to determine the initial intensity at the origin of each trace. The initial ROI-cyto value was subtracted from the initial ROI-focus value to give the I-focus value, which was then divided by the calculated unitary step size to give the number of steps (molecules) per focus. This procedure was repeated for 45 traces.
For further description and discussion of stepwise photobleaching techniques, see Text S1 in the supplemental material.
Expression and localization of Pol IV and RecA after the induction of DNA double-strand breaks.
Strains carrying the lacO array and the I-SceI recognition site on the chromosome, plasmid pPFB1035 (carrying the DNA encoding the I-SceI endonuclease under the tet promoter and the LacI-ECFP fusion protein under the arabinose promoter), and plasmids carrying the DNA encoding fusion proteins were grown overnight in LB broth plus appropriate antibiotics. The cultures were diluted 1:1,000 in the same medium and grown with aeration at 37°C, or 30°C if a strain carried the recA4155-gfp allele. At an OD600 of 0.1, 0.02% l-arabinose was added to induce LacI-ECFP. At an OD600 of 0.2, 400 μg/ml AHT was added to induce I-SceI endonuclease. The cultures were incubated for an additional 90 min, and 1-ml aliquots were centrifuged, the cells were washed 2 times with 1× PBS and resuspended in 100 μl 1× PBS. A 3-μl aliquot of the cell suspension was spotted on a glass slide and immobilized with a poly-l-lysine-treated coverslip for microscopy.
Statistical analyses.
Standard statistical methods were used. Calculation of the expected values and variances of the ratios of variables was as previously described (59).
RESULTS
Activities of fluorescently tagged Pol IV fusions in vivo.
To follow the localization of Pol IV in vivo, we created three fluorescent DinB fusions expressed under the control of the native dinB promoter. First, EYFP was fused to the C terminus of DinB with a 12-amino-acid linker between the proteins and 100 bp upstream of the dinB start codon as the promoter (DinB-12L-EYFP). A second construct fused mCherry to the N terminus of DinB with a 10-amino-acid linker and the same 100 bp upstream of the dinB start codon as the promoter (mCh-DinB). Finally, the DinB-EYFP fusion was modified by increasing the linker to 20 amino acids and using a 450-bp region upstream of the dinB start codon as the promoter (DinB-20L-EYFP). All three constructs were expressed from the low-copy-number plasmid pWSK29 (41).
Because Pol IV efficiently bypasses the N2-dG adducts produced by NQO, Pol IV's translesion activity can be assayed by sensitivity to NQO (8, 50). As shown in Fig. 1A, all three tagged Pol IV proteins on pWSK29 complement the NQO sensitivity of a ΔdinB mutant and thus are proficient in translesion activity; however, the mCh-10L-DinB fusion appeared somewhat less active (Fig. 1A).
FIG 1.

In vivo activity and expression of fluorescence-tagged Pol IV. (A) NQO sensitivities of strains carrying fluorescence-tagged Pol IV. For each strain, 10-μl aliquots of 10−1 to 10−6 dilutions of saturated cultures were spotted on an LB agar plate containing NQO; the 10−6 dilutions spotted on a plate containing no NQO are shown at the right. WT (wild type), FC40; ΔdinB, PFB243; ΔdinB/pDinB-12L-EYFP, PFB243/pPFB913; ΔdinB/pDinBE104A-12L-EYFP, PFB243/pPFB1173; ΔdinB/pmCh-DinB, PFB243/pPFV407; WT/VC (vector control), FC40/pPFB1179; ΔdinB/VC, PFB243/pPFB1179; ΔdinB/pDinB+, PFB243/pPYG768; ΔdinB/pDinB-20L-EYFP, PFB243/pPFB1188. (B) Growth-dependent mutation rates of strains carrying fluorescence-tagged Pol IV. Shown are mutation rates to Tcr ± 95% confidence intervals as determined by fluctuation tests in two experiments with 40 cultures per strain (see Materials and Methods). WT, FC1373; ΔdinB, PFB1284; ΔdinB/pDinB+, PFB1284/pPYG768; ΔdinB/pDinB-20L-EYFP, PFB1284/pPFB1188; ΔdinB/pDinBE104A-12L-EYFP, PFB1284/pPFB1173; ΔdinB/pmCh-10L-DinB, PFB1284/pPFV407. (C) Adaptive-mutation activity of DinB-20L-EYFP+. Shown are the mean cumulative numbers of Lac+ colonies appearing on days 3 to 5 on lactose minimal medium during a small-scale adaptive-mutation assay (see Materials and Methods). Each bar is the mean of four cultures; the error bars are SEM. WT/VC, FC40/pPFB1179; ΔdinB/VC, B243/pPFB1179; ΔdinB/pDinB+, PFB243/pPYG768; ΔdinB/pDinB-20L-EYFP, PFB243/pPFB1188. (D) Expression of fluorescence-tagged Pol IV proteins compared to wild-type Pol IV. Shown are immunoblots of DinB+, DinB-20L-EYFP, and DinBE104A-12L-EYFP before and after induction with Nal. The blots were probed with anti-Pol IV antibody, stripped, and then reprobed with anti-GroEL antibody to show GroEL as a loading control. The band intensities were quantified and normalized to the GroEL bands. The numbers below the lanes give the induced levels of DinB+ and DinB fusion proteins relative to the uninduced levels. The strains used were as follows: DinB+, PFB243/pPYG768; VC, pFB243/pPFB1172; DinB-20L-EYFP, PFB243/pPFB1188; DinBE104A-12L-EYFP, PFB243/pPFB1173.
Overexpression of Pol IV in growing cells is a powerful mutator (18), which can be assayed by reversion to Tcr of a tetA allele with a +1 frameshift mutation (51). As shown in Fig. 1B, the mutation rate of a ΔdinB mutant strain carrying the mutant tetA allele on its chromosome and Pol IV alleles on pWSK29 was increased 9-fold by untagged Pol IV (pDinB+), 6-fold by DinB-20L-EYFP, and 3.5-fold by mCh-DinB. Thus, the EYFP-tagged Pol IV retains most of the activity and mCherry-tagged DinB retains partial activity for growth-dependent mutagenesis.
A third activity of Pol IV is promotion of stationary-phase, or “adaptive,” mutation, which is assayed by reversion of the Lac− strain, FC40, to Lac+ during lactose selection (15, 21, 60). As shown in Fig. 1C, the DinB-20L-EYFP fusion on pWSK29 exhibited partial adaptive-mutation activity, restoring 60% of the adaptive mutation to a ΔdinB mutant strain. However, DinB-12L-EYFP and mCh-10L-DinB did not complement the ΔdinB mutant strains for adaptive mutation (data not shown). Nonetheless, we found no consistent differences between the three DinB fusion proteins in the localization experiments described below.
We also created other tagged Pol IV proteins that had less activity, which are described in Text S2 in the supplemental material.
Fluorescent fusions to an active-site mutant of Pol IV are inactive.
Changing the glutamine at position 104 in Pol IV to arginine creates an inactive protein (12). We placed this mutation in the Pol IV-EYFP fusion, creating DinBE104A-12L-EYFP carried on pWSK29. This catalytically dead DinB fusion failed to complement ΔdinB mutants for NQO resistance (Fig. 1A), for the rate of mutation to Tcr (Fig. 1B), or for adaptive mutation (data not shown).
SOS-induced levels of Pol IV fusions are equivalent to those of wild-type Pol IV.
Immunoblot analysis showed that the basal levels of the Pol IV fusion proteins were about the same as those of wild-type Pol IV when all were expressed from pWSK29. As shown in Fig. 1D, after exposure to Nal, DinB+ and DinB-20L-EYFP were induced 10-fold and 12-fold, respectively, but the active-site mutant protein, DinBE104A-12L-EYFP, was induced only 4.5-fold, although this may underestimate the actual level of induction, as DinBE104A-12L-EYFP appeared to be more readily degraded than the other fusion proteins.
Pol IV localizes as foci in response to DNA damage.
We examined the localization of Pol IV fusion proteins after cells were exposed to DNA-damaging agents. No DinB-EYFP foci were observed in unexposed cells, but after exposure to 40 μg/ml Nal for 1 h, ∼80% of the cells had at least one Pol IV focus, and many had multiple foci. All the Pol IV foci were associated with the nucleoid (Fig. 2A). Similar results were obtained with NQO exposure and with cells expressing the mCh-DinB fusion (data not shown).
FIG 2.
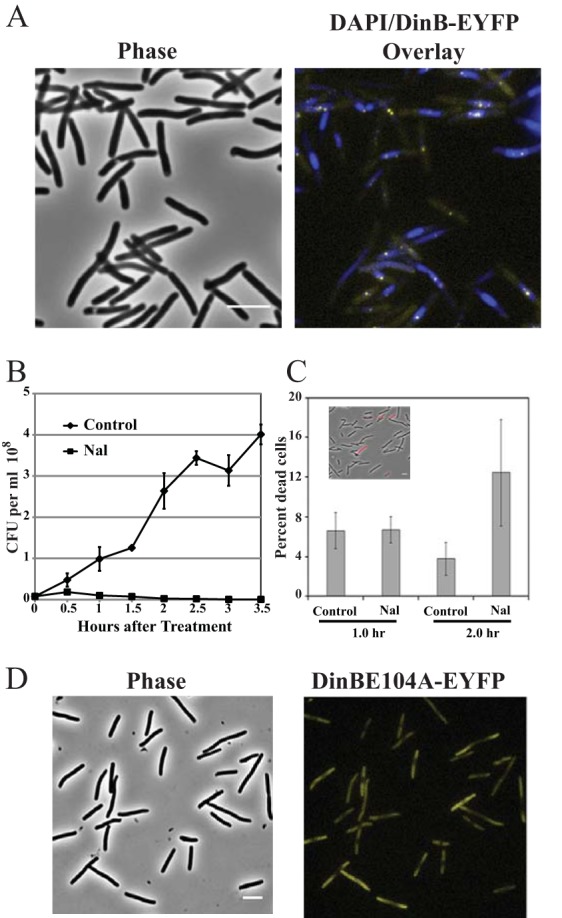
Localization of Pol IV and cell survival after Nal exposure. (A) Phase-contrast and phase plus fluorescence overlay images of DinB-12L-EYFP localization. Strain PFB243/pPFB913 was grown in LB broth plus antibiotics at 37°C to mid-exponential phase (OD600 = 0.5), treated with 40 μg/ml Nal, and incubated for a further 1 h. The cells were then stained with DAPI and visualized for DinB-EYFP foci. The DinB-EYFP (yellow) foci were associated with DAPI-stained nucleoids (blue). (B) Colony-forming ability of Nal-treated cells. Strain PFB243/pPFB913 was grown as described for panel A, and half the culture was treated with Nal. Samples were withdrawn from the cultures at the indicated time points and plated for CFU. Shown are the means ± SEM of 3 cultures (some error bars are smaller than the symbols). (C) Propidium iodide staining of Nal-treated cells to estimate cell viability based on membrane integrity. After exposure to Nal as for panel A, samples were washed and suspended at 10× in saline, stained with propidium iodide, and imaged. Shown are the mean percentages ± SEM of the cells stained with propidium iodide based on 2 or 3 fields examined with 125 to 710 cells counted per field; the inset shows a phase-contrast image overlaid with a fluorescence image (red). (D) Failure of an active-site mutant Pol IV (DinBE104A-EYFP) to form fluorescent foci. Strain PFB243/pPFB1173 was treated with Nal as for panel A. Shown are the phase-contrast and fluorescence images of DinBE104A-EYFP localization. Scale bars = 5 μm.
Nal interacts with DNA gyrase, capturing the DNA-gyrase complex after strand cleavage but before ligation, resulting in DSBs held together by gyrase complexes covalently linked to the 5′ ends of the cleaved DNA (61). These lesions block replication and, if not removed, lead to cell death. As shown in Fig. 2B, upon Nal addition, the number of CFU ceased to increase and then slowly declined (Fig. 2B). To determine if Nal-treated cells were actually dead, we exposed them to the nucleic-acid-binding fluorescent dye propidium iodide, which can penetrate only bacteria with damaged membranes. As shown in Fig. 2C, after 2 h of Nal treatment, 88% ± 5% of the cells were impervious to propidium iodide. Thus, over the period of our microscopy experiments, Nal-treated cells retained intact membranes and were presumably still alive, even though their capacity to reproduce was severely impaired.
Pol IV polymerase activity is required for localization after DNA damage.
Cells with the catalytically dead Pol IV fusion pDinBE104A-12L-EYFP showed no fluorescent foci after treatment with Nal (Fig. 2D). Thus, Pol IV polymerase activity is required for focus formation after Nal-induced DNA damage.
Determination of the number of Pol IV molecules present in Nal-induced foci.
To estimate the number of molecules of Pol IV that localize as foci in response to DNA damage, we subjected Nal-treated cells of a ΔdinB mutant strain carrying pDinB-20L-EYFP (PFB243/pPFB1188) to a stepwise photobleaching protocol (62). We photobleached entire cells in the imaging field using epifluorescence microscopy and measured the loss of intensity over 120 s. For analysis, intensity measurements were made in an ROI-focus and from a nearby ROI-cyto. We modified previously published methods to obtain the intensity drop that corresponds to the photobleaching of a single DinB-EYFP molecule (the unitary step size) (see Materials and Methods) (Fig. 3A to E; see Table S1 in the supplemental material). The initial intensity of the focus itself (I-focus) was obtained by subtracting the ROI-cyto from the ROI-focus. The I-focus was then divided by the calculated unitary step size to give the number of steps (molecules) per focus. This procedure was repeated for 45 traces, and the resulting values are given in Fig. 3F.
FIG 3.

Analysis of the numbers of DinB-EYFP molecules per fluorescent focus by photobleaching. Strain PFB243/pPFB1188 (ΔdinB/pDinB-20L-EYFP) was grown and exposed to Nal as for Fig. 2. The cells were then photobleached by exposure to 460- to 500-nm epifluorescent light. (A) Photobleaching trace for a 7- by 7-pixel area containing a DinB-EYFP focus. The x axis shows the number of frames of image capture, with each frame image taken after 300 ms of exposure. The y axis shows the arbitrary intensity values. The gray line shows the values after applying the Chung-Kennedy (C-K) filter (see Materials and Methods). (B) Histogram showing the distribution of PDDF values [ΔI(t) = I(ta) − I(tb) for all data pairs for which the time ta is greater than tb] for the trace shown in panel A. (C) Fourier transform of the PDDF bin values shown in panel B. The dominant peak indicates that the number of steps in the original trace was seven. (D) Histogram showing the distribution of initial focus intensities (the initial value of ROI-focus minus the initial value of ROI-cyto [see Materials and Methods and Table S1 in the supplemental material]) for the 45 photobleaching traces analyzed. Two normal curves were fitted to the data; normal fit 1 encompassed the lower 26 values with a mean of 2,562 and a standard deviation of 791 arbitrary intensity values, and normal fit 2 encompassed the higher 19 values with a mean of 6,556 and a standard deviation of 1,619 arbitrary intensity values. (E) Histogram showing the distribution of intensity drops corresponding to the unitary step size (photobleaching of one DinB-EYFP molecule) for the 45 traces analyzed (see Table S1 in the supplemental material). The normal-fit curve has a mean of 1,076 and a standard deviation of 263 arbitrary intensity values. (F) Histogram showing the distribution of the numbers of DinB-EYFP molecules per focus for the 45 foci analyzed (see Table S1 in the supplemental material). Two normal curves were fitted to the data; normal fit 1 encompassed the lower 26 values with a mean of 2.7 and a standard deviation of 0.8 DinB-EYFP molecules per focus, and normal fit 2 encompassed the higher 19 values with a mean of 5.5 and a standard deviation of 0.7 DinB-EYFP molecules per focus.
The calculated unitary step size had a 3.2-fold range (Fig. 3E; see Table S1 in the supplemental material), which is an improvement over ranges that have been reported in previous studies (56, 57). The focus intensity varied 8-fold (Fig. 3D; see Table S1 in the supplemental material) and was highly correlated with the computed numbers of molecules per focus (Pearson's coefficient = 0.88; P < 0.0001). As shown in Fig. 3F, the computed numbers of molecules per focus had a bimodal distribution, with the lowest 26 values having a mean of 2.7 and a mode of 3 and the highest 19 values having a mean of 5.5 and a mode of 6. These results suggest that about 60% of the foci have 3 Pol IV molecules and 40% have twice as many.
Pol IV and Rep colocalize after DNA damage.
We previously reported that Pol IV and the replicative helicase, Rep, interact physically and functionally in vitro (32). Thus, during DNA repair in vivo, Rep and Pol IV could colocalize to the sites of DNA lesions. To test this hypothesis, we constructed a fusion of mCherry to the C terminus of Rep. This fusion protein (Rep-mCh) restored viability to a rep uvrD double-mutant strain, which is normally nonviable (63), thus demonstrating that Rep-mCh retains Rep activity (data not shown). When Rep-mCh was expressed in a ΔdinB mutant strain, fluorescent foci were observed in only 0.1 to 0.2% of the cells whether or not they were exposed to Nal (see Fig. S1 in the supplemental material). However, Rep-mCh foci appeared when both DinB-EYFP and Rep-mCh were present in Nal-exposed cells of the ΔdinB mutant strain, and formation of Pol IV foci preceded that of Rep foci (Fig. 4). An hour after Nal exposure, 80% ± 6% of the cells had DinB-EYFP foci, 2% ± 0.1% of the cells had Rep-mCh foci, and all of the Rep foci colocalized with Pol IV. After 2 h, the number of cells with Rep-mCh foci had increased to 22% ± 2%, and again, nearly all (103/104) Rep foci colocalized with Pol IV (means ± standard errors of the mean [SEM] of two experiments, with ∼200 cells counted at each time point). We conclude that Rep colocalizes with Pol IV, most likely to sites of DNA damage, and that Pol IV is required for the recruitment of Rep to these sites. These results support the hypothesis that the interaction between Rep and Pol IV that was observed in vitro (32) also takes place in vivo.
FIG 4.

Colocalization of Rep with Pol IV after Nal-induced DNA damage. (A) Phase-contrast, fluorescence, and overlay images of DinB-12L-EYFP (yellow) and Rep-mCh (red) focus formation and colocalization with time after Nal exposure. Scale bar = 5 μm. Arrows indicate representative colocalized foci. (B) Percentages of cells containing DinB-EYFP or Rep-mCh foci and percentages of Rep-mCh foci colocalizing with DinB-EYFP foci with time after Nal exposure. Shown are means ± SEM based on ∼200 cells (range = 186 to 274) counted at each time point in two independent experiments. Strain FC40 carrying plasmids pPFB913 and pPFB914 was grown in LB broth plus antibiotics at 37°C until mid-exponential phase (OD600 = 0.5), treated with 40 μg/ml Nal, and incubated for a further 2 h. Samples were withdrawn at 30-min intervals and visualized.
Pol IV colocalizes with RecA foci following DNA damage.
RecA, the E. coli recombinase, plays key roles in homologous recombination, DNA repair, and induction of the SOS response (64, 65). To visualize RecA localization after DNA damage, we replaced the chromosomal recA gene with the recA4155-gfp901 allele. This allele encodes a GFP fusion to the C terminus of RecA and has an R28A mutation in recA (48). Unlike the GFP fusion to wild-type RecA, RecAR28A-GFP does not form DNA-less aggregates but retains 10% to 30% of normal RecA activity and is competent for recombination, DNA repair, and SOS induction (48). We studied the induction and localization of RecAR28A-GFP, here designated RecA-GFP, after exposing the cells to Nal or NQO. To minimize toxicity due to expression of the GFP fusion protein, experiments were done at 30°C. In agreement with previous reports (48), in the absence of DNA damage, most cells showed uniform fluorescence, but 10% ± 1% of the cells had 1 or 2 fluorescent foci (mean ± SEM of three experiments, with 323, 342, and 131 cells counted). For 30 to 60 min after Nal or NQO addition, RecA-GFP appeared as filamentous threads throughout the cells; these then slowly progressed to form RecA-GFP foci (Fig. 5A). Within 60 to 180 min, almost all of the cells showed multiple RecA-GFP foci localized to the nucleoid regions (Fig. 5A).
FIG 5.

Colocalization of Pol IV with RecA foci after Nal and NQO exposure. (A) Merged phase-contrast and fluorescence images showing RecA-GFP (green) expression after exposure to Nal and NQO. Cells of strain PFB1137 (recA4155-gfp901) were grown in LB broth at 30°C until mid-exponential phase (OD600 = 0.50), treated with 40 μg Nal or 20 μM NQO, and incubated for a further 3 h. Samples were withdrawn every 30 min and visualized. (B) Percentages of cells containing RecA-GFP and mCh-10L-DinB foci and percentages of cells with mCh-10L-DinB foci colocalizing with RecA-GFP foci after 120 min exposure to Nal or NQO. Shown are means ± SEM. The Nal results are from four independent experiments with three different strains carrying recA4115-gfp901 and pPFV407: PFB1137 (ygaD1::Kn), PFB1220 (ygaD+), and PFB1224 (I-SceI lacZo::Kn) (two experiments); the total numbers of cells examined were 65, 130, 66, and 87, respectively. The NQO results are from two independent experiments, one with PFB1137/pPFV407 and one with PFB1224/pPFV407; the total numbers of cells examined were 141 and 60, respectively. The error bars on the last bar are 0. (C) Phase-contrast, fluorescence, and overlay images of RecA-GFP (green) and mCh-10L-DinB (red) foci and colocalization after Nal exposure for 2 h. Scale bars = 5 μm. Arrows indicate representative colocalized foci.
With both mCh-DinB and RecA-GFP present in the cells, for 30 to 60 min after Nal addition, the RecA-GFP threads appeared with no mCh-DinB foci. After 120 min of exposure, 97% ± 3% of the cells had discrete RecA-GFP foci and 41% ± 10% had mCh-DinB foci, and in 96% ± 3% of the cells with DinB foci, the foci colocalized with RecA (means ± SEM of 4 experiments) (Fig. 5B and C). We observed similar numbers of RecA- and mCh-DinB-colocalized foci after NQO exposure (Fig. 5B).
We did the same experiment with a ΔdinB mutant strain exposed to Nal and to NQO. Although RecA-GFP foci formed in the majority of the cells, we failed to observe consistent mCh-DinB focus formation and colocalization. The mCh-DinB fusion retains only partial Pol IV activity and is less active than the DinB-EYFP fusion proteins (Fig. 1). Thus, it appears that mCh-DinB alone is not proficient in colocalizing with RecA but requires the presence of wild-type Pol IV to do so.
The catalytically dead Pol IV fusion protein did not colocalize with RecA after Nal exposure. After 120 min of Nal exposure of strain PFB1137/pPFB1162 (RecA-GFP/pmCh-DinBE104A), 100% of the cells were filamentous and 85% of them had RecA-GFP foci; however, only 4 of the 1,392 cells examined and only 3 of the 1,180 filamentous cells had mCh-DinBE104A foci, and these foci were not colocalized with RecA.
Because the RecA-GFP foci are so intense, it is possible that the RecA signal could bleed into the red channel, giving false colocalization images. However, we did not observe RecA-GFP foci with the Texas Red filter cube used to visualize the mCherry signal (see Fig. S2 in the supplemental material).
Pol IV localizes to sites of double-strand breaks in a subpopulation of cells.
To study the localization of Pol IV when DSBs occur, we introduced a double-strand cut site at a specific, marked location on the chromosome. An array of 250 lacO sequences (44) and a single recognition site for the I-SceI endonuclease 161 kb from the lacO array were inserted into the chromosome of our parental strain. The strain was transformed with plasmid pPFB1035 carrying the gene encoding the I-SceI endonuclease under the control of the tet promoter (inducible by the tetracycline analog AHT) (46) and a gene encoding LacI-ECFP (44) under the control of an arabinose-inducible promoter. Upon the addition of arabinose, the induced LacI-ECFP molecules bind to the lacO array, fluorescently labeling the double-strand break region. This strain was then transformed with the plasmids carrying the genes encoding fluorescently labeled DinB.
We first studied a dinB+ strain carrying the plasmid-borne DinB-12L-EYFP fusion protein. Within 60 to 90 min after I-SceI endonuclease induction, half (52% ± 3%) of the cells were filamentous, indicating that they were responding to a DSB by inducing the SOS response, and 90% ± 12% of the filamentous cells had 1 to 5 LacI foci (Fig. 6A). The presence of multiple LacI foci probably indicated that the filamentous cells had multiple chromosomes. About half of the filamentous cells had 1 to 4 DinB foci (Fig. 6A), and in the majority (62% ± 13%) of these cells, the DinB foci were colocalized with LacI (Fig. 6B). DinB colocalized with LacI in very few (3% ± 3%) nonfilamentous cells. In cells with multiple DinB foci, only 1 or 2 were colocalized with LacI, while the rest were located elsewhere (Fig. 6C).
FIG 6.

Pol IV localization after double-strand break induction in exponentially growing cells. (A) Phase-contrast, fluorescence, and overlay images of LacI-ECFP and DinB-EYFP foci in cells carrying an I-SceI recognition site 161 kb from the lacO array. The cells were grown in LB broth at 37°C to early exponential phase (OD600, ∼0.1), induced for LacI-ECFP and I-SceI endonuclease, incubated for an additional 1.5 h, and then visualized (see Materials and Methods). Scale bar = 5 μm. Arrows indicate representative colocalized foci. (B) Percentages of cells with LacI-ECFP and DinB-EYFP foci. Shown are means ± SEM. (C) Percentages of cells with DinB-EYFP foci colocalizing with LacI-ECFP. Shown are means ± SEM. WT, PFB1081 carrying plasmids pPFB1035 and pPFB913 (DinB-12L-EYFP); the data are from three independent experiments with 108, 160, and 101 cells observed. ΔdinB, PFB1103 carrying plasmids pPFB1035 and pPFB913 (DinB-12l-EYFP); the data are from three independent experiments with 101, 85, and 69 cells observed. ΔumuDC, PFB1195 carrying plasmids pPFB1035 and pPFB1188 (DinB-20L-EYFP); the data are from two independent experiments with 198 and 207 cells observed. ΔdinB ΔumuDC, PFB1201 carrying plasmids pPFB1035 and pPFB1188 (DinB-20L-EYFP); the data are from three independent experiments with 146, 230, and 189 cells observed.
Not all the LacI and DinB foci appeared to be coincident: a variable number were 0.2 to 0.5 μm apart. These adjacent foci are included in the colocalization results; the fraction that they represented and our attempts to determine why not all foci were coincident are described in the supplemental material (see Text S3 and Table S2 in the supplemental material).
Wild-type Pol IV may interfere with the localization of the Pol IV fusion protein to DSBs.
To determine if wild-type Pol IV affected the localization of the Pol IV fusion proteins, we repeated the experiments described above with a ΔdinB mutant strain. As described above, after induction of LacI-ECFP and I-SceI endonuclease, the majority (68% ± 11%) of the cells were filamentous and ∼90% of the filamentous cells had LacI foci (Fig. 6A). Over half (59% ± 11%) of the filamentous cells had DinB foci, and in most of these (81% ± 20%), the DinB foci colocalized with LacI (Fig. 6A and B). Again, DinB and LacI foci were colocalized in only a few nonfilamentous cells (12% ± 5%). Thus, in the absence of wild-type Pol IV, the number of filamentous cells with DinB foci localizing to DSBs increased by only 20%, suggesting that wild-type Pol IV interferes slightly, if at all, with DinB-EYFP localization to DSBs.
Pol V appears to have no effect on Pol IV localization.
E. coli has two SOS-inducible error-prone DNA polymerases, Pol IV and Pol V (encoded by the umuDC genes). To test if Pol V affects localization of Pol IV to DSBs, we studied DinB-20L-EYFP colocalization with LacI in an umuDC mutant strain (Fig. 6A and B). As before, after induction of LacI and I-SceI endonuclease, most of the cells (80% ± 3%) were filamentous, most (81% ± 5%) of the filamentous cells had LacI foci, a majority (56% ± 3%) of the filamentous cells had DinB foci, and in 63% ± 8% of those cells, the DinB foci were colocalized with LacI (Fig. 6A and B). The umuDC dinB double-mutant strain behaved similarly (Fig. 6A and B). Thus, loss of Pol V did not interfere with the localization of Pol IV to DSBs in dinB+ cells and, in fact, appeared to decrease it slightly in ΔdinB mutant cells. We also found no effect of Pol V on Pol IV focus formation after exposure to Nal or NQO (see Fig. S3 in the supplemental material).
Overall, the results indicate that fluorescently tagged Pol IV is expressed upon induction of DSBs in cells and, in a subpopulation of cells, is recruited to DSBs. Wild-type Pol IV may interfere slightly with localization of the Pol IV fusion protein to DSBs, but Pol V does not.
Pol IV localizes to DSBs in stationary-phase cells.
Pol IV is upregulated in late-stationary-phase cells under the control of the general stress response sigma factor RpoS (13, 49). To test if localization of Pol IV to DSBs was altered under these conditions, we induced DSBs in stationary-phase cells. For these experiments, we used a dinB mutant strain carrying DinB-20L-EYFP. Unlike exponential-phase cells, the stationary-phase cells did not become filamentous after I-SceI endonuclease induction, consistent with the fact that they were not actively dividing. After LacI-ECFP and I-SceI induction, less than half (43% ± 12%) of the cells had LacI foci, only about a third (28% ± 7%) had DinB foci (Fig. 7A), and in only about a third (30% ± 7%) of these was DinB colocalized with LacI (Fig. 7B and C). We obtained similar results with a ΔdinB ΔumuDC strain. When both LacI and I-SceI were induced, 56% ± 10% of the cells had LacI foci, 35% ± 5% of the cells had DinB foci, and in 43% ± 6% of the cells with DinB foci, DinB colocalized with LacI. Thus, induction of DSBs in stationary-phase cells resulted in a weaker response than in exponentially growing cells, with about half the number of Pol IV foci localizing to DSBs.
FIG 7.

Pol IV focus localization after double-strand break induction in stationary-phase cells. (A) Phase-contrast, fluorescence, and overlay images of LacI-ECFP and DinB-EYFP foci following DSB induction in stationary-phase cells. The cells were grown in LB broth at 30°C until early stationary phase (OD600, ∼1.0), induced for LacI-ECFP and I-SceI endonuclease, incubated for another 2.5 h, and then visualized (see Materials and Methods). Scale bar = 5 μm. Arrows indicate representative colocalized foci. (B) Percentages of cells with LacI and DinB foci in stationary-phase cells with and without induction of LacI-ECFP and I-SceI endonuclease from the experiment described in the legend to panel A. Shown are means ± SEM. (C) Percentages of cells with DinB foci colocalizing with LacI. Shown are means ± SEM. ΔdinB, PFB1271 carrying plasmids pPFB1035 and pPFB1188 (DinB-20L-EYFP); the control data are from two independent experiments with 226 and 320 cells observed; the Ara plus AHT data are from three independent experiments with 405, 430, and 394 cells observed. ΔdinB ΔumuDC, PFB1279 carrying plasmids pPFB1035 and pPFB1188 (DinB-20L-EYFP); the control data are from two independent experiments with 178 and 244 cells observed; the Ara plus AHT data are from three independent experiments with 272, 318, and 499 cells observed.
Pol IV colocalizes with RecA after DSB induction.
In E. coli, DNA double-strand breaks are repaired by homologous recombination via the RecA-RecBCD pathway (66). It is hypothesized that error-prone DNA synthesis by Pol IV during DSB repair is the source of adaptive mutations (67). Thus, we examined whether Pol IV localizes with RecA after DSB induction. We first used a strain with the chromosomal RecA-GFP fusion (see above) and the I-SceI recognition site, carrying plasmids with the I-SceI endonuclease and mCh-DinB. Without DSB induction, 40% ± 6% of the cells had RecA foci and 7% ± 2% had DinB foci; in 11% ± 1% of the cells with DinB foci, DinB colocalized with RecA (means ± SEM from three independent experiments, with 99, 126, and 821 cells examined). We assume these localizations to be at sites of spontaneous DNA damage or DSBs induced by low-level expression of I-SceI. After I-SceI endonuclease induction, for the first 30 to 45 min, RecA-GFP appeared as filaments, as was observed after exposure to Nal and NQO. By 90 min after I-SceI endonuclease induction, 43% ± 6% of the cells were filamentous and, of these, 98% ± 8% had RecA-GFP foci; 71% ± 10% of the filamentous cells had DinB foci, and in 83% ± 7% of these cells, the DinB foci colocalized with RecA (Fig. 8). These results support the hypothesis that Pol IV participates with RecA during DSB repair. In a ΔdinB strain, induction of DSBs also resulted in RecA-GFP foci, but we failed to see consistent mCh-DinB foci (data not shown); we hypothesize that the partial functionality of mCh-DinB precludes it from interacting with RecA without the presence of wild-type Pol IV.
FIG 8.

RecA-GFP and mCh-DinB colocalization after double-strand break induction. (A) Phase-contrast, fluorescence, and overlay images of RecA-GFP and mCh-DinB focus localization after I-SceI endonuclease induction. Strain PFB1137 carrying plasmids pPFB1035 and pPFV407 was grown at 30°C until mid-exponential phase (OD600 = 0.2), induced for I-SceI endonuclease, incubated another 1.5 h, and visualized (see Materials and Methods). Scale bar = 5 μm. Arrows indicate representative colocalized foci. (B) Distribution of RecA-GFP, mCh-DinB, and colocalizing foci in cells after DSB induction in the experiment described in the legend to panel A. Shown are means ± SEM from three independent experiments with 144, 161, and 156 cells observed.
After replacing ygaD1::Kn with ygaD+, we were able to place both RecA-GFP and the lacO array in the same strain with the plasmid-borne mCh-DinB. Ninety minutes after induction of LacI and I-SceI endonuclease, 49% ± 1% of the cells were filamentous; of these, 81% ± 10% had LacI foci and 59% ± 7% had RecA foci, and in 67% ± 3% of the cells with RecA foci, RecA colocalized with LacI (means ± SEM of 2 experiments, with 167 and 133 cells observed). Of the filamentous cells, 36% ± 4% had DinB foci, and in 90% ± 10% of these cells, DinB colocalized with both RecA and LacI (Fig. 9). As observed above, no mCh-DinB foci were detected after induction of LacI and I-SceI endonuclease in a dinB mutant strain (data not shown), again suggesting that the formation of mCh-DinB foci requires the presence of wild-type Pol IV. Taken together, these results strongly suggest that Pol IV participates with RecA and is recruited to sites of double-strand breaks during repair.
FIG 9.
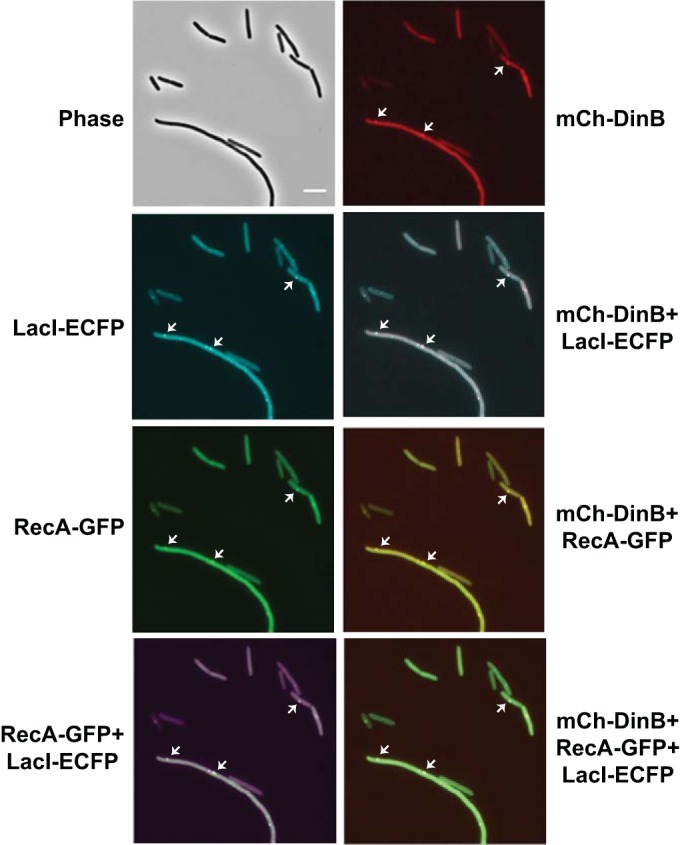
LacI-ECFP, RecA-GFP, and mCh-DinB localization after double-strand break induction. The phase-contrast and fluorescence images show mCh-DinB (red), LacI-ECFP (cyan), and RecA-GFP (green) focus localizations after induction of LacI-DCFP and I-SceI endonuclease. The overlay images show false-colored focus colocalizations; mCh-DinB (red) and LacI-ECFP (cyan); mCh-DinB (red) and RecA-GFP (green); RecA-GFP (green) and LacI-ECFP (magenta); and mCh-DinB (red), RecA-GFP (green), and LacI-ECFP (cyan). Strain PFB1224 carrying plasmids pPFB1035 and pPFV407 was grown in LB broth at 30°C to an OD600 of 0.2, induced for LacI-ECFP and I-SceI endonuclease, incubated for an additional 1.5 h, and visualized. Scale bar = 5 μm. Arrows indicate representative colocalized foci.
DISCUSSION
In the present study, we demonstrated that E. coli DNA Pol IV localizes to the nucleoid as foci in response to DNA damage. Using stepwise photobleaching, we estimate that these foci contain 3 or 6 molecules of Pol IV. After focus formation, Pol IV recruits its interacting partner, Rep helicase. Pol IV also colocalizes with RecA after DNA damage and, specifically, to sites of DNA double-strand breaks. These findings support the hypotheses that Pol IV participates in translesion DNA synthesis, that it is aided at some lesions by the Rep helicase, and that Pol IV also participates with RecA in reestablishing normal replication after the replication fork is blocked or collapses.
Pol IV readily formed foci in SOS-induced cells after exposure to Nal (Fig. 2A). The foci were associated with the nucleoids, suggesting that Pol IV localizes to the Nal-induced DNA lesions. Since an active-site mutant of Pol IV, DinBE104A, did not form foci after DNA damage, Pol IV polymerization activity apparently is required for focus formation. In addition to its translesion synthesis activity, Pol IV is proposed to aid in restarting stalled and collapsed replication forks (68, 69). As mentioned above, by interacting with DNA gyrase and trapping the DNA-gyrase complex after strand cleavage but before ligation, Nal results in double-strand breaks held together by gyrase complexes covalently linked to the 5′ ends of the cleaved DNA; these lesions themselves block replication and also progress to true DSBs (61, 70). Our results indicate that Pol IV accumulates at these sites where DNA forks are stalled and/or at the resulting DSBs.
Previously, we reported that the interaction of Pol IV and Rep helicase stimulates both the polymerase activity of Pol IV and the helicase activity of Rep in vitro (32). Here, we demonstrated that Rep colocalizes with Pol IV foci in response to DNA damage and that Pol IV recruits Rep to these sites (Fig. 4), suggesting that Rep and Pol IV interact in vivo to process DNA damage. However, only 20% of the Pol IV foci had colocalizing Rep foci (Fig. 4B), indicating that not all DNA lesions are processed in this way. We hypothesize that when the replication fork stalls at a site of DNA damage, Pol IV recruits Rep and stimulates its helicase activity to remove the DNA obstructions near the replication fork junction. DNA synthesis by Pol IV then extends the junction and allows the replicative helicase, DnaB, to load for the reassembly of the replication complex.
Given Rep's known ability to remove Okazaki fragments (36), an alternative model is that a complex of Rep, PriC, and Pol IV allows bypassing of lesions on the leading strand via fork regression. Rep would facilitate this process by unwinding the nascent DNA from the lagging-strand template and providing it as a template for Pol IV DNA synthesis, which, when the fork was reestablished, would extend past the lesion. The reestablishment of the fork and resolution of the resulting Holiday junctions would then be accomplished by the RecA-RuvABC pathway. Assuming that adaptive mutations are due to errors made by Pol IV during this synthesis, this alternative model reconciles the requirements for Pol IV, Rep, and recombination functions for maximum levels of adaptive mutation (32).
Because Pol IV is postulated to participate with RecA in DSB repair, we studied RecA-GFP localization after DNA damage. Following DNA damage, RecA-GFP appeared as filaments throughout the cells that progressed to distinct foci over the course of an hour (Fig. 5A). We assume that these filaments are the same as the RecA-GFP “threads” observed by others after UV exposure and during DSB repair (71–73). These threads have been hypothesized to be either the RecA/single-stranded DNA (ssDNA) nucleoprotein filaments that initiate recombination and/or RecA bound to ssDNA gaps left when replication reinitiates downstream of a DNA lesion (71–73).
Pol IV colocalized with RecA after the RecA filaments coalesced into foci (Fig. 5B and C). Although these results do not prove a direct physical interaction between RecA and Pol IV, they support previous reports that RecA and Pol IV form a complex in vitro (30). Indeed, in vitro evidence suggests that RecA is a switch at the replication fork, inhibiting replication by replisomes containing Pol III and activating replisomes containing Pol II, IV, or V (74). Interestingly, mCh-DinB foci did not colocalize with RecA-GFP in a ΔdinB strain but did when wild-type Pol IV was present in the cells. Since mCh-DinB is less active than the DinB-EYFP fusions (Fig. 1), this result suggests that full Pol IV polymerase activity is required for focus formation, a conclusion supported by the fact that active-site mutants of Pol IV also did not form foci.
By inducing DSBs at a specific, labeled chromosome site, we further demonstrated that Pol IV colocalizes with RecA at the sites of DSBs. RecA-GFP formed foci at the DSB site in nearly 100% of the SOS-induced cells (Fig. 8A). Since RecA processes DSBs, we assume, as have others (48, 72), that the RecA-GFP foci are actually at the DSBs. During normal cell growth, DSBs are common and, if unrepaired, are lethal. Estimates of the number of spontaneous DSBs per generation in a growing E. coli cell range from 0.01 to 0.3 (75, 76). As mentioned above, most DSBs are repaired by the RecA-RecBCD homologous-recombination pathway, followed by recruitment of primasome proteins to restart replication (76). Both DNA Pol II and IV have both been implicated in the restart mechanism (69, 76, 77), but the high levels of Pol IV in stressed cells allow it to displace Pol II (69, 78). Nonetheless, since 10 to 20% of the RecA foci were not associated with Pol IV, at some DSB repair sites Pol II apparently is recruited to restart DNA synthesis and repair.
We attempted to determine the number of Pol IV molecules that are present in DNA damage-induced foci by stepwise photobleaching (56, 57). Although there is clearly noise in the data, given the limitations of our methods the results indicate a bimodal distribution with 60% of the foci comprised of 3 and 40% of the foci comprise of 6 Pol IV molecules (Fig. 3E). Using similar techniques, the Pol III replisome was found to contain three Pol III molecules (79). One interpretation of our results is that Pol IV displaces all three Pol III molecules in the replisome during lesion repair or bypass, and the more intense foci may be at sites where two replication forks have encountered a lesion. Alternatively, the foci may be a cloud of Pol IV molecules that are exchanging with molecules bound to the beta-clamp or to the DNA, and the number of molecules may be not of biological significance.
We constructed both C-terminal (DinB-12L-EYFP and DinB-20L-EYFP) and N-terminal (mCh-10L-DinB) fluorescent fusions to Pol IV. All three of the Pol IV fusions retained translesion synthesis activity and showed varying degrees of mutagenesis activity in growing cells, but only the DinB-20L-EYFP construct was proficient in adaptive mutation (Fig. 1A to D). These results support the hypothesis that these three Pol IV-dependent phenotypes are mechanistically and genetically distinct (30, 80, 81). Furthermore, our results predict that fusions to the C terminus of Pol IV with long linkers are more likely to retain full functionality than fusions to the N terminus of Pol IV with short linkers. We note, however, that an N-terminal STREP-FLAG-Pol IV construct that we used in a previous study is fully active for adaptive mutation when expressed from a high-copy-number plasmid (32), and so, the more weakly active fluorescent fusions may also be fully competent if expressed at higher levels than in the experiments reported here.
Supplementary Material
ACKNOWLEDGMENTS
We thank D. J. Sherratt, V. Sourjik, S. J. Sandlers, D. B. Kearns, B. Michel, J. H. Miller, and the National BioResource Project (Japan) for bacterial strains and plasmids and H. Ohmori for reagents. We are grateful to D. B. Kearns for the use of his microscope and to D. T. Kysela, H. C. Tsui, J. A. Powers, and J. M. Murray for technical advice and assistance. We also thank the Indiana University Bloomington Light Microscopy Imaging Center and the IUB Center for Genomics and Bioinformatics. We thank the past and present members of our laboratory for their encouragement, advice, and technical help. We are particularly grateful to former laboratory member T. E. Sladewski, who initiated this project and performed some of the preliminary experiments and constructions. Finally, we thank the reviewers of this paper for helpful suggestions.
This work was supported by USPHS NIH grant GM065175 to P.L.F. and by a grant from the Indiana University Metabolomics and Cytomics Initiative (METACyt) program, which was financed, in part, by a major endowment from the Lilly Foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00101-15.
REFERENCES
- 1.Janion C. 2008. Inducible SOS response system of DNA repair and mutagenesis in Escherichia coli. Int J Biol Sci 4:338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al Mamun AA, Humayun MZ. 2006. Escherichia coli DNA polymerase II can efficiently bypass 3,N(4)-ethenocytosine lesions in vitro and in vivo. Mutat Res 593:164–176. doi: 10.1016/j.mrfmmm.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 3.Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. 2000. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J 19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rangarajan S, Woodgate R, Goodman MF. 1999. A phenotype for enigmatic DNA polymerase II: a pivotal role for pol II in replication restart in UV-irradiated Escherichia coli. Proc Natl Acad Sci U S A 96:9224–9229. doi: 10.1073/pnas.96.16.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman MF, Woodgate R. 2013. Translesion DNA polymerases. Cold Spring Harb Perspect Biol 5:a010363. doi: 10.1101/cshperspect.a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuchs RP, Fujii S. 2013. Translesion DNA synthesis and mutagenesis in prokaryotes. Cold Spring Harb Perspect Biol 5:a012682. doi: 10.1101/cshperspect.a012682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yuan B, Cao H, Jiang Y, Hong H, Wang Y. 2008. Efficient and accurate bypass of N2-(1-carboxyethyl)-2′-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc Natl Acad Sci U S A 105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jarosz DF, Godoy VG, Delaney JC, Essigmann JM, Walker GC. 2006. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature 439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 9.Kim SR, Matsui K, Yamada M, Gruz P, Nohmi T. 2001. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics 266:207–215. doi: 10.1007/s004380100541. [DOI] [PubMed] [Google Scholar]
- 10.Furukohri A, Goodman MF, Maki H. 2008. A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J Biol Chem 283:11260–11269. doi: 10.1074/jbc.M709689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikeda M, Furukohri A, Philippin G, Loechler E, Akiyama MT, Katayama T, Fuchs RP, Maki H. 2014. DNA polymerase IV mediates efficient and quick recovery of replication forks stalled at N2-dG adducts. Nucleic Acids Res 42:8461–8472. doi: 10.1093/nar/gku547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner J, Gruz P, Kim SR, Yamada M, Matsui K, Fuchs RP, Nohmi T. 1999. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol Cell 4:281–286. doi: 10.1016/S1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 13.Layton JC, Foster PL. 2003. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol 50:549–561. doi: 10.1046/j.1365-2958.2003.03704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuban W, Jonczyk P, Gawel D, Malanowska K, Schaaper RM, Fijalkowska IJ. 2004. Role of Escherichia coli DNA polymerase IV in in vivo replication fidelity. J Bacteriol 186:4802–4807. doi: 10.1128/JB.186.14.4802-4807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenzie GJ, Lee PL, Lombardo MJ, Hastings PJ, Rosenberg SM. 2001. SOS mutator DNA polymerase IV functions in adaptive mutation and not adaptive amplification. Mol Cell 7:571–579. doi: 10.1016/S1097-2765(01)00204-0. [DOI] [PubMed] [Google Scholar]
- 16.Strauss BS, Roberts R, Francis L, Pouryazdanparast P. 2000. Role of the dinB gene product in spontaneous mutation in Escherichia coli with an impaired replicative polymerase. J Bacteriol 182:6742–6750. doi: 10.1128/JB.182.23.6742-6750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolff E, Kim M, Hu K, Yang H, Miller JH. 2004. Polymerases leave fingerprints: analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J Bacteriol 186:2900–2905. doi: 10.1128/JB.186.9.2900-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim SR, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, Nohmi T, Ohmori H. 1997. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc Natl Acad Sci U S A 94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stumpf JD, Poteete AR, Foster PL. 2007. Amplification of lac cannot account for adaptive mutation to Lac+ in Escherichia coli. J Bacteriol 189:2291–2299. doi: 10.1128/JB.01706-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tompkins JD, Nelson JL, Hazel JC, Leugers SL, Stumpf JD, Foster PL. 2003. Error-prone polymerase, DNA polymerase IV, is responsible for transient hypermutation during adaptive mutation in Escherichia coli. J Bacteriol 185:3469–3472. doi: 10.1128/JB.185.11.3469-3472.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster PL. 2000. Adaptive mutation in Escherichia coli. Cold Spring Harbor Symp Quant Biol 65:21–29. doi: 10.1101/sqb.2000.65.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ponder RG, Fonville NC, Rosenberg SM. 2005. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell 19:791–804. doi: 10.1016/j.molcel.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 23.Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM. 2011. Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci U S A 108:13659–13664. doi: 10.1073/pnas.1104681108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeiser B, Pepper ED, Goodman MF, Finkel SE. 2002. SOS-induced DNA polymerases enhance long-term survival and evolutionary fitness. Proc Natl Acad Sci U S A 99:8737–8741. doi: 10.1073/pnas.092269199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stumpf JD, Foster PL. 2005. Polyphosphate kinase regulates error-prone replication by DNA polymerase IV in Escherichia coli. Mol Microbiol 57:751–761. doi: 10.1111/j.1365-2958.2005.04724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Layton JC, Foster PL. 2005. Error-prone DNA polymerase IV is regulated by the heat shock chaperone GroE in Escherichia coli. J Bacteriol 187:449–457. doi: 10.1128/JB.187.2.449-457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams AB, Foster PL. 2007. The Escherichia coli histone-like protein HU has a role in stationary phase adaptive mutation. Genetics 177:723–735. doi: 10.1534/genetics.107.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner J, Fujii S, Gruz P, Nohmi T, Fuchs RP. 2000. The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep 1:484–488. doi: 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cafarelli TM, Rands TJ, Godoy VG. 2014. The DinB·RecA complex of Escherichia coli mediates an efficient and high-fidelity response to ubiquitous alkylation lesions. Environ Mol Mutagen 55:92–102. doi: 10.1002/em.21826. [DOI] [PubMed] [Google Scholar]
- 30.Godoy VG, Jarosz DF, Simon SM, Abyzov A, Ilyin V, Walker GC. 2007. UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol Cell 28:1058–1070. doi: 10.1016/j.molcel.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen SE, Godoy VG, Walker GC. 2009. Transcriptional modulator NusA interacts with translesion DNA polymerases in Escherichia coli. J Bacteriol 191:665–672. doi: 10.1128/JB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sladewski TE, Hetrick KM, Foster PL. 2011. Escherichia coli Rep DNA helicase and error-prone DNA polymerase IV interact physically and functionally. Mol Microbiol 80:524–541. doi: 10.1111/j.1365-2958.2011.07590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guy CP, Atkinson J, Gupta MK, Mahdi AA, Gwynn EJ, Rudolph CJ, Moon PB, van Knippenberg IC, Cadman CJ, Dillingham MS, Lloyd RG, McGlynn P. 2009. Rep provides a second motor at the replisome to promote duplication of protein-bound DNA. Mol Cell 36:654–666. doi: 10.1016/j.molcel.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boubakri H, de Septenville AL, Viguera E, Michel B. 2010. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J 29:145–157. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michel B, Ehrlich SD, Uzest M. 1997. DNA double-strand breaks caused by replication arrest. EMBO J 16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heller RC, Marians KJ. 2007. Non-replicative helicases at the replication fork. DNA Repair 6:945–952. doi: 10.1016/j.dnarep.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 37.Heller RC, Marians KJ. 2005. Unwinding of the nascent lagging strand by Rep and PriA enables the direct restart of stalled replication forks. J Biol Chem 280:34143–34151. doi: 10.1074/jbc.M507224200. [DOI] [PubMed] [Google Scholar]
- 38.Sandler SJ. 2000. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics 155:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 40.Kalinin Y, Neumann S, Sourjik V, Wu M. 2010. Responses of Escherichia coli bacteria to two opposing chemoattractant gradients depend on the chemoreceptor ratio. J Bacteriol 192:1796–1800. doi: 10.1128/JB.01507-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang RF, Kushner SR. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195–199. doi: 10.1016/0378-1119(91)90366-J. [DOI] [PubMed] [Google Scholar]
- 42.Ling H, Boudsocq F, Woodgate R, Yang W. 2001. Crystal structure of a Y-family DNA polymerase in action: a mechanism for error-prone and lesion-bypass replication. Cell 107:91–102. doi: 10.1016/S0092-8674(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 43.Chang AC, Cohen SN. 1978. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134:1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau IF, Filipe SR, Soballe B, Okstad OA, Barre FX, Sherratt DJ. 2003. Spatial and temporal organization of replicating Escherichia coli chromosomes. Mol Microbiol 49:731–743. [DOI] [PubMed] [Google Scholar]
- 45.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blank K, Hensel M, Gerlach RG. 2011. Rapid and highly efficient method for scarless mutagenesis within the Salmonella enterica chromosome. PLoS One 6:e15763. doi: 10.1371/journal.pone.0015763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renzette N, Gumlaw N, Nordman JT, Krieger M, Yeh SP, Long E, Centore R, Boonsombat R, Sandler SJ. 2005. Localization of RecA in Escherichia coli K-12 using RecA-GFP. Mol Microbiol 57:1074–1085. doi: 10.1111/j.1365-2958.2005.04755.x. [DOI] [PubMed] [Google Scholar]
- 49.Storvik KA, Foster PL. 2010. RpoS, the stress response sigma factor, plays a dual role in the regulation of Escherichia coli's error-prone DNA polymerase IV. J Bacteriol 192:3639–3644. doi: 10.1128/JB.00358-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams AB, Hetrick KM, Foster PL. 2010. Interplay of DNA repair, homologous recombination, and DNA polymerases in resistance to the DNA damaging agent 4-nitroquinoline-1-oxide in Escherichia coli. DNA Repair 9:1090–1097. doi: 10.1016/j.dnarep.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foster PL. 1997. Nonadaptive mutations occur on the F' episome during adaptive mutation conditions in Escherichia coli. J Bacteriol 179:1550–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarkar S, Ma WT, Sandri GH. 1992. On fluctuation analysis: a new, simple and efficient method for computing the expected number of mutants. Genetica 85:173–179. doi: 10.1007/BF00120324. [DOI] [PubMed] [Google Scholar]
- 53.Hall BM, Ma CX, Liang P, Singh KK. 2009. Fluctuation analysis CalculatOR: a Web tool for the determination of mutation rate using Luria-Delbrück fluctuation analysis. Bioinformatics 25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Foster PL, Trimarchi JM, Maurer RA. 1996. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics 142:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. 1988. Current protocols in molecular biology. John Wiley & Sons, New York, NY. [Google Scholar]
- 56.Leake MC, Chandler JH, Wadhams GH, Bai F, Berry RM, Armitage JP. 2006. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 443:355–358. doi: 10.1038/nature05135. [DOI] [PubMed] [Google Scholar]
- 57.Lenn T, Gkekas CN, Bernard L, Engl C, Jovanovic G, Buck M, Ying L. 2011. Measuring the stoichiometry of functional PspA complexes in living bacterial cells by single molecule photobleaching. Chem Commun 47:400–402. doi: 10.1039/C0CC01707H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chung SH, Kennedy RA. 1991. Forward-backward non-linear filtering technique for extracting small biological signals from noise. J Neurosci Methods 40:71–86. doi: 10.1016/0165-0270(91)90118-J. [DOI] [PubMed] [Google Scholar]
- 59.Rice JA. 1995. Mathematical statistics and data analysis. Wadsworth Publishing Company, Belmont, CA. [Google Scholar]
- 60.Cairns J, Foster PL. 1991. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drlica K, Malik M, Kerns RJ, Zhao X. 2008. Quinolone-mediated bacterial death. Antimicrob Agents Chemother 52:385–392. doi: 10.1128/AAC.01617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coffman VC, Wu JQ. 2012. Counting protein molecules using quantitative fluorescence microscopy. Trends Biochem Sci 37:499–506. doi: 10.1016/j.tibs.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petit MA, Ehrlich D. 2002. Essential bacterial helicases that counteract the toxicity of recombination proteins. EMBO J 21:3137–3147. doi: 10.1093/emboj/cdf317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Little JW. 1991. Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie 73:411–421. doi: 10.1016/0300-9084(91)90108-D. [DOI] [PubMed] [Google Scholar]
- 65.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis, 2d ed. ASM Press, Washington, DC. [Google Scholar]
- 66.Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72:642–671. doi: 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foster PL. 2007. Stress-induced mutagenesis in bacteria. Crit Rev Biochem Mol Biol 42:373–397. doi: 10.1080/10409230701648494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goodman MF. 2002. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem 71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 69.Lovett ST. 2006. Replication arrest-stimulated recombination: dependence on the RecA paralog, RadA/Sms and translesion polymerase, DinB. DNA Repair 5:1421–1427. doi: 10.1016/j.dnarep.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 70.Cheng G, Hao H, Dai M, Liu Z, Yuan Z. 2013. Antibacterial action of quinolones: from target to network. Eur J Med Chem 66:555–562. doi: 10.1016/j.ejmech.2013.01.057. [DOI] [PubMed] [Google Scholar]
- 71.Kidane D, Graumann PL. 2005. Dynamic formation of RecA filaments at DNA double strand break repair centers in live cells. J Cell Biol 170:357–366. doi: 10.1083/jcb.200412090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lesterlin C, Ball G, Schermelleh L, Sherratt DJ. 2014. RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature 506:249–253. doi: 10.1038/nature12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Simmons LA, Grossman AD, Walker GC. 2007. Replication is required for the RecA localization response to DNA damage in Bacillus subtilis. Proc Natl Acad Sci U S A 104:1360–1365. doi: 10.1073/pnas.0607123104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Indiani C, Patel M, Goodman MF, O'Donnell ME. 2013. RecA acts as a switch to regulate polymerase occupancy in a moving replication fork. Proc Natl Acad Sci U S A 110:5410–5415. doi: 10.1073/pnas.1303301110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu LY, Magnan D, Halliday JA, Frisch RL, Gibson JL, Nehring RB, Do HG, Hernandez M, Li L, Herman C, Hastings P, Bates D, Harris RS, Miller KM, Rosenberg SM. 2013. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. eLife 2:e01222. doi: 10.7554/eLife.01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cox MM. 1999. Recombinational DNA repair in bacteria and the RecA protein. Prog Nucleic Acid Res Mol Biol 63:311–366. [DOI] [PubMed] [Google Scholar]
- 77.Rangarajan S, Woodgate R, Goodman MF. 2002. Replication restart in UV-irradiated Escherichia coli involving pols II, III, V, PriA, RecA and RecFOR proteins. Mol Microbiol 43:617–628. doi: 10.1046/j.1365-2958.2002.02747.x. [DOI] [PubMed] [Google Scholar]
- 78.Pomerantz RT, Kurth I, Goodman MF, O'Donnell ME. 2013. Preferential D-loop extension by a translesion DNA polymerase underlies error-prone recombination. Nat Struct Mol Biol 20:748–755. doi: 10.1038/nsmb.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reyes-Lamothe R, Sherratt DJ, Leake MC. 2010. Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328:498–501. doi: 10.1126/science.1185757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Storvik KA, Foster PL. 2011. The SMC-like protein complex SbcCD enhances DNA polymerase IV-dependent spontaneous mutation in Escherichia coli. J Bacteriol 193:660–669. doi: 10.1128/JB.01166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wagner J, Etienne H, Fuchs RP, Cordonnier A, Burnouf D. 2009. Distinct beta-clamp interactions govern the activities of the Y family PolIV DNA polymerase. Mol Microbiol 74:1143–1151. doi: 10.1111/j.1365-2958.2009.06920.x. [DOI] [PubMed] [Google Scholar]
- 82.Borden A, O'Grady PI, Vandewiele D, Fernandez de Henestrosa AR, Lawrence CW, Woodgate R. 2002. Escherichia coli DNA polymerase III can replicate efficiently past a T-T cis-syn cyclobutane dimer if DNA polymerase V and the 3′ to 5′ exonuclease proofreading function encoded by dnaQ are inactivated. J Bacteriol 184:2674–2681. doi: 10.1128/JB.184.10.2674-2681.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maenhaut-Michel G, Janel-Bintz R, Fuchs RP. 1992. A umuDC-independent SOS pathway for frameshift mutagenesis. Mol Gen Genet 235:373–380. doi: 10.1007/BF00279383. [DOI] [PubMed] [Google Scholar]
- 84.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.