

ABSTRACT
The Epstein-Barr virus (EBV) capsid contains a major capsid protein, VCA; two minor capsid proteins, BDLF1 and BORF1; and a small capsid protein, BFRF3. During the lytic cycle, these capsid proteins are synthesized and imported into the host nucleus for capsid assembly. This study finds that EBV capsid proteins colocalize with promyelocytic leukemia (PML) nuclear bodies (NBs) in P3HR1 cells during the viral lytic cycle, appearing as nuclear speckles under a confocal laser scanning microscope. In a glutathione S-transferase pulldown study, we show that BORF1 interacts with PML-NBs in vitro. BORF1 also colocalizes with PML-NBs in EBV-negative Akata cells after transfection and is responsible for bringing VCA and the VCA-BFRF3 complex from the cytoplasm to PML-NBs in the nucleus. Furthermore, BDLF1 is dispersed throughout the cell when expressed alone but colocalizes with PML-NBs when BORF1 is also present in the cell. In addition, this study finds that knockdown of PML expression by short hairpin RNA does not influence the intracellular levels of capsid proteins but reduces the number of viral particles produced by P3HR1 cells. Together, these results demonstrate that BORF1 plays a critical role in bringing capsid proteins to PML-NBs, which may likely be the assembly sites of EBV capsids. The mechanisms elucidated in this study are critical to understanding the process of EBV capsid assembly.
IMPORTANCE Capsid assembly is an important event during the Epstein-Barr virus (EBV) lytic cycle, as this process is required for the production of virions. In this study, confocal microscopy revealed that the EBV capsid protein BORF1 interacts with promyelocytic leukemia (PML) nuclear bodies (NBs) in the host nucleus and is responsible for transporting the other EBV capsid proteins, including VCA, BDLF1, and BFRF3, to these subnuclear locations prior to initiation of capsid assembly. This study also found that knockdown of PML expression by short hairpin RNA significantly reduces EBV capsid assembly capabilities. This enhanced understanding of capsid assembly offers potential for the development of novel antiviral strategies and therapies that can prevent the propagation and spread of EBV.
INTRODUCTION
The Epstein-Barr virus (EBV) capsid is icosahedral in structure, with a triangulation number of 16 (1–4). Capsomers primarily consist of the major capsid protein, VCA, and are held together via 320 triplexes formed by two minor capsid proteins, BDLF1 and BORF1 (2, 5, 6). In addition, a small capsid protein, BFRF3, sits atop the hexameric capsomers (7–9). Since EBV assembles its capsids in the host nucleus (10, 11), these capsid components are transported into the nucleus after synthesis. In herpes simplex virus 1 (HSV-1), a member of the same Herpesviridae family as EBV, the VP5 major capsid protein does not enter the nucleus alone; its nuclear translocation depends on its interaction with VP19C and the pre-VP22a scaffold protein (12, 13), homologs of EBV BORF1 and BVRF2, respectively. VP19C is also responsible for the nuclear translocation of VP23, a homolog of BDLF1 (13). Once these capsid proteins are transported into the nucleus, capsids begin to form, but the exact nuclear assembly sites remain unknown as yet (14, 15).
The human promyelocytic leukemia (PML) protein is a constituent of PML nuclear bodies (NBs), otherwise known as nuclear domain 10 (ND10) or PML oncogenic domains (PODs) (16–19). PML-NBs are present in the interchromosomal space in the nucleus, and each contains up to 160 proteins, including Sp100, p53, CBP/p300, Daxx, BLM, HDAC7, small ubiquitin-related modifier (SUMO), and Rb (19–23). PML-NBs have a wide range of functions and are known to be involved in transcription, DNA synthesis, DNA repair, cell cycle control, apoptosis, telomere lengthening, and degradation of misfolded proteins (18, 20, 24–26). Moreover, PML-NBs are known to influence the lytic cycle of herpesviruses. For example, the immediate early genes of herpesviruses, including ICP0 of HSV-1, IE1 of human cytomegalovirus (HCMV), and Zta of EBV, have been found to disperse PML-NBs (26–33). Furthermore, the dispersion of PML-NBs by arsenic treatment was shown to activate the expression of EBV lytic proteins (34), suggesting that PML-NB dispersion promotes the EBV lytic cycle. It is known that PML-NBs are dispersed after EBV lytic induction, as evidenced by the transfection of plasmids overexpressing Zta into cells latently infected with EBV (30); the dispersion requires expression of a substantial amount of Zta (35–37). The dispersion of PML-NBs by viral regulatory proteins may depend on posttranslational modification of the PML protein, such as by sumoylation and ubiquitination (38–40). Furthermore, the expression of EBV latent membrane protein 1 (LMP1) promotes the expression of PML and increases the immunofluorescence intensity of PML-NBs (41). The increase may be due to LMP1 CTAR3 enhancement of sumoylation to increase PML-NB stability (42). Recently, PML-NBs in infected cell nuclei were reported to sequester varicella-zoster virus (VZV) nucleocapsids and significantly inhibit virion development by preventing viral particle formation and nuclear egress (43), suggesting that PML-NBs may play a critical role in cellular antiviral defense (44, 45).
Other studies have shown that the viral genomes of herpesviruses, adenoviruses, simian virus 40, papillomaviruses, and polyomaviruses are recruited to PML-NBs, and this recruitment is known to be important for viral genome replication (35, 46–49). Although the EBV genome in latently infected cells does not appear to associate with PML-NBs, association during the lytic phase is required for viral DNA replication (35, 50), indicating that PML-NBs are important to EBV lytic development. This study further demonstrates that PML-NBs are essential for EBV capsid assembly.
MATERIALS AND METHODS
Cell lines and EBV lytic activation.
P3HR1, Akata, and EBV-negative Akata cells were cultured in RPMI 1640 medium. 293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM). These media were supplemented with 10% fetal calf serum. P3HR1 and Akata cells were treated with 3 ng/ml of 12-O-tetradecanoylphorbol-13-acetate (TPA) and 3 mM sodium butyrate to activate the EBV lytic cycle.
Plasmids.
Plasmids pcDNA-VCA, pcDNA-BORF1, pcDNA-BDLF1, pGEX-VCA, pGEX-BORF1, and pGEX-BDLF1, which express, respectively, hemagglutinin (HA)-tagged VCA (HA-VCA), HA-BORF1, HA-BDLF1, glutathione S-transferase (GST)-tagged VCA (GST-VCA), GST-BORF1, and GST-BDLF1, have been described elsewhere (5). A DNA fragment containing the entire BORF1 sequence was amplified by PCR, using primers borf1-F (5′-CGCGGATCCGACGCCATGAAGGTCCAG) and borf1-R (5′-GGCCTCGAGCCTCCTCCTCCTGTTTCC). The fragment was cut by BglII and SalI and then inserted at the BglII and SalI sites of pEGFP-C1 (Clontech), pTag2B (Invitrogen), or pHcRed (Evrogen) to generate, respectively, pEGFP-BORF1, pTag-BORF1, and pHcRed-BORF1. The BOFR1 sequence containing a mutated nuclear localization signal (NLS) was amplified using primers borf1mut-F (5′-CGCGGATCCATGAAGGTCCAGGGGTCCGTCGATCGCCGCCGTCTGCAAGCCGCAATCGCGGGGCTGCTG) and borf1-R. The fragment was cut by BglII and SalI before it was inserted at the BglII and SalI sites of pEGFP-C1 to generate pEGFP-mBORF1. A BDLF1 fragment was amplified by PCR, using primers bdlf1-F (5′-CCGGAATTCATGGATTTGAAAGTGGTA) and bdlf1-R (5′-CCGCTCGAGTTATCTTAACCAGCAAGT), digested with EcoRI and XhoI, and inserted at the EcoRI and SalI sites in pEGFP-C2 (Clontech). The BFRF3 fragment was amplified by PCR, using primers bfrf3-F (5′-TTGAATTCATGGCACGCCGGCTGCCCAAG) and bfrf3-R (5′-CCGCTCGAGCTACTGTTTCTTACGTGCCCCG), digested with EcoRI and XhoI, and inserted at the EcoRI and XbaI sites in pcDNA-HA, pcDNA 3.1, and pEGFP-C2 to express, respectively, HA-BFRF3, Myc-BFRF3, and green fluorescent protein (GFP)-tagged BFRF3 (GFP-BFRF3). Plasmid pEGFP-PML was kindly provided by Roeland W. Dirks (48). The PML fragment was amplified by PCR, using primers pml-F (5′-CCGGAATTCCGATCTCCGAGGCCCCAGCAGGAC) and pml-R (5′-CCCCGCGGTACCGTCGACTGCAG), digested with EcoRI and SalI, and inserted at the EcoRI and XhoI sites in pGEX to generate pGEX-PML, which expresses GST-PML. The BVRF2 sequence was amplified using primers bvrf2-F (5′-CCGGAATTCATGGTGCAGGCACCATCT) and bvrf2-R (5′-ACGCGTCGACTCAAGCCACGCGTTTATT), digested with EcoRI and SalI, and inserted at the EcoRI and SalI sites of pEGFP-c2 to generate pEGFP-BVRF2, which expresses GFP-BVRF2. BVRF2(S116C) was generated by inverse PCR (49), using primers BV116-F (5′-GCCTGGCCTCCCTCCAC) and BV116-R (5′-GCCAGGCACAGTGAGGGGAGCCAG), which contain mutated sequences. A plasmid expressing ataxin-3-79Q-HA (pAtaxin3-79Q-HA) was obtained from Hung-Li Wang (51). A plasmid expressing GFP-RNF4 was reported earlier (52).
Antibodies.
Monoclonal mouse anti-VCA antibody was purchased from Argene (Verniolle, France). Rabbit anti-VCA antibody was kindly provided by Yasushi Kawaguchi (53). Anti-BDLF1, anti-BORF1, and anti-BFRF3 antibodies were produced in rabbits using GST-BDLF1, GST-BORF1, and GST-BFRF3 proteins, respectively. Anti-HA and anti-Myc antibodies were purchased from Sigma-Aldrich. Anti-His and anti-GST antibodies were purchased from Loh Tsui Kweh BioLaboratories (Taipei, Taiwan). Rabbit and monoclonal mouse anti-PML antibodies (PG-M3 and H-238, respectively), which recognize all PML isoforms, were purchased from Santa Cruz Biotechnology. Rabbit anti-GFP antibody was purchased from Abcam. Goat anti-BFRF3 antibody (PAI-73003) was purchased from Pierce Biotechnology.
Immunofluorescence analysis.
P3HR1 cells and EBV-negative Akata cells were plated on a poly-l-lysine-coated coverslip, fixed, and immunostained with the appropriate antibody, according to a method described elsewhere (54). Cells were also treated with secondary antibodies conjugated with Alexa Fluor 488, Alexa Fluor 594, or Alexa Fluor-Cy5 (Invitrogen). Nuclei were visualized via staining with 5 mg/ml 4′-6-diamidino-2-phenylindole (DAPI). After staining, the cells were observed under a confocal laser scanning microscope (LSM 510; Zeiss, Oberkochen, Germany).
GST pulldown assay.
The GST pulldown assay was performed using a method described elsewhere (5). GST fusion proteins were expressed in Escherichia coli BL21(DE3) and bound to glutathione-Sepharose 4B beads (GE Healthcare Bio-Sciences). Proteins pulled down by the beads were separated by SDS-PAGE and subjected to immunoblot analysis (55).
ELISA.
Goat anti-BFRF3 antibody (10 μg/ml; Thermo Scientific) in 100 μl was added to coat the surface of each well of a 96-well microtiter plate overnight. A blocking solution containing 3% bovine serum albumin in phosphate-buffered saline (PBS) was added to the wells, and the plate was incubated for 3 h at 25°C. Culture supernatant (100 μl) containing EBV capsids was treated with 0.5% NP-40 for 30 min, added to each well, and then incubated at 37°C for 90 min. After washing with PBS, rabbit anti-BDLF1 antibody (1:100 dilution) and anti-rabbit horseradish peroxidase (HRP)-labeled secondary antibody were added to each well, and the plate was incubated for 2 h at room temperature. After washing with PBS, 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was added to the wells to detect HRP. The A450s of the colors produced by the reaction were determined using an enzyme-linked immunosorbent assay (ELISA) reader (Sunrise, Tecan, Australia).
Enumerating nucleocapsids by PCR.
The number of nucleocapsids was determined by a real-time PCR method described elsewhere using an iCycler iQ multicolor real-time PCR detection system (Bio-Rad) with primers and a probe that were specific to the BKRF1 gene (56).
shRNA and lentiviral infection.
Lentiviral vectors for PML-short hairpin RNA (shRNA) (accession number TRCN0000003865) and control shRNA (accession number TRCN0000072224) were obtained from the National RNAi Core Facility Platform, Academia Sinica, Taiwan. Lentiviral supernatant was prepared using a previously described method (57). P3HR1 cells (3 × 105 cells/ml) were mixed with viral supernatant containing 5 μg/ml Polybrene and then centrifuged at 450 × g for 90 min. After culturing of the cells in medium containing 0.5 μg/ml puromycin for 5 days, the cells were treated with TPA and sodium butyrate to induce the EBV lytic cycle.
RESULTS
Localization of EBV capsid proteins after lytic induction.
An indirect immunofluorescence study was conducted to identify the locations of EBV capsid proteins following lytic induction. The immunofluorescence results revealed that after P3HR1 cells were treated with TPA and sodium butyrate for 3 days, VCA, BDLF1, BORF1, and BFRF3 were present in the nucleus as speckles under a confocal laser scanning microscope (Fig. 1A). We also examined 100 cells that expressed these capsid proteins after lytic induction and found that all of them were present as speckles in the nucleus.
FIG 1.
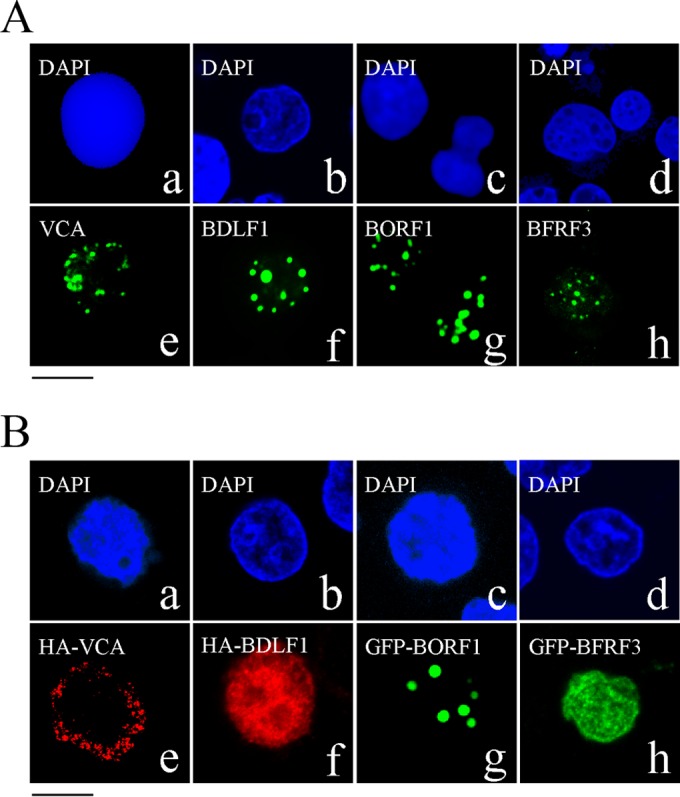
Nuclear entry of EBV capsid proteins. (A) P3HR1 cells were treated with TPA and sodium butyrate for 48 h and then incubated with monoclonal anti-VCA (a, e), rabbit anti-BDLF1 (b, f), rabbit anti-BORF1 (c, g), or rabbit anti-BFRF3 (d, h) antibodies. (B) EBV-negative Akata cells were transfected with pcDNA-VCA (a, e), pcDNA-BDLF1 (b, f), pEGFP-BORF1 (c, g), or pEGFP-BFRF3 (d, h). HA-tagged proteins were detected using rabbit anti-HA antibody and goat anti-rabbit IgG conjugated with Alexa Fluor 594. Cells were stained with DAPI to reveal the nucleus. Cells were examined using a confocal laser scanning microscope (LSM 510 META; Zeiss). Bar, 10 μm.
Transport of EBV capsid proteins into the nucleus.
We transfected EBV-negative Akata cells with plasmids expressing HA-VCA, HA-BDLF1, GFP-BORF1, or GFP-BFRF3 to examine the cellular distribution of these proteins. We found that HA-VCA was present only in the cytoplasm following transfection (Fig. 1Be), demonstrating that VCA cannot enter the nucleus without assistance from other EBV proteins. HA-BDLF1 and GFP-BFRF3 were found to diffuse throughout the cell (Fig. 1Bf and h), rather than aggregating as dots in the nucleus. Only GFP-BORF1 displayed a punctate pattern in the nucleus (Fig. 1Bg), similar to that observed in P3HR1 cells after lytic induction (Fig. 1A). We also cotransfected EBV-negative Akata cells with plasmids expressing GFP-BDLF1 and HA-VCA but found that these two proteins did not influence each other's distribution (Fig. 2Aa to d). However, HA-VCA colocalized with GFP-BORF1 in the nucleus as speckles, following cotransfection of the cells with pcDNA-VCA and pEGFP-BORF1 (Fig. 2Ae to h). Cotransfection of EBV-negative Akata cells with plasmids expressing GFP-BORF1 and HA-BDLF1 also resulted in the colocalization of GFP-BORF1 and HA-BDLF1 in the nucleus as speckles (Fig. 2Ai to l), indicating that the proper localization of BDLF1 in the nucleus relies on BORF1. We also found that the sequence LQRRIA, located at amino acids 12 to 17, is a nuclear localization signal (NLS). After this sequence was mutated to LQAAIA in GFP-BORF1, the mutant protein, GFP-mBORF1, was found to be present in the cytoplasm and did not enter the nucleus in EBV-negative Akata cells (Fig. 2Ba to c). HA-VCA and HA-BDLF1 also colocalized with GFP-mBORF1 in the cytoplasm after cotransfection (Fig. 2Bd to g and h to k), demonstrating that interaction with GFP-mBORF1 does not allow HA-VCA or HA-BDLF1 to enter the nucleus. These results confirm that entry of BDLF1 and VCA into the nucleus depends on their interaction with BORF1. Additionally, we cotransfected EBV-negative Akata cells with three plasmids expressing, respectively, red fluorescent protein (RFP)-tagged BORF1 (RFP-BORF1), GFP-BDLF1, and HA-VCA. Under a confocal laser scanning microscope, these three proteins colocalized and exhibited punctate structures in the nucleus (Fig. 2C), suggesting that EBV capsid proteins are transported to the same locations in the nucleus.
FIG 2.
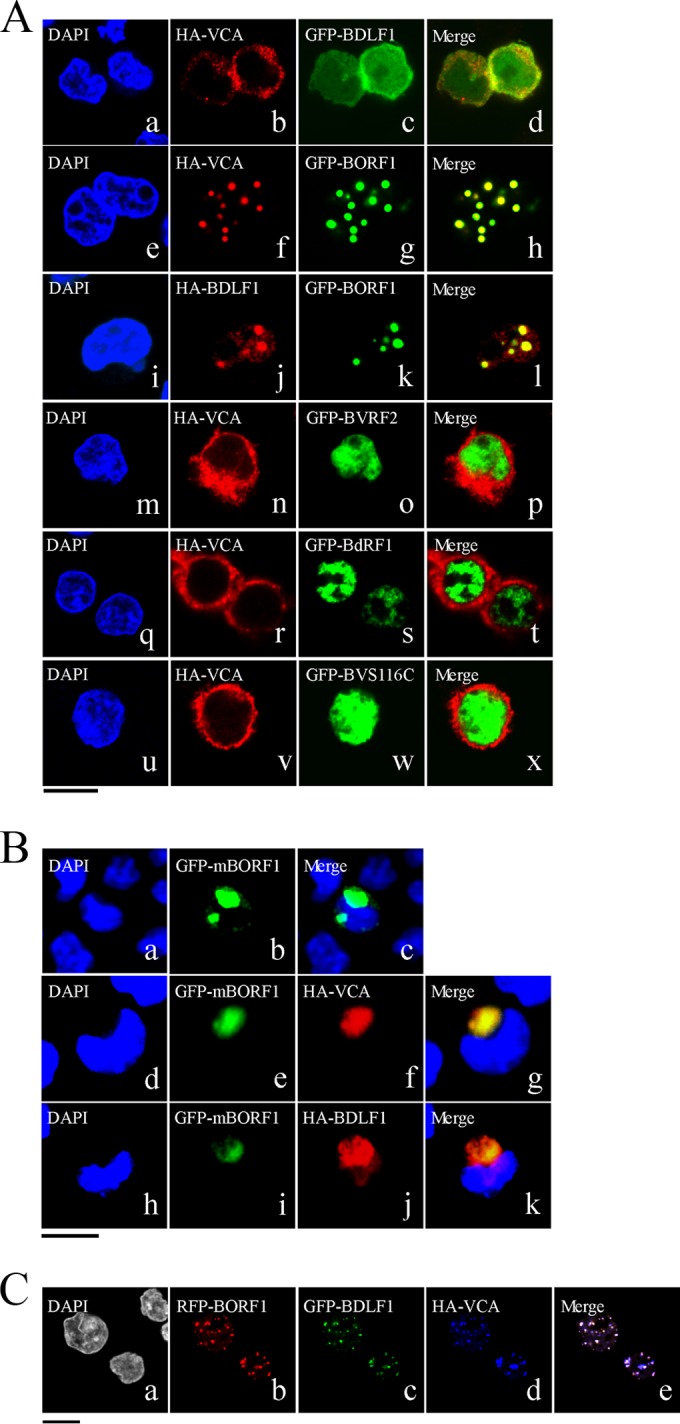
Involvement of BORF1 in the nuclear entry of BDLF1 and VCA. (A) EBV-negative Akata cells were cotransfected with pcDNA-VCA and pEGFP-BDLF1 (a to d), pcDNA-VCA and pEGFP-BORF1 (e to h), pcDNA-BDLF1 and pEGFP-BORF1 (i to l), pcDNA-VCA and pEGFP-BVRF2 (m to p), pcDNA-VCA and pEGFP-BdRF1 (q to t), or pcDNA-VCA and pEGFP-BVRF2(S116C) (GFP-BVS116C) (u to x). (B) EBV-negative Akata cells were transfected with pEGFP-mBORF1 (a to c) or cotransfected with either pEGFP-mBORF1 and pcDNA-VCA (d to g) or pEGFP-mBORF1 and pcDNA-BDLF1 (h to k). (C) EBV-negative Akata cells were cotransfected with pHcRed-BORF1 (b), pEGFP-BDLF1 (c), and pcDNA-VCA (d). (e) Merged image of the images in panels b, c, and d. HA-VCA and HA-ORF1 were detected using rabbit anti-HA antibody and goat anti-rabbit IgG conjugated with Alexa Fluor 594. Cells were examined using a confocal laser scanning microscope (LSM 510 META NLO; Zeiss). Bars, 10 μm.
Earlier studies showed that the nuclear translocation of the HSV-1 and HCMV major capsid proteins depends on interaction with their respective scaffold proteins (12). Therefore, this study investigated whether the EBV scaffold proteins, BVRF2 and BdRF1, convey VCA into the nucleus. We cotransfected EBV-negative Akata cells with plasmids that express HA-VCA and Flag-BVRF2. Confocal laser scanning microscopy revealed that BVRF2 was present in the nucleus and VCA was present in the cytoplasm (Fig. 2Am to p), showing that BVRF2 does not facilitate the nuclear entry of VCA. Cotransfection of the cells with plasmids that express HA-VCA and GFP-BdRF1 yielded similar results that showed that HA-VCA was present in the cytoplasm and GFP-BdRF1 was present in the nucleus (Fig. 2Aq to t). BVRF2 has a proteolytic domain that can cleave 38 amino acids from the C termini of BVRF2 and BdRF1 to yield mature scaffold proteins (58). This study replaced the serine residue at amino acid position 116 with cysteine to derive BVRF2(S116C), removing BVRF2 protease activity in the process (58). Immunoblotting revealed that BVRF2(S116C) had a size of 120 kDa, which was larger than that of cleaved BVRF2 (data not shown). Cotransfection of the cells with plasmids that expressed HA-VCA and GFP-BVRF2(S116C) revealed that GFP-BVRF2(S116C) did not affect the localization of HA-VCA (Fig. 2Au to x).
Nuclear entry of BFRF3.
BFRF3 nuclear entry mechanisms were also investigated. After cotransfecting EBV-negative Akata cells with plasmids expressing HA-VCA and Myc-BFRF3, confocal laser scanning microscopy revealed that both proteins were present in the cytoplasm, although some Myc-BFRF3 appeared to be in the nucleus (Fig. 3a to d). Following cotransfection with plasmids expressing GFP-BORF1 and Myc-BFRF3, GFP-BORF1 was seen to form dots in the nucleus but did not change the distribution of Myc-BFRF3 in the cell (Fig. 3e to h); these two proteins did not appear to colocalize in the nucleus (Fig. 3h). However, when cells were cotransfected with plasmids expressing HA-VCA, GFP-BORF1, and Myc-BFRF3, these three proteins colocalized in the nucleus to form a punctate pattern (Fig. 3Bi to m), demonstrating that GFP-BORF1 causes Myc-BFRF3 to form dots only when HA-VCA is present and indicating that nuclear entry and the proper nuclear localization of the VCA-BFRF3 complex depend upon BORF1.
FIG 3.
Nuclear entry of BFRF3. EBV-negative Akata cells were cotransfected with pcDNA-VCA and pcDNA-myc-BFRF3 (a to d), pEGFP-BORF1 and pcDNA-myc-BFRF3 (e to h), or pcDNA-VCA, pEGFP-BORF1, and pcDNA-myc-BFRF3 (i to m). HA-VCA was detected using anti-HA antibody and goat anti-rabbit IgG antibody conjugated with Alexa Fluor 594. (i to m) Myc-BFRF3 was detected using mouse anti-Myc antibody and goat anti-mouse IgG antibody conjugated with Cy5. Bar, 10 μm.
Colocalization of PML-NBs and EBV capsid proteins.
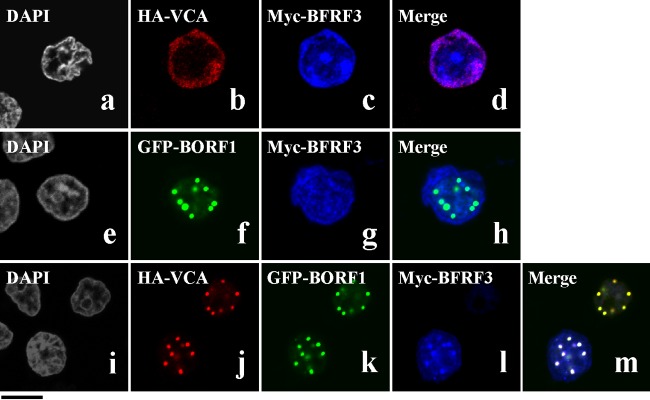
Confocal laser scanning microscopy of P3HR1 cells that had been treated with TPA and sodium butyrate and then immunostained using rabbit anti-BORF1 antibody revealed that EBV capsids colocalized with PML-NBs in the nucleus (Fig. 4A). We further transfected EBV-negative Akata cells with pEGFP-BORF1 and found that GFP-BORF1 colocalized with PML-NBs (Fig. 4Ba to d), indicating that BORF1 targets PML-NBs after entering the nucleus. We further cotransfected EBV-negative Akata cells with pEGFP-BORF1 and pcDNA-VCA and found that GFP-BORF1 and HA-VCA colocalized with PML-NBs (Fig. 4Be to i). A similar experiment also showed that GFP-BORF1 and HA-BDLF1 colocalized with PML-NBs (Fig. 4Bj to n). In addition, Myc-BFRF3 was found to colocalize with PML-NBs after the cells were cotransfected with plasmids expressing HA-VCA, Myc-BFRF3, and Flag-BORF1 (Fig. 4Bo to r). We further examined how a mutation in BORF1 would affect the localization of VCA and BDLF1. We found that BORF1, VCA, and BDLF1 colocalized with PML-NBs in 293 cells carrying bacmid 2089 after lytic induction (Fig. 4Ca to o). However, VCA and BDLF1 in a mutant EBV strain with a defective BORF1 (bacmid M68) (56) did not form punctate structures in the nucleus, nor did they colocalize with PML-NBs (Fig. 4Cp to y). Additionally, we cotransfected 293T cells with pEGFP-PML and pHcRed-BORF1 and then treated the cells with 0.02% methyl methanesulfonate (MMS) at 24 h after transfection to disperse the PML-NBs (59). In a live cell expressing GFP-PML and RFP-BORF1, these two proteins initially colocalized in the nucleus (Fig. 4D, 0 min). When this cell was treated with MMS for 80 min, PML-NBs were considerably dispersed. At 140 min after treatment, only a few PML-NBs were evident in the nucleus (Fig. 4D). The disappearance of RFP-BORF1 dots was found to coincide with the dispersion of PML-NBs following treatment (Fig. 4D), further verifying the association between BORF1 and PML-NBs. This study also cotransfected P3HR1 cells with plasmids expressing GFP-BORF1 and RFP-BORF1. We found that across 98 cells, the GFP-BORF1 dots colocalized with the RFP-BORF1 dots (data not shown), indicating that the GFP tag in GFP-BORF1 did not affect BORF1 migration to PML-NBs. We subsequently treated P3HR1 cells with TPA and sodium butyrate to induce the EBV lytic cycle (Fig. 4A), and among the 342 cells examined by confocal microscopy, 104 cells expressed BORF1. In these 104 cells, we found 243 BORF1 dots and 255 PML-NB dots; all the BORF1 dots colocalized with PML-NBs.
FIG 4.

Colocalization of EBV capsid proteins at PML-NBs. (A) P3HR1 cells were treated with TPA and sodium butyrate for 72 h. BORF1 and PML-NBs were stained using rabbit anti-BORF and mouse anti-PML antibodies, respectively. Goat anti-rabbit IgG antibody conjugated with Alexa Fluor 488 and goat anti-mouse IgG antibody conjugated with Alexa Fluor 594 were used as secondary antibodies. (B) EBV-negative Akata cells were transfected with pEGFP-BORF1 (a to d) or cotransfected with pcDNA-VCA and pEGFP-BORF1 (e to i), pcDNA-BDLF1 and pEGFP-BORF1 (j to n), or pcDNA-VCA, pTag-BORF1, and pcDNA-myc-BFRF3 (o to r). HA-VCA and HA-BDLF1 were detected using mouse anti-HA antibody and goat anti-mouse IgG antibody conjugated with Alexa Fluor 594. PML-NBs were detected using rabbit anti-PML antibody and goat anti-rabbit IgG antibody conjugated with Alexa Fluor-cy5 or Alexa Fluro-594. (C) 293 cells carrying bacmid 2089 (a to o) and 293 cells carrying bacmid M68 (p to ad) were treated with TPA and sodium butyrate for 3 days. VCA, BDLF1, and BORF1 were detected using rabbit anti-VCA, rabbit anti-BDLF1, and rabbit anti-BORF1 antibodies, respectively. Goat anti-rabbit IgG antibody conjugated with Alexa Fluor 594 was used as a secondary antibody. PML was detected using mouse anti-PML antibody and goat anti-mouse IgG antibody conjugated with Alexa Fluor 488. Cell nuclei were stained with DAPI. (D) 293T cells were cotransfected with pEGFP-PML and pHcRed-BORF1 for 24 h and then treated with 0.02% MMS. Images of a live cell were captured at 20-min intervals under a confocal laser scanning microscope, following MMS treatment (LSM 510 META NLO; Zeiss). Bars, 10 μm.
RNF4 and the stability of overexpressed BORF1.
An earlier study showed that ataxin-1-82Q, a misfolded protein, is conjugated by small ubiquitin-like modifiers (SUMOs) in PML-NBs. After SUMO conjugation, the protein is ubiquitinated by RNF4 and then subjected to proteasome degradation (25). Overexpressed proteins are often degraded in PML-NBs by this mechanism (25) as well, and this raises the possibility that overexpressed BORF1 is conveyed to PML-NBs for proteasomal degradation. We therefore cotransfected 293T cells with plasmids expressing GFP-BORF1 and RNF4, but immunoblot analysis revealed that RNF4 expression did not destabilize BORF1 (Fig. 5A). As a control, we cotransfected the cells with plasmids expressing ataxin-3-79Q-HA, a misfolded protein (51), and RNF4. We found that the expression of RNF4 decreased the amount of ataxin-3-79Q-HA in a dose-dependent manner (Fig. 5B). These results suggest that overexpressed GFP-BORF1 is neither misfolded nor conveyed to PML-NBs for the purpose of degradation.
FIG 5.
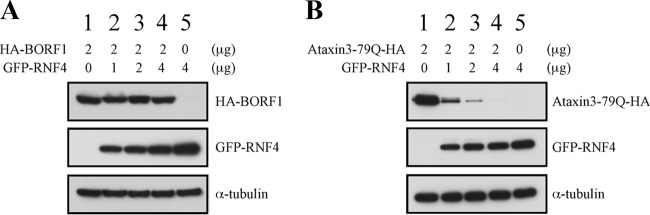
RNF4 and degradation of misfolded proteins. 293T cells were cotransfected with pcDNA-BORF1 and pEGFP-RNF4 (A) or pAtaxin3-79Q-HA and pCMV-RNF4 (B). Proteins in the lysates were detected by immunoblot analysis using anti-HA, anti-GFP, and anti-α-tubulin antibodies.
Interaction between PML and EBV capsid proteins.
To elucidate how EBV capsid proteins interact with PML, we expressed GST, GST-BDLF1, GST-BFRF3, GST-BORF1, and GST-VCA in E. coli. These proteins were bound to glutathione-Sepharose beads (Fig. 6A, lanes 7 to 11), which were then added to the lysates of 293T cells transfected with pEGFP-PML to study their interaction with GFP-PML. Immunoblot analysis revealed that GST-BORF1, but not the other GST fusion proteins or GST, pulled down GFP-PML (Fig. 6A, lanes 2 to 6). The results also showed that the inability to pull down GFP-PML was not attributable to a lack of GST proteins bound to the beads (Fig. 6A, lanes 7 to 11). A similar experiment was conducted using bacterially expressed GST-PML, and the results showed that GST-PML-glutathione-Sepharose beads did not pull down His-BORF1 from the E. coli BL21(DE3)(pET32a-BORF1) lysate (Fig. 6B, lane 4); nonpulldown was not due to a lack of GST-PML on the beads (Fig. 6B, lane 8). The lack of pulldown was also not attributable to the absence of His-BORF1, as GST-BDLF1 pulled down His-BORF1 from the same lysate (Fig. 6B, lane 3). We also found that GST-PML-glutathione-Sepharose beads pulled down His-BORF1 if a 293T cell lysate was added to the reaction mixture (Fig. 6B, lane 5). These results suggest that BORF1 likely interacts with a protein other than PML at PML-NBs.
FIG 6.
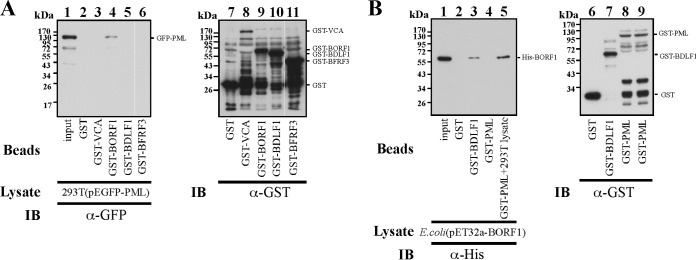
Interaction between PML and BORF1. (A) Bacterially expressed GST (lane 2), GST-VCA (lane 3), GST-BORF1 (lane 4), GST-BDLF1 (lane 5), and GST-BFRF3 (lane 6) were bound to glutathione-Sepharose beads and mixed with the lysates of 293T cells transfected with pEGFP-PML (lanes 2 to 6). Lane 1 was loaded with 1% of the lysate. Proteins pulled down by glutathione-Sepharose beads were analyzed via immunoblotting (IB) with anti-GFP antibody (lanes 1 to 6). Proteins on the beads were analyzed using anti-GST antibody (lanes 7 to 11). (B) The E. coli BL21(DE3)(pET32a-BORF1) lysate was mixed with glutathione-Sepharose beads labeled with GST (lane 2), GST-BDLF1 (lane 3), or GST-PML (lane 4). The lysate from 293T cells was also added to the reaction mixture for reactions involving GST-PML-glutathione-Sepharose beads (lane 5). Proteins on the beads were analyzed by immunoblotting, using rabbit anti-His antibody (lanes 2 to 5) and rabbit anti-GST antibody (lanes 6 to 9). Input lanes were loaded with 1% of the bacterial lysate.
PML-NBs and EBV capsid assembly.
We sought to understand how the reduction of PML expression by shRNA would affect EBV capsid assembly. EBV virions are commonly quantified using quantitative PCR (qPCR) methods (56). However, these methods could not be utilized in this study, as EBV lytic DNA replication occurs at PML-NBs (35, 50); any changes in EBV lytic DNA replication due to a reduction in PML-NBs would affect the production of encapsidated viral particles, ultimately distorting the qPCR results. Accordingly, an ELISA method was developed to determine capsid assembly, on the basis of the principle that on each capsid, VCA interacts with both BFRF3 and BDLF1 (5, 8) but BFRF3 does not interact directly with BDLF1 (8). The method involved the use of plates coated with anti-BFRF3 antibody to capture EBV capsids, followed by detection of these capsids with anti-BDLF1 antibody. Through this method, we found that EBV in induced P3HR1 cells produced significantly more capsids after lytic induction than EBV in untreated cells (Fig. 7A). This method also found that latently infected P3HR1 cells transfected with plasmids expressing either BDLF1 or BFRF3 or cotransfected with plasmids expressing these two proteins produced relatively small quantities of capsids (Fig. 7A). Although accurate estimation of viral capsid numbers produced by the cells was difficult to achieve, based on qPCR analysis results, we found that about 1.1 × 108 encapsidated viral particles were produced by 1 × 106 P3HR1 cells after lytic induction, while a similar number of latently infected cells produced only about 2 × 104 particles. Therefore, the ELISA data in Fig. 7A likely correspond to EBV particle numbers. Our results revealed that P3HR1 cells expressing control shRNA had an optical density (OD) reading at 450 nm of 0.15, which increased to 1.0 in cells treated with TPA and sodium butyrate (Fig. 7B), demonstrating that EBV capsids are produced after lytic induction. Expression of PML-shRNA in uninduced P3HR1 cells led to background ELISA levels (Fig. 7B); however, upon expression of PML-shRNA and induction of the lytic cycle, the OD reading was only about 50% of that for control shRNA-expressing cells subjected to lytic induction (Fig. 7B), indicating that the reduction in the level of PML expression negatively impacts capsid assembly. We also determined the number of nucleocapsids produced from 1 × 105 P3HR1 cells in 30 ml of culture medium by qPCR. We found that after transfecting control shRNA for 5 days, the cells produced about 8 × 106 nucleocapsids. Meanwhile, transfection of the cells with PML-specific shRNA reduced the number to 1.8 × 106 (Fig. 7C), showing that a reduction in the level of PML expression also reduces the number of nucleocapsids. An immunoblot study showed that after infecting P3HR1 cells with a lentivirus that expresses PML-specific shRNA, followed by treatment with TPA and sodium butyrate, PML expression levels were reduced, but the levels of Rta, VCA, BDLF1, BORF1, and α-tubulin (Fig. 7D) were not affected, thus demonstrating that the decrease in capsid production could not be attributed to the reduced expression of capsid proteins.
FIG 7.
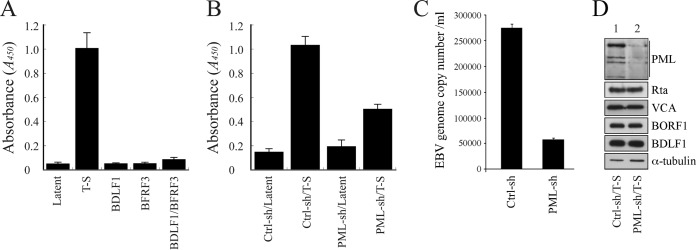
PML and capsid assembly by EBV. (A) P3HR1 cells were untreated (Latent) or treated with TPA and sodium butyrate (T-S). After treatment, cells were transfected with pCMV-BDLF1 or pCMV-BFRF3 or cotransfected with these two plasmids to express BDLF1 and BFRF3. (B) Cells were infected with lentivirus expressing control shRNA (Ctrl-sh) or PML-specific shRNA (PML-sh) for 5 days and then treated with TPA and sodium butyrate or left untreated. Three days after lytic induction, the quantity of capsids produced by the cells was analyzed via ELISA. EBV capsids were captured by anti-BFRF3 antibody and detected using rabbit anti-BDLF1 antibody and goat HRP-conjugated anti-rabbit immunoglobulin antibodies. (C) P3HR1 cells (1 × 105) were transfected with control shRNA and PML-specific shRNA. Cells were treated with TPA and sodium butyrate and cultured in 30 ml medium for 5 days. EBV particles were collected from the culture medium by centrifugation. The EBV genome in the viral particles was measured by real-time qPCR. (D) The PML, Rta, VCA, BORF1, BDLF1, and α-tubulin expressed by the cells were analyzed via immunoblotting 3 days after lytic induction.
DISCUSSION
Capsid assembly is an important event for EBV since during capsid assembly all capsid proteins must be transported to the sites of assembly in the nucleus (10, 11). In this study, we show that BORF1, which contains an NLS and is capable of entering the nucleus by itself (Fig. 1Bg and 2Bb), is required for transporting VCA into the nucleus (Fig. 2Ah). This is similar to the nuclear entry process of the HSV-1 major capsid protein, VP5; its transport depends on the interaction with the homolog of EBV BORF1, VP19C (13). Interestingly, nuclear transport of the HSV-1 and HCMV major capsid proteins depends on interaction with their respective scaffold proteins (12, 60); however, the EBV scaffold protein BVRF2 does not seem to be involved in the nuclear transport of VCA (Fig. 2Am to p). The expression of BVRF2(S116C), which lacks the autocleavage activity of BVRF2 (data not shown) (52), or BdRF1 also does not affect the localization of VCA in the cell (Fig. 2Aq to x), demonstrating that, unlike pre-VP22a of HSV-1, EBV scaffold proteins are not responsible for the nuclear translocation of VCA. We also found that BFRF3 forms dots in the nucleus only when BORF1 and VCA are both present (Fig. 3), suggesting that BORF1 conveys the VCA-BFRF3 complex to the host nucleus. As for BDLF1, a small protein of 33.6 kDa, it is probably able to enter the nucleus alone due to its small size, which explains its presence in both the cytoplasm and the nucleus (Fig. 1Bf), yet BDLF1 forms dots in the nucleus only when BORF1 is present (Fig. 2A), suggesting that BDLF1 is not properly localized in the absence of BORF1. Moreover, after cotransfecting EBV-negative Akata cells with plasmids that express GFP-BDLF1, RFP-BORF1, and HA-VCA, these three proteins colocalized in the nucleus (Fig. 2Ca to e), indicating that EBV capsid proteins are transported to the same nuclear locations. Additionally, we cotransfected EBV-negative Akata cells with plasmids that express GFP-BORF1 and RFP-BORF1 and found that they colocalized in the nucleus as dots (data not shown), indicating that the GFP tag in GFP-BORF1 does not affect the function of BORF1.
The capsid of the human polyomavirus JC virus (JCV) is assembled at PML-NBs (48). In addition, the major and minor capsid proteins of human papillomavirus are assembled into virus-like particles at PML-NBs, with the L2 capsid protein previously being shown to be essential for virion assembly, as it recruits other capsid proteins to PML-NBs (61). L2-dependent colocalization probably serves as a mechanism to promote the assembly of papillomaviruses, either by increasing the local concentration of virion constituents or by providing the physical architecture necessary for efficient packaging and assembly. This study reveals a similar mechanism for EBV, with the minor capsid protein BORF1 being responsible for mediating the nuclear entry of capsid proteins BDLF1 and VCA to PML-NBs for EBV capsid assembly.
Under a confocal laser scanning microscope, we observed that EBV capsids in P3HR1 cells colocalize with PML-NBs (Fig. 4A), implying that PML-NBs are important to EBV capsid assembly. Our experiments in EBV-negative Akata cells further reveal that BORF1 is required to convey BDLF1, BFRF3, and VCA to PML-NBs (Fig. 4B and C). We also examined the localization of VCA and BDLF1 in 293 cells carrying bacmid 2089 and found that they form dots in the nucleus. However, in 293 cells carrying bacmid M68 in which the EBV strain possesses a mutant BORF1, VCA is present in the cytoplasm (Fig. 4Cp to t) and BDLF1 is distributed throughout the cell (Fig. 4Cu to y), indicating that BORF1 is responsible for conveying VCA and BDLF1 into the nucleus. A recent study showed that in Kaposi's sarcoma-associated herpesvirus, the expression of the homologs of EBV BFRF3 and BdRF1 is required for the nuclear transport of its major capsid protein (62). However, this appears to not be the case in EBV, as the VCA expressed by 293 cells carrying bacmid M68 is present in the cytoplasm rather than in the nucleus (Fig. 4C). A GST pulldown study also confirmed that BORF1, but not BDLF1 or VCA, interacts with PML-NBs (Fig. 6), demonstrating that BORF1 not only is responsible for nuclear entry but also transports BDLF1 and the VCA-BFRF3 complex to PML-NBs. Since EBV-negative Akata cells lack the EBV genome and do not contain all the proteins necessary for capsid assembly, the presence of EBV capsid proteins at PML-NBs hints at a process of direct transport to this subnuclear structure for capsid assembly. We found that the expression of PML-specific shRNA substantially reduces the numbers of capsids produced by EBV (Fig. 7). Electron microscopy results also showed that EBV capsids are present in nuclear areas stained by anti-PML antibody (data not shown), suggesting that PML-NBs are crucial to capsid assembly. Although it is known that misfolded proteins are transported to PML-NBs for degradation by proteasomes after ubiquitination by the ubiquitin E3 ligase RNF4 (25), we found that after cotransfecting plasmids that express RNF4 and BORF1, the stability of BORF1 remained unchanged (Fig. 5), demonstrating that the BORF1 localized at PML-NBs is likely not misfolded.
Capsid assembly is an important process in EBV lytic development. This study elucidated the mechanisms by which EBV capsid proteins are transported into the nucleus and revealed the involvement of PML-NBs in capsid assembly. It is known that PML-NBs act as cellular restriction factors that prevent the release of VZV (43). However, in the case of EBV, PML-NBs are important to viral lytic DNA replication (20, 50). Although the expression of Zta causes PML-NB dispersion, Adamson and Kenney (30) showed that PML-NBs are only partially dispersed and some structures remain visible under a fluorescence microscope. This partial dispersion has also been documented in other studies (35, 50), and it was recently found that the comprehensive dispersion of PML-NBs in cells occurs only at extremely high levels of Zta expression (36, 37). This study showed that BORF1 colocalizes with PML-NBs in P3HR1 cells after lytic induction (Fig. 4A), suggesting that some PML-NBs remain intact and support capsid assembly at this stage. To the best of our knowledge, this is the first study to show that herpesviruses assemble their capsids at PML-NBs. These results advance the current understanding of EBV capsid protein assembly and may have a bearing on the development of novel antiviral therapies in the future.
ACKNOWLEDGMENTS
This work was supported by grants from the National Health Research Institute of the Republic of China (NHRI-EX103-10135BI), the Ministry of Science and Technology of the Republic of China (NSC101-2320-B-182-014-MY3), and Chang Gung Memorial Hospital (CMRPD1C0571-2, CMRPD1B0371-3).
We also thank Roeland W. Dirks for providing the pEGFP-PML plasmid and Hung-Li Wang for providing pAtaxin-3-79Q-HA.
REFERENCES
- 1.Baker TS, Newcomb WW, Booy FP, Brown JC, Steven AC. 1990. Three-dimensional structures of maturable and abortive capsids of equine herpesvirus 1 from cryoelectron microscopy. J Virol 64:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newcomb WW, Trus BL, Booy FP, Steven AC, Wall JS, Brown JC. 1993. Structure of the herpes simplex virus capsid. Molecular composition of the pentons and the triplexes. J Mol Biol 232:499–511. [DOI] [PubMed] [Google Scholar]
- 3.Trus BL, Newcomb WW, Booy FP, Brown JC, Steven AC. 1992. Distinct monoclonal antibodies separately label the hexons or the pentons of herpes simplex virus capsid. Proc Natl Acad Sci U S A 89:11508–11512. doi: 10.1073/pnas.89.23.11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Germi R, Effantin G, Grossi L, Ruigrok RW, Morand P, Schoehn G. 2012. Three-dimensional structure of the Epstein-Barr virus capsid. J Gen Virol 93:1769–1773. doi: 10.1099/vir.0.043265-0. [DOI] [PubMed] [Google Scholar]
- 5.Wang WH, Chang LK, Liu ST. 2011. Molecular interactions of Epstein-Barr virus capsid proteins. J Virol 85:1615–1624. doi: 10.1128/JVI.01565-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spencer JV, Newcomb WW, Thomsen DR, Homa FL, Brown JC. 1998. Assembly of the herpes simplex virus capsid: preformed triplexes bind to the nascent capsid. J Virol 72:3944–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wingfield PT, Stahl SJ, Thomsen DR, Homa FL, Booy FP, Trus BL, Steven AC. 1997. Hexon-only binding of VP26 reflects differences between the hexon and penton conformations of VP5, the major capsid protein of herpes simplex virus. J Virol 71:8955–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henson BW, Perkins EM, Cothran JE, Desai P. 2009. Self-assembly of Epstein-Barr virus capsids. J Virol 83:3877–3890. doi: 10.1128/JVI.01733-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai P, Akpa JC, Person S. 2003. Residues of VP26 of herpes simplex virus type 1 that are required for its interaction with capsids. J Virol 77:391–404. doi: 10.1128/JVI.77.1.391-404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson W. 1996. Structure and assembly of the virion. Intervirology 39:389–400. [DOI] [PubMed] [Google Scholar]
- 11.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J Virol 76:1537–1547. doi: 10.1128/JVI.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholson P, Addison C, Cross AM, Kennard J, Preston VG, Rixon FJ. 1994. Localization of the herpes simplex virus type 1 major capsid protein VP5 to the cell nucleus requires the abundant scaffolding protein VP22a. J Gen Virol 75:1091–1099. doi: 10.1099/0022-1317-75-5-1091. [DOI] [PubMed] [Google Scholar]
- 13.Rixon FJ, Addison C, McGregor A, Macnab SJ, Nicholson P, Preston VG, Tatman JD. 1996. Multiple interactions control the intracellular localization of the herpes simplex virus type 1 capsid proteins. J Gen Virol 77:2251–2260. doi: 10.1099/0022-1317-77-9-2251. [DOI] [PubMed] [Google Scholar]
- 14.Preston VG, al-Kobaisi MF, McDougall IM, Rixon FJ. 1994. The herpes simplex virus gene UL26 proteinase in the presence of the UL26.5 gene product promotes the formation of scaffold-like structures. J Gen Virol 75:2355–2366. doi: 10.1099/0022-1317-75-9-2355. [DOI] [PubMed] [Google Scholar]
- 15.Homa FL, Brown JC. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev Med Virol 7:107–122. doi:. [DOI] [PubMed] [Google Scholar]
- 16.Spector DL. 1990. Higher order nuclear organization: three-dimensional distribution of small nuclear ribonucleoprotein particles. Proc Natl Acad Sci U S A 87:147–151. doi: 10.1073/pnas.87.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Salomoni P, Ferguson BJ, Wyllie AH, Rich T. 2008. New insights into the role of PML in tumour suppression. Cell Res 18:622–640. doi: 10.1038/cr.2008.58. [DOI] [PubMed] [Google Scholar]
- 19.Hodges M, Tissot C, Howe K, Grimwade D, Freemont PS. 1998. Structure, organization, and dynamics of promyelocytic leukemia protein nuclear bodies. Am J Hum Genet 63:297–304. doi: 10.1086/301991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lallemand-Breitenbach V, de The H. 2010. PML nuclear bodies. Cold Spring Harb Perspect Biol 2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L, van Driel R. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J Cell Sci 101:773–784. [DOI] [PubMed] [Google Scholar]
- 22.Reineke EL, Kao HY. 2009. Targeting promyelocytic leukemia protein: a means to regulating PML nuclear bodies. Int J Biol Sci 5:366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Damme E, Laukens K, Dang TH, Van Ostade X. 2010. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci 6:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi Y, Lallemand-Breitenbach V, Zhu J, de The H. 2004. PML nuclear bodies and apoptosis. Oncogene 23:2819–2824. doi: 10.1038/sj.onc.1207533. [DOI] [PubMed] [Google Scholar]
- 25.Guo L, Giasson BI, Glavis-Bloom A, Brewer MD, Shorter J, Gitler AD, Yang X. 2014. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol Cell 55:15–30. doi: 10.1016/j.molcel.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nisole S, Maroui MA, Mascle XH, Aubry M, Chelbi-Alix MK. 2013. Differential roles of PML isoforms. Front Oncol 3:125. doi: 10.3389/fonc.2013.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korioth F, Maul GG, Plachter B, Stamminger T, Frey J. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res 229:155–158. doi: 10.1006/excr.1996.0353. [DOI] [PubMed] [Google Scholar]
- 28.Ahn JH, Hayward GS. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39–55. doi: 10.1006/viro.2000.0448. [DOI] [PubMed] [Google Scholar]
- 29.Boutell C, Orr A, Everett RD. 2003. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J Virol 77:8686–8694. doi: 10.1128/JVI.77.16.8686-8694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adamson AL, Kenney S. 2001. Epstein-Barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J Virol 75:2388–2399. doi: 10.1128/JVI.75.5.2388-2399.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bailey D, O'Hare P. 2002. Herpes simplex virus 1 ICP0 co-localizes with a SUMO-specific protease. J Gen Virol 83:2951–2964. [DOI] [PubMed] [Google Scholar]
- 32.Scherer M, Klingl S, Sevvana M, Otto V, Schilling EM, Stump JD, Muller R, Reuter N, Sticht H, Muller YA, Stamminger T. 2014. Crystal structure of cytomegalovirus IE1 protein reveals targeting of TRIM family member PML via coiled-coil interactions. PLoS Pathog 10:e1004512. doi: 10.1371/journal.ppat.1004512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuchet-Lourenco D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J Virol 86:11209–11222. doi: 10.1128/JVI.01145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sides MD, Block GJ, Shan B, Esteves KC, Lin Z, Flemington EK, Lasky JA. 2011. Arsenic mediated disruption of promyelocytic leukemia protein nuclear bodies induces ganciclovir susceptibility in Epstein-Barr positive epithelial cells. Virology 416:86–97. doi: 10.1016/j.virol.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell P, Lieberman PM, Maul GG. 2000. Lytic but not latent replication of Epstein-Barr virus is associated with PML and induces sequential release of nuclear domain 10 proteins. J Virol 74:11800–11810. doi: 10.1128/JVI.74.24.11800-11810.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Everett RD. 2001. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20:7266–7273. doi: 10.1038/sj.onc.1204759. [DOI] [PubMed] [Google Scholar]
- 37.Salsman J, Zimmerman N, Chen T, Domagala M, Frappier L. 2008. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog 4:e1000100. doi: 10.1371/journal.ppat.1000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748–2752. [PubMed] [Google Scholar]
- 39.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147:221–234. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muller S, Matunis MJ, Dejean A. 1998. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J 17:61–70. doi: 10.1093/emboj/17.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sides MD, Block GJ, Chadwick RW, Shan B, Flemington EK, Lasky JA. 2011. Epstein-Barr virus latent membrane protein 1 suppresses reporter activity through modulation of promyelocytic leukemia protein-nuclear bodies. Virol J 8:461. doi: 10.1186/1743-422X-8-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bentz GL, Whitehurst CB, Pagano JS. 2011. Epstein-Barr virus latent membrane protein 1 (LMP1) C-terminal-activating region 3 contributes to LMP1-mediated cellular migration via its interaction with Ubc9. J Virol 85:10144–10153. doi: 10.1128/JVI.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reichelt M, Wang L, Sommer M, Perrino J, Nour AM, Sen N, Baiker A, Zerboni L, Arvin AM. 2011. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog 7:e1001266. doi: 10.1371/journal.ppat.1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glass M, Everett RD. 2013. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol 87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu Y, Everett RD. 2015. Analysis of the functional interchange between the IE1 and pp71 proteins of human cytomegalovirus and ICP0 of herpes simplex virus 1. J Virol 89:3062–3075. doi: 10.1128/JVI.03480-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM, Maul GG. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev 10:196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- 47.Jul-Larsen A, Visted T, Karlsen BO, Rinaldo CH, Bjerkvig R, Lonning PE, Boe SO. 2004. PML-nuclear bodies accumulate DNA in response to polyomavirus BK and simian virus 40 replication. Exp Cell Res 298:58–73. doi: 10.1016/j.yexcr.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 48.Shishido-Hara Y, Ichinose S, Higuchi K, Hara Y, Yasui K. 2004. Major and minor capsid proteins of human polyomavirus JC cooperatively accumulate to nuclease domain 10 for assembly into virions. J Virol 78:9890–9903. doi: 10.1128/JVI.78.18.9890-9903.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Erickson KD, Bouchet-Marquis C, Heiser K, Szomolanyi-Tsuda E, Mishra R, Lamothe B, Hoenger A, Garcea RL. 2012. Virion assembly factories in the nucleus of polyomavirus-infected cells. PLoS Pathog 8:e1002630. doi: 10.1371/journal.ppat.1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amon W, White RE, Farrell PJ. 2006. Epstein-Barr virus origin of lytic replication mediates association of replicating episomes with promyelocytic leukaemia protein nuclear bodies and replication compartments. J Gen Virol 87:1133–1137. doi: 10.1099/vir.0.81589-0. [DOI] [PubMed] [Google Scholar]
- 51.Chou AH, Chen YL, Hu SH, Chang YM, Wang HL. 2014. Polyglutamine-expanded ataxin-3 impairs long-term depression in Purkinje neurons of SCA3 transgenic mouse by inhibiting HAT and impairing histone acetylation. Brain Res 1583:220–229. doi: 10.1016/j.brainres.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 52.Yang YC, Yoshikai Y, Hsu SW, Saitoh H, Chang LK. 2013. Role of RNF4 in the ubiquitination of Rta of Epstein-Barr virus. J Biol Chem 288:12866–12879. doi: 10.1074/jbc.M112.413393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asai R, Kato A, Kato K, Kanamori-Koyama M, Sugimoto K, Sairenji T, Nishiyama Y, Kawaguchi Y. 2006. Epstein-Barr virus protein kinase BGLF4 is a virion tegument protein that dissociates from virions in a phosphorylation-dependent process and phosphorylates the viral immediate-early protein BZLF1. J Virol 80:5125–5134. doi: 10.1128/JVI.02674-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang LK, Chuang JY, Nakao M, Liu ST. 2010. MCAF1 and synergistic activation of the transcription of Epstein-Barr virus lytic genes by Rta and Zta. Nucleic Acids Res 38:4687–4700. doi: 10.1093/nar/gkq243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang LK, Lee YH, Cheng TS, Hong YR, Lu PJ, Wang JJ, Wang WH, Kuo CW, Li SS, Liu ST. 2004. Post-translational modification of Rta of Epstein-Barr virus by SUMO-1. J Biol Chem 279:38803–38812. doi: 10.1074/jbc.M405470200. [DOI] [PubMed] [Google Scholar]
- 56.Chiu YF, Tung CP, Lee YH, Wang WH, Li C, Hung JY, Wang CY, Kawaguchi Y, Liu ST. 2007. A comprehensive library of mutations of Epstein-Barr virus. J Gen Virol 88:2463–2472. doi: 10.1099/vir.0.82881-0. [DOI] [PubMed] [Google Scholar]
- 57.Hung CH, Chen LW, Wang WH, Chang PJ, Chiu YF, Hung CC, Lin YJ, Liou JY, Tsai WJ, Hung CL, Liu ST. 2014. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J Virol 88:12133–12145. doi: 10.1128/JVI.02033-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donaghy G, Jupp R. 1995. Characterization of the Epstein-Barr virus proteinase and comparison with the human cytomegalovirus proteinase. J Virol 69:1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conlan LA, McNees CJ, Heierhorst J. 2004. Proteasome-dependent dispersal of PML nuclear bodies in response to alkylating DNA damage. Oncogene 23:307–310. doi: 10.1038/sj.onc.1207119. [DOI] [PubMed] [Google Scholar]
- 60.Plafker SM, Gibson W. 1998. Cytomegalovirus assembly protein precursor and proteinase precursor contain two nuclear localization signals that mediate their own nuclear translocation and that of the major capsid protein. J Virol 72:7722–7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Day PM, Roden RB, Lowy DR, Schiller JT. 1998. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J Virol 72:142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Capuano CM, Grzesik P, Kreitler D, Pryce EN, Desai KV, Coombs G, McCaffery JM, Desai PJ. 2014. A hydrophobic domain within the small capsid protein of Kaposi's sarcoma-associated herpesvirus is required for assembly. J Gen Virol 95:1755–1769. doi: 10.1099/vir.0.064303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]